

Abstract
Purpose:
Intratumoral injection of oncolytic adenovirus Delta-24-RGDOX induces efficacious anti-glioma immunity in syngeneic glioma mouse models. We hypothesized that localized treatment with the virus is effective against disseminated melanomas.
Experimental Design:
We tested the therapeutic effect of injecting Delta-24-RGDOX into primary subcutaneous (s.c.) B16-Red-FLuc tumors in s.c./s.c. and s.c./intracranial (i.c.) melanoma models in C57BL/6 mice. Tumor growth and in vivo luciferase-expressing ovalbumin-specific (OT-I/Luc) T cells were monitored with bioluminescence imaging. Cells were profiled for surface markers with flow cytometry.
Results:
In both s.c./s.c. and s.c./i.c. models, 3 injections of Delta-24-RGDOX significantly inhibited the growth of both the virus-injected s.c. tumor and untreated distant s.c. and i.c. tumors, thereby prolonging survival. The surviving mice were protected from rechallenging with the same tumor cells. The virus treatment increased the presence of T cells and the frequency of effector T cells in the virus-injected tumor and mediated the same changes in T cells from peripheral blood, spleen, and brain hemispheres with untreated tumor. Moreover, Delta-24-RGDOX decreased the numbers of exhausted T cells and regulatory T cells in the virus-injected and untreated tumors. Consequently, the virus promoted the in situ expansion of tumor-specific T cells and their migration to tumors expressing the target antigen.
Conclusions:
Localized intratumoral injection of Delta-24-RGDOX induces an in situ antovaccination of the treated melanoma, the effect of which changes the immune landscape of the treated mice, resulting in systemic immunity against disseminated s.c. and i.c. tumors.
Keywords: oncolytic adenovirus, Delta-24-RGDOX, immunity, melanoma, metastasis
Introduction
Because of its propensity to metastasize throughout the body, melanoma is a deadly tumor, accounting for the majority (60–80%) of deaths from skin cancer (1, 2). Ninety-five percent of melanoma patients with 3 or more sites of metastatic disease die within 1 year (3). Over the past decade, immune checkpoint blockade (ICB) has significantly improved the clinical outcomes of melanoma patients. However, the clinical benefit of ICB is preferentially achieved in only a subset of patients with a pre-existing T-cell response against the tumor (“hot” tumor) (4–8). Moreover, because checkpoint receptors play important regulatory roles in autoimmunity, the toxicity of ICB therapies causes immune-related adverse events (IRAEs). The IRAE incidence ranges from 70% in patients treated with αPD-1/αPD-L1 antibodies to as high as 90% in patients treated with αCTLA-4 (9). Therefore, safe and efficacious therapies are imperatively needed.
Oncolytic viruses are a new class of immunotherapy drugs (10). They are genetically modified or naturally occurring viruses that selectively replicate in and disrupt cancer cells (11–13). In 2015, talimogene laherparepvec (T-VEC), a modified herpes simplex virus type 1 with cancer-selective replication and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression, became the first oncolytic virus approved by U.S. Food and Drug Administration (FDA) for the treatment of surgically unresectable skin and lymph node lesions in patients with advanced melanoma (14, 15). In a randomized open-label phase III trial, those in the T-VEC arm achieved a better durable response rate than those in the control arm did (16% versus 2%; median overall survival duration, 23.3 versus 18.9 months) (14). Thus, even though T-VEC has not dramatically improved melanoma therapy, it marks the beginning of a new era in which metastatic cancers can be treated with localized intratumoral injections of oncolytic viruses, which induced the regression of both the virus-injected and distant tumors, including visceral metastases (16).
In a recent phase I clinical trial based on our long-term effort to develop oncolytic adenoviruses for cancer therapy, an intratumoral injection of Delta-24-RGD (DNX-2401, Tasadenoturev), a second-generation oncolytic adenovirus that is derived from human adenovirus serum type 5 and targets tumor cells with an aberrant RB pathway (17–19), elicited inflammation within the injected tumors, leading to a durable complete response in 12% of patients with recurrent glioblastoma (20). To further increase the efficacy of the virus, we developed a next-generation oncolytic adenovirus, Delta-24-RGDOX, which expresses the immune stimulatory OX40 ligand (OX40L). Compared with its predecessor, this new virus elicits a more potent autologous cancer vaccination in situ, resulting in an efficacious, tumor-specific, long-lasting therapeutic effect in immunocompetent mouse glioma models (21). Because the virus preferentially expresses OX40L on the surface of infected tumor cells, thereby stimulating activated T cells that recognize the antigens presented on the tumor cells (21), Delta-24-RGDOX may be more cancer-specific and have a better safety profile in patients than T-VEC, which secrets GM-CSF in the tumor milieu (16). A clinical trial of Delta-24-RGDOX in glioma patients has been approved in Spain and is pending approval by the U.S. FDA.
In our experience, Delta-24-RGD and Delta-24-RGDOX induce T-cell infiltration in the virus-injected glioma and immune memory against the treated tumor (20–22), suggesting that the viruses are able to turn immune-suppressive (“cold”) tumors into immune-active (“hot”) tumors. We hypothesized that Delta-24-RGDOX has the same effect in melanoma and that the virus-mediated therapeutic effect could extend to untreated disseminated/metastatic tumors. To test this hypothesis, we examined the virus in subcutaneous (s.c.)/s.c. and s.c./i.c. syngeneic melanoma models in immunocompetent mice. We found that injections of Delta-24-RGDOX not only induced immunity against the virus-injected s.c. tumor but also affected distant untreated tumors and changed the immune landscape in the treated mice. For the first time, we demonstrated this type of virotherapy induced efficacious immunity against melanoma in brain. Our findings suggest that localized treatment with Delta-24-RGDOX can be an effective therapeutic agent for patients with metastatic tumors, including brain metastasis.
Materials and Methods
Cell lines and culture conditions
Human lung carcinoma A549 cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium nutrient mixture F12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Inc.), 100 μg/ml penicillin, and 100 μg/ml streptomycin. The mouse melanoma cell lines B16-F10 (ATCC), B16F10 Red-FLuc (PerkinElmer), B16F10 Red-FLuc-3 (a subclone of B16F10 Red-FLuc with slower growing s.c. and i.c. tumors), and B16-ovalbumin (OVA; a kind gift from Dr. Hong Qin, MD Anderson Cancer Center) (23, 24) were grown in RPMI 1640 medium supplemented with 10% FBS and antibiotics. Human embryonic kidney 293 cells (QBioGene, Inc.) human melanoma A375 cells (ATCC) and mouse lung carcinoma CMT64 cells (Culture Collections, Public Health England, UK) were maintained in DMEM supplemented with 10% FBS and antibiotics. All cells were kept at 37°C in a humidified atmosphere containing 5% CO2. Experiments were carried out within 6 months after the cell lines were obtained from a cell bank or within 15 passages in the laboratory. All cell lines were tested for and found to be free of mycoplasma.
B16-F10–enhanced green fluorescent protein (EGFP) cells
First, EGFP cDNA (Clontech) was subcloned into the lentivector plasmid pCDH-CMV-MCS-EF2-Puro (System Biosciences). The plasmid was transduced into B16-F10 cells with X-tremeGENE HP DNA Transfection Reagent (Millipore Sigma), and a stable cell line was established by selection with 1 μg/ml puromycin.
Viruses
The Delta-24-RGD and Delta-24-RGDOX viruses were propagated in A549 cells and titrated in 293 cells as described previously (21).
Mice
C57BL/6 mice were provided by MD Anderson’s Mouse Resource Facility. OT-I/Luc mice (a kind gift from Dr. J.J. Moon, University of Michigan) were bred at MD Anderson’s animal facility as described previously (25).
