

Abstract
Background
Approximately one-third of cases of dilated cardiomyopathy (DCM) are caused by genetic mutations. With new sequencing technologies, numerous variants have been associated with this inherited cardiomyopathy, however the prevalence and genotype-phenotype correlations in different ethnic cohorts remain unclear. This study aimed to investigate the variants in Chinese DCM patients and correlate them with clinical presentations and prognosis.
Methods and Results
From September 2013 to December 2016, 70 index patients underwent DNA sequencing for 12 common disease-causing genes with next generation sequencing. Using a bioinformatics filtering process, 12 rare truncating variants (7 nonsense variants, 4 frameshift variants, and 1 splice site variant) and 29 rare missense variants were identified. Of these, 3 patients were double heterozygotes and 10 patients were compound heterozygotes. Overall, 47.1% (33/70) of the index patients had the seputatively pathogenic variants. The majority (33/41, 80.4%) of these variants were located in titin (TTN). More than 80% of the TTN variants (27/33, 81.8%) were distributed in the A band region of the sarcomere. Patients carrying these variants did not have a different phenotype in disease severity, clinical outcome and reversibility of ventricular function compared with non-carriers.
Conclusions
Several new rare variants were identified in a Chinese population in this study, indicating that there are ethnic differences in genetic mutations in DCM patients. TTN remains the major disease-causing gene. Our results could be a reference for future genetic tests in Chinese populations. No specific genotype-phenotype correlations were found, however a prospective large cohort study may be needed to confirm our findings.
Keywords: Dilated cardiomyopathy, Genetic mutation, Next generation sequencing
INTRODUCTION
Dilated cardiomyopathy (DCM) is characterized by left ventricular dilation and systolic dysfunction,1 and has a prevalence of 1 in 250 the general population worldwide.2,3 DCM patients require frequent hospitalizations for symptomatic heart failure and ventricular arrhythmia in early adulthood.4-6 Diverse etiologies have been reported to cause DCM, including drugs, toxins, nutritional deficiency, endocrine disorders, and immune or infectious diseases. Approximately 30-48% of all cases of DCM are caused by genetic mutations.4,7 Currently, more than 30 genes, encoding a variety of proteins from sarcomere, nuclear envelope, ion channels and cytoskeleton complexes,8 have been reported to be associated with DCM. Moreover, these identified genetic variants are usually scattered across a gene, making sequencing and analysis challenging and time-consuming. With the introduction of the massive parallel sequencing technique, genetic testing has become more comprehensive and efficient. Multiple disease-associated genes can now be targeted and sequenced simultaneously using next-generation sequencing (NGS) technology, providing greater coverage for a targeted disease.9 However, along with the advance in sequencing technology, a rapidly growing number of disease-associated variants have been identified.7,10 Clarifying the clinical significance of variants has become a challenge and requires replication and validation in clinical DCM cohorts.11
Several studies have reported improved ventricular function with an early diagnosis and treatment.12 Patients with inherited cardiomyopathy usually have a long clinically indolent period before symptoms emergence. The early detection and initiation of interventions in the pre-symptomatic stage of probands and their relatives are expected to ameliorate disease progression and prevent major adverse events.13,14 Since the report from Herman et al.15 and subsequent studies, titin (TTN) truncating variants have been recognized in 25% of familial DCM cases and in 18% of sporadic cases.15-19 TTN encodes the almost 4 million Dalton sarcomeric protein titin, which has several biologic functions including maintenance of resting tension and elasticity, stabilization of contractile proteins, and force transmission.20,21 Recently, Jansweijer et al.22 reported that patients with TTN truncating variants had a relatively mild phenotype and were more amenable to therapy than those with LMNA (lamin A/C) mutations. These results were in contrast to early reports which reported that TTN truncating variant carriers had a more severe19 or similar15,18 prognosis than non-carriers. On the other hand, earlier atrial and ventricular arrhythmias have been reported in TTN truncating variant carriers, which may suggest that different treatment strategies are needed to prevent cardiac morbidity and mortality in these patients. Genotype-phenotype correlations remain controversial, and racial differences in DCM genomics have yet to be explored. In order to establish a DCM genetic database in a Chinese population, we designed a target gene panel with 12 common DCM-associated genes to analyze genetic information in patients and controls. The correlations between genetic variants and clinical outcomes in these patients were also assessed.
MATERIAL AND METHODS
Study design and population
In this study, we prospectively screened patients admitted for heart failure symptoms at Chang Gung Memorial Hospital from September 2013 to December 2016. After a comprehensive workup, patients who were > 20 years old and had a clinical diagnosis of DCM were enrolled in the study. DCM was diagnosed according to the European Society of Cardiology criteria, with dilatation of the left ventricle and a left ventricular ejection fraction (LVEF) < 45%.23 Patients with ischemic, hypertensive, significant valvular, and congenital heart diseases were excluded by history, physical examination, echocardiography, 24-hour Holter electrocardiography monitoring and coronary angiography. Patients with cardiotoxic drug exposure, chronic alcohol usage, severe concomitant diseases or systemic inflammatory diseases were also excluded. Healthy participants with no history of heart diseases and with a normal LVEF and electrocardiography results served as the normal control subjects. Blood samples from the participants were stored for genetic sequencing. Clinical information including family history, clinical symptoms, echocardiography evaluation, and laboratory results were collected.
The clinical endpoints were defined as all-cause mortality, cardiovascular mortality, non-fatal stroke, heart failure re-hospitalization, or ventricular arrhythmia requiring an implantable cardioverter defibrillator (ICD). Heart failure re-hospitalization was defined as a hospital admission within 1 year after the first visit, in which the patients presented with heart failure symptoms and required diuretics, vasodilators, or inotropic agents. The reversibility of ventricular systolic function was defined as at least a 10-point increase in LVEF 1 year after standard therapy. When the patients had multiple events, the time to the first event was counted as the censored outcome. All participants were followed up in outpatient clinics at 3, 6, 9, 12, 24, and 36 months after study enrollment. Major cardiac events occurring during the first visit to enrollment were also included. All patients provided written informed consent. This study complied with the declaration of Helsinki, and the study protocol was approved by the Institutional Review Board of Chang Gung Memorial Hospital.
