

Abstract
Background:
The heterogeneity of androgen receptor (AR) activity is well characterized in heavily treated metastatic castration-resistance prostate cancer (mCRPC). However, the diversity and clinical implications of AR-activity in treatment naïve primary prostate cancer is largely unknown. We sought to characterize AR-activity in localized prostate cancer and understand its molecular and clinical implications.
Methods:
Genome-wide expression profiles from prostatectomy or biopsy samples from 19,470 patients were used, all with independent pathology review. This was comprised of prospective discovery (n=5,239) and validation (n=12,728) cohorts, six retrospective institutional cohorts with long-term clinical outcomes data (n=1,170), and TCGA (n=333).
Results:
A low AR-active subclass was identified, which comprised 9-11% of each cohort, and was characterized by increased immune signaling, neuroendocrine expression, and decreased DNA repair. These tumors were predominantly ERG- and basal subtype. Low AR-active tumors had significantly more rapid development of recurrence or metastatic disease across cohorts, which was maintained on multivariable analysis (HR 2.61, 95%CI 1.22-5.60, p=0.014). Low AR-active tumors were predicted to be more sensitive to poly (ADP-ribose) polymerase inhibition, platinum chemotherapy, and radiotherapy, and less sensitive to docetaxel and androgen-deprivation therapy. This was validated clinically in that low AR-active tumors were less sensitive to androgen-deprivation therapy (OR 0.41, 95%CI 0.21-0.80, p=0.008).
Conclusions:
Leveraging large-scale transcriptomic data allowed the identification of an aggressive subtype of treatment-naïve primary prostate cancer that harbors molecular features more analogous to mCRPC. This suggests that a pre-existing subgroup of patients may have tumors that are predisposed to fail multiple current standard of care therapies and warrant dedicated therapeutic investigation.
Keywords: Prostate cancer, gene expression, androgen receptor, neuroendocrine
Introduction:
Although the genomic diversity and clinical relevance of androgen receptor (AR) activity (AR-A) is well established in heavily pretreated metastatic castration-resistant prostate cancer (CRPC), in treatment naïve primary prostate cancer (pPCa) it is less well characterized(1-3). Recently, The Cancer Genome Atlas (TCGA) demonstrated heterogeneity in AR and AR-A expression within 333 pPCa tumors. Although provocative, the biologic and clinical implications of these findings are unknown due to the small sample size, short follow-up (<36 months), and lack of information on metastatic progression(3). Nonetheless, the well documented interplay between AR-signaling and numerous biological processes within mCRPC suggests that these interactions could potentially impact therapeutic response in even earlier disease states. Additionally, it is unknown if the variability in AR-A in mCRPC is solely treatment induced, or if it may exist de novo in the treatment naïve setting.
We hypothesized that understanding the implications of AR-A diversity within treatment naïve pPCa would provide important prognostic and predictive information that could explain the observed clinical heterogeneity in response to standard treatments. In addition, we sought to explore if the observed biologic phenotypes of heavily pretreated mCRPC tumors (e.g. increased neuroendocrine differentiation) could be appreciated in treatment naïve pPCa. To test these hypotheses with a high degree of granularity, we performed the largest transcriptomic study to date, 40 times the size of the TCGA prostate adenocarcinoma dataset, wherein full transcriptomic profiles from 19,470 pPCa tumors were comprehensively analyzed, including patients with long-term detailed clinical follow-up.
Methods
Clinical samples and microarray processing
Genome-wide expression profiles of prostate adenocarcinoma (small cell and neuroendocrine prostate cancer were excluded) from radical prostatectomy (RP) or biopsy tumor samples for a total of 19,470 pPCa patients were used. This was comprised from two prospective population-based registry cohorts (discovery n=5,239 and validation n=12,728), six retrospective institutional cohorts with long-term clinical outcomes (n=1,170), and the TCGA (n=333). The discovery cohort was exploratory in nature, and the validation cohorts were used to independently confirm any findings. The prospective cohorts were comprised of anonymized genome-wide expression profiles of formalin-fixed paraffin-embedded samples from clinical use of the Decipher test between February 2014 to August 2017 retrieved from the Decipher GRID™ (). Individual patient genomic and clinicopathologic data were gathered from each study after institutional review boards at the participating institutions approved the research protocol under which the data were collected. Informed consent was not necessary to conduct this study, and thus was not obtained.
Basic demographic and pathological data, but not longitudinal clinical outcomes, were available. Data from the six retrospective cohorts (Table 1, Supplementary Table 1) of men treated with RP at Johns Hopkins Medical Institution (JHMI, n=355), Mayo Clinic (MC, n=235), Thomas Jefferson University (TJU, n=139), Durham VA (DVA, n=117), Kaiser Permanente Northwest (KPN, n=224) or external beam radiotherapy (EBRT) at Brigham & Women’s Hospital (BWH, n=100). Local institutional review boards (IRB) approved all data collection. The TCGA PRAD dataset (n=333) was also used and is publicly available(3).
Table 1.
Cohort Characteristics of prospective and pooled retrospective samples
| Variables | Prospective Discovery |
Prospective Validation | Retrospective Institutional Cohorts |
TCGA |
|---|---|---|---|---|
| No. (%); Median (IQR) | No. (%); Median (IQR) | No. (%); Median (IQR) | No. (%); Median (IQR) | |
| Total | 5,239 (100%) | 12,728 (100%) | 1170 (100%) | 333 (100%) |
| Age (years) | 65.5 (60, 69.2) | 65(59,69) | 60 (55-65) | 61(56-66) |
| PSA at diagnosis (ng/mL) | 6.5 (4.8, 9.7) | 6.6(4.9-10) | 7.7 (5.3,12.4) | 7.4(5.1-11.9) |
| <10 ng/mL | 1886 (36%) | 5528 (43%) | 684 (58%) | 127 (38%) |
| 10-20 ng/mL | 441 (8.4%) | 1433(11%) | 261 (22%) | 36 (11%) |
| >20 ng/mL | 166 (3.1%) | 449(3.5%) | 117 (10%) | 23(7%) |
| Gleason Grade group (Bx or post-RP) | ||||
| Group 1 (GS 3+3) | 271 (5%) | 826(6%) | 121 (10%) | 65(19%) |
| Group 2 (GS 3+4) | 1769 (34%) | 5469(43%) | 452 (38%) | 102(30%) |
| Group 3 (GS 4+3) | 1209 (23%) | 3650(28%) | 246 (21%) | 78(23%) |
| Group 4 (GS 8) | 396 (7%) | 1038(8%) | 143 (12%) | 45(13%) |
| Group 5 (GS 9-10) | 554 (11%) | 1582(12%) | 211 (18%) | 43(13%) |
| SM | ||||
| Positive | 2099 (40%) | 6231(49%) | 581 (49%) | 69(20.7%) |
| EPE | ||||
| Present | 2092 (40%) | 6698(52%) | 505 (43%) | 110 (33.0%) |
| SVI | ||||
| Present | 781 (15%) | 2236(18%) | 318 (27%) | 82 (25%) |
| LNI | ||||
| Positive | 195 (4%) | 617(5%) | 97 (8%) | NA |
| Median follow-up (months) | 48 | 36 | 104 | 28 |
Abbreviations: RP, radical prostatectomy; Bx, biopsy; PSA, prostate specific antigen; IQR, interquartile range; SM, surgical margins; SVI, seminal vesicle invasion (pT3b); EPE, extraprostatic extension (pT3a); LNI, lymph node invasion (pN1); ADT, androgen deprivation therapy; AR, androgen receptor; HR, hazard ratio; CI, confidence interval.