Animal studies
Six- to ten-week-old mice were used. B16-F10 cells and their derivatives (5 × 105 cells/mouse) were grafted subcutaneously onto the backs of the mice. B16F10 Red-Fluc-3 (2,000 cell/mouse) were grafted into the caudate nucleus of the mice using a guide-screw system as described previously (18). One day before virus treatment, the mice with implanted tumors were evaluated with bioluminescent imaging to assess tumor growth. Mice with off-size tumors were eliminated from the study, and the rest of the mice were randomly assigned to experimental groups. Delta-24-RGDOX (2 × 108 plaque-forming units [PFUs]/mouse) was injected into the first implanted s.c. tumor. For rechallenging the surviving mice, B16F10 Red-FLuc (5 × 105 cells /mouse) or CMT64 (1× 106 cells /mouse) cells were implanted s.c.; or B16F10 Red-FLuc-3 (2,000 cells /mouse) cells were implanted in the contralateral hemisphere of the mouse brain. For monitoring tumor-specific T cells in vivo, 1× 106 OT-I/Luc CD8+ T cells /mouse were injected into the primary s.c. tumor one day before the virotherapy. In the s.c./s.c. model, mice were euthanized when the sum of the longer diameters of the two s.c. tumors was ≥ 2 cm. In the s.c./i.c. model, mice were euthanized when either the longer diameter of s.c. tumor was ≥ 2 cm or the mice showed neurological disorders, such as hunched posture, loss of weight and slow movement. All animal studies were conducted in C57BL/6 mice. All experimental procedures involving the use of mice were done in accordance with protocols approved by MD Anderson’s Animal Care and Use Committee and followed U.S. National Institutes of Health and Department of Agriculture guidelines.
Bioluminescence imaging of tumor-bearing mice
Following implantation of the bioluminescent cells, the luminescence from the growing tumors in the mice was detected with the IVIS Spectrum imaging system (PerkinElmer). Briefly, each mouse was injected subcutaneously with 200 μl D-luciferin (Gold Biotechnology; 15 mg/ml in phosphate-buffered saline [PBS]). Ten minutes later, the animals were anesthetized by placing them in a chamber with 2% isoflurane gas in O2 until they were unresponsive. The anesthetized animals were moved to the imaging chamber to acquire images of the tumors. The luminescence in the mice was assessed with the Living Image software program (PerkinElmer).
Immunofluorescence staining
Paraffin-embedded tumor tissue was cut into 5-μm sections and processed for immunofluorescence staining with standard procedures. The primary antibodies were anti-E1A (MA1-91861; Invitrogen); the secondary antibodies were anti-mouse IgG Alexa Fluor (A11001). Finally, the slides were mounted with cover slips using a ProLong Gold antifade reagent (Invitrogen) containing DAPI for nuclei staining. Images were viewed and assessed using a FluoView confocal laser scanning microscope (Olympus).
Enrichment of leukocytes from tumors
The enrichment of leukocytes from s.c. tumors was conducted as described previously with modification (26). Briefly, the tumors were collected, placed in a 100-μm cell strainer set in petri dishes with RPMI 1640 medium, and then processed through the cell strainer into the dish. The mixture in the dish was gently pipetted up and down and brought up to 20 ml/g of tumor. After centrifugation for 5 min at 500×g, the pellet was resuspended in 10 ml of medium and loaded on top of a discontinuous Percoll gradient medium (44%:67%). After centrifugation at 800×g for 30 minutes at 4°C, the cells were retrieved from the 44%-67% interphase.
Brain-infiltrating leukocytes from mouse brain hemispheres were separated from myelin debris using Percoll gradient medium (GE Healthcare Bio-Sciences) and centrifuged as described previously (27).
Preparation of splenocytes, lymph node cells, and CD8+ T lymphocytes
Mouse spleens and lymph nodes were collected and processed as described previously, except that the lymph node cells were not subjected to red blood cell lysis (21). CD8+ T cells were enriched from the splenocytes with a mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotec, Inc.).
IFNγ ELISPOT assay
The ELISPOT assay was performed with Mouse IFNγ ELISpot kit from R&D systems. Briefly, splenocytes (5 × 105 per well) were stimulated with 1 μg/ ml CD8+ T cell-specific OVA (257-264) (Invitrogen) or GFP (118-126) peptide (Biomatik) for 21 hrs. IFNγ spot-forming cells were determined according to the instructions from the manufacturer.
Flow cytometry analysis
To analyze cell surface protein expression, we incubated cells (2-5 × 105) in 100 μl of primary antibody solution diluted in PBS plus 3% bovine serum albumin and 1 mM ethylenediaminetetraacetic acid (EDTA). After incubation at 4°C in the dark for 30 minutes, the cells were washed once with 1 ml of cold PBS. Then, the cells were resuspended in 0.5 ml of PBS. The stained cells were analyzed using flow cytometry. The antibodies used in the studies were to the following mouse proteins: CD252 (OX40L) allophycocyanin (APC; 17-5905-80), CD3e phycoerythrin (PE; 12-0031-81), CD4 APC (17-0041-81), CD8a PE-Cyanine5.5 (35-0081-80), PD-1 fluorescein isothiocyanate (11-9981-81), TIM3 PE-Cyanine7 (25-5870-80), CD44 super bright 600 (63-0441-80), CD62L APC-eFlour 780 (47-0621-80), CD25 PE-Cyanine5.5 (35-0251-80), FR4 PE-Cyanine7 (25-5445-80), MHC Class I (H-2Kb) APC (17-5958-80 ), Mouse IgG2a kappa Isotype Control APC (17-4724-81), Rat IgG2b kappa Isotype Control (14-4031-82), MHC Class II (I-A/I-E, 14-5321-81), from eBioscience; and CD45 BV786 (564225), from BD Biosciences. The goat anti-rat IgG F(ab’)2-APC (sc-3832) was from Santa Cruz Biotechnology, Inc.. Tetramer/PE - H-2 Kb OVA (SIINFEKL) (OVA-Tet; 14087-PE Kb/OVA) was from the MHC Tetramer Production Facility, Baylor College of Medicine. Cell counts were obtained using 123count eBeads (01-1234, eBioscience) as a control for calculating processed sample volumes. Dead cells were excluded from the analysis through staining with Ghost Dye Violet 510 (13-0870, Tonbo Biosciences).
Stimulation of immune cells
For preparation of the target cells, B16F10 Red-FLuc cells were infected with the virus (100 PFU/cell). Four hours later, 50 units/ml mouse interferon gamma (IFNγ; Prospec Protein Specialists) was added to the culture. Forty-eight hours after viral infection, the cells were fixed with 1% paraformaldehyde and washed before added to the co-culture. For immune cell stimulation, pre-fixed target cells (2 × 104 cells/well) were incubated with splenocytes or lymph node cells (5 × 105 cells/well). Forty hours after the co-culture in a round-bottom 96-well plate, the concentration of IFNγ in the medium was assessed with a Mouse IFNγ DuoSet ELISA kit (R&D Systems).
Statistical analysis
In quantitative studies of cultured cells and flow cytometry, each group consisted of triplicate samples. Each study was repeated at least once. Differences between groups were evaluated using a 2-tailed Student t-test. The animal survival curves were plotted according to the Kaplan–Meier method. Survival rates in the different treatment groups were compared using the log-rank test. Waterfall charts were plotted with R from http://www.R-project.org. P values < 0.05 were considered significant.
