

Summary
Inhibition of immune cell trafficking to the pancreatic islets during type 1 diabetes (T1D) has therapeutic potential, since targeting of T cell and B cell trafficking has been clinically effective in other autoimmune diseases. Trafficking to the islets is characterized by redundancy in adhesion molecule and chemokine usage, which has not enabled effective targeting to date. Additionally, cognate antigen is not consistently required for T cell entry into the islets throughout the progression of disease. However, myeloid cells are required to enable T cell and B cell entry into the islets, and may serve as a convergence point in the pathways controlling this process. In this review we describe current knowledge of the factors that mediate immune cell trafficking to pancreatic islets during T1D progression.
Keywords: autoimmunity, adhesion molecules, cell trafficking, chemokines, diabetes
Immune cell migration to the pancreatic islets during type 1 diabetes occurs by transendothelial migration, through a series of steps including rolling, arrest, and crawling on the vascular wall culminating with diapedesis through the endothelial barrier. Migration to the islets is characterized by redundancy in adhesion molecule and chemokine usage, and variable requirement for cognate antigen. However, myeloid cells are required to enable T cell and B cell entry into the islets and may serve as a convergence point in the pathways controlling this process.
![]()
Introduction
Type 1 diabetes (T1D) is a T cell‐mediated autoimmune disease that affects more than 19 million people worldwide. It is driven by activated autoreactive T cells within the islets of Langerhans causing the destruction of the insulin‐producing β cells of the pancreas. T1D is characterized by glucose dysregulation and if left untreated is lethal 1. Although insulin supplementation works well to prevent mortality, curative therapies that are effective in the inhibition and reversal of T1D progression are lacking. Furthermore, at the time of diagnosis of T1D, not all the patients’ islets have been destroyed, and maintaining the remaining β cell mass may be critical for preventing complications that occur as a result of T1D. One potential strategy for the treatment of T1D is inhibiting trafficking of immune cells to the islets, as targeting of T cell and B cell trafficking has been effective in other autoimmune diseases such as multiple sclerosis 2. In this review we describe what is known about immune cell trafficking to pancreatic islets during T1D progression.
Progression of T1D within the islets
In order to mediate β cell destruction, T cells must first traffic to the islets and exit the blood stream. Autoreactive T cells initially escape negative selection in the thymus, enter the periphery and traffic to the pancreatic lymph node (PLN) 3. Initiation of T1D is then probably driven by an event of β cell death that activates islet‐resident antigen‐presenting cells (APCs) in a pathogenic manner to cause inflammatory cytokine production and trafficking of β cell antigens to the PLN 3, 4, 5. Under homeostatic conditions in mice there are, on average, seven resident myeloid APCs within each islet 6. While islet antigens can be carried to the PLN by APCs from inflamed islets, it is unclear if islet resident APCs carry islet antigen to the PLN prior to inflammation or if antigens are passively transported via pancreatic lymphatics 5, 6. After T cells become activated in the PLN, islet‐specific T cells recirculate through the lymphatics and the thoracic duct into the bloodstream, and can then infiltrate into the islets by extravasation.
Once T cells infiltrate the islet, additional lymphocytes are constantly being recruited 7. Following establishment of islet inflammation, islet infiltration and destruction is self‐sustaining and does not require the presence of the PLN to prime additional autoreactive T cells 8. The levels of individual islet infiltration have been shown to be highly heterogeneous in both non‐obese diabetic (NOD) mice and T1D patients 9, 10. Within a single pancreas, islets can range from infiltrated to destroyed, and the infiltration state of each islet determines the characteristics of immune cell recruitment to and behavior in that islet 11, 12, 13. We determined that, at early stages of islet infiltration, T cells are restimulated by sustained interactions with CD11c‐expressing APCs, leading to increased interferon (IFN)‐γ production 4, 11, 12. Production of IFN‐γ within the islet creates an inflammatory milieu that causes up‐regulation of vascular adhesion molecules on the islet endothelium and increased chemokine production (see sections below on adhesion molecules and chemokines) 4, 14. These inflammatory changes within the islet allow for increased recruitment of immune cells, including but not limited to T cells, mononuclear phagocytic cells (MNPs) and B cells 4, 15, 16.
MNPs within the islets are made up of dendritic cells (DCs), macrophages and monocytes that have been shown to express the surface marker CD11c within the islets 6. Notably, we showed that CD11c+ cells in previously infiltrated islets are required for effective lymphocyte trafficking to the islets 13. It is likely that CD11c+ cells within the islets regulate multiple pathways to facilitate extravasation into islets with established infiltration. Once β cells within an islet have been destroyed, immune cells leave the islet and may traffic to other β cell‐containing islets or secondary lymphoid organs.
CD11c+ cells serve as gatekeepers for lymphocyte entry into the islets
Our work showed that islet CD11c+ cells have a gatekeeper function and are required for the recruitment of both T cells and B cells to infiltrated islets, probably through regulation of multiple chemotactic and inflammatory cues 13. CD11c+ cells within the islets produce more than 20 different chemokines that can bind chemokine receptors expressed on islet T cells 13. The precise mechanism by which CD11c+ cells facilitate the recruitment of lymphocytes remains unclear; however, during extravasation into the islets, T cells that adhered to the islet vasculature did so in close proximity to vascular‐associated CD11c+ cells within the islets 13. It is probable that CD11c+ cells are a convergence point for multiple redundant mechanisms to allow for proper immune cell trafficking within infiltrated islets. Perhaps therapeutic targeting to deplete subsets or inhibit the function of islet CD11c+ cells may allow for inhibition of lymphocyte trafficking to previously infiltrated islets.
Role of antigen in T cell trafficking to the islets
The role played by antigen in T cell trafficking to the islets has been controversial. This could be due to differences in timing and status of islet inflammation between studies when analyzed. Although still controversial, multiple studies conclude that islet antigen presentation by the islet vascular endothelium is a requirement for initial CD8 T cell trafficking to the islets, but this has not been shown for CD4 T cell trafficking 17, 18. APCs in the islet could also present antigen to intravascular T cells through their ability to periscope into the islet vasculature 15. Initial T cell trafficking to the islets is thought to be dependent on antigen. In NOD.β2M–/– mice that lack a critical component for major histocompatibility complex (MHC) class I expression, T cell trafficking to the islets and insulitis is significantly delayed 19, 20. However, it is difficult to deduce the role of antigen presentation in trafficking from these studies, as MHC class I expression is also required for CD8 T cell survival and activation.
With respect to CD4 T cell trafficking to the islets, non‐islet antigen hen egg lysozyme (HEL)‐specific 3A9 transgenic T cells do not traffic to the islets unless HEL is expressed in the islets using B10.BR.RIP‐mHEL mice 15. However, it is likely that factors other than antigen are also involved in trafficking to the islets, as antibody blockade of MHC class II only leads to a slight impairment in initial CD4 T cell trafficking to the islets 15. In the B10.BR.RIP‐HEL model, once islets become inflamed changes in expression of proinflammatory cytokines, endothelial vascular adhesion molecules and chemokines allow for non‐islet antigen‐specific T cell trafficking 14. However, an elegant study using NOD retrogenic bone marrow chimeric mice that expressed islet antigen‐specific or non‐specific T cell receptors (TCRs) concluded that antigen is required for long‐term accumulation of T cells within the islets 21, which could result from a combination of trafficking, expansion, retention and survival of T cells.
