

Abstract
5-methylcytosine (m5C) is one of the most common and abundant post-transcriptional modifications (PTCMs) in RNA. Recent studies showed that m5C plays important roles in many biological functions such as RNA metabolism and cell fate decision. Because most experimental methods that determine m5C sites across the transcriptome are time-consuming and expensive, it is urgent to develop accurate computational methods to identify m5C sites effectively. A benchmark dataset is important for developing and evaluating computational methods. In this work, we constructed four different datasets according to the data redundancy and imbalance. Based on these datasets, we generated three different kinds of features, i.e., KNFs (K-nucleotide frequencies), KSNPFs (K-spaced nucleotide pair frequencies), and pseDNC (pseudo-dinucleotide composition), and then used a support vector machine (SVM) to build our models. Based on the imbalanced and nonredundant dataset, Met935, we extensively studied the three kinds of features and determined an optimal combination of the features. Based on the feature combination, we built models on the three different datasets and compared them with state-of-the-art models. According to the predictive results of the stringent jackknife test, the models based on the three features, 4NF, 1SNPF, and pseDNC, are superior or comparable to other methods. To determine the best model between the models based on the imbalanced dataset Met935 and the balanced dataset Met240, we further evaluated the two models on an independent test set Test1157. Our results demonstrate that the model based on the balanced dataset Met240 achieved the highest recall (68.79%) and the highest Matthews correlation coefficient (MCC) (0.154). In addition, the model is also superior to other state-of-the-art methods according to the integrated parameter MCC on the independent test set. Thus, we selected the model based on Met240 as our final model, which was named RNAm5CPred. In addition, a web server for RNAm5CPred (http://zhulab.ahu.edu.cn/RNAm5CPred/) has been provided to facilitate experimental research.
Keywords: 5-methylcytosine site, post-transcriptional modification, support vector machine, nucleotide composition, prediction
Introduction
5-methylcytosine (m5C) is a highly abundant post-transcriptional modification (PTCM) of RNA, which has been discovered in various organisms.1 Under the catalysis of RNA methyltransferase, m5C occurs on carbon atoms in the fifth position of cytosine. m5C has been widely studied because of its crucial role in many different biological processes, such as secondary structure stabilization of tRNA, aminoacylation and codon identification, and stress response regulation, among others.2, 3, 4, 5, 6, 7 Therefore, it is of great importance to develop efficient methods to locate m5C sites in RNA sequences for understanding its mechanism and function.
Several experimental techniques such as bisulfite sequencing, m5C-RNA immunoprecipitation (m5C-RIP), 5-azacytidine-mediated RNA immunoprecipitation (Aza-IP), and N-methyladenosine (m6A) individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) have been developed to identify m5C sites.4,8, 9, 10, 11 However, these techniques are time-consuming and expensive. Furthermore, the explosive increase of RNA sequences revealed by the rapid development of sequencing technology will require faster and more cost-efficient analyzing methods.
Thanks to their high speed and low cost, accurate computational methods provide an alternative way to identify m5C sites in RNA sequences. To the best of our knowledge, at least three research groups have made their efforts to predict m5C sites of RNA by the submission time of our paper.12, 13, 14 Feng et al.12 developed a model to predict m5C sites of RNA in Homo sapiens. The model was built based on a balanced dataset with 120 positive and 120 negative examples. Support vector machine (SVM) was employed as the classifier, and the pseudo-dinucleotide composition (PseDNC) that incorporates three RNA physicochemical properties was used as the feature to encode the RNA sequences. Another model, iRNAm5C-PseDNC, proposed by Qiu et al.,13 was built based on an imbalanced dataset with 475 positive and 1,425 negative examples. This model also used SVM as the classifier but with a modified PseDNC with more properties to encode the RNA sequences. More recently, Zhang et al.14 proposed a method called M5C-HPCR (m5C-heuristic nucleotide physicochemical property reduction). In this method, a heuristic algorithm was introduced to select a part of PseDNC features, and then the model was built by an ensemble method. M5C-HPCR was validated on both the balanced and imbalanced datasets used by Feng et al. and Qiu et al., respectively.
Although the three methods mentioned above have achieved promising predictive results for m5C sites identification, the benchmark datasets used to validate these methods contain crucial flaws. The imbalanced dataset used by Qiu et al.,13 which we denoted as Met1900, is severely redundant, so that the generalization will be overestimated based on the dataset.14 The balanced dataset used by Feng et al.,12 which we denoted as Met240, is obtained by using a culling cutoff of 70% in CD-HIT.15 However, realistically, the number of m5C sites is substantially less than the number of normal cytosine residues in RNA, which indicated that using a balanced dataset would overestimate the sensitivity.
In addition, all three of these methods used only PseDNC to encode the RNA sequences. Other types of features that have been used to predict other kinds of modifications such as m6A,16,17 including K-nucleotide frequencies (KNFs)16 and K-spaced nucleotide pair frequencies (KSNPFs),16 have not been extensively tested and evaluated in predicting m5C.
In this work, we first constructed a new benchmark dataset Met935. Then, we encoded RNA segments by using KNF, KSNPF, and PseDNC features, and the performances of these features were extensively tested and compared. By combining these three different kinds of features, we were able to build models on three different benchmark datasets, i.e., Met1900, Met240, and Met935. The cross-validation results of the models based on the three features are superior or comparable to the existing methods. The two models based on Met240 and Met935 were further tested on an independent dataset Test1157, the results of which indicate that the model based on Met240 outperforms the other methods according to the Matthews correlation coefficient (MCC).