Results
Localized treatment with Delta-24-RGDOX inhibits treated primary and untreated distant s.c. melanomas and induces immune memory
It has been reported that oncolytic adenoviruses have shown oncolytic efficacy in melanomas (28, 29). Thus, first we tested Delta-24-RGDOX in cultured human and mouse melanoma cells. The virus infected both human and mouse melanoma cells and expressed OX40L efficiently in these cells (Supplementary Fig. S1A). The B16F10 and its derivatives used in our studies are comparably sensitive to Delta-24-RGDOX infection. 48 hours after viral infection at 30 pfu/cell, the percentage of OX40L positive cells ranged from 50% to 96% among these cell lines (Supplementary Fig. S1B). Three days after the virus was injected into a s.c. melanoma derived from B16F10-EGFP cells, OX40L expression was detected in about 11% of the tumor cells but not in the PBS-treated tumor (P = 0.0002; Supplementary Fig. S1C). This virus replicated in human melanoma A375 cells, albeit not as efficiently as in human lung cancer A549 cells, which are optimal hosts for adenovirus replication. The replication of Delta-24-RGDOX was detectable in mouse melanoma B16F10 Red-FLuc cells (Supplementary Fig. S1D) and the virus lysed the cells efficiently (Supplementary Fig. S1E).
Next, we examined the anti-melanoma activity of the virus in an s.c./s.c. syngeneic melanoma model, derived from B16F10 Red-FLuc cells, in immunocompetent C57BL/6 mice (Fig. 1A). Monitoring of tumor growth with bioluminescence imaging ((Fig. 1B) revealed that 3 injections of Delta-24-RGDOX into the primary s.c. tumor inhibited both the treated tumor and untreated distant tumors (Fig. 1C, Supplementary Fig. S2), thereby prolonging survival and resulting in a long-term survival rate of 44% in the tumor-bearing mice (median survival durations: 36 vs 16 days, P = 0.005, Fig. 1D). The survivors of the virus treatment were protected from rechallenging with s.c. injection of the same tumor cells but not CMT64 cells of lung carcinoma (Fig. 1E), indicating that the treatment induced immune memory specifically against the virus-treated tumor.
Figure 1. Injection of Delta-24-RGDOX into the primary s.c. melanoma inhibits distant untreated s.c. melanoma and induces immune memory.
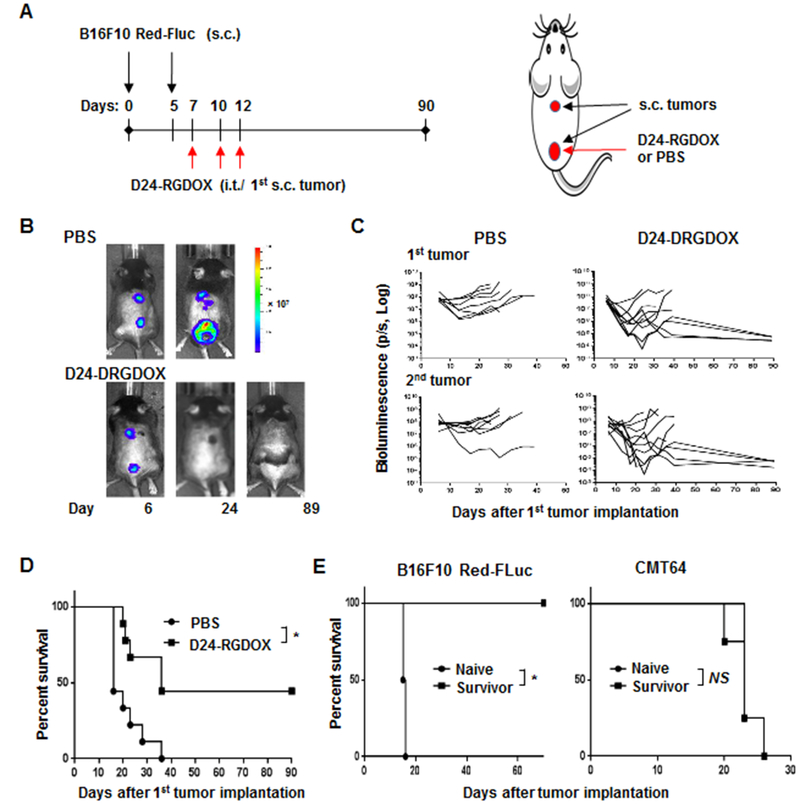
A, A cartoon depiction of the treatment scheme for the schedule (left) and position of implantation and viral injection (right). s.c.: subcutaneously; i.t.: intratumorally. B, Representative bioluminescent images of mice treated with PBS or Delta-24-RGDOX at indicated time points. C, Spider plots of the tumor bioluminescence in the mice from the indicated treatment groups. D, Survival plots of the treatment groups (n = 9). E, Survival plots Delta-24-RGDOX treatment survivors after being first re-challenged with B16F10 Red-FLuc (left panel, n = 4) and then CMT64 (right panel, n = 4) cells. D24-RGDOX: Delta-24-RGDOX; NS: not significant (P ≥ 0.05); *P < 0.01, log-rank test.
Localized treatment with Delta-24-RGDOX inhibits treated s.c. primary and untreated distant i.c. melanomas
Consistently, in the s.c./i.c. melanoma model derived from B16F10 Red-FLuc-3 cells, a subclone of B16F10 Red-FLuc cells that grow slower than the parental cells in the i.c. tumor and give enough window period for us to evaluate the effect of Delta-24-RGDOX in this model (Fig. 2A), viral injections into the primary s.c. tumor inhibited not only the treated tumor but also the untreated i.c. tumor (Fig. 2B and C, Supplementary Fig. S3), thereby prolonging survival and resulting in a long-term survival rate of 40% in the tumor-bearing mice (median survival durations: 58 vs 27 days, P = 0.005, Fig. 2D). Since the s.c. tumor grew faster, all the mice in PBS group were euthanized because of s.c. tumor burden and 6 of the brains showed visible tumor. Because the virus inhibited s.c. tumor growth, in the D24-RGDOX group, of the six euthanized mice, 4 died of brain tumor, 2 died of s.c. tumor. The survivors of the virus treatment were resistant to rechallenging with the same type of tumor cells in the contralateral hemisphere of the brain (Fig. 2E and F), indicating that the treatment induced immune memory against the virus-treated tumor. Moreover, we found no pathological changes in the brains from the survivors (Figure 2F, lower panel).
Figure 2. Injection of Delta-24-RGDOX into the s.c. melanoma inhibits distant untreated i.c. melanoma and induces immune memory.
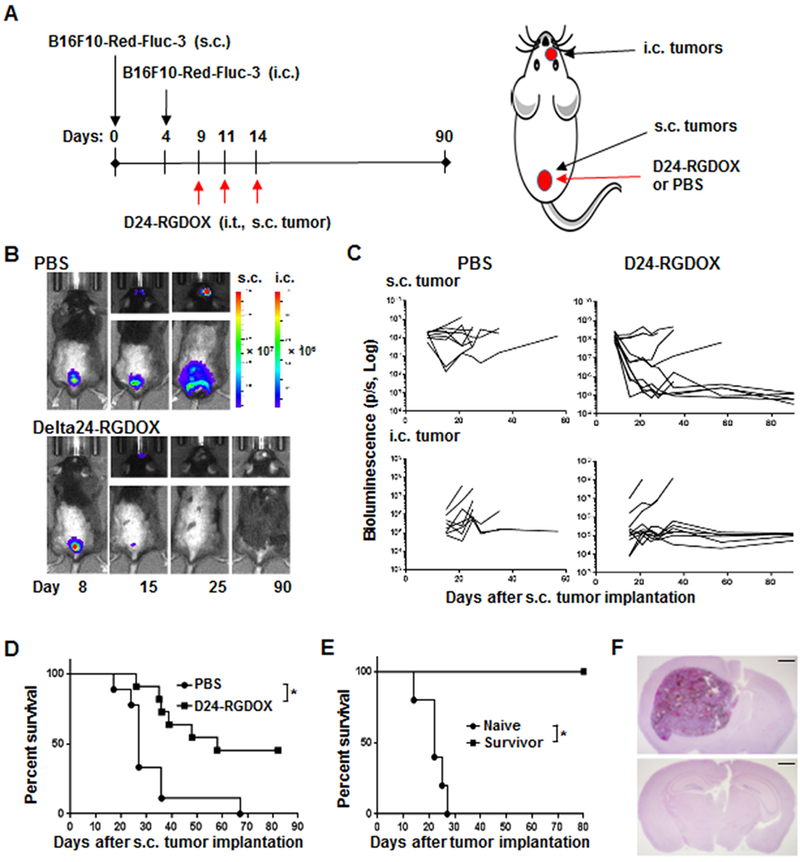
A, A cartoon depiction of the treatment scheme for the schedule (left) and position of implantation and viral injection (right). s.c.: subcutaneously; i.c.: intracranially; i.t.: intratumorally. B, Representative bioluminescent images of mice treated with PBS or Delta-24-RGDOX at indicated time points. C, Spider plots of the tumor bioluminescence in the mice from the indicated treatment groups. D, Survival plots of the treatment groups. PBS: n = 8; D24-RGDOX: n = 11. E, Survival plots for re-challenging survivors from Delta-24-RGDOX treatment with B16F10 Red-FLuc-3 (n = 5) cells in the contralateral hemisphere. F, Shown are representative hematoxylin-and-eosin-stained, whole-mount coronal sections of the mouse brains from Group Naïve (upper panel, euthanized on Day 26) or Survivor (lower panel, euthanized on Day 95) from E. Scale bars: 1 mm. D24-RGDOX: Delta-24-RGDOX; NS: not significant (P ≥ 0.05); * P = 0.005, log-rank test.