Once islet infiltration is established, redundant pathways for T cell recruitment allow both islet antigen‐specific and non‐specific T cell trafficking to inflamed islets. To this end, our work clearly demonstrated this to be true by using NOD.C6 mice, which establish islet infiltration and T1D progression equivalent to wild‐type (WT) NOD mice, but lack the antigen for T cells expressing the BDC‐6.9 TCR 22, 23. T cells from NOD.BDC‐6.9 transgenic mice trafficked similarly to the islets of NOD.C6 and WT NOD mice, proving that antigen is not required for T cell trafficking to previously infiltrated islets 13. This is further highlighted by the observation that the majority of T cells that traffic to inflamed islets have a naive phenotype 7, 24. The trafficking of naive cells may be due in part to the presence of tertiary lymphoid organization within inflamed islets in NOD mice 25, 26, 27. Interestingly, non‐islet antigen‐specific T cells within the islets may be suppressive, as these non‐specific T cells lead to decreased activation of islet antigen‐specific T cells and lower disease progression 24. Antigen is not required for the entry of T cells into the inflamed islets, but instead is probably required for the long‐term retention and restimulation of pathogenic T cells within the islets. The requirements for retention of immune cells within inflamed tissues also remain unclear.
Process of lymphocyte extravasation
One of the key regulators of immune cell function is the ability to traffic to sites of inflammation and extravasate from the vasculature into the tissue parenchyma. The process of lymphocyte extravasation into an inflamed non‐lymphoid tissue is a highly regulated process governed by many extracellular and intracellular cues. Prior to trafficking to inflamed non‐lymphoid tissue, lymphocytes are normally activated within the lymph nodes causing up‐regulation of integrins and chemokine receptors that, in many cases, are specific to the sites to which they will home 28, 29, 30. Extravasation of T cells has been studied to a greater extent than that of B cells, but both types of lymphocytes have been shown to use similar mechanisms to traffic to sites of inflammation 31. Extravasation can be broken down into three separate processes: (1) rolling on the vascular wall; (2) firm adhesion and crawling on the endothelium; and finally (3) diapedesis (the process of exiting through the vascular wall) (Fig. 1) 32.
Figure 1.
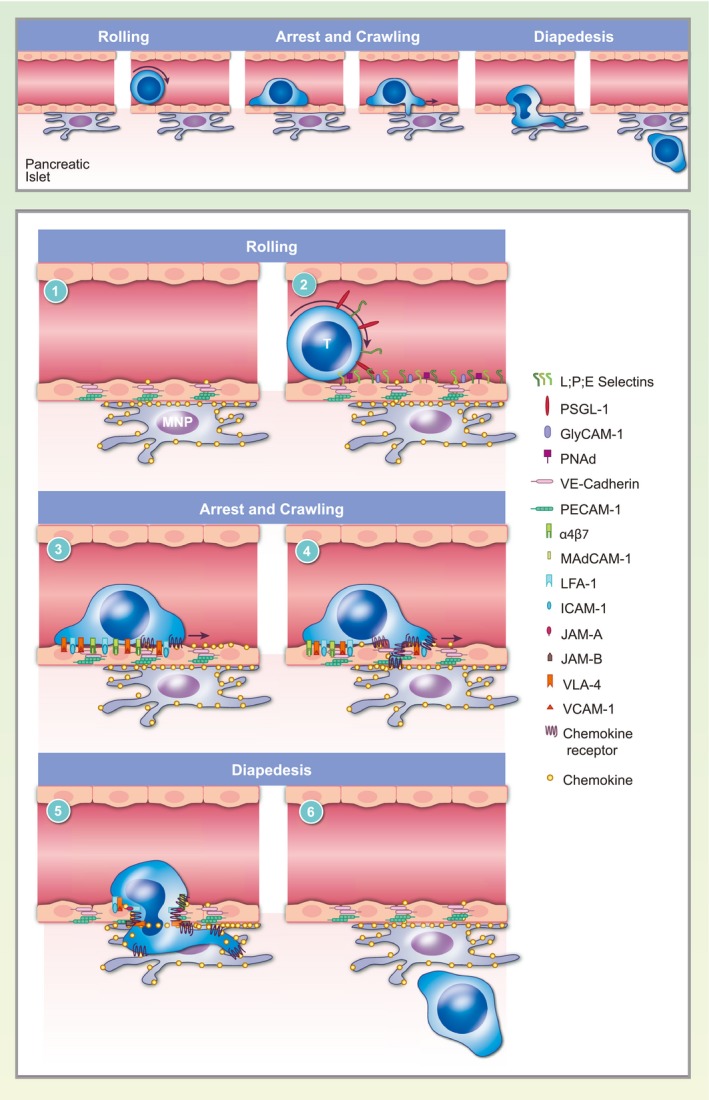
T cell extravasation into the pancreatic islets. The process of extravasation can be broken down into rolling on the vascular wall, firm adhesion and crawling and diapedesis. T cell extravasation is mediated by molecular interactions between the T cells and the vascular endothelium. (1) Interactions between vascular adhesion molecules, including vascular endothelial (VE)‐cadherin, platelet and endothelial cell adhesion molecule 1 (PECAM‐1) and junctional adhesion molecule (JAM) proteins, maintain tight junctions between endothelial cells forming the vascular barrier. Chemokine‐producing mononuclear phagocytes associate with the islet vasculature. (2) T cells roll on the vascular endothelium by tethering to P‐selectin glycoprotein ligand 1 (PSGL‐1) and L‐selectin through low affinity interactions between T cell‐expressed selectins and cell adhesion molecules on the vasculature. (3) Chemokine signaling causes T cells to arrest on the vascular endothelium by promoting the high‐affinity conformation of integrins such as lymphocyte function‐associated antigen 1 (LFA‐1), very late antigen 4 (VLA‐4) and integrin alpha 4 beta 7 (LPAM‐1), which bind cellular adhesion molecules intercellular adhesion molecule 1 (ICAM‐1), vascular cell adhesion molecule 1 (VCAM‐1) and mucosal vascular addressin cell adhesion molecule 1 (MadCAM‐1), respectively. (4) T cells crawl on the vasculature following chemokine gradients to find a permissive site of extravasation in proximity to mononuclear phagocytes. (5) Integrins on T cells interact with endothelial junction proteins to disrupt the vascular barrier and facilitate diapedesis. Chemokine and integrin signaling also lead to rearrangement of the actin cytoskeleton, which allows the T cell to squeeze through the endothelial junctions. (6) T cells complete extravasation to enter the islet parenchyma and the endothelial barrier is restored.