Results and Discussion
Extensive Study of the Two Kinds of Sequence Features on Met935
Dataset Met935
To overcome the shortcomings of the datasets (i.e., Met240 and Met1900) proposed in previous works,12, 13, 14 a new benchmark dataset, Met935, was constructed in the present study. First, all RNA segments with a center m5C site recorded in RMBase7 were collected as positive samples. Second, the 1,425 negative samples of Met1900 were collected as negative samples. Finally, the redundancy among those samples was removed by using the CD-HIT15 program. The resulting 127 positive segments and 808 negative segments constitute the dataset Met935. The details about the benchmark datasets can be found in the Materials and Methods section.
Performances of KNF with Different Ks
As a classical sequence-encoding feature, KNF (sometimes also called NC), has been extensively used to build bioinformatics models.16, 17, 18, 19, 20 In this study, the performances of KNF in predicting m5C of RNAs were evaluated with different K values on the dataset Met935. Met935 is a nonredundant dataset consisting of 127 positive samples and 808 negative samples.
The value of K was set from 1 to 4 to encode RNA segments of Met935, respectively, to avoid the “dimension disaster.” Then, the 10-fold cross-validation test was performed to evaluate the performance of each kind of KNF. For Met935 as an imbalanced dataset, we used MCC as a fair index to compare different models.
As shown in Table 1, with the increasing of K value, the values of MCC are also increasing overall. More specifically, the models based on 3NF and 4NF both achieved the highest MCC of 0.696. Then, strict jackknife tests were employed to compare the performances between 3NF and 4NF in a more cautious way. Table 2 shows that the cross-validation MCC (0.694) based on 4NF is slightly higher than the MCC (0.688) based on 3NF.
Table 1.
Prediction Results of KNF with Different K Values on Met935 over 10-Fold Cross Validation
| KNF | Feature Dimension | Sen (%) | Spe (%) | Pre (%) | Acc (%) | MCC |
|---|---|---|---|---|---|---|
| 1NF | 4 | 20.55 | 87.15 | 20.09 | 78.11 | 0.076 |
| 2NF | 16 | 51.26 | 98.39 | 83.4 | 91.99 | 0.615 |
| 3NF | 64 | 66.14 | 97.59 | 81.19 | 93.32 | 0.696 |
| 4NF | 256 | 63.07 | 98.19 | 84.61 | 93.42 | 0.696 |
Sen, sensitivity; Spe, specificity; Pre, precision; Acc, accuracy.
Table 2.
Prediction Results of 3NF and 4NF on Met935 over Jackknife Test
| KNF | Feature Dimension | Sen (%) | Spe (%) | Pre (%) | Acc (%) | MCC |
|---|---|---|---|---|---|---|
| 3NF | 64 | 65.35 | 97.52 | 80.58 | 93.16 | 0.688 |
| 4NF | 256 | 63.78 | 98.02 | 83.51 | 93.37 | 0.694 |
Sen, sensitivity; Spe, specificity; Pre, precision; Acc, accuracy.
To conclude, our experimental results showed that the performance of KNF improved with an increasing K value. However, the difference between 3NF and 4NF is small.
Performances of KSNPF with Different Ks
KSNPF provided another way to encode the nucleotide composition of RNA segments, and it has been used to build models to predict the m6A sites of RNA.16,21 In this study, we also evaluated the performances of KSNPF in predicting m5C of RNAs with different K values. Table 3 lists the corresponding prediction results. We found that the results based on different K values did not differ much, among which the best results came from 1SNPF, for which the MCC is 0.300.
Table 3.
Prediction Results of KSNPF with Different K Values on Met935 over 10-Fold Cross-Validation
| KSNPF | Dimension | Sen (%) | Spe (%) | Pre (%) | Acc (%) | MCC |
|---|---|---|---|---|---|---|
| 1SNPF | 16 | 20.79 | 97.80 | 59.75 | 87.34 | 0.300 |
| 2SNPF | 16 | 11.50 | 98.84 | 61.24 | 86.97 | 0.224 |
| 3SNPF | 16 | 19.37 | 96.89 | 49.51 | 86.36 | 0.248 |
| 4SNPF | 16 | 23.78 | 95.68 | 46.47 | 85.91 | 0.262 |
| 5SNPF | 16 | 21.26 | 92.44 | 30.59 | 82.77 | 0.160 |
Sen, sensitivity; Spe, specificity; Pre, precision; Acc, accuracy.
The performances of PseDNC for predicting m5C of RNAs have been evaluated in several works.12, 13, 14 Because we intended to compare our models with these methods, in this study, we did not test the performances of different forms of PseDNC. According to the results shown in Tables 1 and 2, we found that the performance of KNF is generally better than KSNPF, especially when K is set to 3 or 4.
The Optimal Feature Combination Based on Met935
Considering the complementarity between different kinds of features, we combined the best KNF (4NF) and the best KSNPF (1SNPF) features with the PseDNC feature. Table 4 summarizes the performances of different combinations of these three different features on Met935 over the jackknife tests.