Next generation sequencing of customized targeted gene panel
Genomic DNA from peripheral venous blood from the subjects was extracted using a QIAamp DNA blood mini kit (Qiagen, Taiwan) and sequenced on an Ion PGMTM System (Ion torrent) using NGS. A customized panel of 12 associated genes (ACTC1, LDB3, MYBPC3, MYH6, MYH7, MYL2, MYL3, TTN, TNNC1, TPM1, TNNI3, TNNT2) was designed using Ion AmpliseqTM Designer (Version 2.2, Applied Biosystems, Life Technologies, Carlsbad, CA) (Supplementary Table 1). We constructed sequencing libraries using the target exon amplification method for the protein-coding portions of the 12 disease-associated genes, composed of 1102 amplicons with a total size of 157.66 kilobases and a coverage of 98.46% of the targeted genes. More than 95% of the targeted bases were sequenced to a read depth of more than 20 times.
Supplementary Table 1. Twelve targeted genes sequencing for dilated cardiomyopathy in the study.
| Gene | Description | CDS (bps) | # of Exons |
| ACTC1 | Actin α, cardiac muscle | 3693 | 7 |
| LDB3 | Lim domain-binding 3 | 4978 | 13 |
| MYBPC3 | Cardiac myosin-binding protein C | 4217 | 35 |
| MYH6 | Myosin, heavy chain 6, cardiac muscle, a | 5941 | 39 |
| MYH7 | β - Myosin heavy chain 7, cardiac muscle | 6044 | 39 |
| MYL2 | Regulatory myosin light chain 2, cardiac, slow | 855 | 7 |
| MYL3 | Myosin light chain 3, alkali; ventricular, skeletal, slow | 942 | 7 |
| TNNC1 | Troponin C, type 1 | 705 | 6 |
| TNNI3 | Troponin I type 3, cardiac | 866 | 8 |
| TNNT2 | Troponin T type 2, cardiac | 1195 | 16 |
| TPM1 | Tropomyosin 1, α | 1797 | 9 |
| TTN | Titin | 281434 | 367 |
Bioinformatic filtering process of genetic variants
The sequencing results were aligned to the human reference genome assembly (Feb. 2009, GRCh37/hg19) from the National Center for Biotechnology Information (NCBI) using Torrent Mapping Alignment. Variants were identified by the Torrent Variant Caller and were annotated with wANNOVAR.24 Rare genetic variants were defined as an allele frequency < 1% in the 1000 Genomes Project and Exome Aggregation Consortium (ExAC) project.25,26 Variants present in the in-house genetic database of 299 healthy controls were also excluded (Cardiovascular Laboratory, Linkou Chang Gung Memorial Hospital and Whole-Genome Research Core Laboratory of Human Diseases, Keelung Chang Gung Memorial Hospital). Nonsynonymous mutations of missense mutations were predicted with bioinformatics programs, including PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org/). We divided these variants into two major types: truncating variants and missense variants (Supplementary Figure 1). All variants were confirmed using traditional Sanger sequencing.
Supplementary Figure 1.
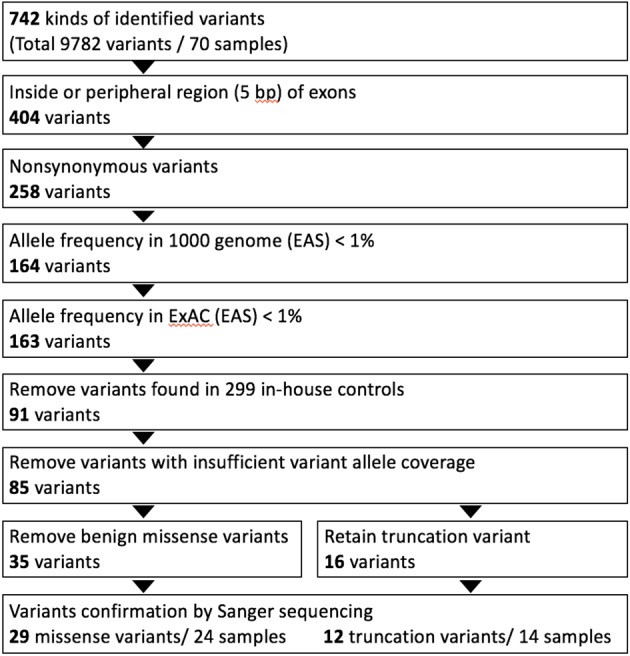
The diagram of genetic variants analyzing and filtering workflow in this study. A customized dilated cardiomyopathy (DCM) panel and Ion torrent PGM sequencer were used for genetic screening in 70 DCM patients. Residual variant numbers after each step of analyzing ad filtering processes were listed in the figure. Identified variants were classified as missense and truncation variants (nonsense, splice site, insertion-deletion).
Statistical analysis
Statistics were presented as mean ± standard deviation (SD) or median (interquartile range, IQR) depending on value distribution. Differences between subgroups were assessed using the Student t-test for normally distributed values and the Mann-Whitney U test for values with skewed distribution. Pearson’s chi-squared test was used for comparisons of categorical data. Event-free survival curves for major cardiac events were estimated and plotted using the Kaplan-Meier method. The log-rank test was used to investigate differences in event-free survival. All statistical analyses were performed using Statistical Package for Social Sciences V.18 software (IBM Corp., Armonk, NY, USA). Two-sided probability values were considered significant at p < 0.05.
RESULTS
Clinical characteristics of the study population
A total of 70 patients with DCM were recruited at our hospital. Of these patients, 18.6% were female, 12.8% had familial DCM, 24.3% had heart failure functional class III or IV, 25.7% had hypertension, 21.4% had diabetes mellitus and 28.6% had atrial fibrillation. The mean age of the patients was 55 ± 13 years. The mean LVEF at diagnosis was 28.0 ± 11.1%, and the mean left ventricular end diastolic volume index was 130.5 ± 43.6 ml. The mean serum B-type natriuretic peptide level was 1015.9 ± 429.3 pg/ml. With regards to medications, 84.2% of the patients received angiotensin-converting-enzyme inhibitors or angiotensin receptor blockers, and 81.4% received beta-blockers. The clinical characteristics of the patients are summarized in Table 1.