Our laboratory recently characterized the process of T cell extravasation into the islets using intravital 2‐photon imaging 13. We found that the majority of T cells that adhere to the islet vasculature failed to complete the process of diapedesis within our 2‐h imaging period, with a subset releasing back into the bloodstream. T cells that completed diapedesis into the islet parenchyma took more than an hour to complete the process. Furthermore, within our 2‐h imaging window, completion of extravasation was only observed in islets with advanced levels of T cell infiltration, suggesting that the process of extravasation takes longer than 2 h in islets that are at early stages of infiltration 13. As a point of comparison, timing of T cell extravasation into the islets is more similar to extravasation into the highly restrictive central nervous system than into the permissive sites such as the lymph nodes, which takes only 5–10 min 33, 34. These data suggest that the islet vasculature is a highly restrictive site for T cell entry.
In the vasculature at the site of inflammation, T cells are captured by vascular adhesion molecules and roll on the vascular lumen. This tethering and rolling is caused by interactions of P‐selectin glycoprotein ligand 1 (PSGL1) or L‐selectin on lymphocytes binding P‐ and E‐selectins or mucosal vascular addressin cell adhesion molecule 1 (MAdCAM1), respectively, on the inflamed endothelium while under shear force 32 (Fig. 1). Chemokine receptors on activated T cells then bind chemokines bound to the inflamed vascular endothelium. Chemokine receptor signaling in activated T cells causes up‐regulation of integrin affinity through a process referred to as ‘inside‐out signaling’ 35, 36. High‐affinity integrins on activated T cells bind to the endothelial expressed cellular adhesion molecules (CAMs) 32. The high‐affinity integrin interaction with their ligands causes T cells to arrest on the vascular endothelium (Fig. 1). T cells then crawl on the vascular endothelium following chemokine gradients to find a permissive site of entry before they undergo diapedesis 32, 36. T cell diapedesis involves integrin‐ and chemokine‐driven cytoskeletal rearrangements within the T cells, loosening of the endothelial junctions of the vascular wall and breakdown of the endothelial basement membrane to successfully extravasate into the inflamed tissues 32, 37 (Fig. 1). These processes can vary greatly, depending on the site of T cell priming, T cell subset polarization, endothelial adhesion molecule expression and the site of inflammation. Although there is a high potential for therapeutic intervention by inhibiting T cell entry into inflamed islets, the mechanisms of T cell trafficking and extravasation into the islets during T1D are still not well understood.
Role of vascular adhesion molecules in T cell trafficking to the islets
Vascular adhesion molecules are involved in leukocyte adhesion to the vasculature within sites of inflammation, which is necessary for extravasation 32. The initial step of tethering, or leukocyte capture by the vascular endothelium, during leukocyte extravasation is mediated by lower‐affinity interactions of selectins with their ligands causing rolling on the vascular endothelium 38. These interactions include: (1) L‐selectin (CD62L) on leukocytes binding to the mucosal adhesion molecule MAdCAM or to the high endothelial venule (HEV) adhesion molecules glycosylation‐dependent cell adhesion molecule‐1 (GlyCAM) and peripheral node addressin (PNAd) on the vasculature; (2) PSGL‐1 on leukocytes binding vascular P‐selectin, L‐selectin or E‐selectin; and (3) homotypic binding of E‐selectin on both lymphocytes and the vasculature 30, 32 (Fig. 1, Table 1).
Table 1.
T cell adhesion molecules in T1D
| Receptor | Ligands | Ligand expression in islets | Role in T1D | |
|---|---|---|---|---|
| Animal models | Human | |||
| L‐selectin | MAdCAM1 | Low–high 41, 44 | Low–high 42, 43 | |
| GlyCAM | ||||
| PNAd | ||||
| PSGL‐1 | Selectins | High 45 | ? |
|
| LFA‐1 | ICAM‐1 | Mid 14, 48, 50, 108 | + 42, 46, 100 | |
| VLA‐4 | VCAM‐1 | High 14, 48 | + 42, 100 |
|
| LPAM‐1 | MAdCAM‐1 | High 39, 51, 52 | + 42, 100 | |
T1D = type 1 diabetes; MAdCAM‐1 = mucosal vascular addressin cell adhesion molecule 1; GlyCAM = glycosylation‐dependent cell adhesion molecule‐1; PNAd = peripheral node addressin; PSGL‐1 = P‐selectin glycoprotein ligand 1; LFA‐1 = lymphocyte function‐associated antigen 1; VLA‐4 = very late antigen 4; LPAM‐1 = integrin alpha 4 beta 7; NOD = non‐obese diabetic.
Although islet‐infiltrating lymphocytes express a variety of selectins and the islet vasculature expresses their corresponding ligands, none of these initial tethering molecules are required for T cell homing to the islets during T1D 4, 39. Combinatorial blockade of L‐ and P‐selectin does not inhibit T1D in transfer models, and there is no literature implicating E‐selectin in lymphocyte trafficking to inflamed islets 40. Furthermore, NOD mice deficient in L‐selectin progress normally to T1D 41. In patients with T1D, serum levels of L‐selectin are elevated and memory T cells have decreased L‐selectin, suggesting increased activation of memory T cells during T1D 42, 43. Interestingly, CD4 T cells that express high levels of L‐selectin and traffic to the islets have been shown to have regulatory function within the islets 44. Blockade of PSGL‐1 inhibits T1D progression but not through blockade of islet trafficking. Instead, crosslinking of PSGL‐1 was shown to cause lymphocyte apoptosis 45. Although these interactions are probably involved in lymphocyte trafficking to the islets, it seems that there must be alternative or redundant pathways that allow for lymphocyte tethering and rolling in the islets.
The second step of extravasation is integrin‐mediated adhesion to the vasculature. Chemokine signaling in T cells rolling on the vasculature drives increased integrin affinity leading to integrin ligation, T cell arrest and crawling on the vasculature. Within the serum of T1D patients and islets of NOD mice, T cells have been shown to express the integrins lymphocyte function‐associated antigen 1 (LFA‐1) (αLβ2), very late antigen 4 (VLA‐4) (α4β1) and integrin alpha 4 beta 7 (LPAM‐1) (α4β7), and the islet vasculature expresses their respective ligands intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1) and MAdCAM1 4, 39. Cytokines produced locally in the islets or present systemically in the serum of T1D patients and NOD mice concurrently drive the expression of the integrin ligands, ICAM1, VCAM1 and MAdCAM1 in the islet vasculature 4, 14, 15, 46. Although multiple integrins seem to play a necessary role in initial T cell trafficking, once infiltration is established even these adhesion molecules can sometimes become redundant. NOD mice that are deficient in αL or β2, the two chains of LFA‐1, or treated with antibody blockade of α4 integrins prior to islet infiltration do not progress to T1D 47, 48, 49. Notably, in the context of T1D, the role of αL and β2 integrins appear different, with αL deficiency probably having a dominant effect on T cell activation and β2 deficiency directly affecting adhesion to islet vasculature 47. These differences may be due to the ability of β2 integrin to form dimers with other α chains to form additional integrin pairs. ICAM‐1, the ligand for LFA‐1, also has a dominant role early in T1D progression, as NOD mice deficient in ICAM‐1 do not develop T1D, but this may be caused by an impairment of T cell activation 50. Once islet infiltration is established, blockade of α4 integrins or ICAM‐1 strongly reduces T1D progression, but approximately 20–40% of mice still progress to T1D 14, 49. Antibody blockade of MAdCAM‐1 prior to islet infiltration also strongly reduces T1D disease incidence, although some mice were still able to progress to T1D, and once islet infiltration occurred it no longer inhibited disease progression 51, 52. While these studies are important in understanding the requirements for T cell trafficking to the islets, blockade of all of these selectins and integrins would be impractical and probably lead to global immunosuppression.