Table 4.
Prediction Performances of Different Feature Combinations on Met935 over Jackknife Test
| Feature | Sen (%) | Spe (%) | Pre (%) | Acc (%) | MCC |
|---|---|---|---|---|---|
| 1SNPF | 20.47 | 97.90 | 60.47 | 87.38 | 0.300 |
| PseDNC | 48.82 | 98.64 | 84.93 | 91.87 | 0.606 |
| 4NF | 63.78 | 98.02 | 83.51 | 93.37 | 0.694 |
| 1SNPF + pseDNC | 52.76 | 98.76 | 87.01 | 92.51 | 0.642 |
| 1SNPF + 4NF | 64.57 | 99.13 | 92.13 | 94.44 | 0.744 |
| pseDNC + 4NF | 62.20 | 98.27 | 84.95 | 93.37 | 0.692 |
| 1SNPF + pseDNC + 4NF | 62.99 | 99.50 | 95.24 | 94.55 | 0.749 |
Sen, sensitivity; Spe, specificity; Pre, precision; Acc, accuracy.
As shown in Table 4, for single feature-based models, the two features, 4NF and PseDNC, performed significantly better than 1SNPF. Then, the performances of binary feature combinations were also evaluated. Table 4 shows that the two combinations including 1SNPF exhibit improved performances compared with single features, which proved the complementarity between different features. Finally, the triple feature combination 1SNPF + pseDNC + 4NF showed the highest MCC (0.749) compared with the values of other combinations.
In addition, we plotted the receiver operating characteristic (ROC) curves to provide an intuitive illustration of the performances of different feature combinations. The area under the ROC curve (AUC) is another useful index to evaluate different methods. Being within the range of 0 and 1, the larger the AUC is, the better the model will be. As shown in Figure 1, The AUCs of models based on 1SNPF + pseDNC + 4NF, pseDNC + 4NF, and 1SNPF + 4NF are 0.935, 0.936, and 0.937, respectively, on Met935. All of these values are close to 1, which indicates that all feature combinations have good predictability. Finally, the feature combination of 1SNPF + pseDNC + 4NF was selected for building our model considering both MCC and AUC values.
Figure 1.
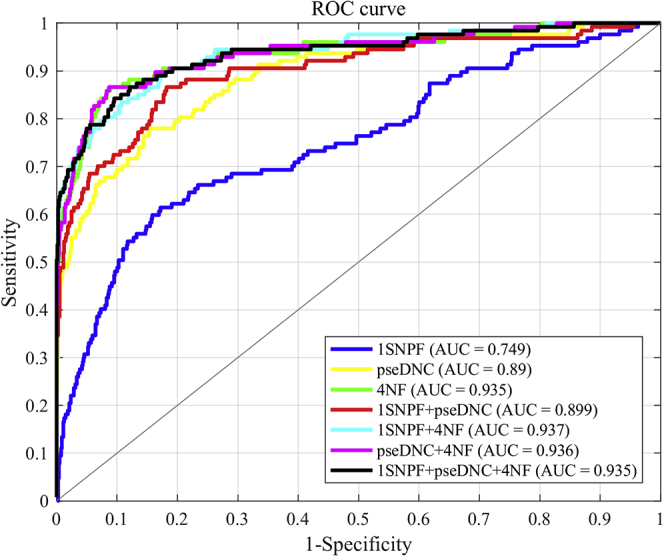
The ROC Curves for Different Feature Combinations on Met935 Over Jackknife Test
Comparisons with Existing Predictors on Different Datasets
In this section, we compare our models with other existing methods for m5C site prediction, including M5C-PseDNC,12 iRNAm5C-PseDNC,13 M5C-HPCS, and M5C-HPCR.14 Note that our models based on different benchmark datasets were all built using the selected feature combination 1SNPF + pseDNC + 4NF to ensure fair comparison with other methods.
Comparison on Met240
First, we compared different m5C site predictive models on the Met240 dataset. Table 5 summarizes the results based on the jackknife test. We found that the results of our model are on par with the best performing model, M5C-HPCR. MCC and AUC of M5C-HPCR are 0.859 and 0.962, respectively, and the corresponding values of our model are 0.850 and 0.957.
Table 5.
Comparison between M5C-PseDNC, iRNAm5C-PseDNC, M5C-HPCR, M5C-HPCS, and Our Model on Met240 Dataset over Jackknife Test
| Predictor | Sen (%) | Spe (%) | Acc (%) | MCC | AUC |
|---|---|---|---|---|---|
| M5C-PseDNCa | 85.00 | 95.83 | 90.42 | 0.810 | 0.950 |
| iRNAm5C-PseDNCa | 81.70 | 95.00 | 88.33 | 0.774 | 0.934 |
| M5C-HPCSa | 90.83 | 92.50 | 91.67 | 0.833 | 0.956 |
| M5C-HPCR | 90.83 | 95.00 | 92.92 | 0.859 | 0.962 |
| Our model | 90.83 | 94.17 | 92.50 | 0.850 | 0.957 |
Sen, sensitivity; Spe, specificity; Acc, accuracy.