For all cases (except TCGA), tumor RNA was extracted from FFPE blocks or unstained slides after macrodissection guided by histologic review of the tumor lesion by a genitourinary pathologist. All cases had central pathology review prior to sampling for the Decipher assay, at least 0.5 cm2 of tumor with ≥60% tumor cellularity was required for RNA extraction and microarray hybridization (Human Exon 1.0 ST GeneChips), which were performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory facility (GenomeDx, San Diego, CA, USA)(4). Quality control was performed using Affymetrix Power Tools, and normalization was performed using the Single Channel Array Normalization (SCAN) algorithm(5). This study was conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects.
AR expression and AR Activity (AR-A) scores
AR gene expression was determined by summarizing 72 intronic and exonic probe sets within the AR gene. This AR-activity signature was derived from prior work(6). The finalized AR-A signature used in this study was defined a priori, and the AR and TMPRSS2 genes were excluded from the original gene model that was selected to allow comparison of AR-A to AR expression and to not bias AR-activity with ERG status. AR-A score was taken as a weighted linear sum of 9 canonical AR transcriptional target genes (KLK3, KLK2, FKBP5, STEAP1, STEAP2, PPAP2A, RAB3B, ACSL3, NKX3-1).(6) Gene weights were based on their distribution skewness in a subset of the prospective cohort using “robustbase” R package. Patients with outlier AR-A score (less than mean (AR-A) – 1 * standard deviation (AR-A)) were classified as low AR-A. Using the prospective discovery cohort, low AR-A was defined and locked as a score of 11 or less and then applied to the prospective validation and retrospective institutional cohorts to define AR-A low. For TCGA, which used an RNAseq platform, the same methodology was utilized to as the discovery cohort to define AR-A low.
Gene Expression Analyses and Tumor Purity Assessment
The Molecular Signatures Database (MSigDB) was queried for 37 oncology related hallmark gene sets(7). Hallmark gene set scores were computed by taking the mean expression of each gene in the set. For immune cell quantification, we used immunophenoscores to measure suppressor immune cells (T regulatory, MDSC) infiltration from gene expression data(8), and CIBERSORT tool to measure immune infiltration of 22 immune cells from gene expression data as previously described(9). FGFR activity score was calculated using zscore method in GSVA R package using 9 FGF and 4 FGFR genes. Decipher and cell cycle activity scores were extracted from the GRID as previously described(10). We investigated the expression of 39 NEPC markers(11) in our large cohort of histologically confirmed adenocarcinomas. Stromal infiltration score was calculated by averaging 141 stromal genes previously reported(12). Additionally, tumor purity based on consensus purity scores within TCGA for both gene expression and immunohistochemistry (IHC) were calculated(13).
Treatment Sensitivity Analyses
Radiation sensitivity score was calculated using a gene expression signature developed and validated to predict response to radiotherapy(14). Drug sensitivity was calculated using in vitro drug sensitivity and microarray data to generate gene signatures predicting tumor sensitivity to 89 oncology drugs(15,16). For each drug, CellMiner tool(15) was used to identify drug response related genes and their correlations to the IC50 value. Most significantly correlated genes were selected, and the expression of the corresponding genes were extracted for drug response score (DRS) calculations. A patient specific drug response score (DRS) was calculated using these correlation coefficients (Cor) as weighting factors of the corresponding gene expression normalized by the sum of Cor.
Immunohistochemistry
P53 missense mutation was detected using IHC assay as previously described(17). Each tissue microarray spot containing tumor cells was visually dichotomously scored for presence or absence of nuclear p53 signal by a urologic pathologist blinded to the gene expression data (TLL). ERG and PTEN IHC was performed as described previously(4),(18).
Statistical analysis and endpoints
Pearson correlation was used to assess correlation coefficients. Euclidean distance and ward linkage function was used for hierarchical clustering. We used FDR to adjust for multiple testing when we looked at association between low AR-A and gene expression and signature activity. Recurrence was defined per TCGA dataset, and metastatic disease for the retrospective cohorts was defined by radiographic evidence of metastatic disease. Development of CRPC was assessed within the JHMI cohort, and was defined as radiographic or biochemical progression in the setting of castrate levels of testosterone. Cumulative incidence curves were constructed using Fine-Gray competing risks analysis(19) to estimate the risk of metastasis over time with deaths from other causes as a competing risk. Additionally, Kaplan Meier analyses with log-rank test for recurrence (TCGA) or metastasis (retrospective cohorts) was performed. Time to distant metastasis from initial local therapy was modeled using multivariable competing risks regression analysis. Univariable logistic regression was used to associate AR-A with CRPC endpoint. Statistical analyses were performed in R v3.3.1, and all tests were using a 5% significance level.