Role of chemokines in leukocyte trafficking to the islets
Chemokines are cytokines that are responsible for directing migration, extravasation and positioning of immune cells throughout the body. The chemokine superfamily is made up of 46 members in humans, many of which have homologs in mice 53. Chemokines are named and grouped together based on the position of conserved cysteine residues into CC and CXC ligands and receptors, with a few outliers such as XC and CX3C motifs 54. Between mice and humans, more than half the chemokine ligands and receptors have been implicated to have a role in T1D 55 (Table 2). Many of the chemokines produced within the islets are IFN‐stimulated genes driven by IFN‐γ 4, 14, 55, 56, 57, 58, 59, 60. Our laboratory and others have shown that chemokine receptor signaling on T cells is necessary for T cell recruitment to previously infiltrated islets 13, 15. Furthermore, the role of chemokines in the progression of T1D was elegantly highlighted by expressing the gammaherpesvirus‐68 chemokine decoy receptor, M3, within the islets. The M3 protein broadly blocks binding to multiple CC chemokines inhibiting their chemotactic function 61, 62. NOD.M3 mice exhibited inhibited immune trafficking to the islets and did not progress to T1D 63. Unfortunately, while targeting of individual chemokine pathways has been effective at delaying trafficking to uninfiltrated islets, it does not seem to be effective in inhibiting trafficking once infiltration is established 13, 57, 64, 65, 66. This is due probably to the fact that chemokines are highly redundant and promiscuous, being able to bind multiple chemokine receptors on a variety of cell types 4, 14, 55, 56, 57, 58, 59, 60.
Table 2.
Roles of islet‐expressed chemokines in type 1 diabetes
| Receptor | Ligands | Ligand expression in islets | Role in T1D | |
|---|---|---|---|---|
| Animal models | Human | |||
| CCR2 | CCL2 | Mid 4 | Mid 60, 102, 109 | |
| CCR5 | CCL3, | Mid 63 | Low/neg 60 | |
| CCL4, | Mid 63 | Low/neg 60 | ||
| CCL5 | Mid 55 | Mid 55 | ||
| CCR7 | CCL19, | Mid 110 |
|
|
| CCL21 | Mid 110 | |||
| CXCR3 | CXCL9, | Mid 13, 55, 57 | Mid 55, 60 |
|
| CXCL10 | High 13, 55, 57, 64, 82 | High 55, 60 | ||
| CXCR4 | CXCL12 | Mid 89, 90, 91 | Mid 60 | |
| CXCR5 | CXCL13 | Mid 25, 111 | ||
| CXCR6 | CXCL16 | High 13 | ||
| M3 viral decoy receptor | CC chemokine ligands |
|
||
CCR = chemokine receptor; CCL = CC chemokine ligand; NOD = non‐obese diabetic; T1D = type 1 diabetes; CXCR = C‐X‐C chemokine receptor; CXCL = C‐X‐C motif) ligand 1; Idd = insulin‐dependent diabetes.
Chemokines in MNP trafficking to the islets
Chemokine‐driven recruitment of MNPs to the islets has a complex role in T1D progression. MNPs within the islets are highly heterogeneous, with both pathogenic and tolerogenic populations 6, 11, 12, 67, 68. Two of the major chemokine receptors that are responsible for MNP recruitment are chemokine receptor (CCR)2 and CCR5. CCR2 binds the chemokine CC chemokine ligand (CCL)2, whereas CCR5 ligands include CCL3, CCL4 and CCL5 53. Interestingly, deficiencies in CCR2 or CCR5 in NOD mice have countervailing effects during T1D progression. CCR2 deficiency inhibits T1D progression, whereas CCR5 deficiency leads to increased MNP recruitment and accelerated T1D progression 69. This regulation becomes even more complicated when also considering the role of the chemokine ligands, as well as mouse strain susceptibility to T1D. CCL2 expression within the islets of diabetes‐resistant mice (C57BL/6 × DBA.RIP‐CCL2) caused the recruitment of MNPs, insulitis and T1D progression, and these effects were lost with CCR2 deficiency 70. Conversely, CCL2 expression in the islets of NOD mice (NOD.RIP‐CCL2) resulted in recruitment of tolerogenic MNPs leading to lowered insulitis and inhibited T1D progression 71. Furthermore, the deletion of CCL3 in NOD mice led to decreased insulitis and delayed T1D, which contradicts the data on the deletion of its receptor CCR5 69, 72. These studies highlight the complexity of the MNP populations and their recruitment to the islets. Notably, as MNP populations in the islets are required for T cell entry into the islets 13, recruitment of the islet MNP populations may, in turn, affect T cell recruitment to the islets.
Chemokines in T cell trafficking to the islets
The chemokine receptor CCR7 and its ligands CCL19 and CCL21 are known to be important for immune cell recruitment to, and localization within, the lymph nodes 73, 74, 75, 76. However, during T1D in NOD mice, CCL19 and CCL21 are also expressed around prediabetic islets 77. This could be due in part to the presence of tertiary lymphoid organs (TLOs) within the islets. This expression of CCL21 around the islets has been shown to be important for homing of islet antigen‐specific T cells to the islets 17. The receptor CCR7 has also been associated with the insulin‐dependent diabetes (Idd)9 T1D risk allele in NOD mice, which is involved with homing of T cells to the islets 78. Furthermore, NOD mice deficient in CCR7 do not develop T1D 79, highlighting its role in the initiation of islet‐specific disease probably through its function in both the lymph nodes and the islets.