Table 1. Baseline characteristics of enrolled patients with dilated cardiomyopathy.
| Characteristics | (number = 70) |
| Age, years | 55 ± 13 (27-84) |
| Female gender, n (%) | 13 (18.6%) |
| BMI, kg/m2 | 24.4 ± 5.6 |
| Hypertension, n (%) | 18 (25.7%) |
| Diabetes mellitus, n (%) | 15 (21.4%) |
| Atrial fibrillation, n (%) | 20 (28.6%) |
| Systolic blood pressure, mmHg | 118 ± 20 |
| Heart rate, bpm | 81 ± 16 |
| NYHA III or IV, n (%) | 17 (24.3%) |
| BNP, pg/mL | 1015.9 ± 429.3 |
| Creatinine | 1.35 ± 1.58 |
| eGFR, mL/min/1.73 m2 | 81.5 ± 30.5 |
| Cholesterol, mg/dl | 157 ± 42 |
| HbA1C, % | 5.9 ± 0.98 |
| LVEF, % | 28.0 ± 11.1 |
| LVEDD, mm | 66.0 ± 9.8 |
| LVEDV index, ml/m2 | 130.5 ± 43.5 |
| LV mass index, g/m2 | 179.6 ± 61.2 |
| LA diameter, mm | 46.0 ± 10.7 |
| Mitral E/A ratio | 1.5 ± 1.3 |
| ACEI or ARB, n (%) | 59 (84.2%) |
| Beta-blockers, n (%) | 57 (81.4%) |
| Spironolactone, n (%) | 40 (57.1%) |
| Loop diuretics, n (%) | 46 (65.7%) |
| Digoxin, n (%) | 21 (30.0%) |
All numeric data were presented with means ± standard deviation.
All nominal data were presented with numbers (percentage).
ACEI/ARB, angiotensin I-converting enzyme inhibitor/angiotensin II receptor antagonists; BMI, body mass index; BNP, B-type natriuretic peptides; eGFR, estimated glomerular filtration rate; HbA1C, hemoglobin A1c; LA, left atrium; LV, left ventricular; LVEDD, left ventricular end-diastolic dimension; LVEDV, left ventricular end-diastolic volume; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association functional classification.
Genetic variants in the DCM patients
A total of 742 variants were identified from the DNA samples from the 70 patients (Supplementary Figure 1). Among these variants, 404 were located in the exon or splicing regions and 258 were protein affecting variants. These variants were then selected for further analysis if they had (1) an allele frequency < 1% in the 1000 genome project and ExAC projects; and (2) were not present in the 299 in-house Chinese normal controls. After confirmation using traditional Sanger sequencing, 29 missense variants and 12 truncating variants were considered to be putatively pathogenic variants in these index patients (Figure 1). Of the truncating variants, 8 were nonsense variants, 3 were frameshift variants, and 1 was a splice site variant. The majority of these genetic variants (33/41, 80.4%) were located in TTN (10 truncating variants, 23 missense variants). The distribution of other genetic variants included 1 nonsense variant and 4 missense variants in MYBPC3, 1 nonsense variantin MYH6, 1 missense variant in MYH7, and 1 missense variant in ACTC1. No rare variants were identified in the other DCM-associated genes (TNNT2, TPM1, LDB3, MYL2, MYL3, TNNC1 and TNNI3) in the study (Supplementary Figure 2). The detailed information of genetic variants is summarized in Table 2 and the Supplementary Tables 2, 3.
Figure 1.

The spatial distribution of genetic variants in titin protein. The canonical titin molecule of human (UniProtKB entry Q8WZ42-1) have 34350 amino acids. The whole titin protein extends from Z-disk, I-band, A-band to M-band. All TTN variants were shown as vertical bars with different colors, which represented of frameshift (red), splice site (green), nonsense (yellow), missense (blue) variants. Thicker bars indicated that variants located on the same or close sites.
Supplementary Figure 2.
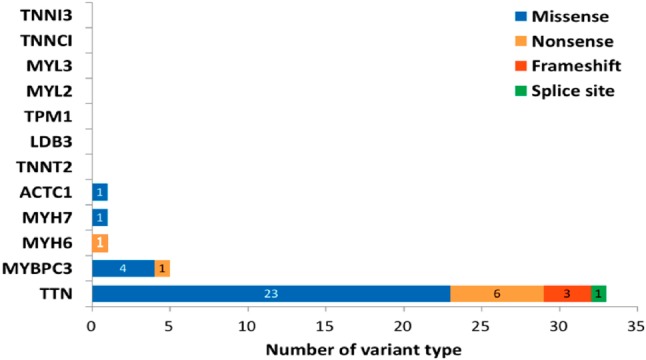
The bar diagram demonstrated the numbers and distribution of genetic variants in twelve disease associated genes. The number of selected variant types was counted and their distribution in associated genes are shown in the graph.
Table 2. List of TTN truncating variants identified in the study group.
| Patient ID | Mutation type | AA change (Q8WZ42-1) | Exon | Domain | Nucleotide change (NM_001256850) | dbSNP | ClinVar | AMA and ACMG guidelines | SIFT | Polyphen2 | Mutation taster | 1000 Genomes ASN | ExAC ASN | Normal control (allele fre, %) | Other mutation |
| Z-disk | |||||||||||||||
| NGS008 | Nonsense | p.Q10X | 2 | I1 Ig | c.C28T | rs1309114959 | Pathogenic | D | - | A | 0 | 0 | 0 | - | |
| NGS036 | Frameshift | p.A475fs | 9 | Z-repeat 2 | c.1425delT | - | Pathogenic | - | - | D | 0 | 0 | 0 | - | |
| NGS067 | Frameshift | p.A475fs | 9 | Z-repeat 2 | c.1425delT | - | Pathogenic | - | D | 0 | 0 | 0 | - | ||
| A-Band | |||||||||||||||
| NGS047 | Frameshift | p.P12945fs | 187 | I86 Ig | c.38834delC | - | Pathogenic | - | - | D | 0 | 0 | 0 | - | |
| NGS009 | Nonsense | p.R14191X | 203 | A2 FN3 | c.C42571T | rs751746401 | Likely pathogenic | Pathogenic | D | - | A | 0 | 0 | 0 | TN: p.V11879I |
| NGS013 | Nonsense | p.R14191X | 203 | A2 FN3 | c.C42571T | rs751746401 | Likely pathogenic | Pathogenic | D | - | A | 0 | 0 | 0 | TTN: p.Y24227H |
| NGS032 | Splice site | - | 204 | Unique sequence | c.42650-2A>G | - | Pathogenic | - | - | D | 0 | 0 | 0 | - | |
| NGS033 | Nonsense | p.R16810X | 236 | I101 Ig | c.C50428T | rs1440093502 | Pathogenic | D | - | A | 0 | 0 | 0 | - | |
| NGS005 | Frameshift | p.P18143fs | 251 | I105 Ig | c.54428_54429del | rs886039027 | Likely pathogenic | Pathogenic | - | - | D | 0 | 0 | 0 | - |
| NGS066 | Nonsense | p.S20300X | 263 | I111 Ig | c.C60899A | - | Pathogenic | D | - | A | 0 | 0 | 0 | TTN: p.R15445H; p.R25922C | |
| NGS069 | Nonsense | p.G23394X | 276 | A70 FN3 | c.G70180T | - | Pathogenic | D | - | A | 0 | 0 | 0 | TTN: p.H29228Y | |
| NGS068 | Nonsense | p.Q23503X | 276 | I119 Ig | c.C70507T | - | Pathogenic | D | - | A | 0 | 0 | 0 | - |
A, Disease_causing_automatic; D,Deleterious/Disease_causing; FN3, fibronectin type III; Ig, immunoglobulin.