Results
Heterogeneity of AR expression and AR-Activity
Cohort and tumor characteristics are summarized in Table 1 and how each cohort was used in Figure 1. Leveraging the prospective discovery pPCa samples, we first characterized the population-level variability in the distribution of the expression of 72 AR exon and intron probe sets (Figure 2A), demonstrating remarkable heterogeneity in AR expression. Next we rank ordered the expression of nine canonical transcriptional AR targets(6) (Figure 2B). After generation of a composite AR-A score, unsupervised hierarchical clustering revealed a distinct subclass of tumors with low AR-A expression, which was confirmed in the validation cohort (Supplementary Figure 1). Importantly, analyses were conducted and confirmed that the subset of low AR-A pPCa was not impacted by either stromal contamination or tumor purity (Supplementary Figure 2).
Figure 1.

CONSORT Diagram.
Figure 2.
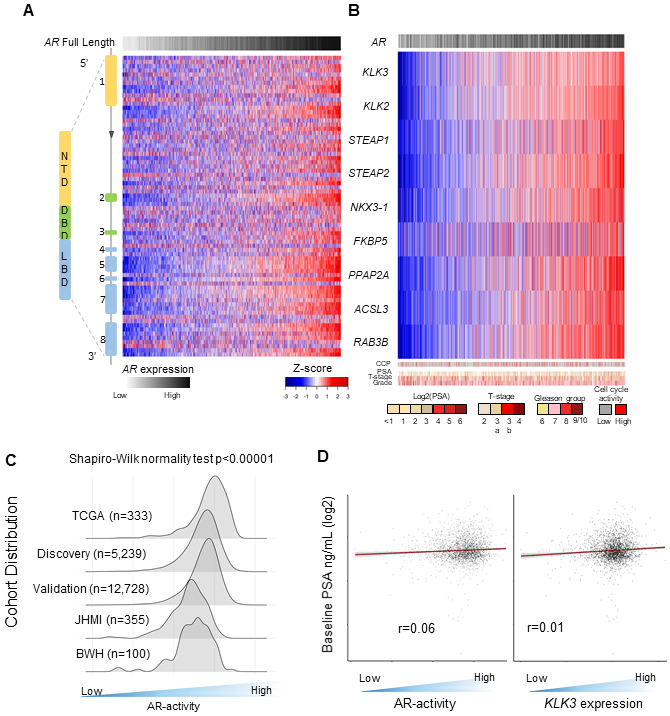
Transcriptomic profiling of treatment naïve primary prostate cancers demonstrates significant inter-individual diversity of AR gene and AR-activity expression. A. Heatmap representing the gene expression over the eight exons and intronic region probe sets of the AR, as well as summarized full length AR using the prospective discovery cohort. B. Heatmap of the gene expression of 9 canonical AR-target genes using the prospective discovery cohort. C. Distribution of AR-activity across five independent cohorts (TCGA, prospective discovery and validation cohorts, JHMI, and BWH; total n=19,470). D. Heat scatter plot of the relationship between serum pre-treatment PSA that is log2 transformed, to the AR-activity score for each tumor and the gene for PSA, KLK3.
Abbreviations: PSA, prostate-specific antigen; TCGA, The Cancer Genome Atlas; JHMI, Johns Hopkins Medical Institute; BWH, Brigham and Women Hospital
Overall, low AR-A pPCa was uncommon (approximately 10% across all cohorts, Figure 2C). The distribution of AR-A is a skewed distribution (Shapiro-Wilk normality test p<0.0001), rather than a normal distribution, with a significant tail that captures the low AR-A subclass (Figure 2C). Notably, the AR-A signature utilized in this study is not unique to capture AR-signaling and has a high correlation to other AR-A signatures, including Kumar et al(2) (r=0.82) or AR-response from Hallmarks of cancer signatures(7) (r=0.74) (Supplementary Figure 3). Interestingly, baseline pre-treatment serum PSA is often suggested to be a clinical surrogate for AR-A. However, serum PSA had no correlation to either intratumoral AR-A (r=0.06) or KLK3 expression (r=0.01) (Figure 2D, Supplementary Figure 4). Low AR-A tumors were enriched in higher grade tumors (10%, 14%, and 22% for Grade group 1-3, 4, and 5, respectively; Supplementary Figure 5).
Biology of Low AR-Active Prostate Cancer
To better understand the biologic phenotype of low AR-A tumors, a series of gene expression and IHC analyses were conducted. Low AR-A tumors were more likely to be triple negative (ERG-, ETS-, and SPINK-), and resemble a basal (or non-luminal) subtype (Figure 3A, Supplementary Figure 6). High AR-A tumors were more likely to ERG+, which was confirmed by IHC for ERG staining in a subset of patients from the JHMI cohort. Low AR-A tumors had more p53 mutations in TCGA, which was confirmed in the JHMI cohort in that by IHC p53 staining assay(17) was found only in lower AR-A tumors (Supplementary Figure 6).
Figure 3:
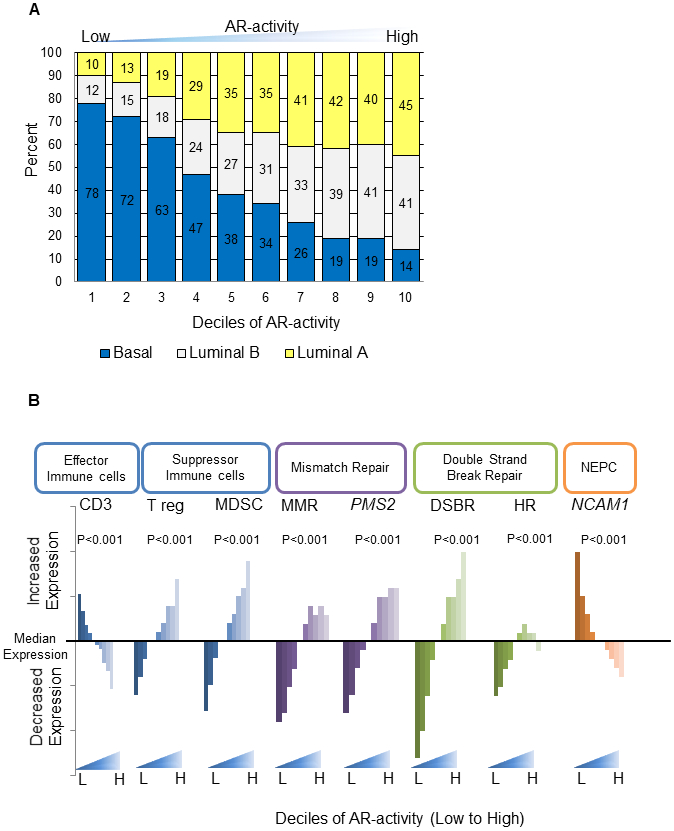
Biologic landscape in primary prostate cancer of low AR-active tumors. A. PAM50 subtypes of prostate cancer (Zhao et al; Basal, Luminal A, and Luminal B) by decile of AR-A. B. Analysis of AR-A decile and distribution of immune cell content, DNA repair, and neuroendocrine marker expression. Additional neuroendocrine markers shown in supplementary table S3.