Collectively, these data demonstrate that the anti-cancer effect of intratumoral injection of Delta-24-RGDOX could extend from the treated tumor to the distant untreated s.c. tumor and could even pass the blood-brain barrier and reach the brain tumor, meaning that localized treatment of a primary s.c. tumor with the virus can have a efficacious therapeutic effect on disseminated/metastatic tumors.
Intratumoral injection of Delta-24-RGDOX increases the presence of T cells and activates these cells at the virus-injected tumor
To study the effect induced by Delta-24RGDOX within the injected tumor, we first checked the tumor 24 h after viral injection. Immunostaining revealed that the virus expressed the early protein E1A in the infected tumor cells at this time (Fig. 3A). Two days after the viral injections, we also observed virus-mediated upregulation of MHC I (about 6-fold increase) and MHC II (almost doubled) expression on melanoma cells (Fig. 3B), indicating the tumor cells are more active in presenting tumor-associated antigens to the immune cells. Three days after the viral injections, the number of leukocytes (CD45+ cells) nearly doubled in the treated tumor (Fig. 3C). The frequency of T cells (CD45+ CD3+) tripled in the virus-treated tumors (from 20.7% to 63.4%), while the frequency of cytotoxic T cells (CD8+) increased by 23% (P < 0.0001) and that of T helper cells (CD4+) showed little change (P = 0.9; Fig. 3D). However, owing to the increase of the leukocytes in the tumor, the numbers of T cells, cytotoxic T cells, and helper T cells all increased dramatically in the tumors (Fig. 3E). Further analysis of the T-cell populations revealed that the virus activated T cells, thereby increasing the frequency and number of effector (CD44+ CD62L−) CD8+ or CD4+ T cells within the virus-treated tumor (Fig. 3F). Thus, intratumoral injection of Delta-24-RGDOX caused immune activation in the tumor.
Figure 3. Effect of Delta-24-RGDOX on tumor cells and lymphocytes at the injected tumor.
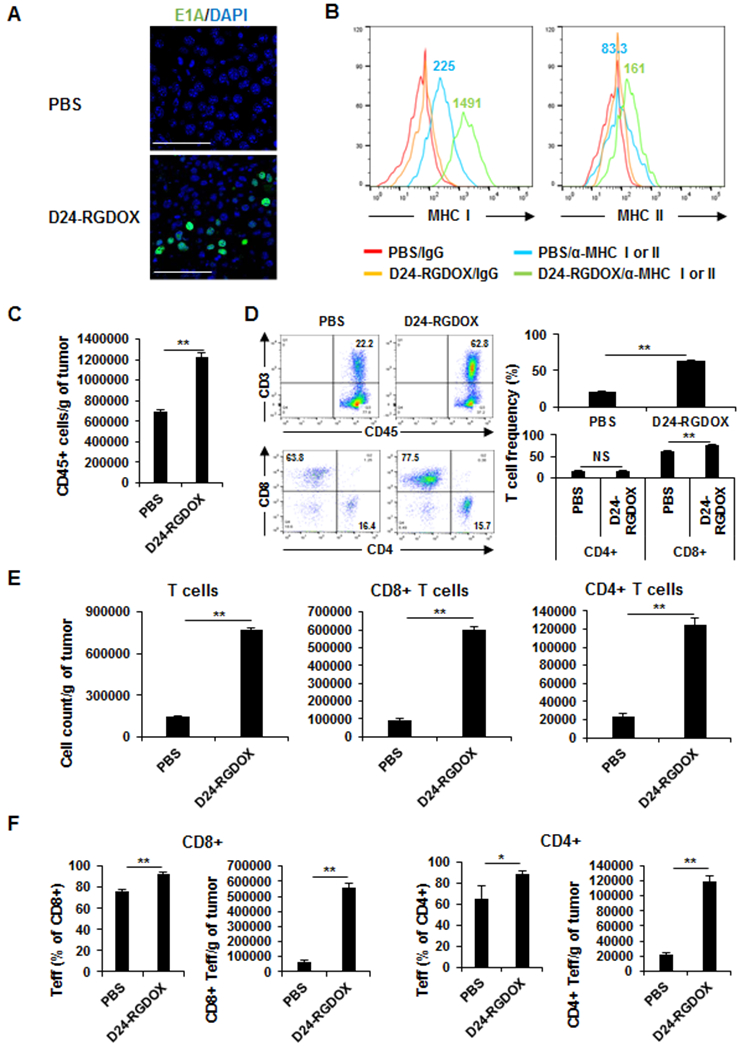
A, Fourteen days after s.c. tumor implantation, Delta-24-RGDOX was injected intratumorally. 24 h later, the tumor was fixed and processed for immunofluorescent staining for E1A. Representative images are shown. Scale bars: 100 μm. B, MHC I and MHC II expression in melanoma cells from implanted tumors. After the implantation of B16F10-EGFP, Delta-24-RGDOX was injected intratumorally on days 7. After 48 hours, the tumors (taken from 3-4 mice/group) were harvested, dissociated, and analyzed with flow cytometry for MHC I and MHC II expression. Tumor cells were gated for EGFP+. IgG staining was used as a negative control. The colored numbers indicate the MFI for the curve of the same color in the graphs. C-F, The mice with 1 s.c. tumor were treated as depicted in Figure 1A. Three days after the last dose of viral injection, tumors were collected (4-5 mice/group), and the leukocytes in the tumor were enriched and analyzed with flow cytometry to assess the number of leukocytes (CD45+) (C); the frequency (D) and number (E) of T cells (CD3+), cytotoxic T cells (CD3+ CD8+) and T helper cells (CD3+ CD4+); and the frequency and number of effector T cells (F). D, Left panel: Representative flow cytometry plots; the numbers are the frequency of the gated cell populations. Right panel: quantification of the cell populations in left panel. D24-RGDOX: Delta-24-RGDOX. Teff: effector T cells. Values represent means ± standard deviations (n = 3). *P = 0.03; **P < 0.0002; NS: not significant (P ≥ 0.05); 2-tailed Student t-test.