Supplementary Table 2. List of TTN missense variants identified in the study group.
| Patient ID | Mutation type | AA change (Q8WZ42-1) | Exon | Domain | Nucleotide change (NM_001256850) | dbSNP | ClinVar | AMA and ACMG recommended guidelines | SIFT | Polyphen2 | Mutation taster | 1000 Genomes ASN | ExAC ASN | Normal control (allele fre, %) | Non-TTN gene mutation |
| I-band | |||||||||||||||
| NGS072 | Missense | p.A9522V | 103 | Unique sequence | c.C28565T | rs578069922 | Uncertain significance | D | D | D | 0.001 | 0 | 0 | ||
| NGS009 | Missense | p.V11879I | 169 | PEVK 27 | c.G35635A | rs587780488 | Likely benign | Uncertain significance | D | D | D | 0 | 0 | 0 | |
| NGS059 | Missense | p.R12167H | 176 | I81 Ig | c.G36500A | - | Uncertain significance | D | D | D | 0 | 0.0002 | 0 | ||
| NGS064 | Missense | p.P13572S | 197 | I92 Ig | c.C40714T | rs867112278 | Uncertain significance | D | P | D | 0 | 0 | 0 | ||
| A-band (D-zone) | |||||||||||||||
| NGS066 | Missense | p.R15445H | 221 | A12 FN3 | c.G46334A | rs72632860 | - | Uncertain significance | D | D | D | 0 | 0.0002 | 0 | |
| NGS014 | Missense | p.V17255M | 241 | A25 FN3 | c.G51763A | rs370629962 | Likely benign; Uncertain significance | Uncertain significance | D | P | D | 0 | 0.0012 | 0 | |
| NGS041 | Missense | p.I17637V | 245 | A27 FN3 | c.A52909G | rs56025724 | - | Uncertain significance | D | D | D | 0 | 0 | 0 | |
| A-band (C-zone) | |||||||||||||||
| NGS031 | Missense | p.F21385S | 274 | A113 Ig | c.T64154C | rs375365023 | - | Uncertain significance | D | P | D | 0 | 0 | 0 | |
| NGS017 | Missense | p.P22706L | 276 | A65 FN3 | c.C68117T | rs762412998 | - | Uncertain significance | D | D | D | 0 | 0 | 0 | |
| NGS011 | Missense | p.N23202D | 276 | A68 FN3 | c.A69604G | rs373527654 | Uncertain significance | Uncertain significance | D | D | D | 0 | 0.0005 | 0 | |
| NGS013 | Missense | p.Y24227H | 276 | A121 Ig | c.T72679C | rs755691336 | - | Uncertain significance | D | D | D | 0 | 0.0007 | 0 | |
| NGS018 | Missense | p.G24255V | 276 | A76 FN3 | c.G72764T | rs766283033 | - | Uncertain significance | D | D | D | 0 | 0.0006 | 0 | MYBPC3: p.P186L; p.Q508X |
| NGS059 | Missense | p.L24309F | 276 | A76 FN3 | c.C72925T | rs376814602 | Uncertain significance | Uncertain significance | D | D | D | 0.003 | 0.0022 | 0 | |
| NGS039 | Missense | p.C25096S | 276 | A82 FN3 | c.T75286A | rs566764105 | Uncertain significance | Uncertain significance | D | P | D | 0.001 | 0.0001 | 0 | |
| NGS066 | Missense | p.R25922C | 276 | A88 FN3 | c.C77764T | rs72648214 | Uncertain significance | Uncertain significance | D | P | D | 0 | 0.0001 | 0 | |
| NGS027 | Missense | p.P26910T | 276 | Unique sequence | c.C80728A | rs142478636 | Likely benign | Uncertain significance | D | P | D | 0 | 0.0001 | 0 | |
| NGS038 | Missense | p.R27734C | 280 | A102 FN3 | c.C83200T | rs368439674 | Uncertain significance | Uncertain significance | D | P | D | 0 | 0.0006 | 0 | |
| NGS064 | Missense | p.S28585P | 285 | A108 FN3 | c.T85753C | rs1334122653 | - | Uncertain significance | D | D | D | 0 | 0 | 0 | |
| NGS069 | Missense | p.H29228Y | 289 | A113 FN3 | c.C87682T | - | - | Uncertain significance | D | P | D | 0 | 0 | 0 | |
| NGS022 | Missense | p.P29444L | 289 | A114 FN3 | c.C88331T | rs549841864 | Likely benign; Uncertain significance | Uncertain significance | D | P | D | 0.001 | 0.0015 | 0 | |
| NGS075 | Missense | p.P29444L | 289 | A114 FN3 | c.C88331T | rs549841864 | Likely benign; Uncertain significance | Uncertain significance | D | P | D | 0.001 | 0.0015 | 0 | |
| NGS020 | Missense | p.T30729N | 298 | A138 Ig | c.C92186A | rs146098114 | - | Uncertain significance | D | D | D | 0 | 0.0007 | 0 | |
| NGS018 | Missense | p.E31056D | 301 | A125 FN3 | c.A93168C | rs748057839 | - | Uncertain significance | D | D | D | 0 | 0.0005 | 0 | MYBPC3: p.P186L; p.Q508X |
| M-band | |||||||||||||||
| NGS011 | Missense | p.K31616T | 305 | Unique sequence | c.A94847C | rs766867347 | - | Uncertain significance | D | P | D | 0 | 0.0008 | 0 |
D, Deleterious/Disease causing; P, probably damaging.