Abbreviations: DSBR, double strand break repair; MDSC, Myeloid derived suppressor cells; MMR, mismatch repair; NEPC, neuroendocrine prostate cancer; Treg, Regulatory T-cell
Molecular Signatures Database analyses demonstrated that high AR-A tumors were associated with androgen response, DNA repair, and cell cycle (Supplementary Figure 7.A). Low AR-A tumors were significantly associated with multiple immune response gene sets. CD3 expression, which is specific for effector immune cells, was significantly overexpressed in low AR-A tumors (Figure 3B). Additionally, immune suppressor regulatory T-cells and myeloid-derived suppressor cell signature scores(8) were lower in low AR-A tumors. Low AR-A tumors showed higher activity of Interferon alpha, gamma, TNF signaling and higher activity of genes needed for immune cell recruitment and lower activity of cytosolic nucleic acid sensing pathways (Supplementary Figure 7.B). Furthermore, Gene Set Enrichment Analyses demonstrated increased enrichment of signatures for chemokine-chemokine interactions, PD1 signaling, and CD3 phosphorylation (Supplementary Figure 8). Additionally, using CIBERSORT, low AR-A tumors are estimated to have increased neutrophils, B cells, activated mast cells, gamma delta T cells, activated dendritic cells, eosinophils, and activated memory CD4 T cells. Collectively this data suggests low AR-A tumors may have enhanced immunogenicity.
Low AR-A tumors had significantly decreased DNA repair pathway expression, including individual genes of mismatch repair (PMS2 and MLH1), as well as mismatch repair pathway gene sets (Figure3B, Supplementary Figure 9, p<1e−20). Low AR-A tumors also had significantly lower pathway expression of homologous recombination (p<1e−20) (Figure 3B).
Given the increased understanding and recognition of the spectrum from adenocarcinoma to neuroendocrine PCa (NEPC)(20), we investigated the expression of 39 NEPC markers(11) in the prospective cohorts of histologically confirmed adenocarcinomas. After adjusting for false discovery rate, low AR-A tumors had significantly higher neuroendocrine marker expression of 32 of the 39 neuroendocrine biomarkers, including NCAM1, ENO2, and SYP (Figure 3B,Supplementary Figure 10, Supplementary Table 2) (p<1e−10). Recent, work has also demonstrated that in AR-null/NEPC-null mCRPC tumors FGF pathway activation is utilized to bypass AR-dependence(21). We found that FGF-activity is significantly increased in low AR-A pPCa (p<1e−10), suggesting this bypass event may begin in primary tumor before androgen-ablation (Supplementary Figure 11). Additionally, low AR-A pPCa had significantly higher expression of alternative nuclear hormone receptors, including PGR, NR3C1, and ESR1 (p<1e−10) (Supplementary Figure 12) consistent with observations in mCRPC(2).
Prognostic Impact of AR-Activity
Given low AR-A tumors were more likely to harbor a more aggressive biologic phenotype, we next assessed the prognostic difference of low vs high AR-A tumors. Across the prospective discovery, prospective validation, JHMI, and BWH cohorts, lower AR-A patients were significantly more likely to have higher Decipher scores, a biomarker of metastatic potential (Supplementary Figure 13). To clinically validate this, we analyzed AR-A in the JHMI natural history cohort(22) for the cumulative incidence of metastases by both AR-A quartile and by a 4-tiered system of ≤10%, 10-50%, >50%-90%, and >90% AR-A expression. Lower AR-A in both analyses was associated with worse metastatic outcome (p<0.001), and the patients with tumors having the lowest decile of AR-A had the worst metastatic outcome (Supplementary Figure 14). This was confirmed when analyzing patients with low AR-A tumors versus all others in three independent cohorts; TCGA (HR 2.00 (95%CI 1.02-3.57), p=0.04, Figure 4A), JHMI (HR 1.82 (95%CI 1.27-2.63), p<0.001, Figure 4B), and BWH (HR 5.26 (95%CI 1.75-14.29), p<0.001, Figure 4C). The median time to event for AR-A low patients in TCGA, JHMI, and BWH was 82, 96, and 76 months, respectively. The median time to event for AR-A high patients was not reached in any cohort.
Figure 4:
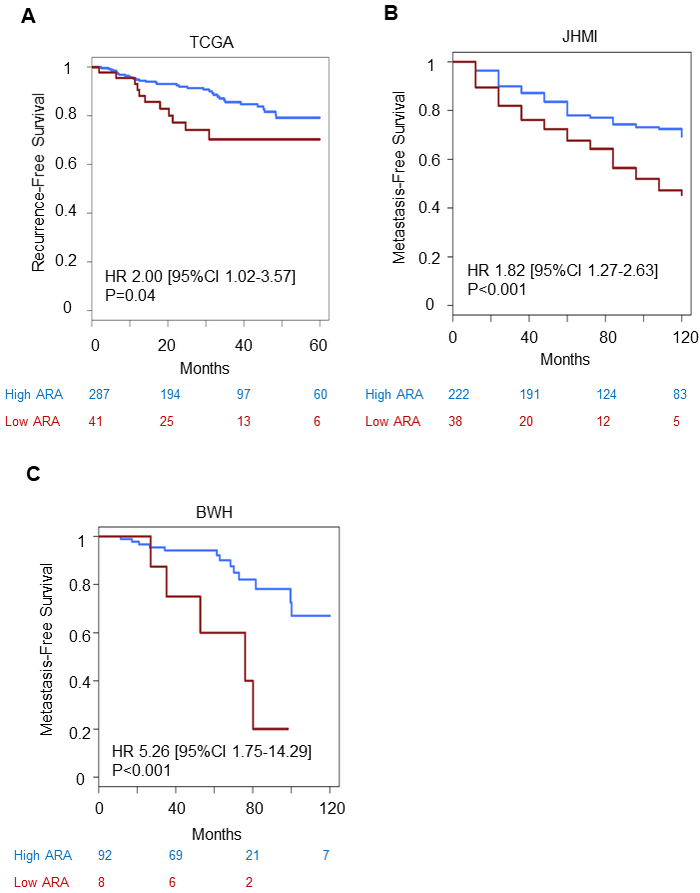
Association of AR-activity with recurrence and metastases. Kaplan-Meier curves by AR-activity for A) recurrence-free survival within TCGA, B) metastasis-free survival within JHMI cohort and C) BWH cohort.