Results excerpted from Zhang et al.14
Comparison on Met1900
Then, we compared different models on dataset Met1900. As shown in Table 6, the values of MCC and AUC of our model are the highest among these five predictive models. The sensitivity, specificity, accuracy, MCC, and AUC of our model are 91.58%, 99.51%, 97.53%, 0.934, and 0.991, respectively. The value of AUC is very close to 1, and all other values are greater than 90% or 0.9, which demonstrated that our model is superior to other methods in m5C site prediction on this dataset.
Table 6.
Comparison Between M5C-PseDNC, iRNAm5C-PseDNC, M5C-HPCR, M5C-HPCS, and Our Model on Met1900 Dataset over Jackknife Test
| Predictor | Sen (%) | Spe (%) | Acc (%) | MCC | AUC |
|---|---|---|---|---|---|
| M5C-PseDNCa | 84.21 | 94.88 | 92.21 | 0.792 | 0.960 |
| iRNAm5C-PseDNCa | 69.89 | 99.86 | 92.37 | 0.794 | 0.963 |
| M5C-HPCSa | 83.37 | 96.84 | 93.47 | 0.823 | 0.968 |
| M5C-HPCRa | 88.42 | 97.33 | 95.11 | 0.868 | 0.977 |
| Our model | 91.58 | 99.51 | 97.53 | 0.934 | 0.991 |
Sen, sensitivity; Spe, specificity; Acc, accuracy.
Results excerpted from Zhang et al.14
Comparison on the Independent Test Test96
Table 4 shows the performances of our model on the new dataset Met935. Our model achieved an MCC of 0.749 over the jackknife test; however, it is not fair for other methods when we compared the predictive results of our model with the predictive results of other models directly on Met935, as our model was built on the dataset. To have a fair comparison of our model with M5C-HPCR on this dataset, two datasets derived from Met935, Train839 and Test96, were used. Our model was first trained with the dataset Train839 using the three features and then tested with the independent test set Test96. Because the test dataset Test96 was also an independent test set of M5C-HPCR, it will be fair to compare our model with M5C-HPCR on the dataset.
Table 7 and Figure 2 show the predictive performances of M5C-HPCR and our model on the test dataset Test96. All of the metrics, except sensitivity, of our model are superior to those of M5C-HPCR, which means that our model has better generalization performance than does M5C-HPCR. More specifically, the precision of our model is 100%, which will be very helpful for experimental researchers since they can have high confidence that the predicted m5C sites are actual m5C sites.
Table 7.
Prediction Results of M5C-HPCS and Our Model on Test96
| Predictor | Sen (%) | Spe (%) | Pre (%) | Acc (%) | MCC |
|---|---|---|---|---|---|
| M5C-HPCRa | 100.00 | 62.65 | 29.55 | 67.71 | 0.430 |
| Our model | 84.62 | 100.00 | 100.00 | 97.92 | 0.909 |
Sen, sensitivity; Spe, specificity; Pre, precision; Acc, accuracy.
Results obtained by using M5C-HPCS web server14 on Test96.
Figure 2.
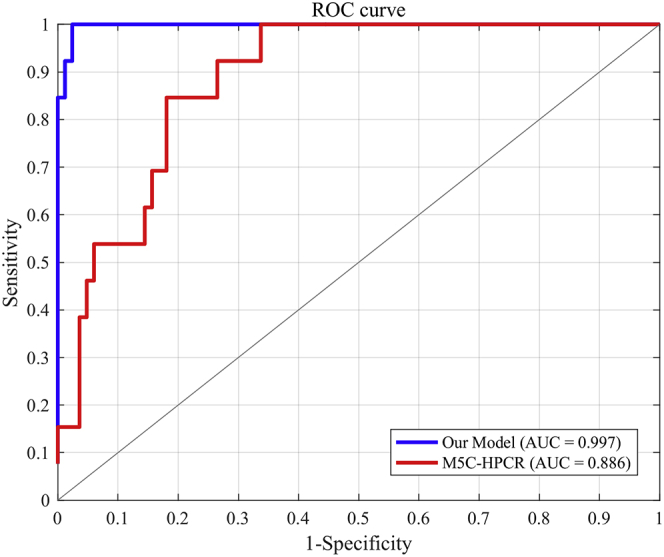
The ROC Curve Shows the Performances of Our model and M5C-HPCR on Test96
Evaluation of Different Models on the Independent Test Test1157
To further compare our models based on the three kinds of nucleotide compositions with other methods, we tested different models on a new independent test set Test1157. The dataset contains 157 high-threshold m5C sites and 1,000 non-m5C sites, which were collected from the GEO database (GEO: GSE90963). All sequences of Test1157 are nonredundant to the sequences in Met935. In Table 8, four models were compared with each other. iRNA-m5C22 is a method that was published during the revision of this manuscript. The results in Table 8 indicate that our model based on Met240 achieves the highest sensitivity (68.79%) and highest MCC (0.154%), and our model based on Met935 achieves the highest specificity (93.00%), highest precision (19.54%), and highest accuracy (81.85%). As Test1157 is an imbalanced dataset, MCC is a fair parameter for comparing different methods, and thus we selected the model based on Met240 as our final model (RNAm5CPred).
Table 8.