Another major chemokine receptor‐ligand pathway that has been well studied in the progression of T1D is C‐X‐C chemokine receptor (CXCR)3 binding to C‐X‐C motif ligand (CXCL)9 and CXCL10 produced within the islets. CXCR3 also binds CXCL11, which is not functional in C57Bl/6 mice, but may be carried forward in backcrosses from the 129 background, potentially complicating interpretations of chemokine knockout analyses 80. CXCR3 is expressed on T helper type 1 (Th1) CD4 and effector CD8 T cells, and expression of its ligands is driven by IFN‐γ 81. The receptor CXCR3 is important for T cell trafficking to a variety of sites of anti‐viral and autoimmune inflammation 81. CXCL9 is produced within the islets of NOD mice, and β cells have been shown to be the major producer of CXCL10 in the islets 57, 82. Neutralization of CXCL10 in NOD mice can delay and sometimes reverse T1D progression 65, 83. Furthermore, expression of CXCL10 in the beta cells of T1D‐resistant mice can drive insulitis and cause accelerated virally induced T1D 84. Also, during virally induced T1D, mice deficient in CXCR3 had reduced disease onset 82. Conversely, NOD mice deficient in CXCR3 exhibited an accelerated rate of T1D, due probably to the inhibition of regulatory T cell trafficking to the islets 66, 85, 86. Our work has shown that T cell‐specific deficiency of CXCR3 is not sufficient to prevent T cell trafficking to inflamed islets in the RIP‐mOva model 13. CXCR3 and its ligands may be involved in early T cell trafficking to the islets as well as trafficking of Tregs, but are not necessary for effector T cell trafficking and T1D progression.
Another chemokine that has both tolerogenic and pathogenic roles in T1D pathogenesis is CXCL12 and its receptor CXCR4. The receptor CXCR4 is highly expressed on most immune cells and has an important role in the maintenance of hematopoietic stem cells within the bone marrow 87, 88. In T1D, CXCR4+ T cells have been shown to be regulatory and can inhibit disease transfer into NOD mice. In these disease transfer models, blockade of CXCL12 led to increased insulitis and accelerated T1D progression 89. CXCL12 has been shown to be chemorepulsive to diabetogenic T cells in vitro by blocking up‐regulation of high affinity integrins and strong binding to the islet endothelium 90. However, CXCL12 expression in the islets of C57BL/6 mice protects β cell survival after streptozotocin treatment 91. Additionally, long‐term blockade of CXCL12 in NOD mice reduced T1D disease progression 92. The differences in these results are likely be due to differential recruitment of effector and regulatory T cells to the islets at different stages of T1D progression.
There are many other chemokines that probably play redundant roles in the recruitment of T cells to the islets, including CXCL16 and its receptor CXCR6. This chemokine ligand‐receptor pair is of particular interest in T1D, as CXCL16 is a potential candidate gene for the Idd4 T1D risk locus in mice, and CXCR6 is located within the IDDM22 T1D risk locus in humans 93, 94, 95, 96. In our work, CXCR6 was the highest chemokine receptor mRNA transcript expressed in islet T cells, and CXCL16 was the third highest expressed chemokine transcript in islet CD11c+ cells 13. CXCL16 protein is selectively expressed in the islets by CD11c+ cells 13. Although this chemokine ligand‐receptor pair probably contributes to recruitment of T cells by islet CD11c+ cells, NOD.CXCR6–/– mice progressed to T1D similarly to WT NOD mice 13. NOD.CXCR6–/– T cells also trafficked normally to the islets of WT NOD mice with established islet infiltration. Surprisingly, even C57Bl/6.CXCR3–/–CXCR6–/– T cells trafficked normally to the islets of C57Bl/6.RIP‐mOVA mice with established islet infiltration 13. These data highlight the high level of redundancy in chemokines that are able to promote T cell trafficking to the islets once islet inflammation is established.
Chemokines in B cell trafficking to the islets
B cell recruitment to the islets during T1D is also probably driven by chemokines. The chemokine CXCL13 binds the receptor CXCR5, which is highly expressed on most B cells 53. Expression of CXCL13 in diabetes‐resistant mice causes insulitis and B cell‐driven TLO formation within the islets 97. Antibody blockade of CXCL13 in NOD mice disrupts TLO formation in the islets but does not affect T1D disease progression 25. This shows both that B cells can be recruited to the islets by chemokine expression and that one of the roles that B cells play during T1D progression is maintenance of TLOs in the islets.
Biomarkers of islet trafficking in T1D
Understanding biomarkers of T1D progression has also been of great scientific interest. Molecules involved in leukocyte trafficking, including chemokines and elevated serum levels of soluble adhesion molecules, may represent promising biomarkers to understand immune cell activation and progression of islet infiltration during T1D 42, 43, 98, 99, 100, 101. Patients with T1D have been shown to have elevated serum levels of inflammatory chemokines, including CCL2 and CXCL10 98, 102, 103, 104. Upon early onset of T1D, there is also a reduction in peripheral blood leukocytes expressing Th1 chemokine receptors, such as CCR5 and CXCR3 105. This reduction is thought to be due to the recruitment of peripheral lymphocytes to the islets during disease onset. Decreased levels of L‐selectin on memory T cells and increased serum levels of cleaved sL‐selectin in T1D patient serum could be biomarkers of increased T cell activation 42, 43. Serum chemokine levels may be useful in understanding the subtle changes of the immune response during clinical trials in conjunction with other accepted biomarkers for disease progression. Some of these readouts could potentially be added to established prognostic biomarkers for disease progression and response to interventions such as serum c‐peptide levels, islet autoantibody expression, T cell phenotype, HbA1c, and serum blood sugar 98, 101.
Concluding remarks
The only current treatment for T1D is insulin replacement. While insulin replacement is effective in treating T1D symptoms, it does not cure the underlying autoimmunity that drives the disease. Inhibition of immune cell trafficking to diabetic islets has the potential to intervene in the underlying immune dysfunction that leads to T1D, but this strategy has not yet been effective.
Multiple redundant pathways, particularly with relation to chemokines, are involved in the trafficking of immune cells to diabetic islets, as well as in normal immune cell homeostasis and function during inflammation and infection. Many chemokines and chemokine receptors are located within T1D risk alleles 56, 93, 106. Several chemokines that have been shown to be elevated during T1D progression have not yet been well studied 14, 15, 55. Also, some of these chemokines, such as CCL22, are thought to be more involved in trafficking to the PLN than in trafficking to the islets 107. Other chemokines still have unknown or redundant functions. Despite the therapeutic potential of targeting chemokines and their receptors, the high level of redundancy, as well as the important role of chemokines in normal immune function, make this a challenging avenue of research.
Effectively targeting these redundant islet homing pathways while avoiding the induction of broad immunosuppression caused by non‐specific inhibition of lymphocyte trafficking is a major unaddressed challenge. Our work identifying CD11c+ cells as gatekeepers for lymphocyte trafficking to the islets may provide a convergence point for targeting multiple pathways utilized for lymphocyte entry into the islets, but would require further research to enable specific targeting of pathogenic CD11c+ populations without driving systemic immunosuppression. While of great interest, much work must still be conducted in order to understand the mechanisms that are required for immune cell trafficking to the islets during T1D.
Acknowledgements
This work was supported by NIH 1R01DK111733‐01 (RSF), JDRF #5‐2013‐200 (RSF & JJ), and NIH 1R21AI119932‐01 (RSF).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Historical and new insights into pathogenesis of type 1 diabetes. Clinical and Experimental Immunology 2019, 198: 292–293.
Birth and coming of age of islet autoantibodies. Clinical and Experimental Immunology 2019, 198: 294–305.
HIPs and HIP-reactive T cells. Clinical and Experimental Immunology 2019, 198: 306–313.