Localized treatment of the tumor with Delta-24-RGDOX causes systemic activation of T cells in tumor-bearing mice
Since the injection of Delta-24-RGDOX in the primary s.c. tumor mediated the regression of both the treated tumor and disseminated untreated tumors (Fig. 1 and 2), we hypothesized that the virus could change the entire immune landscape in the treated mice while inducing the same effect in untreated tumors. Indeed, we found that in peripheral blood, the frequency of T cells and cytotoxic T cells increased after virus treatment (P ≤ 0.001), whereas the frequency of T helper cells declined slightly (P = 0.02; Fig. 4A). The frequency of T effector CD8+ T cells almost doubled (from 12.9% to 25.1%), whereas that of CD4+ T cells barely changed (P = 0.08; Fig. 4B). Consistent with these findings, in the hemispheres with implanted tumors, Delta-24-RGDOX induced the following changes: the frequency of T cells more than doubled after virus injections in the s.c. tumor; the frequency of CD8+ T cells increased by 28% (P = 0.0001), whereas the frequency of CD4+ T cells remained almost the same (P = 0.1; Fig. 4C); the numbers of total T cells, CD4+ T cells, and CD8+ T cells increased by 85%−137% (Supplementary Fig. S4); and the numbers of effector T cells in both the CD8+ and CD4+ T-cell populations nearly doubled (Fig. 4D). As in peripheral blood, after virus treatment, the frequency of effector CD8+ T cells increased 2-fold in splenocytes and increased by 60% in cells from inguinal lymph nodes (the tumor-draining lymph nodes [TDLNs] for s.c. tumors), whereas the frequency of CD4+ T cells increased only slightly in splenocytes and declined in TDLN cells (Supplementary Fig. S5). When splenocytes or inguinal lymph node cells were co-cultured with melanoma cells with or without viral infection, by assessing the IFNγ secretion into the medium, we found that the immune cells from the virus-treated group demonstrated higher activity against the tumor cells (Fig. 4E). Furthermore, Delta-24-RGDOX-infected melanoma cells stimulated immune cell activity more potently than Delta-24-RGD-infected cells or cells without viral infection did (Fig. 4E), suggesting that OX40L expression on the cell surface enhances the capability of tumor cells to induce anti-cancer immunity, which is consistent with what we observed previously in glioma virotherapy (21).
Figure 4. Systemic immune activation induced by localized Delta-24-RGDOX treatment in tumor-bearing mice.
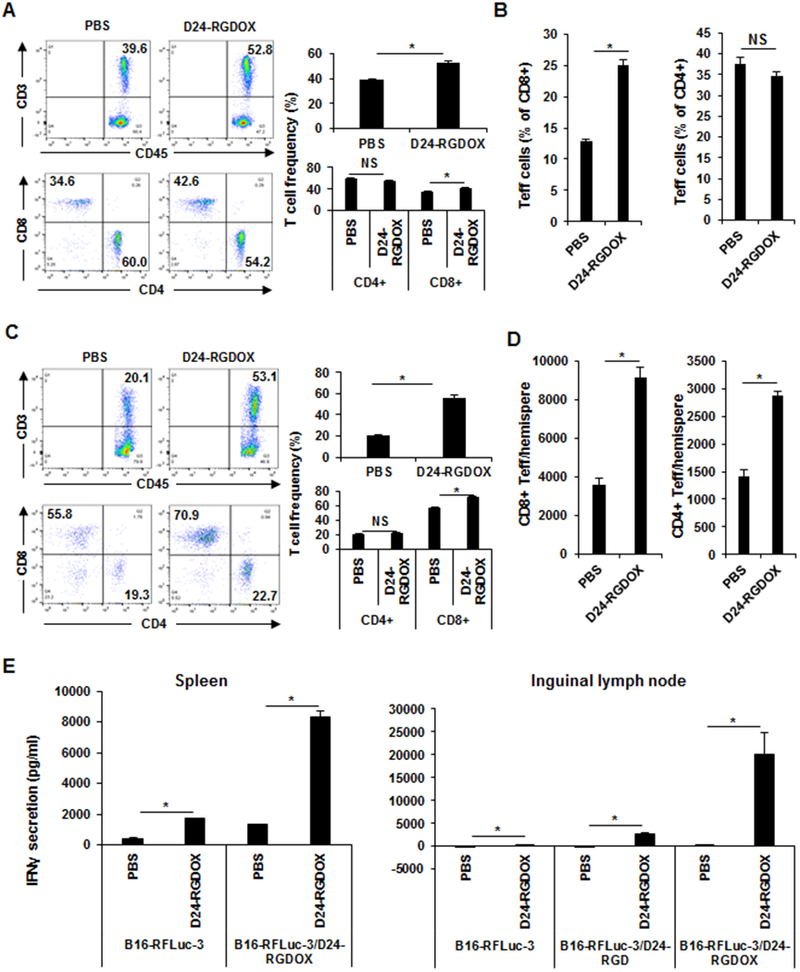
The mice with s.c. and i.c. tumors were treated as depicted in Figure 2A. Three days after the last dose of viral injection, tissues were collected from the mice (4-5 mice/group) for immunological analysis. A-D, The leukocytes from the blood (A and B) and brain hemispheres with tumor implantation (C and D) were analyzed with flow cytometry to assess the frequency of T cells (CD3+), cytotoxic T cells (CD3+ CD8+), and T helper cells (CD3+ CD4+) (A and C); and the frequency (B) and number (D) of effector T cells. A and C, Left panel: Representative flow cytometry plots; the numbers are the frequency of the gated cell populations. Right panel: quantification of the cell populations in left panel. E, Splenocytes and cells from inguinal lymph nodes (tumor-draining lymph node of the s.c. tumor) were co-cultured with B16F10 Red-FLuc-3 cells with or without the infection of the indicated viruses. The amount of IFNγ secreted into the medium was assessed with ELISA forty hours later. D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX. Teff: effector T cells. Values represent means ± standard deviations (n = 3). NS: not significant (P ≥ 0.05); *P < 0.05, 2-tailed Student t-test.
Delta-24-RGDOX disrupts immune suppression in both treated primary and untreated distant tumors
To better understand the effect of virotherapy on immunity, we further examined the status of exhausted T cells (PD-1+ TIM3+) (30) and regulatory T cells (CD4+ CD25+ FR4+) (31). First, we analyzed these cell populations in the tumors. We found that Delta-24-RGDOX reduced the frequency of exhausted CD8+ T cells in both virus-injected s.c. tumors (from 37% to 10.5%, P = 0.000001) and untreated i.c. tumors (from 30.1% to 14.4%, P = 0.001; Fig. 5A). The frequency of exhausted CD4+ T cells was also reduced in these 2 tumor locations, albeit to a lesser extent (P < 0.005; Fig. 5A). As reported previously (30), all the TIM3+ T cells were PD-1+ (Fig. 5A). Like its effect on T-cell exhaustion, Delta-24-RGDOX decreased the frequency of regulatory T cells in both the virus-injected s.c. tumors (from 20.3% to 5.6%, P = 0.00004) and untreated i.c. tumors (from 30.7% to 14.0%, P = 0.00001) (Fig. 5B). However, exhausted T cells were barely detectable in peripheral blood and inguinal lymph nodes (Supplementary Fig. S6A), and regulatory T-cell frequency was hardly detected in blood (Supplementary Fig. S4B, left panel). Although we observed a regulatory T-cell frequency of 8-9% in inguinal lymph nodes, the virus treatment had little effect on the frequency of this cell population in the tissue (Supplementary Fig. S6B, right panel). Interestingly, in all the tissues we examined, we observed the virus-mediated increase of PD-1+ cytotoxic T cell frequency with the highest in untreated i.c. tumors (Fig. 5C).
Figure 5. Disruption of immune suppression in both treated and untreated tumors by Delta-24-RGDOX.