Prediction Results of Different Models on Test1157
Effects of the Dataset Redundancy and Imbalance
With the same feature combination, 1SNPF+pseDNC+4NF, a jackknife test has been implemented on the three different datasets: Met240, Met1900, and Met935. The corresponding MCCs are 0.850, 0.934, and 0.749, respectively, and the corresponding AUCs are 0.957, 0.991, and 0.935, respectively. Because Met1900 is a redundant dataset, it is expected that the model based on this dataset would give a better performance. However, its performance is actually overestimated. The performance differences of the models based on Met240 and Met935 may be partially affected by the composition of the datasets. The balanced dataset Met240 achieved sensitivity and specificity of 90.83% and 94.17%, respectively, while the imbalanced dataset Met935 achieved sensitivity and specificity of 62.99% and 99.50%, respectively. To further evaluate the effects of the dataset imbalance on the generalization, the two models built on Met240 and Met935 with the same feature combination 1SNPF+pseDNC+4NF were tested on the independent dataset Test96 (note that the model for Met935 was first trained on dataset Train839). For the model M5C-HPCR, which was based on Met240, the sensitivity and the specificity evaluated with Test96 are 92.31% and 56.63%, which indicated that the generalization for negative examples was not good, especially when compared with the sensitivity and specificity of 90.83% and 94.17% achieved by the cross-validation test. One possible reason is that the distribution of the negative examples in Met240 deviates from the distribution of all negative examples. On the contrary, the sensitivity and the specificity of the model based on Met935 are 84.62% and 100%, indicating that the generalization for the negative examples is good. Further evaluation of different methods on a new independent test set Test1157 shows the similar phenomenon.
Web Implementation
To facilitate the access of our model for the vast majority of experimental researchers, a web server has been established online at http://zhulab.ahu.edu.cn/RNAm5CPred/. Note that the final online RNAm5CPred predictive model was trained on Met240, which contains 240 RNA segments with a length of 41. The instructions for using the RNAm5CPred method online for the prediction of m5C sites are discussed below.
First, the user can submit a query RNA sequence in FASTA format, which should be longer than 41 bp. After the submission, the user only needs to wait for the result, and the predictive model will complete all of the prediction works. The first thing that the predictive model does is to find the cytosine in the query sequence. As mentioned earlier, we need a cytosine-centric RNA fragment. If a cytosine is not in the center of the query sequence, a corresponding length of the cytosine-centric RNA segment will be constructed by placing a sliding window centered at the cytosine, and the missing nucleotides will be filled by the previously proposed “mirror image” technique.19 There may be many cytosines in a sequence, and our predictive model will reconstruct the sequence separately for each of them. Then, based on these reconstructed 41-bp RNA segments, the feature vector will be extracted and fed to the SVM classification engine to complete the prediction. Finally, the user will get the prediction results for every cytosine in the query sequence that was submitted.
Conclusions
In this study, we established a novel predictive model RNAm5CPred for accurate identification of the m5C sites in RNA sequences. Considering the imbalance between the occurrences of m5C and normal cytosine sites in the realistic RNA sequences, we built a new imbalanced dataset Met935, and the performances of two kinds of nucleotide composition features (KNF and KSNPF) were extensively studied based on this dataset. Based on three selected features (4NF, 1SNPF, and PseDNC), three models were built on three benchmark datasets, i.e., Met1900, Met240, and Met935, respectively. The three models were then compared with other available m5C site predictive models by performing stringent jackknife tests. The comparison results showed that our models achieved better or comparable prediction performance on different datasets. The models were further evaluated on an independent test set Test1157, which showed that the model based on Met240 (RNAm5CPred) achieved the best performances according to MCC. More specifically, our model RNAm5CPred has good generalization and can be a practically useful model for experimental researchers. To facilitate the accessibility of our predictive model RNAm5CPred, a web server has been provided online at http://zhulab.ahu.edu.cn/RNAm5CPred/.
Materials and Methods
Benchmark Datasets
In this study, we used three benchmark datasets, i.e., Met935, Met240, and Met1900, to train and validate our models. In addition, two other datasets, Train839 and Test96, which were derived from Met935, were also used. All of these datasets consist of a positive subset and a negative subset. The positive subset contains RNA sequences with a center cytosine that can be modified as m5C, while the negative subset contains RNA sequences with a center cytosine that cannot be modified as m5C.
Met240 comes from the dataset Met1320 constructed by Feng et al.12 The Met1320 consists of a positive subset (S+) and 10 negative subsets (S0− ∼ S0−). Met240 was built by combining the positive subset (S+) and the first negative subset (S0−), which was also used by Feng et al.12 and Zhang et al.14 in their works. Met240 contains 120 positive and 120 negative samples. Sequence similarity between any two segments is less than 70%.
Met1900 was constructed by Qiu et al.13 and contains 475 positive samples and 1,425 negative samples. The sequence similarity between sequences is greater than 90%. For more details on the building of Met1900, please refer to Qiu et al.13
Both Met240 and Met1900 have their own shortcomings. Although the redundancy of Met240 is well under control, the size of this dataset is too small. In addition, realistically, there are fewer m5C sites than normal C sites in RNA sequences. Using the balanced dataset would make the model biased to the positive samples. As for Met1900, it has a serious redundancy problem. To build a reliable and practical predictive model, in this study, a new benchmark dataset Met935 was constructed according to the following procedures. (1) From RMBase,7 we downloaded all m5C sequences. Importantly, note that the downloaded sequences are DNA fragments. Therefore, we have to change the code T to U for all sequences to convert them into RNA sequences. (2) For the data processed in the first step, we used the CD-HIT15 program to reduce their redundancy and homologous bias, which resulted in a <70% similarity between the sequences. In this way, 127 positive samples were successfully obtained. (3) The 1,425 negative samples of Met1900 were utilized for the acquisition of negative samples. The CD-HIT program was used again to remove the redundancy by setting the sequence similarity cutoff at 70%, and finally 808 negative samples were successfully obtained. Finally, these 127 positive samples and 808 negative samples were combined to form the dataset Met935. Figure 3 shows the flowchart for generating the dataset Met935.