Islet‐immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clinical and Experimental Immunology 2019, 198: 326–340.
References
- 1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014; 383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vajkoczy P, Laschinger M, Engelhardt B. Alpha4‐integrin‐VCAM‐1 binding mediates G protein‐independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest 2001; 108:557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wallberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends Immunol 2013; 34:583–91. [DOI] [PubMed] [Google Scholar]
- 4. Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PLOS ONE 2013; 8:e59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self‐reactive T cells by dendritic cells in a type‐1 diabetes model. J Exp Med 2003; 198:1527–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melli K, Friedman RS, Martin AE et al Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol 2009; 182:2590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Magnuson AM, Thurber GM, Kohler RH, Weissleder R, Mathis D, Benoist C. Population dynamics of islet‐infiltrating cells in autoimmune diabetes. Proc Natl Acad Sci USA 2015; 112:1511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med 2002; 196:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makino S, Kunimoto K, Muraoka Y, et al Breeding of a non‐obese, diabetic strain of mice. Jikken Dobutsu 1980; 29:1–13. [DOI] [PubMed] [Google Scholar]
- 10. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lindsay RS, Corbin K, Mahne A et al Antigen recognition in the islets changes with progression of autoimmune islet infiltration. J Immunol 2015; 194:522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedman RS, Lindsay RS, Lilly JK et al An evolving autoimmune microenvironment regulates the quality of effector T cell restimulation and function. Proc Natl Acad Sci USA 2014; 111:9223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sandor AM, Lindsay RS, Dyjack N et al CD11c+ cells are gatekeepers for lymphocyte trafficking to infiltrated islets during type 1 diabetes. Front Immunol 2019; 10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calderon B, Carrero JA, Miller MJ, Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci USA 2011; 108:1567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci USA 2011; 108:1561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohan JF, Kohler RH, Hill JA et al Imaging the emergence and natural progression of spontaneous autoimmune diabetes. Proc Natl Acad Sci USA 2017; 114:E7776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin‐specific CD8+ T cells. J Exp Med 2003; 197:643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lozanoska‐Ochser B, Peakman M. Level of major histocompatibility complex class I expression on endothelium in non‐obese diabetic mice influences CD8 T cell adhesion and migration. Clin Exp Immunol 2009; 157:119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serreze DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major histocompatibility complex class I‐deficient NOD‐B2mnull mice are diabetes and insulitis resistant. Diabetes 1994; 43:505–9. [DOI] [PubMed] [Google Scholar]
- 20. Kay TW, Parker JL, Stephens LA, Thomas HE, Allison J. RIP‐beta 2‐microglobulin transgene expression restores insulitis, but not diabetes, in beta 2‐microglobulin null nonobese diabetic mice. J Immunol 1996; 157:3688–93. [PubMed] [Google Scholar]
- 21. Lennon GP, Bettini M, Burton AR et al T cell islet accumulation in type 1 diabetes is a tightly regulated, cell‐autonomous event. Immunity 2009; 31:643–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pauza ME, Dobbs CM, He J et al T‐cell receptor transgenic response to an endogenous polymorphic autoantigen determines susceptibility to diabetes. Diabetes 2004; 53:978–88. [DOI] [PubMed] [Google Scholar]
- 23. Wiles TA, Delong T, Baker RL et al An insulin‐IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non‐obese diabetic mouse. J Autoimmun 2017; 78:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, von Herrath MG. Suppression of diabetes by accumulation of non‐islet‐specific CD8(+) effector T cells in pancreatic islets. Sci Immunol 2018; 3:eaam6533. [DOI] [PubMed] [Google Scholar]
- 25. Henry RA, Kendall PL. CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J Immunol 2010; 185:1460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol 2007; 178:5643–51. [DOI] [PubMed] [Google Scholar]
- 27. Penaranda C, Tang Q, Ruddle NH, Bluestone JA. Prevention of diabetes by FTY720‐mediated stabilization of peri‐islet tertiary lymphoid organs. Diabetes 2010; 59:1461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol 1999; 72:209–53. [DOI] [PubMed] [Google Scholar]
- 29. Fu H, Wang A, Mauro C, Marelli‐Berg F. T lymphocyte trafficking: molecules and mechanisms. Front Biosci (Landmark edn) 2013; 18:422–40. [DOI] [PubMed] [Google Scholar]
- 30. Mora JR, von Andrian UH. T‐cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol 2006; 27:235–43. [DOI] [PubMed] [Google Scholar]
- 31. Schnoor M, Alcaide P, Voisin MB, van Buul JD. Crossing the vascular wall: common and unique mechanisms exploited by different leukocyte subsets during extravasation. Mediat Inflamm 2015; 2015:946509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007; 7:678–89. [DOI] [PubMed] [Google Scholar]
- 33. Park EJ, Peixoto A, Imai Y et al Distinct roles for LFA‐1 affinity regulation during T‐cell adhesion, diapedesis, and interstitial migration in lymph nodes. Blood 2010; 115:1572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soriano SF, Hons M, Schumann K et al In vivo analysis of uropod function during physiological T cell trafficking. J Immunol 2011;187:2356–64. [DOI] [PubMed] [Google Scholar]
- 35. Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol 2005; 5:546–59. [DOI] [PubMed] [Google Scholar]
- 36. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol 2008; 9:953–9. [DOI] [PubMed] [Google Scholar]
- 37. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity 2014; 41:694–707. [DOI] [PubMed] [Google Scholar]
- 38. Abadier M, Pramod AB, McArdle S et al Effector and regulatory T cells roll at high shear stress by inducible tether and sling formation. Cell Rep 2017; 21:3885–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hänninen A, Taylor C, Streeter PR et al Vascular addressins are induced on islet vessels during insulitis in nonobese diabetic mice and are involved in lymphoid cell binding to islet endothelium. J Clin Invest 1993; 92:2509–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hänninen A, Nurmela R, Maksimow M, Heino J, Jalkanen S, Kurts C. Islet beta‐cell‐specific T cells can use different homing mechanisms to infiltrate and destroy pancreatic islets. Am J Pathol 2007; 170:240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mora C, Grewal IS, Wong FS, Flavell RA. Role of L‐selectin in the development of autoimmune diabetes in non‐obese diabetic mice. Int Immunol 2004; 16:257–64. [DOI] [PubMed] [Google Scholar]