Abbreviations: TCGA, The Cancer Genome Atlas; JHMI, Johns Hopkins Medical Institute; BWH, Brigham and Women Hospital
These findings were further confirmed in a multivariable competing risks regression analysis adjusting for age, PSA, Gleason grade, surgical margin status, extracapsular extension, seminal vesicles invasion, lymph node invasion, adjuvant and salvage treatment, and low AR-A remained significantly associated with an increased risk for developing metastatic disease (HR 2.61(95%CI 1.22-5.60), p=0.01, Figure 5A). To account for any potential batch effects, batch was included in the model, and low AR-A remained significant (HR 2.69 (95% CI 1.21-5.96), p=0.02, Supplementary Table 3).
Figure 5:
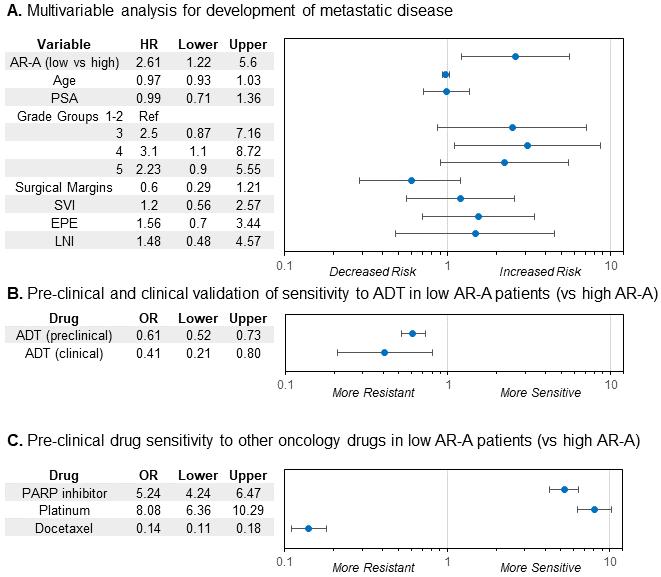
Prognostic and predictive treatment implications of AR-A in primary prostate cancer. A. Multivariable competing risk analysis for the development of metastasis within the JHMI cohort. B. Logistic regression for pre-clinical in vitro drug sensitivity analysis and clinical validation using the JHMI cohort for treatment sensitivity to ADT by AR-A status. C. Logistic regression for pre-clinical in vitro drug sensitivity analysis to PARP inhibitor therapy, platinum chemotherapy, and taxane chemotherapy by AR-A.
Abbreviations: ADT, androgen-deprivation therapy; AR-A, androgen receptor-activity; JHMI, Johns Hopkins Medical Institute
AR-A as a Predictive Biomarker of Response to ADT
The most common treatment for prostate cancer are targeted to the AR or elicit DNA damage using radiotherapy or chemotherapy, and thus treatment sensitivity may differ by AR-A. To test potential differing pharmacologic sensitivities, we used in vitro drug sensitivity and microarray data to generate drug response scores for 89 oncology drugs (Supplementary Figure 15). Low AR-A tumors had significantly lower predicted sensitivity than AR-A high tumors for response to ADT (OR 0.61 (95%CI 0.52-0.73, p<0.001) (Figure 5B). This was then validated clinically using the JHMI cohort to determine in patients that developed metastatic disease who were receiving chemical castration if low AR-A predicted development of CRPC (e.g. resistance to ADT). Again, low AR-A tumors were significantly less likely to be sensitive to ADT and more likely to develop CRPC (OR 0.41 (95%CI 0.21-0.80), p=0.008) (Figure 5B,Supplementary Figure 16). Furthermore, we demonstrate that clinically low AR-A pPCa tumors had similar AR-A scores as AR-independent tumors from more advanced or aggressive disease states (CRPC, neuroendocrine, and/or small cell prostate cancer, Supplementary Figure 17).
AR-A as a Predictive Biomarker to Other Oncology Therapies
Within the prospective cohort, tumors with low AR-A had increased sensitivity scores for platinum chemotherapy (OR 8.08 (95%CI 6.36-10.29)) and PARP inhibitors (OR 5.24 (95%CI 4.24-6.47)), and predicted worse response to taxane chemotherapy (OR 0.14 (95%CI 0.11-0.18), Figure 5C). Notably, the cisplatin and PARP inhibitor signatures have <5% gene overlap, and independently correlated with AR-A.
Lastly, we investigated AR-A as a predictive biomarker of treatment response to radiotherapy. Utilizing a 24-gene signature developed by Zhao et al that predicts radiation response (PORTOS)(14), we demonstrated that low AR-A tumors have significantly higher PORTOS scores, denoting increased potential radiation sensitivity (p<1e−20, Supplementary Figure 18).
Discussion
In the largest full transcriptomic analysis of primary prostate cancer, comprised of nearly 20,000 patients, we have performed a detailed series of analyses to gain an understanding of the biological and clinical relevance of AR-A. We provide robust clinical results from a large prospective cohort, with multiple forms of independent validation, of a low AR-A subclass of prostate adenocarcinomas that has high metastatic potential and distinct therapeutic sensitivities. Furthermore, we identify a subset of treatment naïve pPCa that has comparable AR-A to heavily pretreated mCRPC, and similar biological phenotypic characteristics (e.g. increased NEPC markers and FGF signaling, decreased DNA repair expression, and predominately basal subtype). Our findings suggest that a portion of the observed differences in biology between pPCa and mCRPC may be an expansion of a pre-existing subset of low AR-A tumors rather than solely therapy induced changes, consistent with other work(23,24). Our work suggests that these men can and should be identified prior to initial therapy.