Figure 3.

The Flowchart for Generating Dataset Met935
Two other datasets, i.e., Train839 and Test96 (an independent validation dataset), were derived from Met935 following the procedures below. Among the 127 positive RNA segments in Met935, 13 segments that are not included in Met240 were selected as the positive samples of Test96. Then, from all of the negative samples of Met935 that are not included in Met240, 83 samples were randomly selected and used as the negative subset of Test96. Then, the remaining 114 (127 – 13) positive samples and 725 (808 – 83) negative samples in Met935 were combined to form Train839.
To further evaluate the performances of different methods, we also built another dataset, Test1157. The dataset was constructed according to the following steps. (1) We obtained the “high-threshold” m5C sites information from “GSE90963_Table_S1-m5C_candidate_sites.xlsx,” which was downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE90963). (2) According to the high-threshold m5C sites23 information from the first step, we collected all RNA segments of 41-tuple nt with the m5C sites at the center position by sliding a flexible window along each RNA sequences transcribed from the human genome. This set of segments was named P1. (3) All negative 41-tuple RNA segments were generated by excluding the possible m5C site recorded in the file GSE90963_Table_S1-m5C_candidate_sites.xlsx. This set of segments was named N1. (4) CD-HIT-2D was used to remove similar sequences of P1 to the positive examples of Met935 and the similar sequences of N1 to the negative examples of Met935, respectively, with a cutoff of 0.7. Thus, a positive dataset P2 and a negative dataset N2 were obtained. (5) The CD-HIT was used to remove redundant sequences in P2 and N2, respectively, with a cutoff of 0.7. In this manner, we generated a positive dataset P3 and a negative dataset N3. (6) A subset of N3 with 1,000 RNA segments was randomly selected as N4. (7) P3 with 157 positive examples and N4 were combined as the independent test set Test1157. Figure 4 shows the flowchart to generate Test1157.
Figure 4.

The Flowchart for Generating Dataset Test1157
Each of these RNA segments in all datasets is 41 bp long, according to the sequential scheme by Chou.24, 25, 26 We can express each RNA sample (segment) that has a potential m5C site at the center as follows:
| (Equation 1) |
where N−ζ represents the ξth upstream nucleotide from the central cytosine and N+ζ represents the ξth downstream nucleotide.
To further simplify the description, Equation 1 can be rewritten in the following form:
| (Equation 2) |
where Ni (i = 1, 2, … 20, 21 … 41) represents the nucleotide at the ith position of the RNA segment and can be any one of the four nucleotide bases in RNA, i.e., Ni ∈{A (adenine), C (cytosine), G (guanine), U (uracil)}. The detailed sequence information for all the aforementioned datasets is given in Table 9. All six datasets used in this study are included in Data S1, S2, S3, S4, S5, and S6 or can be freely downloaded from http://zhulab.ahu.edu.cn/RNAm5CPred/.
Table 9.
The Information of the Six Datasets
| Dataset | Length (bp) | Positive Subset | Negative Subset | Total |
|---|---|---|---|---|
| Met240 | 41 | 120 | 120 | 240 |
| Met1900 | 41 | 475 | 1425 | 1900 |
| Met935 | 41 | 127 | 808 | 935 |
| Train839 | 41 | 114 | 725 | 839 |
| Test96 | 41 | 13 | 83 | 96 |
| Test1157 | 41 | 157 | 1000 | 1157 |
Feature Representation of the RNA Segments
Encoding RNA segments into feature vectors with highly discriminative information plays pivotal roles in the building of a machine learning model to predict m5C sites. Among all of the existing features, KNFs, KSNPFs, and PseDNC27 have been used to determine whether a cytosine can be modified. In this study, we explored the predictive power of these three features to predict the m5C sites. Details about feature encoding are described as below.
KNFs
KNFs are a classical method to represent nucleotide sequence features.16 For a given K value, KNF means the frequency of occurrence for each K-mer nucleotide component in a nucleotide sequence. In this way, an RNA segment can be represented as a 4K-dimensional feature vector. The following formula was used to calculate KNF:
| (Equation 3) |
where n1n2 … nK indicates a K-mer nucleotide component, and nK can be any one of the four nucleotide bases in RNA, i.e., nK ∈{A (adenine), C (cytosine), G (guanine), U (uracil)}. N(n1n2 … nK) represents the number of occurrences of n1n2 … nK in a nucleotide sequence, and L indicates the length of the nucleotide segment (L = 41). For instance, when K = 2 there are 16 types of dinucleotide, and the RNA segment can be encoded as:
| (Equation 4) |
Clearly, as K increases, the dimension of the feature vector increases exponentially. To avoid the dimension disaster, the maximum K used in this study is set at 4.