Supplementary Table 3. List of rare variants in ACTC1, MYBPC3, MYH6 and MYH7 genes.
| Patient ID | Gene | Mutation type | AA change | Exon | Domain | Nucleotide change | dbSNP | ClinVar | AMA and ACMG recommended guidelines | SIFT | Polyphen2 | Mutation taster | 1000 Genomes ASN | ExAC ASN | Normal control (allele fre, %) |
| NGS056 | ACTC1 | Missense | p.A274P | 6 | Actin | c.G820C | - | - | Uncertain significance | D | D | D | 0 | 0 | 0 |
| NGS024 | MYBPC3 | Missense | p.R238H | 6 | IgC-1 | c.G713A | rs727504396 | Uncertain significance | Uncertain significance | D | D | D | 0 | 0 | 0.0004 |
| NGS018 | MYBPC3 | Nonsense | p.Q508X | 16 | IgC-3 | c.C1522T | rs730880544 | Pathogenic | Likely pathogenic | T | - | A | 0 | 0 | 0 |
| NGS070 | MYBPC3 | Missense | p.G835E | 24 | IgC-3 | c.2504_2505TT | - | - | Uncertain significance | - | - | D | 0 | 0 | 0 |
| NGS028 | MYBPC3 | Missense | p.R470Q | 15 | IgC-3 | c.G1409A | rs776734314 | Uncertain significance | Uncertain significance | D | D | D | 0 | 0 | 0 |
| NGS018 | MYBPC3 | Missense | p.P186L | 5 | IgC-1 | c.C557T | rs727503216 | Uncertain significance | Uncertain significance | D | P | D | 0 | 0.0003 | 0 |
| NGS021 | MYH6 | Nonsense | p.R1923X | 38 | Myosin tail | c.C5767T | rs765861895 | Uncertain significance | Likely pathogenic | T | - | A | 0 | 0 | 0 |
| NGS015 | MYH7 | Missense | p.P527S | 16 | Myosin motor | c.C1579T | rs1437377039 | - | Uncertain significance | D | P | D | 0 | 0 | 0 |
Truncating variants in the DCM cohort
Among the 70 DCM patients, 10 heterozygous TTN truncating variants in 12 subjects (17.1%, 12/70) were identified. These TTN truncating mutations included 6 nonsense, 3 frameshift mutations, and 1 splice variant. Two variants (p.R14191X and p.A475fs) were identified in familial DCM patients. In segregation analysis, the nonsense variant (p.R14191X) showed concordant segregation among the family members, in whom two affected individuals carried the variant and two unaffected individuals did not carry the variant (Supplementary Figure 3). All of these TTN variants were located in symmetric exons. Eight TTN variants (80%) were localized in the sarcomeric A-band region, and the distribution of these variants is shown in Figure 1. According to the TTN transcript annotations at http://cardiodb.org/titin, none of these 10 TTN truncating variants have been reported before. The other two nonsense variants in MYH6 and MYBPC3, respectively, have also not been reported before.
Supplementary Figure 3.
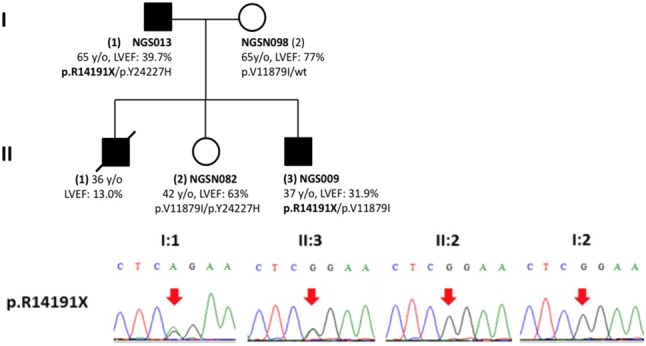
The pedigree of familial dilated cardiomyopathy and their genetic variants in individuals of NGS013, NGS009, NGSN080 and NGSN083. (A) Pedigree of this family. (B) Results of Sanger sequencing of p.R14191X.
Missense variants in the DCM cohort
According to our bioinformatic filtering criteria, 23 rare TTN missense variants in 24 patients were identified (Supplementary Table 2). The distribution of these genetic variants in titin protein is shown in Figure 1. Of them, 18/23 (78.2%) TTN missense variants resided in the C terminal zone of the A-band region, which is organized by numerous repeated fibronectin type III (FN-III) domains and immunoglobulin domains. Other identified missense variants included 4 in MYBPC3, 1 in MYH7 and 1 missense variant in ACTC1, that the distributation of this variants is shown in Supplementary Figure 4. Among these missense variants, familiar segregation analysis for p.P29444L did not show the presentation of a concordant disease phenotype. Two of three carriers who were family members developed the disease phenotype, whereas none of the non-carriers were affected.
Supplementary Figure 4.
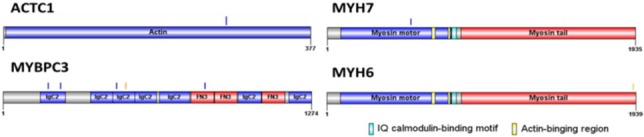
The spatial distribution of variants in ACTC1, MYBPC3, MYH6 and MYH7 proteins. Blue bars indicate the locations of missense variants. Orange color bar indicate the nonsense variant.
Genotype-phenotype analysis
Comparisons of the clinical characteristics of the patients with and without affecting variants are summarized in Table 3 (carriers vs. non-carriers or TTN truncating variants vs. others). There were no significant differences in age at diagnosis, sex, the severity of symptoms, serum B-type natriuretic peptide level, cardiac chamber diameter, and LVEF. There was also no significant difference in the reversibility of left ventricular systolic function after 1-year of standard therapy between the groups. Although patients with these variants tended to have less severe clinical manifestations than those without variants, it did not reach statistical significance. After a median of 44 months clinical follow-up, there were 8 all-cause deaths (5 cardiovascular deaths), 4 non-fatal strokes, 15 ventricular arrhythmias requiring ICD implantation, and 16 re-hospitalizations within 1 year among these DCM patients. There were no significant differences in event rates between the patients with or without these variants, although there were numerically more events in the non-carriers. We defined major adverse cardiac events as all-cause death, non-fatal stroke, and ICD implantation. The Kaplan-Meier survival curve for major adverse cardiac events also did not show a significant difference in the outcomes of patients with and without these variants (log-rank test p = 0.218, Figure 2).