The data presented here offer several additional novel insights. First, we demonstrate that there is marked inter-individual heterogeneity in AR and AR-A expression in pPCa across multiple retrospective and prospective cohorts. Second, we demonstrate that serum PSA, a clinical biomarker often used to infer AR-A, had no correlation to intratumoral AR-A, or even the gene for PSA, KLK3. Thus, PSA should not be used in isolation to clinically assign AR-A in pPCa. We hypothesize that in pPCa PSA is a marker of tumor burden and prostate size, and is unreliable to capture intrinsic AR-A.
Third, the diversity in AR-signaling in pPCa represents important biological heterogeneity that is both prognostic and predictive of treatment response. ADT is commonly delivered as concurrent or adjuvant therapy to radical treatment based on multiple randomized trials demonstrating benefit in unselected populations(25,26). Furthermore, more intensive combinatorial approaches are showing promise, including recent data from STAMPEDE from the addition of abiraterone to standard LHRH agonist therapy(27). These clinical trial results are consistent with our data, as only approximately 10% of pPCa would be classified as low AR-A, and thus the vast majority of patients would be predicted to be sensitive to AR directed therapies. However, despite the general efficacy of combined modality therapy, approximately 10% of high risk men will develop distant metastases 10-years post-treatment(28), and we show these patients are more likely to harbor low AR-A tumors.
Currently, there are no prospectively validated predictive biomarkers in pPCa to help select men towards a specific therapeutic approach. Recently, luminal and basal subtyping of prostate cancer patients using the breast cancer classifier, PAM50, has demonstrated the ability to predict which patients are most likely to respond to post-prostatectomy ADT(29). Our data shed insights into underlying biology for these findings, in that we demonstrate that low AR-A patients are most likely to be of basal subtype, which Zhao et al have shown to have less sensitivity to ADT than luminal B tumors (which more often have higher AR-A). Our study builds upon these efforts by providing sound biologic rationale and provocative results of a distinct subclass with a poor prognosis to current standard of care therapies.
Our study is timely given the greater understanding of DNA repair defects (e.g. ATM and BRCA2), microsatellite instability, and the spectrum of NEPC, which has sparked interest in combined modality therapy with PARP inhibitors (), platinum chemotherapy (), and immunotherapy (). Our data demonstrates that low AR-A tumors are not only more resistant to ADT and potentially docetaxel, but also are more sensitive to radiotherapy and alternative non-standard of care treatment options, including PARP inhibition and platinum chemotherapy. Furthermore, low AR-A tumors have increased immunogenicity and decreased expression of mismatch repair, both potential markers of an ideal population to investigate immunotherapeutic strategies on. Our study will require prospective validation to confirm our drug sensitivity predictions.
Our study has limitations. To minimize potential sources of bias or limitations we performed robust validation of all analyses in at least one if not multiple independent cohorts. Analyses for contamination of stromal content, assessment of tumor purity, and correction for batch effects were performed, which are known potential confounders within gene expression analyses, and we were unable to demonstrate this as an unlikely source of bias. There are known differences in microarray and RNA-seq data, and thus TCGA was used to validate our findings which showed similar distribution of AR-A and a negative prognostic impact of low AR-A status. We demonstrate that our AR-A score is highly correlated to other AR-A scores in the literature. However, AR signaling is complex and context specific, and alternative AR-A models could improve the utility of using AR-A to serve as a predictive biomarker. Finally, time to CRPC analyses were not performed and rather simply the development of CRPC as a binary event was used given exact dates for the formation of CRPC could not be collected.
Conclusion
In summary, our study establishes low AR-A pPCa as a clinically relevant subclass of treatment naïve localized prostate adenocarcinoma that harbors biology more akin to mCRPC. This aggressive subtype of basal-like low AR-A tumors are more likely to develop resistance to ADT and be less responsive to docetaxel, and may have other distinct treatment sensitivities. Thus, dedicated biomarker enhanced clinical trials in earlier stages of the disease are warranted for these patients.
Supplementary Material
Figure S1: Heatmap of the 9 AR-target genes after unsupervised hierarchical clustering, using Euclidean distance and ward function for linkage, demonstrating a subclass of patients (dashed box) with predominately low AR-activity using the prospective discovery and validation cohorts.
Figure S2: Associations between AR-activity and stromal content. A. Tumor purity based on consensus purity estimate is not correlated to AR-activity in TCGA. B. Percentage of low AR-activity is not associated with IHC-based tumor purity (The Cochran-Armitage Trend Test, p=0.56). C. Stromal content based on the average of 141 stromal genes(12)(12)(12) is poorly correlated with AR-activity in prospective discovery cohort. D. Percentage of ERG+, a marker of prostate cancer, is similar across deciles of stromal score indicating that high stromal score is not indicative of tumor purity.
Figure S3: Heat scatter plot of the relationship between AR-activity Score in this work (Spratt) and other AR-activity signatures from Kumar etal and Hallmarks of cancer (MSIGDB).
Figure S4: Heat scatter plot of the relationship between serum pre-treatment PSA that is log2 transformed, to AR-activity (left) and KLK3 expression (right) using the prospective validation cohort.
Figure S5: AR-activity vs Gleason scores. AR-activity is lower in Gleason 8 and 9-10 compared to Gleason 6 and 7. 22% of Gleason9-10 have low AR-A compared to 10% for Gleason 6-7.
Figure S6: A. Tomlins et al subtypes of prostate cancer (Triple negative, SPINK+, ETS+, and ERG+) by decile of AR-activity. B. In the JHMI cohort where p53 mutation, PTEN deletion and ERG fusion data is available, patients with higher AR-A lack p53 mutations compared to low AR-A. However, PTEN deletion and ERG fusion didn’t show any association with AR-A
Figure S7: A.Heatmap generated from the MSigDB cancer hallmark gene set scores based on AR-activity expression utilizing the prospective discovery cohort. Low AR-activity tumors had enrichment of cancer hallmarks of immune signatures.B. Heatmap of innate immune signaling from Atlas of cancer signaling showing low AR-A have higher activity of interferon pathways and higher activity of genes needed for immune cells recruitment.
Figure S8: Gene set enrichment analysis using Reactome gene sets showing that genes negatively correlated to AR-A are enriched with immune signatures (PD1 signaling, immune regulatory interaction)
Figure S9: Low AR-A patients have higher expression of MLH1.