- 42. Glowinska B, Urban M, Peczynska J, Florys B. Soluble adhesion molecules (sICAM‐1, sVCAM‐1) and selectins (sE selectin, sP selectin, sL selectin) levels in children and adolescents with obesity, hypertension, and diabetes. Metabolism 2005; 54:1020–6. [DOI] [PubMed] [Google Scholar]
- 43. Kretowski A, Gillespie KM, Bingley PJ, Kinalska I. Soluble L‐selectin levels in type I diabetes mellitus: a surrogate marker for disease activity? Immunology 2000; 99:320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. You S, Slehoffer G, Barriot S, Bach JF, Chatenoud L. Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc Natl Acad Sci USA 2004; 101(Suppl 2):14580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang CC, Lu YF, Wen SN et al A novel apoptosis‐inducing anti‐PSGL‐1 antibody for T cell‐mediated diseases. Eur J Immunol 2005; 35:2239–49. [DOI] [PubMed] [Google Scholar]
- 46. Vives M, Soldevila G, Alcalde L, Lorenzo C, Somoza N, Pujol‐Borrell R. Adhesion molecules in human islet beta‐cells. De novo induction of ICAM‐1 but not LFA‐3. Diabetes 1991; 40:1382–90. [DOI] [PubMed] [Google Scholar]
- 47. Glawe JD, Patrick DR, Huang M, Sharp CD, Barlow SC, Kevil CG. Genetic deficiency of Itgb2 or ItgaL prevents autoimmune diabetes through distinctly different mechanisms in NOD/LtJ mice. Diabetes 2009; 58:1292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baron JL, Reich EP, Visintin I, Janeway CA Jr. The pathogenesis of adoptive murine autoimmune diabetes requires an interaction between alpha 4‐integrins and vascular cell adhesion molecule‐1. J Clin Invest 1994; 93:1700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang XD, Michie SA, Tisch R, Karin N, Steinman L, McDevitt HO. A predominant role of integrin alpha 4 in the spontaneous development of autoimmune diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA 1994; 91:12604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin S, van Den Engel NK, Vinke A, Heidenthal E, Schulte B, Kolb H. Dominant role of intercellular adhesion molecule‐1 in the pathogenesis of autoimmune diabetes in non‐obese diabetic mice. J Autoimmun 2001; 17:109–17. [DOI] [PubMed] [Google Scholar]
- 51. Hanninen A, Jaakkola I, Jalkanen S. Mucosal addressin is required for the development of diabetes in nonobese diabetic mice. J Immunol 1998; 160:6018–25. [PubMed] [Google Scholar]
- 52. Phillips JM, Haskins K, Cooke A. MAdCAM‐1 is needed for diabetes development mediated by the T cell clone, BDC‐2.5. Immunology 2005; 116:525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014; 32:659–702. [DOI] [PubMed] [Google Scholar]
- 54. International Union of Immunological Sciences (IUIS) World Health Organization (WHO) Subcommittee on Chemokine Nomenclature . Chemokine/chemokine receptor nomenclature. Cytokine 2003; 21:48–9.12668160 [Google Scholar]
- 55. Sarkar SA, Lee CE, Victorino F et al Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes 2012; 61:436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burke SJ, Collier JJ. Transcriptional regulation of chemokine genes: a link to pancreatic islet inflammation? Biomolecules 2015; 5:1020–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN‐gamma‐inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN‐gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol 2003; 171:6838–45. [DOI] [PubMed] [Google Scholar]
- 58. Donath MY, Boni‐Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab 2010; 21:261–7. [DOI] [PubMed] [Google Scholar]
- 59. Fallahi P, Corrado A, Di Domenicantonio A, Frenzilli G, Antonelli A, Martina Ferrari S. CXCR3, CXCR5, CXCR6, and CXCR7 in diabetes. Curr Drug Targets 2016; 17:515–9. [DOI] [PubMed] [Google Scholar]
- 60. Planas R, Carrillo J, Sanchez A et al Gene expression profiles for the human pancreas and purified islets in type 1 diabetes: new findings at clinical onset and in long‐standing diabetes. Clin Exp Immunol 2010; 159:23–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parry CM, Simas JP, Smith VP et al A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J Exp Med 2000; 191:573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Berkel V, Barrett J, Tiffany Hl et al Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J Virol 2000; 74:6741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martin AP, Grisotto MG, Canasto‐Chibuque C et al Islet expression of M3 uncovers a key role for chemokines in the development and recruitment of diabetogenic cells in NOD mice. Diabetes 2008; 57:387–94. [DOI] [PubMed] [Google Scholar]
- 64. Coppieters KT, Amirian N, Pagni PP et al Functional redundancy of CXCR3/CXCL10 signaling in the recruitment of diabetogenic cytotoxic T lymphocytes to pancreatic islets in a virally induced autoimmune diabetes model. Diabetes 2013; 62:2492–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Morimoto J, Yoneyama H, Shimada A et al CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in nonobese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J Immunol 2004; 173:7017–24. [DOI] [PubMed] [Google Scholar]
- 66. Yamada Y, Okubo Y, Shimada A et al Acceleration of diabetes development in CXC chemokine receptor 3 (CXCR3)‐deficient NOD mice. Diabetologia 2012; 55:2238–45. [DOI] [PubMed] [Google Scholar]
- 67. Klementowicz JE, Mahne AE, Spence A et al Cutting edge: origins, recruitment, and regulation of CD11c(+) cells in inflamed islets of autoimmune diabetes mice. J Immunol 2017; 199:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Calderon B, Carrero JA, Ferris ST et al The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med 2015; 212:1497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Solomon M, Balasa B, Sarvetnick N. CCR2 and CCR5 chemokine receptors differentially influence the development of autoimmune diabetes in the NOD mouse. Autoimmunity 2010; 43:156–63. [DOI] [PubMed] [Google Scholar]
- 70. Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin‐producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes 2008; 57:3025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kriegel MA, Rathinam C, Flavell RA. Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c+ CD11b+ dendritic cells. Proc Natl Acad Sci USA 2012; 109:3457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cameron MJ, Arreaza GA, Grattan M et al Differential expression of CC chemokines and the CCR5 receptor in the pancreas is associated with progression to type I diabetes. J Immunol 2000; 165:1102–10. [DOI] [PubMed] [Google Scholar]
- 73. Forster R, Schubel A, Breitfeld D et al CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999; 99:23–33. [DOI] [PubMed] [Google Scholar]
- 74. Hopken UE, Droese J, Li J‐P et al The chemokine receptor CCR7 controls lymph node‐dependent cytotoxic T cell priming in alloimmune responses. Eur J Immunol 2004; 34:461–70. [DOI] [PubMed] [Google Scholar]
- 75. Yoshida R, Nagira M, Kitaura M, Imagawa N, Imai T, Yoshie O. Secondary lymphoid‐tissue chemokine is a functional ligand for the CC chemokine receptor CCR7. J Biol Chem 1998; 273:7118–22. [DOI] [PubMed] [Google Scholar]