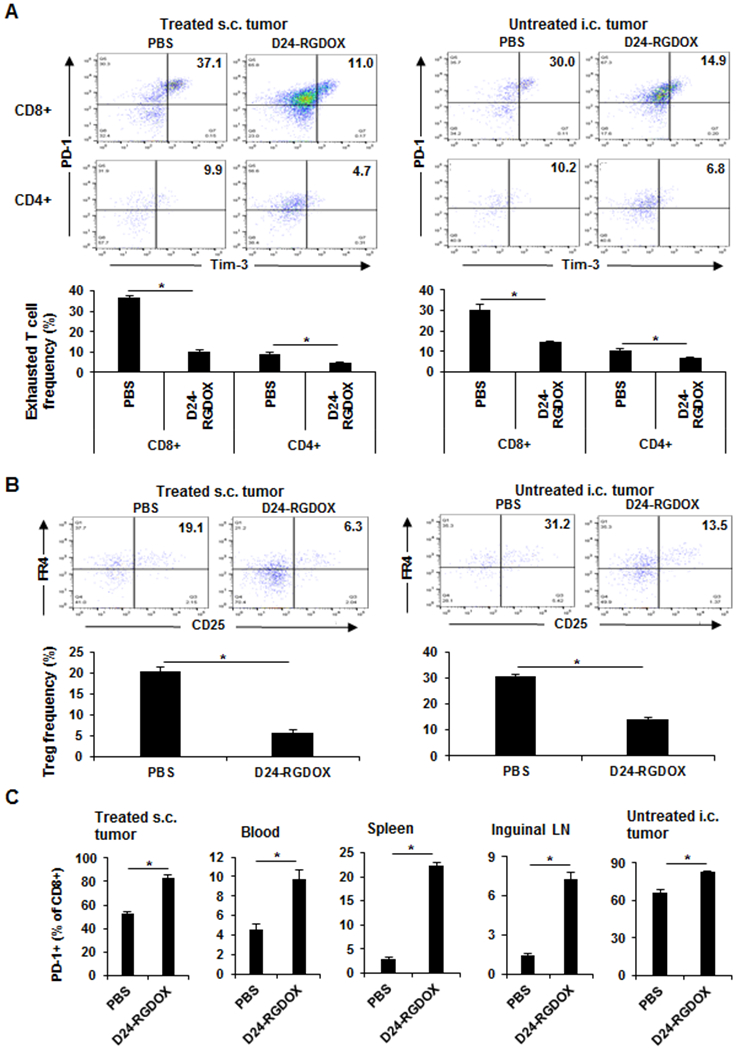
The mice with s.c. and i.c. tumors were treated as depicted in Figure 2A. Three days after the last dose of viral injection, the leukocytes from the virus-injected s.c. tumors, peripheral blood, spleens, inguinal lymph nodes, and brain hemispheres with untreated tumor (4 - 5 mice/group) were analyzed with flow cytometry to assess the frequency of exhausted T cells (PD-1+ TIM3+) (A) and regulatory T cells (B) and the frequency of PD-1+ cells in CD8+ T cells (C). A and B, Upper panel: representative flow cytometry plots; the numbers are the frequency of the gated cell populations. Lower panel: quantification of the cell populations in left panel. Values represent means ± standard deviations (n = 3). *P < 0.005, 2-tailed Student t-test. D24-RGDOX: Delta-24-RGDOX; Treg: regulatory T cell.
Intratumoral injection of Delta-24RGDOX mediates in situ tumor-specific T-cell expansion and migration to distant untreated tumors
To examine the relevance of the virus-mediated effect on immunity to tumor-specific T-cell response, we used the OVA protein as a model tumor-associated antigen. We set up the s.c./s.c. tumors as depicted in Figure 6A. Six days after the implantation of the first tumor and 1 day after the implantation of the second tumor, OVA-specific CD8+ T cells with luciferase expression (OT-I/Luc T cells) were injected into the first tumor, followed by 3 doses of viral injections in the same tumor. As demonstrated by bioluminescence imaging of OT-I/Luc T cells (Fig. 6B), Delta-24-RGDOX treatment promoted the migration of this cell population to distant OVA+ tumors but not OVA- tumors 4 days after the last dose of viral injection (Fig. 6C). Consistent with these findings, 3 days later, quantification of OT-I/Luc T cells in the tumors with flow cytometry analysis revealed that Delta-24-RGDOX treatment increased the number of these cells in the first injected tumor and greatly enhanced the presence of the cells in the second OVA+ tumors but not OVA- tumors (Fig. 6D), suggesting that the virus can activate tumor-specific cytotoxic T cells and increase their in situ presence at the treated tumor and their migration to distant tumors expressing the T-cell target in this artificial antigen system. Consistently, Delta-24-RGDOX treatment mediated the increase of OVA-specific CD8+ T cells frequency in the spleens of the mice bearing s.c. tumors derived from B16-OVA cells (Fig. 6E).
Figure 6. Tumor-specific T-cell expansion and migration to target distant tumors mediated by localized Delta-24RGDOX treatment.
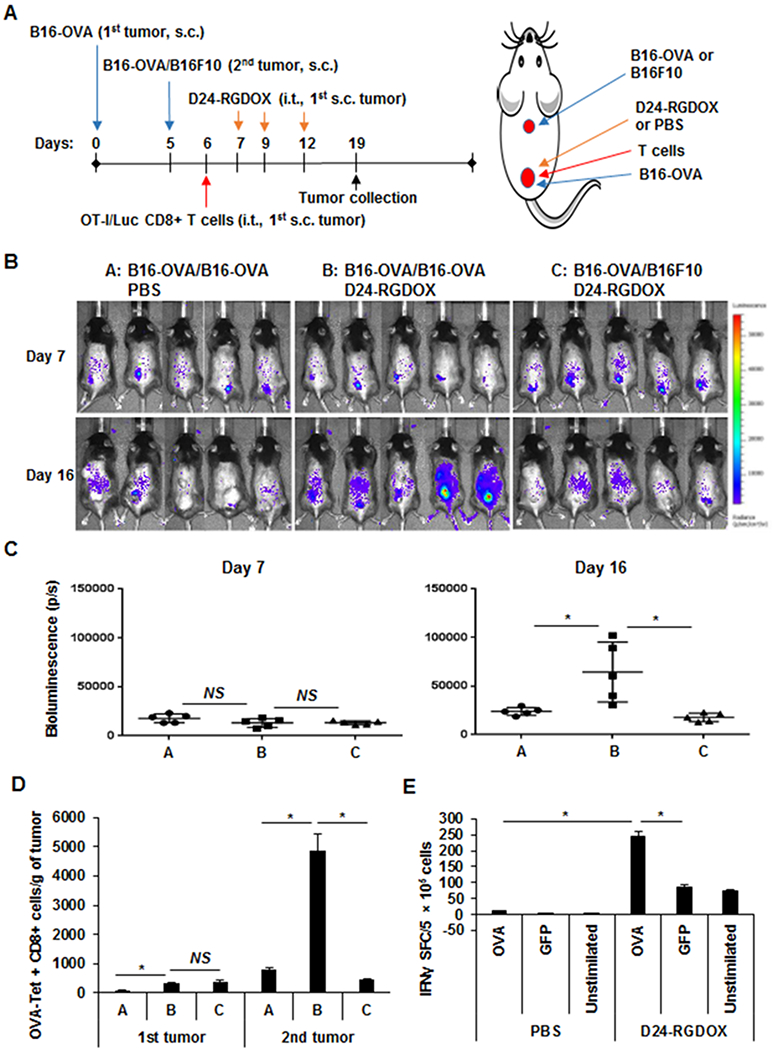
A, A cartoon depiction of the treatment scheme for the schedule (left) and position of implantation and viral injection (right). OVA-specific (OT-I/Luc) T cells (1 × 106) were injected on day 6. s.c.: subcutaneously; i.t.: intratumorally. B, Bioluminescence imaging shows the distribution of tumor-specific T cells after i.t. injection of Delta-24-RGDOX in the first implanted s.c. tumor at the indicated times. C, Quantification of bioluminescence in the second untreated tumors of the mice shown in B on days 7 and 16. D, On day 19, the lymphocytes from the tumors (5 mice/group) were analyzed with flow cytometry to assess OVA-Tet+ CD8+ cells. The quantification of this cell population in the tumors is shown. Values represent means ± standard deviations (n = 3). Groups A, B, and C in C and D are the same as those indicated in B. E, OVA-specific IFNγ-producing cells detected by ELISPOT. After implantation of B16-OVA cells s.c., three doses of Delta-24-RGDOX were injected introtumorally on Days 7, 9, 11. On Day 16, splenocytes were isolated from the mice (3 mice per group) and stimulated with indicated peptides. IFNγ spot-formaing cells (SFC) were measured using an IFNγ ELISPOT assay. D24-RGDOX: Delta-24-RGDOX; NS: not significant (P ≥ 0.05); *P < 0.05, 2-tailed Student t-test.