Table 3. Comparison of clinical outcomes in DCM patients with or without truncating variants.
| Characteristic | Carrier | Non-carrier | p value | Truncation | Non-truncation | p value |
| Patients, n | 33 | 37 | 11 | 59 | ||
| Age of onset, y | 53.4 | 52.7 | 0.838 | 52.0 | 53.2 | 0.788 |
| NYHA III or IV, % | 6 (20.0%) | 9 (30.6%) | 0.333 | 2 (22.2%) | 15 (20.3%) | |
| LVEDD, mm | 62.9 | 68.6 | 0.015 | 62.4 | 66.6 | 0.18 |
| LA, mm | 44.4 | 47.0 | 0.331 | 46.6 | 45.7 | 0.789 |
| LVEF, % | 31.0 | 27.4 | 0.196 | 33.6 | 28.3 | 0.185 |
| BNP, pg/ml | 812 | 1184 | 0.284 | 1121 | 1002 | 0.859 |
| Reversibility, % | 5 (15.2%) | 9 (24.3%) | 0.342 | 1 (9.1%) | 13 (22.0%) | 0.328 |
| All mortality, % | 4 (12.1%) | 4 (10.8%) | 0.361 | 1 (9.1%) | 7 (11.9%) | 0.792 |
| Cardiac mortality, % | 2 (6.1%) | 3 (8.1%) | 0.742 | 1 (9.1%) | 4 (6.8%) | 0.947 |
| Stroke, % | 1 (3.0%) | 3 (8.1%) | 0.361 | 0 (0) | 4 (6.8%) | 0.377 |
| ICD implant, % | 6 (18.2%) | 9 (24.3%) | 0.535 | 3 (27.3%) | 12 (20.3%) | 0.609 |
| HF hospitalization, % | 5 (15.2%) | 11 (29.7%) | 0.150 | 1 (9.1%) | 15 (25.4%) | 0.240 |
BNP, B-type natriuretic peptide; DCM, dilated cardiomyopathy; HF, heart failure; ICD, implantable cardioverter-defibrillator; LA, Left atrium; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; NYHA, New York Heart Failure Association functional class.
Figure 2.
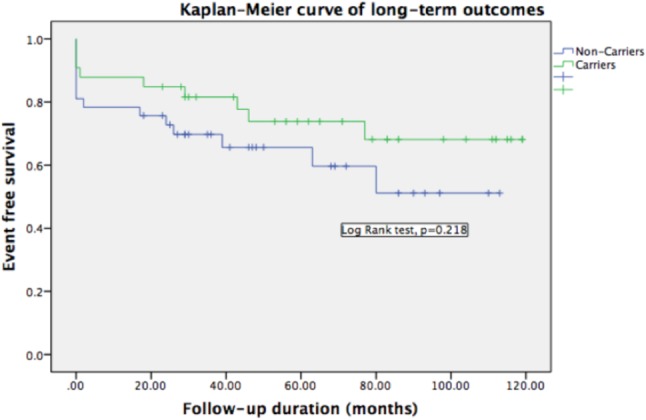
Kaplan-Meier event-free survival curves for death, non-fatal stroke, ventricular arrhythmia requiring ICD implant in variant carriers and non-carriers. During a median follow up of 44 months, patients carrying variants had less adverse events although it did not reach statistical significance for limited patient numbers. ICD, implantable cardioverter-defibrillator.
DISCUSSION
In this study, we used high coverage NGS sequencing and stringent bioinformatic filtering strategies and identified 12 truncating and 29 missense variants in 12 common disease-associated genes, considered to probably be affecting-function variants in a Chinese population. TTN remained the major pathogenic gene in DCM, and approximately 80% of the identified variants in the study were located in TTN. The prevalence of TTN truncating variants in our cohort was 17.1%, which is consistent with previous reports.15-17,27,28 Based on the database of TTN transcript annotations (http://cadiodb.org/titin), in which most of these variants have been reported in cohort studies from North American and European countries, none of the 10 TTN truncating variants identified in the study have been reported before. To date, more than 30 genes and 400 variants have been associated with DCM, however the pathogenicity of most genetic variants is still undetermined.29 Truncating variants of TTN were the most commonly identified variants in this study, and carried the greatest risk of DCM, especially the variants located at the A-band. Most previous studies have also focused on the genetic variants of TTN in DCM, however comparisons of the distribution of variants in DCM-associated genes among ethnic groups has not been reported before. In a review of previous publications, two novel TTN variants (1 nonsense p.R4109X and 1 missense mutation p.G5919R) were reported in two distinct Chinese DCM families,30 however neither were present in our cohort. Furthermore, probably affecting-function variants in 3 candidate genes, tropomyosin (TPM1), cardiac troponin T type-2 (TNNT2), and nuclear lamina protein A/C (LMNA), have been reported in Chinese patients.31-33 However, none of the aforementioned variants in the 3 genes were identified in our DCM cohort. To the best of our knowledge, this is the first panel-based, comprehensive NGS and bioinformatic filtering investigation for DCM genetic variants in a Chinese population. We assumed that disease-associated genetic variants in a Chinese population should be distinct from those in Caucasian and other ethnic groups. With accumulating genetic data in Chinese populations, our study results could be a reference for clinical genetic tests in the future.
Beyond differences in sequencing and bioinformatic filtering methods among studies, we hypothesized that there would be actual racial differences in the complex DCM genomics. This would indicate that certain interactions between environmental and genetic variants play an important role in the pathogenesis and clinical phenotype of DCM. With advances in genomics, several overlapping or crossover genetic variants have been identified in DCM and other inherent cardiomyopathies,34 such as hypertrophy cardiomyopathy,35 long QT syndrome,36 arrhythmogenic right ventricular dysplasia,37 and left ventricular noncompaction.38 Furthermore, a shared common genetic background has also been observed in DCM and peripartum cardiomyopathy.17,39 Currently, information about the mechanisms between genetic mutations and presenting phenotypes is very limited. Moreover, these genetic mutations may behave as pure disease modifiers and clinical phenotypes resulting from a series of maladaptive remolding and interactions with genetic, epigenetic, and environmental factors.