- 76. MartIn‐Fontecha A, Sebastiani S, Höpken UE et al Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 2003; 198:615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bouma G, Coppens JMC, Mourits S et al Evidence for an enhanced adhesion of DC to fibronectin and a role of CCL19 and CCL21 in the accumulation of DC around the pre‐diabetic islets in NOD mice. Eur J Immunol 2005; 35:2386–96. [DOI] [PubMed] [Google Scholar]
- 78. Waldner H, Sobel RA, Price N, Kuchroo VK. The autoimmune diabetes locus Idd9 regulates development of type 1 diabetes by affecting the homing of islet‐specific T cells. J Immunol 2006; 176:5455–62. [DOI] [PubMed] [Google Scholar]
- 79. Martin AP, Marinkovic T, Canasto‐Chibuque C et al CCR7 deficiency in NOD mice leads to thyroiditis and primary hypothyroidism. J Immunol 2009; 183:3073–80. [DOI] [PubMed] [Google Scholar]
- 80. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 2011; 89:207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res 2011; 317:620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Frigerio S, Junt B, Lu B, et al Beta cells are responsible for CXCR3‐mediated T‐cell infiltration in insulitis. Nat Med 2002; 8:1414–20. [DOI] [PubMed] [Google Scholar]
- 83. Homann D. Back from the brink: the uses of targeting the CXCL10:CXCR3 axis in type 1 diabetes. Diabetes 2015; 64:3990–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rhode A, Pauza ME, Barral AM et al Islet‐specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol 2005; 175:3516–24. [DOI] [PubMed] [Google Scholar]
- 85. Tan TG, Mathis D, Benoist C. Singular role for T‐BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc Natl Acad Sci USA 2016; 113:14103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kornete M, Mason ES, Girouard J, Lafferty EI, Qureshi S, Piccirillo CA. Th1‐Like ICOS+ Foxp3+ Treg cells preferentially express CXCR3 and home to beta‐islets during pre‐diabetes in BDC2.5 NOD mice. PLOS ONE 2015; 10:e0126311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kucia M, Jankowski K, Reca R et al CXCR4‐SDF‐1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol 2004; 35:233–45. [DOI] [PubMed] [Google Scholar]
- 88. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12‐CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006; 25:977–88. [DOI] [PubMed] [Google Scholar]
- 89. Aboumrad E, Madec AM, Thivolet C. The CXCR4/CXCL12 (SDF‐1) signalling pathway protects non‐obese diabetic mouse from autoimmune diabetes. Clin Exp Immunol 2007; 148:432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sharp CD, Huang M, Glawe J, Patrick DR, Pardue S, Barlow SC, Kevil CG. Stromal cell‐derived factor‐1/CXCL12 stimulates chemorepulsion of NOD/LtJ T‐cell adhesion to islet microvascular endothelium. Diabetes 2008; 57:102–12. [DOI] [PubMed] [Google Scholar]
- 91. Yano T, Liu Z, Donovan J, Thomas MK, Habener JF. Stromal cell derived factor‐1 (SDF‐1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta‐cell survival by activation of the prosurvival kinase Akt. Diabetes 2007; 56:2946–57. [DOI] [PubMed] [Google Scholar]
- 92. Matin K, Salam MA, Akhter J, Hanada N, Senpuku H. Role of stromal‐cell derived factor‐1 in the development of autoimmune diseases in non‐obese diabetic mice. Immunology 2002; 107:222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ivakine EA, Gulban OM, Mortin‐Toth SM et al Molecular genetic analysis of the Idd4 locus implicates the IFN response in type 1 diabetes susceptibility in nonobese diabetic mice. J Immunol 2006; 176:2976–90. [DOI] [PubMed] [Google Scholar]
- 94. Online Mendelian Inheritance in Man . Diabetes mellitus, insulin‐dependent, 22; iddm22. Available at: https://www.omim.org/entry/612522?search=iddm%26highlight=iddm (2007, January 7) (accessed 28 September 2018).
- 95. Online Mendelian Inheritance in Man . Chemokine, CXC motif, receptor 6; CXCR6 (2018, July 2). Available at https://www.omim.org/entry/605163 (accessed 28 September 2018).
- 96. Chen YG, Mathews CE, Driver JP. The role of NOD mice in type 1 diabetes research: lessons from the past and recommendations for the future. Front Endocrinol (Lausanne) 2018; 9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin‐dependent lymphoid neogenesis. Immunity 2000; 12:471–81. [DOI] [PubMed] [Google Scholar]
- 98. Purohit S, She JX. Biomarkers for type 1 diabetes. Int J Clin Exp Med 2008; 1:98–116. [PMC free article] [PubMed] [Google Scholar]
- 99. Atkinson MA, Wilson SB. Fatal attraction: chemokines and type 1 diabetes. J Clin Invest 2002; 110:1611–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fathollahi A, Massoud A, Amirzargar AA, Aghili B, Nasli Esfahani E, Rezaei N. sICAM‐1, sVCAM‐1 and sE‐selectin levels in type 1 diabetes. Fetal Pediatr Pathol 2018; 37:69–73. [DOI] [PubMed] [Google Scholar]
- 101. Tooley JE, Herold KC. Biomarkers in type 1 diabetes: application to the clinical trial setting. Curr Opin Endocrinol Diabetes Obes 2014; 21:287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Antonelli A, Fallahi P, Ferrari SM et al Serum Th1 (CXCL10) and Th2 (CCL2) chemokine levels in children with newly diagnosed Type 1 diabetes: a longitudinal study. Diabet Med 2008; 25:1349–53. [DOI] [PubMed] [Google Scholar]
- 103. Hanifi‐Moghaddam P, Kappler S, Seissler J et al Altered chemokine levels in individuals at risk of Type 1 diabetes mellitus. Diabet Med 2006; 23:156–63. [DOI] [PubMed] [Google Scholar]
- 104. Shimada A, Morimoto J, Kodama K et al Elevated serum IP‐10 levels observed in type 1 diabetes. Diabetes Care 2001; 24:510–5. [DOI] [PubMed] [Google Scholar]
- 105. Lohmann T, Laue S, Nietzschmann U et al Reduced expression of Th1‐associated chemokine receptors on peripheral blood lymphocytes at diagnosis of type 1 diabetes. Diabetes 2002; 51:2474–80. [DOI] [PubMed] [Google Scholar]
- 106. Grattan M, Mi QS, Meagher C, Delovitch TL. Congenic mapping of the diabetogenic locus Idd4 to a 5.2‐cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes 2002; 51:215–223. [DOI] [PubMed] [Google Scholar]
- 107. Kim SH, Cleary MM, Fox HS, Chantry D, Sarvetnick N. CCR4‐bearing T cells participate in autoimmune diabetes. J Clin Invest 2002; 110:1675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chakrabarti D, Huang X, Beck J et al Control of islet intercellular adhesion molecule‐1 expression by interferon‐alpha and hypoxia. Diabetes 1996; 45:1336–43. [DOI] [PubMed] [Google Scholar]
- 109. Piemonti L, Leone BE, Nano R et al Human pancreatic islets produce and secrete MCP‐1/CCL2: relevance in human islet transplantation. Diabetes 2002; 51:55–65. [DOI] [PubMed] [Google Scholar]
- 110. Shan Z, Xu B, Mikulowska‐Mennis A, Michie SA. CCR7 directs the recruitment of T cells into inflamed pancreatic islets of nonobese diabetic (NOD) mice. Immunol Res 2014; 58:351–7. [DOI] [PubMed] [Google Scholar]
- 111. Leon B, Ballesteros‐Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin‐expressing B cells. Nat Immunol 2012; 13:681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]