Discussion
Our findings demonstrate that localized treatment with Delta-24-RGDOX induces immunity against both virus-injected tumors and untreated distant tumors in syngeneic s.c./s.c. and s.c./i.c. melanoma mouse models. The viral injections into the first s.c. tumor immunologically activate the tumor microenvironment of not only the treated tumor but also the distant untreated tumors, resulting in the regression of both the treated primary s.c. tumor and disseminated untreated s.c. and i.c. tumors. In addition, the virus also causes immune activation in peripheral blood and lymphoid organs. Importantly, the virus treatment stimulates in situ tumor-specific cytotoxic T-cell expansion and migration to distant untreated tumors expressing the T-cell target.
Based on the evidence from preclinical and clinical experience, unlike ICB, oncolytic virotherapies should be effective in patients with “cold” tumors. Moreover, localized treatment of the tumor with cancer-selective oncolytic viruses is expected to cause fewer IRAEs than global immune checkpoint inhibition. In the phase I clinical trial of Delta-24-RGD in patients with recurrent malignant glioma, patients tolerated intratumal injection of the virus (up to 3 × 1010 viral particles) well (20). Further, we reported previously that, through expressing immune stimulator OX40L on cancer cells, Delta-24-RGDOX was more potent than Delta-24-RGD in inducing tumor-specific immunity and immune memory against gliomas in orthotopic mouse models (21). During these studies, we observed no virotherapy toxicities, such as weight loss or neurological disorders, in long-term surviving mice. In our current study of virotherapy with Delta-24-RGDOX in melanoma mouse models, in the long-term surviving mice, we also found no sign of treatment-related toxicity, such as weight loss, neurological disorders, or chronic inflammation indicated by swelling and redness at the injection site. These observations suggest that Delta-24-RGDOX is well-tolerated in tumor-bearing mice.
Tumors escape immune surveillance through inactivating tumor-specific T cells in the tissue microenvironment, posing obstacles in cancer immunotherapy (32). T cells are inactivated through multiple tumor-intrinsic and -extrinsic immunosuppressive mechanisms (33). One such mechanism is tumor cells’ expression of PD-L1, which ligates to immune checkpoint receptor PD-1 on the surfaces of activated T cells, natural killer cells, macrophages, and several subsets of dendritic cells, thereby impairing T-cell receptor (TCR) signaling and CD28 co-stimulation (34–36). Upon TCR activation, PD-1 expression is transiently induced on naïve T cells, and this expression decreases in the absence of TCR signaling but is maintained by chronic activation with a persisting epitope target (34). We observed much higher PD-1 expression on CD8+ T cells from tumors than on T cells from peripheral blood and lymphoid organs (Fig. 5C), which suggests that T cells have a feedback response to continuous stimulation by tumor antigens within the tumor. As in glioma mouse models described previously (21), PD-L1 was highly expressed on cultured melanoma cells with or without Delta-24-RGDOX infection and was greatly upregulated by IFNγ which is a pro-inflammatory cytokine induced by oncolytic adenovirus in vivo (22) (Supplementary Fig. S7A). Through their ligation to PD-1 on T cells, these cells stay at an anergy status and allow the immune escape of tumor cells. Delta-24-RGDOX treatment increased the PD-L1 expression level of melanoma cells from the treated tumors in mice (Supplementary Fig. S7B) and the upregulation of PD-1 expression on CD8+ T cells from treated and untreated tumors, peripheral blood, and lymphoid organs (Fig. 5C). Nonetheless, the virus treatment increased the activity of the immune cells against the tumor cells (Fig. 4E) and mediated the regression of both the treated and untreated tumors (Fig. 1 and 2), suggesting that PD-1 upregulation alone is not sufficient to inhibit T-cell activity. Actually, accumulating evidence indicates that PD-1 expression is an early marker of T-cell activation, allowing the identification of the tumor-reactive CD8+ T-cell fraction in melanoma tumors (37–39). T-cell inactivation needs the collaboration of PD-1 and other immune checkpoint inhibitors, such as TIM3 and TIGIT (40, 41). Consistent with the immune activation by the virus, in the present study, Delta-24-RGDOX decreased the frequency of PD-1+ TIM3+ T cells in treated and untreated tumors despite the increase in PD-1+ T cell frequency (Fig 5A and C). One study of PD-1-targeted therapy in lung cancer patients revealed that the upregulation of PD-1 expression on CD8+ T cells from peripheral blood within 4 weeks of treatment initiation correlates with clinical benefit (42). Thus, the early Delta-24-RGDOX-induced upregulation of PD-1 on CD8+ T cells from peripheral blood might serve as a biomarker to predict patients’ responses to the virotherapy. In addition, regulatory T cells selectively accumulate in the tumor microenvironment, suppressing T cell immune responses and activities of antigen-presenting cells, including DCs and macrophages (43). Consistent with the role of OX40 in inhibiting Foxp3 expression and regulatory T cell induction (44), Delta-24-RGDOX downregulated the frequency of regulatory T cells in treated and untreated tumors (Fig. 5B), which contributed to the immune activation within the tumors (Figs. 3, 4C and D).
Melanoma has the potential to metastasize to virtually any anatomical site. Compared with other types of cancers, melanoma is more likely to metastasize to the brain, and brain metastasis occurs in more than 50% of patients with advanced disease (45). The median survival time after the detection of brain metastasis is 17–22 weeks (45). The prognosis for patients with melanoma brain metastasis is dismal despite current chemotherapy, surgery, and radiation options (46). The blood-brain barrier (BBB) limits systemic chemotherapies’ penetration of the central nervous system (CNS), and the negative side effects of radiotherapy pose challenges to therapeutic success, and some lesions are not accessible for surgery (46). As with T-VEC virotherapy in melanoma patients (16), we observed that intratumoral injection of Delta-24-RGDOX into the first s.c. tumor led to immune activation in distant untreated melanomas (Fig. 4C and D), resulting in the regression of both treated and untreated i.c. tumors (Fig. 2). Meanwhile, we found T-cell activation in the peripheral blood, spleen, and inguinal lymph nodes after injection of Delta-24-RGDOX into the first s.c. tumor (Fig. 4A, B, and E). During brain metastasis, the BBB is selectively disrupted (47). In addition, recent studies indicate that, instead of being immune-privileged, the CNS actively communicates with the immune system through the lymphatic vascular system (48, 49). These contribute to the interaction between the CNS and immune system. Thus, the immune factors induced by localized treatment with Delta-24-RGDOX, including pro-inflammatory cytokines and immune cells, may reach the i.c. melanoma through BBB leakage or lymphatic circulation. In our study, the anti-tumor immunity mediated by localized virus administration is strong enough to have an abscopal anti-melanoma effect in the brain (Fig. 2).