KSNPFs
KSNPFs have also been used to encode RNA sequences.16 A K-spaced nucleotide pair is a nucleotide pair separated by K arbitrary nucleotides; e.g., UxxxG is a three-spaced nucleotide pair in which three arbitrary nucleotides are between the nucleotides U and G. For the sake of illustration, we use N1x{K}N2 (N1, N2, and x∈{A, C, G, U}) to express a K-spaced nucleotide pair. Clearly, for a nucleotide pair consisting of N1 and N2, there will be 16 (i.e., 4 × 4) possible combinations. In other words, there will be 16 K-spaced nucleotide pairs for a fixed K. For example, when K = 3, N1x{K}N2 may be AxxxA, AxxxC, …, or UxxxU. Similar to KNF, we can use the following formula to calculate KSNPF:
| (Equation 5) |
where N(N1x{K}N2) represents the number of occurrences of N1x{K]N2 in a nucleotide sequence, and L indicates the length of the nucleotide segment (L = 41). In this study, we tried different Ks (i.e., 1, 2, 3, 4, 5). For example, when K = 1, the RNA segment can be encoded as:
| (Equation 6) |
PseDNC
PseDNC is a feature that can incorporate both the local and global sequence pattern information of the RNA segments.27 Each component of PseDNC was derived from a physical-chemical matrix via a series of auto-covariance and cross-covariance transformations. For more details about PseDNC, please refer to Chen et al.27 As far as we know, there are at least 23 kinds of physical-chemical properties that can be used by PseDNC to encode RNA segments.14,28, 29, 30, 31, 32, 33. In this study, we chose 3 from these 23 kinds of physical-chemical properties, i.e., free energy, hydrophilicity, and stacking energy. Tables 10 lists the details.
Table 10.
List of Physicochemical Properties of Dinucleotides in RNA
| Dinucleotide | Free Energy | Hydrophilicity | Stacking Energy |
|---|---|---|---|
| GG | −3.260 | 0.170 | −11.100 |
| GA | −2.350 | 0.100 | −14.200 |
| GC | −3.420 | 0.260 | −16.900 |
| GU | −2.240 | 0.270 | −13.800 |
| AG | −2.080 | 0.080 | −14.000 |
| AA | −0.930 | 0.040 | −13.700 |
| AC | −2.240 | 0.140 | −13.800 |
| AU | −1.100 | 0.140 | −15.400 |
| CG | −2.360 | 0.350 | −15.600 |
| CA | −2.110 | 0.210 | −14.400 |
| CC | −3.260 | 0.490 | −11.100 |
| CU | −2.080 | 0.520 | −14.000 |
| UG | −2.110 | 0.340 | −14.400 |
| UA | −1.330 | 0.210 | −16.000 |
| UC | −2.350 | 0.480 | −14.200 |
| UU | −0.930 | 0.440 | −13.700 |
SVM as Prediction Engine
SVM is a machine learning algorithm based on nonlinear mapping, and it is widely used in various fields of bioinformatics.34, 35, 36, 37 The final decision function of SVM is only determined by a few support vectors. The complexity of the calculation depends on the number of support vectors, rather than the dimension of the sample space, which in some sense is able to avoid the dimension disaster. In this study, we used the MATLAB function FITCSVM to construct our models. There are several issues of SVM to be aware of: (1) kernel function: the most widely used kernel function is the radial basis kernel function (RBF), which is also used in this study; (2) parameters: two parameters are considered here: one is the penalty coefficient c, and the other is the radial basis kernel function’s parameter g (gamma), which represents the RBF width (in FITCSVM, they are called BoxContraint and KernelScale respectively); and (3) optimization of parameters: we optimized these two parameters by using a grid search based on the 10-fold cross-validation test. The ranges of the two parameters in the grid search are:
| (Equation 7) |
Performance Evaluation
Cross-validation is widely used to evaluate the performance of predictive models. The jackknife test is a special case of cross-validation, and its evaluation results are often considered to be accurate38. In the jackknife test, each sample in the original dataset is rotated as a testing set, while the remaining samples are used as a training set.26 In this way, the number of samples in the training set is only one less than that of the original dataset, which makes the model that is evaluated in the jackknife test similar to the model that is trained by the original dataset. To ensure the quality of the experimental results, we employed the jackknife test to assess the accuracy of the model constructed with dataset Met935, Met240, and Met1900. However, the complexity of the jackknife test is proportional to the amount of data in the dataset. To reduce the complexity, first a 10-fold cross-validation test was carried out to optimize the SVM parameters and select the features. After that, the jackknife test was implemented to get the unique result that was not affected by the random partition of the samples. For a predictive model, its generalization performance is very important. In this study, we used an independent test to demonstrate the excellent generalization performance of our model.
In our experiment, we employed six frequently used evaluation indexes to check the performance of our method: sensitivity (Sen), specificity (Spe), precision (Pre), accuracy (Acc), and the MCC.39 They are defined as follows:
| (Equation 8) |
where TP, TN, FP, and FN represent the counts of true-positive, true-negative, false-positive, and false-negative predictions, respectively.
Furthermore, to compare different models, the ROC curve and AUC were employed.40 When a ROC curve is completely enveloped by another ROC curve, the latter is regarded as dominant of the former, which means that the latter’s performance is superior to that of the former. However, it is difficult to judge the performance when the two ROC curves cross over each other. In this case, the AUC will be a better choice to judge the performance.