Despite advances in heart failure therapies, DCM is still a leading cause of heart transplantation. Numerous clinical markers have been identified for the early prediction of advanced heart failure with a poor prognostic.40,41 A genetic test is expected to improve the clinical outcomes of probands and their relatives by risk stratification, preventative measures and early treatment. For example, individuals with LMNA mutations invariably have conduction system disorders, atrial or ventricular arrhythmia, and severe ventricular dysfunction at a young age which lowers the threshold for device implantation and anti-arrhythmic treatment. However, the prognostic value of other genetic information in DCM is controversial. In an unselected DCM cohort, patients with TTN truncating variants had more severely impaired left ventricle (LV) function, more sustained ventricular tachycardia, faster disease progression, and earlier events of death, cardiac transplant or left ventricular assist devices.19 The presence of truncating TTN variants was considered to be an indicator of lower LVEF at 1 year follow-up.39 Patients with rare variants in MYH6, MYH7, MYBPC3 and TNNT2 genes have also been reported to have a worse prognosis than noncarriers.42 In contrast, other studies have shown no significant difference in clinical manifestations and outcomes among patients with or without TTN truncating mutations.15,18,27 Two single center cohort studies in Canada and Singapore concluded that patients with or without TTN truncating variants had a similar response to medical therapy14 or left ventricular assist device support.43 In this study, there were no significant differences in clinical manifestations and long-term adverse events between the patients with or without these variants. Although the number of patients in this study may be too small to identify differences between them, a recent, large, well-designed prospective cohort study in London also found no association among titin truncation, cardiac phenotype and outcomes in DCM.44
The molecular mechanisms for the pathogenesis of titin-truncating variants are unclear. About 1-2% of the general population has titin-truncating variants in the absence of DCM. Recently, eccentric remodeling was detected by high-resolution 2D and 3D cardiac magnetic resonance imaging in heathy individuals with titin-truncating variants. Moreover, another study showed that cardiac physiology and performance of rats with titin-truncating variants at baseline were also unremarkable, but became impaired during cardiac stress.45 In metabolic and signaling analyses, rat hearts harboring titin-truncating variants had a shift in metabolism from fatty acids toward glycolysis and elevated mTORC1 signaling at baseline. These changes were commonly seen in failing or pressure-loaded hearts.45 We suggest that individuals with titin-truncating variants may be in a compensated state at baseline, and only develop disease upon an environmental stimulus such as alcohol, pregnancy or chemotherapy. Thus, titin-truncating variant-associated molecular, physiologic, functional and geometric changes may precipitate future cardiac events compared to normal controls. Individuals with titin-truncating variants have a higher risk of disease development than those without variants, however the disease manifestations are similar to heart failure of other etiologies. Another possible explanation is position-dependent effects and penetrance of titin-truncating variants. In observational analysis, distal I-band and A-band of titin-truncating variants were found to be associated with the highest incidence of DCM. Simplified comparisons between all titin-truncating variants with others may attenuate the effects of true pathologic variants in disease manifestations. Further large-scale population studies are needed to elucidate the associations between clinical manifestations and genotypes. Our results provide valuable information for the development of risk stratification tools and disease management strategies in clinical practice.
NGS technologies provide a quicker, cost-effective and comprehensive tool for analyzing genetically heterogeneous disorders. However, the greatest obstacle is to differentiate whether variants are affecting-function or innocent variants. For example, few individuals without overt cardiomyopathy also have titin-truncating variants.46,47 In this study, we used high-coverage NGS sequencing, a stringent bioinformatics approach and accurate variant confirmation to identify relevant pathogenic mutations. Any variants present in the in-house genome database of 299 heathy individuals were excluded from analysis. Results of integrated analysis in more than 5000 individuals have shown that TTN truncating mutations within sarcomeric A band are likely to be pathogenic.19 The A-band region, which provides repetitive binding sites for myosin-associated proteins, is critical for biomechanical sensing and signaling. The TTN truncating mutations (8/10, 80%) identified in this study were non-uniformly distributed in the sarcomeric A band region, which is consistent with previous conclusions. On account of the small number of families and limited availability of DNA samples from affected and unaffected members, we could not validate the pathogenicity of all identified variants in co-segregation analysis. Of the truncating variants, nonsense mutation p.R14191X was present in affected members but not in unaffected members, which increases the pathogenic evidence of this variant.
With regards to missense variants, the relevance in cardiomyopathy is still uncertain. In the Exome Sequencing Project cohort, 23 TTN missense variants per individual on average were found.16 According to another DCM cohort study, more than a half of TTN missense variants, identified by stringent filtering criteria, demonstrated incompatible segregation with clinical phenotypes in their families.48 In the current study, 29 rare missense variants were identified. From available DNA samples in distinct families, 3 variants, p.V11879I, p.P29444L and p.Y24227H revealed discordant segregation with phenotype, and missense variants were also present in the unaffected relatives. However, age-dependent penetrance is commonly observed in genetic DCM, in which a disease-causing mutation manifests an overt phenotype later in life, commonly in the fourth to sixth decades. In the study by Akinrinade et al.,27 the disease penetrance increased with age from 53.8% at 50 years to 84.6% at 60 years and 100% at 70 years. The phenotype could not be fully assessed in the individuals aged < 60 years. Thus, we cannot conclude that these 3 missense variants were not relevant, because the majority of relatives in co-segregation analysis were < 50 years. These genetic variant carriers may still have an increased risk of developing cardiac dysfunction under superimposed stress, such as increased hemodynamic demands during pregnancy or cardiotoxic chemotherapy treatment or vulnerable to viral myocarditis.
CONCLUSIONS
Our study results from a comprehensive target exon sequencing in DCM patients and healthy controls provide a reliable reference for genetic testing of DCM in a Chinese population. Several large families available for co-segregation analyses are needed to increase the strength of evidence in the pathogenicity of these variants. Further functional investigations on these variants may provide a better understanding and new insights into the pathogenesis of DCM.
Limitations
Only 12 pre-specified DCM-associated genes were sequenced in this study. It is possible that some other rare genetic variants also occurred in other genes in our DCM cohort. Because we used inclusive criteria for DCM patient enrollment, there was probably selection bias for the patients with more advanced disease and fewer concomitant systemic diseases. Even with comprehensive family history acquisition, not all genetic materials from the relatives of probands were available for co-segregation analysis. In addition, the limited number of patients made the interpretation of the study results less reliable and confident. Further assessments of mRNA transcripts and protein levels in subjects with these rare variants may help to estimate the probability of the pathogenicity of these genetic variants.
Acknowledgments
J.K.Y. received support from Chang Gung Memorial Hospital (CMRPG3I0061 and CORPG3H0291). C.Y.W. received support from the National Health Research Institute (NHRI-EX106-10617SI), National Science Council (105-2628-B-182-009-MY4), and Chang Gung Memorial Hospital (CMRPG3H0131 and CMRPG3H0271). M.S.W. received support from Chang Gung Memorial Hospital (CMRPG3E1852-3, CMRPG3E1892-3, and CMRPG3C 1391-3).
CONFLICT OF INTEREST
All the authors declare no conflict of interest.