Since mouse cells are not optimal host for human adenovirus type 5 replication (50), our current models have limitations to recapitulate the oncolytic effect of the virus in human melanomas although we detected low level Delta-24-RGDOX replication in B16F10 Red-FLuc cells (Supplementary Fig. S1D). We expect the virus will be more potent to replicate in human melanomas to initiate tumor destruction cascade and consequential anti-melanoma immunity. In addition, although the work has been done in tumor models from B16F10 and its derivative cell lines, we believe the virus should be able to induce similar effect in other solid tumors. We have shown previously this virus is efficacious to inhibit gliomas in mouse (21).
In summary, we report for the first time that oncolytic adenovirus Delta-24-RGDOX effectively inhibits both treated primary s.c. and untreated distant s.c. and i.c. tumors, stimulates systemic immune activation in the treated mice, and promotes tumor-specific cytotoxic T-cell expansion and their targeting of disseminated untreated tumors. Our data provide strong evidence that this strategy can be further developed into an efficacious and safe clinical intervention for patients with advanced melanomas, including those with brain metastasis.
Supplementary Material
Translational Relevance.
Immune checkpoint blockade revolutionized the therapy for melanoma. But the therapeutic benefit is limited to only a subset of patients with immunogenic (“hot”) tumors and is restrained by immune-related adverse events. We tested the effect of injecting oncolytic adenovirus Delta-24-RGDOX into primary subcutaneous melanoma on disseminated subcutaneous and intracranial melanomas in syngeneic mouse models. We observed systemic immune activation in the treated mice, resulting in the regression of both treated and untreated tumors. Thus, the virus is expected to be efficacious in patients with immunosuppressive (“cold”) tumors as well and, given its cancer-selective property and localized application, have better safety profile in cancer patients than immune checkpoint blockade. Importantly, this is the first report to demonstrate that localized treatment with an oncolytic virus in the subcutaneous tumor is able to reject intracranial tumors, suggesting Delta-24-RGDOX can be applied to patients with brain melanoma metastasis.
Acknowledgements
We thank Joe Munch (the Department of Scientific Publications, MD Anderson Cancer Center) for editing the manuscript.
Financial Support: This work was supported in part by NIH/NCI grants P50CA127001 and Cancer Center Support Grant P30CA016672 (Research Animal Support Facility, Small Animal Imaging Facility); the Cancer Prevention and Research Institute of Texas (RP170066); the Department of Defense (Team Science Award CA160525); the Marnie Rose Foundation; the J.P. Harris Brain Tumor Research Fund; the Bradley Zankel Foundation; the Ruth L. Kirschstein National Research Service Award (NRSA) Individual Predoctoral Fellowship (Parent F31: 1 F31 CA228207 01A1), the American Legion Auxiliary Fellowship in Cancer Research; Terry and Janet Klebe Fellowship.
Footnotes
Disclosure of Potential Conflicts of Interest: HJ, FFL, JF, and CGM are reporting ownership interest (including patents). CGM and JF are consultants and shareholders of DNATrix, Inc.. The authors have no other financial interests.
References
- 1.Damsky WE, Theodosakis N, Bosenberg M. Melanoma metastasis: new concepts and evolving paradigms. Oncogene 2014;33: 2413–22. [DOI] [PubMed] [Google Scholar]
- 2.Sadozai H, Gruber T, Hunger RE, Schenk M. Recent Successes and Future Directions in Immunotherapy of Cutaneous Melanoma. Front Immunol 2017;8: 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27: 6199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523: 231–5. [DOI] [PubMed] [Google Scholar]
- 5.Sharma PAllison JP. The future of immune checkpoint therapy. Science 2015;348: 56–61. [DOI] [PubMed] [Google Scholar]
- 6.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 2015;33: 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27: 450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DBSosman JA. Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol 2015;1: 380–6. [DOI] [PubMed] [Google Scholar]
- 9.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54: 139–148. [DOI] [PubMed] [Google Scholar]
- 10.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015;14: 642–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer 2014;14: 559–67. [DOI] [PubMed] [Google Scholar]
- 12.Jiang H, Gomez-Manzano C, Rivera-Molina Y, Lang FF, Conrad CA, Fueyo J. Oncolytic adenovirus research evolution: from cell-cycle checkpoints to immune checkpoints. Curr Opin Virol 2015;13: 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012;30: 658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33: 2780–8. [DOI] [PubMed] [Google Scholar]
- 15.Sheridan C First oncolytic virus edges towards approval in surprise vote. Nat Biotechnol 2015;33: 569–70. [DOI] [PubMed] [Google Scholar]
- 16.Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum Vaccin Immunother 2018;14: 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000;19: 2–12. [DOI] [PubMed] [Google Scholar]
- 18.Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst 2003;95: 652–60. [DOI] [PubMed] [Google Scholar]
- 19.Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst 2007;99: 1410–4. [DOI] [PubMed] [Google Scholar]
- 20.Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol 2018;36: 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res 2017;77: 3894–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One 2014;9: e97407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falo LD Jr., Kovacsovics-Bankowski M, Thompson K, Rock KL. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat Med 1995;1: 649–53. [DOI] [PubMed] [Google Scholar]
- 24.Park HJ, Qin H, Cha SC, Sharma R, Chung Y, Schluns KS, et al. Induction of TLR4-dependent CD8+ T cell immunity by murine beta-defensin2 fusion protein vaccines. Vaccine 2011;29: 3476–82. [DOI] [PubMed] [Google Scholar]
- 25.Ochyl LJMoon JJ. Whole-animal imaging and flow cytometric techniques for analysis of antigen-specific CD8+ T cell responses after nanoparticle vaccination. J Vis Exp 2015: e52771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapp M, Anz D, Schnurr M. Isolation of intratumoral leukocytes of TLR-stimulated tumor-bearing mice. Methods Mol Biol 2014;1169: 175–9. [DOI] [PubMed] [Google Scholar]
- 27.LaFrance-Corey RGHowe CL. Isolation of brain-infiltrating leukocytes. J Vis Exp 2011: e2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schipper H, Alla V, Meier C, Nettelbeck DM, Herchenroder O, Putzer BM. Eradication of metastatic melanoma through cooperative expression of RNA-based HDAC1 inhibitor and p73 by oncolytic adenovirus. Oncotarget 2014;5: 5893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascallo M, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res 2015;21: 1406–18. [DOI] [PubMed] [Google Scholar]
- 30.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010;207: 2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaguchi T, Hirota K, Nagahama K, Ohkawa K, Takahashi T, Nomura T, et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity 2007;27: 145–59. [DOI] [PubMed] [Google Scholar]
- 32.Anderson KG, Stromnes IM, Greenberg PD. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017;31: 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016;44: 1255–69. [DOI] [PubMed] [Google Scholar]
- 34.Simon SLabarriere N PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2018;7: e1364828-1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012;209: 1201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355: 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother 2010;33: 956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124: 2246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 2016;22: 433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010;207: 2175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest 2015;125: 2046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 2017;114: 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Franco F, Ho PC. Metabolic Regulation of Tregs in Cancer: Opportunities for Immunotherapy. Trends Cancer 2017;3: 583–592. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 Costimulation Inhibits Foxp3 Expression and Treg Induction via BATF3-Dependent and Independent Mechanisms. Cell Rep 2018;24: 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 2012;13: 1087–95. [DOI] [PubMed] [Google Scholar]
- 46.Du W, Seah I, Bougazzoul O, Choi G, Meeth K, Bosenberg MW, et al. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc Natl Acad Sci U S A 2017;114: E6157–E6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH. Metastatic Brain Tumors Disrupt the Blood-Brain Barrier and Alter Lipid Metabolism by Inhibiting Expression of the Endothelial Cell Fatty Acid Transporter Mfsd2a. Sci Rep 2018;8: 8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015;212: 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523: 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blair GE, Dixon SC, Griffiths SA, Zajdel ME. Restricted replication of human adenovirus type 5 in mouse cell lines. Virus Res 1989;14: 339–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.