Figure S10: Low AR-A have higher activity of neuroendocrine genes (SYP and ENO2).
Figure S11: Low AR-A have higher activity of FGFR activity.
Figure S12: Low AR-A have higher activity of alternative nuclear hormone receptors, including PGR, NR3C1, and ESR1
Figure S13: High 22-gene genomic classifier scores of metastatic potential (e.g. high Decipher scores) distribution by AR-activity decile across the prospective discovery, prospective validation, JHMI, and BWH cohorts. There was an exponential association (R2 = 0.51-0.95) between lower AR-activity and having a high metastatic potential score
Figure S14: Cumulative incidence curves with competing risk analysis for development of metastatic disease by AR-activity split into four groups based on AR-A (<10%, 10-50%, >50-90%, >90%). These patients were in the JHMI natural history cohort and received radical prostatectomy alone. Patients with the lowest AR-activity tumors developed metastatic disease significantly more frequently than all other groups (p<0.001).
Figure S15: Prospective discovery cohorts gene expression drug response analysis based on 89 drug response scores derived from in vitro NCI-60 drug response, demonstrates that low AR-A tumors have a distinct drug response profile.
Figure S16: Development of CRPC (e.g. hormone therapy resistance) in a cohort treated by radical prostatectomy stratified by AR-activity. Patients that developed CRPC had lower AR-activity (p=0.008)
Figure S17: Normalized AR-activity scores across eight different cohorts dichotomized into primary prostate cancer and advanced prostate cancer (CRPC/mCRPC/neuroendocrine/small cell carcinoma). Orange dashed box denotes low AR-activity primary prostate cancer, which has similar expression to advanced disease states.
Figure S18: Radiation response score by AR-activity decile. The 24-gene PORTOS score was used to calculated predictive radiation sensitivity to post-operative radiation therapy, demonstrating that low AR-active patients have higher predicted radiation sensitivity scores.
Translational Relevance.
Using nearly 20,000 individual patients’ transcriptomic data from localized prostate cancer, we were able to identify a critically important subset of localized treatment naïve primary prostate cancer with low AR-activity that is biologically and clinically behaves very similar to metastatic castration-resistant prostate cancer with unique treatment sensitivities. Low AR-active tumors have unique molecular profile (e.g. increase neuroendocrine expression, immune signaling, and decreased DNA repair), as is associated with an aggressive natural history and show longterm clinical outcomes with validation in triplicate. Furthermore, this sub-class of low AR-active tumors appears through transcriptional analyses to be more sensitive to platinum chemotherapy and PARP inhibition and less sensitive to androgen-deprivation therapy and docetaxel. By unraveling the distinct biology, prognostic, and predictive information that is contained within AR-activity in localized prostate cancer, our work provides strong rational for personalizing treatment based on AR-activity status.
Acknowledgments
This work was funded in part by the Prostate Cancer Foundation Young Investigator Award (to DES), Prostate Cancer Foundation Challenge Award (to EMS), the Department of Defense (to DES; W81XWH-16-1-0571), U01CA196390 (to EMS), P50 CA186786 (to DES and AMC), and generous philanthropic gifts from patients (SEH, MP, PM) and the Ambrose Monell Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest
Y. Liu, E. Davicioni, N. Fishbane, J. Lehrer are employees of and hold ownership interest (including patents) in Decipher Biosciences.
T. Lotan and J. Karnes received research grants from Decipher biosciences.
S.G. Zhao reports receiving commercial research support from and holds ownership interest (including patents) in Decipher Biosciences.
P.L. Nguyen reports receiving commercial research grants from Janssen, Astellas, and Bayer; holds ownership interest (including patents) in Augmenix; and is a consultant/advisory board member for Augmenix, Ferring, Blue Earth, Bayer, Cota, Dendreon, Decipher biosciences, and Nanobiotix. F.Y. Feng is an employee of PFS Genomics, and is a consultant/advisory board member for Sanofi, Janssen, Medivation/Astellas, Dandreon, Ferring, EMD Serono, Bayer, and Clovis.
REFERENCES:
- 1.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A, Coleman I, Morrissey C, Zhang X, True L, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres A, Alshalalfa M, Tomlins S, Erho N, Gibb E, Chelliserry J, et al. Comprehensive Determination of Prostate Tumor ETS Gene Status in Clinical Samples Using the CLIA Decipher Assay. J Mol Diagn. 2017;19:475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccolo SR, Sun Y, Campbell JD, Lenburg ME, Bild AH, Johnson WE. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics. 2012;100:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faisal FA, Sundi D, Tosoian JJ, Choeurng V, Alshalalfa M, Ross AE, et al. Racial Variations in Prostate Cancer Molecular Subtypes and Androgen Receptor Signaling Reflect Anatomic Tumor Location. Eur Urol. 2016;70:14–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liberzon A, Birger C, Thorvaldsd??ttir H, Ghandi M, Mesirov JP, Tamayo P The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–62. [DOI] [PubMed] [Google Scholar]
- 9.Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, et al. The Immune Landscape of Prostate Cancer and Nomination of PD-L2 as a Potential Therapeutic Target. J Natl Cancer Inst. 2019;111:301–10. [DOI] [PubMed] [Google Scholar]
- 10.Spratt D, Yousefi K, Deheshi S, Ross A, Den R, Schaeffer E, et al. Individual Patient-Level Meta-Analysis of the Performance of the Decipher Genomic Classifier in High-Risk Men After Prostatectomy to Predict Development of Metastatic Disease. JCO. 2017;35:1991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai H, Lehrer J, Alshalalfa M, Erho N, Davicioni E, Lotan T. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer. 2017;17:759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao SG, Chang SL, Spratt DE, Erho N, Yu M, Ashab HAD, et al. Development and validation of a 24-gene predictor of response to postoperative radiotherapy in prostate cancer: a matched, retrospective analysis. Lancet Oncol 2016;17:1612–20. [DOI] [PubMed] [Google Scholar]