REFERENCES
- 1.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 2.Taylor MRG, Carniel E, Mestroni L. Cardiomyopathy,familial dilated. Orphanet J Rare Dis. 2006;1:27. doi: 10.1186/1750-1172-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codd MB, Sugrue DD, Gersh BJ, Melton LJ., 3rd Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 4.Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010; 375:752–762. doi: 10.1016/S0140-6736(09)62023-7. [DOI] [PubMed] [Google Scholar]
- 5.Chang HY, Wang CC, Wu YW, et al. One-year outcomes of acute decompensated systolic heart failure in Taiwan: lessons from TSOC-HFrEF Registry. Acta Cardiol Sin. 2017;33:127–138. doi: 10.6515/ACS20170202A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin YS, Liu PH, Chu PH. Obstructive sleep apnea independently increases the incidence of heart failure and major adverse cardiac events: a retrospective population-based follow-up study. Acta Cardiol Sin. 2017;33:656–663. doi: 10.6515/ACS20170825A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 9.Meder B, Haas J, Keller A, et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011;4:110–122. doi: 10.1161/CIRCGENETICS.110.958322. [DOI] [PubMed] [Google Scholar]
- 10.Gerull B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can J Cardiol. 2015;31:1351–1359. doi: 10.1016/j.cjca.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90:49–54. doi: 10.1111/cge.12741. [DOI] [PubMed] [Google Scholar]
- 12.McMurray JJJV. Improving outcomes in heart failure: a personal perspective. Eur Heart J. 2015;36:3467–3470. doi: 10.1093/eurheartj/ehv565. [DOI] [PubMed] [Google Scholar]
- 13.Fatkin D, Johnson R, McGaughran J, et al. Position statement on the diagnosis and management of familial dilated cardiomyopathy. Hear Lung Circ. 2017:1–6. doi: 10.1016/j.hlc.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Luk K, Bakhsh A, Giannetti N, et al. Recovery in patients with dilated cardiomyopathy with loss-of-function mutations in the titin gene. JAMA Cardiol. 2017;2:700. doi: 10.1001/jamacardio.2017.0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norton N, Li D, Rampersaud E, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J. 2014;35:2165–2173. doi: 10.1093/eurheartj/ehu050. [DOI] [PubMed] [Google Scholar]
- 18.Franaszczyk M, Chmielewski P, Truszkowska G, et al. Titin truncating variants in dilated cardiomyopathy - prevalence and genotype-phenotype correlations. PLoS One. 2017;12:1–14. doi: 10.1371/journal.pone.0169007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knöll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 21.Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114:1052–1068. doi: 10.1161/CIRCRESAHA.114.301286. [DOI] [PubMed] [Google Scholar]
- 22.Jansweijer JA, Nieuwhof K, Russo F, et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2017;19:512–521. doi: 10.1002/ejhf.673. [DOI] [PubMed] [Google Scholar]
- 23.Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norton N, Li D, Hershberger RE. Next-generation sequencing to identify genetic causes of cardiomyopathies. Curr Opin Cardiol. 2012;27:214–220. doi: 10.1097/HCO.0b013e328352207e. [DOI] [PubMed] [Google Scholar]
- 26.McVean GA, Altshuler DM, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akinrinade O, Ollila L, Vattulainen S, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36:2327–2337. doi: 10.1093/eurheartj/ehv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135. doi: 10.1093/eurheartj/ehu301. [DOI] [PubMed] [Google Scholar]
- 29.Olivotto I, D’amati G, Basso C, et al. Defining phenotypes and disease progression in sarcomeric cardiomyopathies: contemporary role of clinical investigations. Cardiovasc Res. 2015:409–423. doi: 10.1093/cvr/cvv024. [DOI] [PubMed] [Google Scholar]
- 30.Liu JS, Fan LL, Zhang H, et al. Whole-exome sequencing identifies two novel TTN mutations in Chinese families with dilated cardiomyopathy. Cardiology. 2016;136:10–14. doi: 10.1159/000447422. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Feng Y, Zhang YM, et al. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med. 2015;2015:1–12. doi: 10.3892/ijmm.2015.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li YD, Ji YT, Zhou XH, et al. Significance of sarcomere gene mutation in patients with dilated cardiomyopathy. Genet Mol Res. 2015;2:11200–11210. doi: 10.4238/2015.September.22.14. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Luo R, Gu H, et al. Cardiac troponin T (TNNT2) mutations in Chinese dilated cardiomyopathy patients. Biomed Res Int. 2014;2014:907360. doi: 10.1155/2014/907360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNally EM, Puckelwartz MJ. Genetic variation in cardiomyopathy and cardiovascular disorders. Circ J. 2015;79:1409–1415. doi: 10.1253/circj.CJ-15-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hershberger RE, Cowan J, Morales A, et al. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2:253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Hear Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 37.Elliott P, O’Mahony C, Syrris P, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:314–322. doi: 10.1161/CIRCGENETICS.110.937805. [DOI] [PubMed] [Google Scholar]
- 38.Klaassen S, Probst S, Oechslin E, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. doi: 10.1161/CIRCULATIONAHA.107.746164. [DOI] [PubMed] [Google Scholar]
- 39.Ware JS, Li J, Mazaika E, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374:233–241. doi: 10.1056/NEJMoa1505517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu HP, Jen HL, Yin WH, et al. Circulating adiponectin levels following treatment can predict late clinical outcomes in chronic heart failure. Acta Cardiol Sin. 2017;33:139–149. doi: 10.6515/ACS20160427B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chi PC, Kuo JY, Chen CY, et al. Changes of atrial natriuretic peptides after defibrillation threshold testing predicted future ventricular arrhythmia event. Acta Cardiol Sin. 2017;33:401–409. doi: 10.6515/ACS20161212A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merlo M, Sinagra G, Carniel E, et al. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci. 2013;6:424–428. doi: 10.1111/cts.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Felkin LE, Walsh R, Ware JS, et al. Recovery of cardiac function in cardiomyopathy caused by titin truncation. JAMA Cardiol. 2016;1:234. doi: 10.1001/jamacardio.2016.0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tayal U, Newsome S, Buchan R, et al. Phenotype and clinical outcomes of titin cardiomyopathy. J Am Coll Cardiol. 2017;70:2264–2274. doi: 10.1016/j.jacc.2017.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53. doi: 10.1038/ng.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akinrinade O, Koskenvuo JW, Alastalo TP. Prevalence of titin truncating variants in general population. PLoS One. 2015;10:1–14. doi: 10.1371/journal.pone.0145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Golbus JR, Puckelwartz MJ, Fahrenbach JP, et al. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Begay RL, Graw S, Sinagra G, et al. Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc. 2015;4:e002645. doi: 10.1161/JAHA.115.002645. [DOI] [PMC free article] [PubMed] [Google Scholar]