- 15.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, et al. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: A genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guedes LB, Almutairi F, Haffner MC, Rajoria G, Liu Z, Klimek S, et al. Analytic, preanalytic, and clinical validation of p53 IHC for detection of TP53 missense mutation in prostate cancer. Clin Cancer Res. 2017;23:4693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lotan TL, Wei W, Morais CL, Hawley ST, Fazli L, Hurtado-Coll A, et al. PTEN Loss as Determined by Clinical-grade Immunohistochemistry Assay Is Associated with Worse Recurrence-free Survival in Prostate Cancer. Eur Urol Focus. 2016;2:180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fine J, Gray R. A proportional hazards model for the subdistribution of a competing risk. J AM Stat Assoc. 1999;94. [Google Scholar]
- 20.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bluemn E, Coleman I, Lucas J, Coleman R, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32:474–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–7. [DOI] [PubMed] [Google Scholar]
- 23.Sowalsky AG, Ye H, Bhasin M, Van Allen EM, Loda M, Lis RT, et al. Neoadjuvant-intensive androgen deprivation therapy selects for prostate tumor foci with diverse subclonal oncogenic alterations. Cancer Res. 2018;78:4716–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B, et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur Urol. 2018; [DOI] [PubMed] [Google Scholar]
- 25.Messing E, Manola J, Yao J, Kiernan M, Crawford D, Wilding, G di’SantAgnese, PA Trump D. Immediate versus deferred androgen deprivation treatment in patients with node-positive prostate cancer after radical prostatectomy and pelvic lymphadenectomy. Lancet Oncol. 2006;7:472–9. [DOI] [PubMed] [Google Scholar]
- 26.Chetner MP, Bruner DW, Amin MB, Sandler HM, Husain SM, Rotman M, et al. Radiotherapy and Short-Term Androgen Deprivation for Localized Prostate Cancer. N Engl J Med. 2011;365:107–18. [DOI] [PubMed] [Google Scholar]
- 27.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med [Internet]. 2017;377:338–51. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1702900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spratt DE, Pei X, Yamada J, Kollmeier MA, Cox B, Zelefsky MJ. Long-term survival and toxicity in patients treated with high-dose intensity modulated radiation therapy for localized prostate cancer. Int J Radiat Oncol Biol Phys. 2013;85:686–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao S, Chang S, Erho N, Yu M, Lehrer J, Alshalalfa M, et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017;3:1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Heatmap of the 9 AR-target genes after unsupervised hierarchical clustering, using Euclidean distance and ward function for linkage, demonstrating a subclass of patients (dashed box) with predominately low AR-activity using the prospective discovery and validation cohorts.
Figure S2: Associations between AR-activity and stromal content. A. Tumor purity based on consensus purity estimate is not correlated to AR-activity in TCGA. B. Percentage of low AR-activity is not associated with IHC-based tumor purity (The Cochran-Armitage Trend Test, p=0.56). C. Stromal content based on the average of 141 stromal genes(12)(12)(12) is poorly correlated with AR-activity in prospective discovery cohort. D. Percentage of ERG+, a marker of prostate cancer, is similar across deciles of stromal score indicating that high stromal score is not indicative of tumor purity.
Figure S3: Heat scatter plot of the relationship between AR-activity Score in this work (Spratt) and other AR-activity signatures from Kumar etal and Hallmarks of cancer (MSIGDB).
Figure S4: Heat scatter plot of the relationship between serum pre-treatment PSA that is log2 transformed, to AR-activity (left) and KLK3 expression (right) using the prospective validation cohort.
Figure S5: AR-activity vs Gleason scores. AR-activity is lower in Gleason 8 and 9-10 compared to Gleason 6 and 7. 22% of Gleason9-10 have low AR-A compared to 10% for Gleason 6-7.
Figure S6: A. Tomlins et al subtypes of prostate cancer (Triple negative, SPINK+, ETS+, and ERG+) by decile of AR-activity. B. In the JHMI cohort where p53 mutation, PTEN deletion and ERG fusion data is available, patients with higher AR-A lack p53 mutations compared to low AR-A. However, PTEN deletion and ERG fusion didn’t show any association with AR-A
Figure S7: A.Heatmap generated from the MSigDB cancer hallmark gene set scores based on AR-activity expression utilizing the prospective discovery cohort. Low AR-activity tumors had enrichment of cancer hallmarks of immune signatures.B. Heatmap of innate immune signaling from Atlas of cancer signaling showing low AR-A have higher activity of interferon pathways and higher activity of genes needed for immune cells recruitment.
Figure S8: Gene set enrichment analysis using Reactome gene sets showing that genes negatively correlated to AR-A are enriched with immune signatures (PD1 signaling, immune regulatory interaction)
Figure S9: Low AR-A patients have higher expression of MLH1.
Figure S10: Low AR-A have higher activity of neuroendocrine genes (SYP and ENO2).
Figure S11: Low AR-A have higher activity of FGFR activity.
Figure S12: Low AR-A have higher activity of alternative nuclear hormone receptors, including PGR, NR3C1, and ESR1
Figure S13: High 22-gene genomic classifier scores of metastatic potential (e.g. high Decipher scores) distribution by AR-activity decile across the prospective discovery, prospective validation, JHMI, and BWH cohorts. There was an exponential association (R2 = 0.51-0.95) between lower AR-activity and having a high metastatic potential score
Figure S14: Cumulative incidence curves with competing risk analysis for development of metastatic disease by AR-activity split into four groups based on AR-A (<10%, 10-50%, >50-90%, >90%). These patients were in the JHMI natural history cohort and received radical prostatectomy alone. Patients with the lowest AR-activity tumors developed metastatic disease significantly more frequently than all other groups (p<0.001).
Figure S15: Prospective discovery cohorts gene expression drug response analysis based on 89 drug response scores derived from in vitro NCI-60 drug response, demonstrates that low AR-A tumors have a distinct drug response profile.
Figure S16: Development of CRPC (e.g. hormone therapy resistance) in a cohort treated by radical prostatectomy stratified by AR-activity. Patients that developed CRPC had lower AR-activity (p=0.008)
Figure S17: Normalized AR-activity scores across eight different cohorts dichotomized into primary prostate cancer and advanced prostate cancer (CRPC/mCRPC/neuroendocrine/small cell carcinoma). Orange dashed box denotes low AR-activity primary prostate cancer, which has similar expression to advanced disease states.
Figure S18: Radiation response score by AR-activity decile. The 24-gene PORTOS score was used to calculated predictive radiation sensitivity to post-operative radiation therapy, demonstrating that low AR-active patients have higher predicted radiation sensitivity scores.