Author Contributions
Conceived the study: XZ, YX, BH. Designed the study: TF, XZ, YX. Participate designed the study: ZZ, RS, LZ, JH. Analyzed the data: TF, ZZ, LZ, JH, YX. Wrote the paper: TF, XZ, YX, BH. All authors read and approved the manuscript.
Acknowledgments
This work is supported by the grants from the National Natural Science Foundation of China (21403002, 31601074, 61872094, and 61832019).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.10.008.
Contributor Information
Bei Huang, Email: beihuang@163.com.
Yi Xiong, Email: xiongyi@sjtu.edu.cn.
Xiaolei Zhu, Email: xlzhu_mdl@hotmail.com.
Supplemental Information
References
- 1.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agris P.F. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9:629–635. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexandrov A., Chernyakov I., Gu W., Hiley S.L., Hughes T.R., Grayhack E.J., Phizicky E.M. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 4.David R., Burgess A., Parker B., Li J., Pulsford K., Sibbritt T., Preiss T., Searle I.R. Transcriptome-wide mapping of RNA 5-methylcytosine in arabidopsis mRNAs and noncoding RNAs. Plant Cell. 2017;29:445–460. doi: 10.1105/tpc.16.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motorin Y., Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49:4934–4944. doi: 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 6.Motorin Y., Lyko F., Helm M. 5-Methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 2010;38:1415–1430. doi: 10.1093/nar/gkp1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun W.J., Li J.H., Liu S., Wu J., Zhou H., Qu L.H., Yang J.H. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44(D1):D259–D265. doi: 10.1093/nar/gkv1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Squires J.E., Patel H.R., Nousch M., Sibbritt T., Humphreys D.T., Parker B.J., Suter C.M., Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussain S., Sajini A.A., Blanco S., Dietmann S., Lombard P., Sugimoto Y., Paramor M., Gleeson J.G., Odom D.T., Ule J., Frye M. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013;4:255–261. doi: 10.1016/j.celrep.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoddami V., Cairns B.R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013;31:458–464. doi: 10.1038/nbt.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edelheit S., Schwartz S., Mumbach M.R., Wurtzel O., Sorek R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013;9:e1003602. doi: 10.1371/journal.pgen.1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng P., Ding H., Chen W., Lin H. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. Biosyst. 2016;12:3307–3311. doi: 10.1039/c6mb00471g. [DOI] [PubMed] [Google Scholar]
- 13.Qiu W.R., Jiang S.Y., Xu Z.C., Xiao X., Chou K.C. iRNAm5C-PseDNC: identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget. 2017;8:41178–41188. doi: 10.18632/oncotarget.17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M., Xu Y., Li L., Liu Z., Yang X., Yu D.J. Accurate RNA 5-methylcytosine site prediction based on heuristic physical-chemical properties reduction and classifier ensemble. Anal. Biochem. 2018;550:41–48. doi: 10.1016/j.ab.2018.03.027. [DOI] [PubMed] [Google Scholar]
- 15.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X., Yan R. RFAthM6A: a new tool for predicting m6A sites in Arabidopsis thaliana. Plant Mol. Biol. 2018;96:327–337. doi: 10.1007/s11103-018-0698-9. [DOI] [PubMed] [Google Scholar]
- 17.Li G.Q., Liu Z., Shen H.B., Yu D.J. TargetM6A: identifying N6-methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans. Nanobioscience. 2016;15:674–682. doi: 10.1109/TNB.2016.2599115. [DOI] [PubMed] [Google Scholar]
- 18.Xiang S., Liu K., Yan Z., Zhang Y., Sun Z. RNAMethPre: a Web server for the prediction and query of mRNA m6A sites. PLoS ONE. 2016;11:e0162707. doi: 10.1371/journal.pone.0162707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W., Feng P., Ding H., Lin H., Chou K.C. iRNA-methyl: identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 20.He J., Fang T., Zhang Z., Huang B., Zhu X., Xiong Y. PseUI: pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics. 2018;19:306. doi: 10.1186/s12859-018-2321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lv H., Zhang Z.M., Li S.H., Tan J.X., Chen W., Lin H. Evaluation of different computational methods on 5-methylcytosine sites identification. Brief. Bioinform. 2019:bbz048. doi: 10.1093/bib/bbz048. [DOI] [PubMed] [Google Scholar]
- 23.Khoddami V., Yerra A., Mosbruger T.L., Fleming A.M., Burrows C.J., Cairns B.R. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA. 2019;116:6784–6789. doi: 10.1073/pnas.1817334116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou K.C. Using subsite coupling to predict signal peptides. Protein Eng. 2001;14:75–79. doi: 10.1093/protein/14.2.75. [DOI] [PubMed] [Google Scholar]
- 25.Chou K.C. A sequence-coupled vector-projection model for predicting the specificity of GalNAc-transferase. Protein Sci. 1995;4:1365–1383. doi: 10.1002/pro.5560040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W., Feng P.M., Lin H., Chou K.C. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013;41:e68. doi: 10.1093/nar/gks1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barzilay I., Sussman J.L., Lapidot Y. Further studies on the chromatographic behaviour of dinucleoside monophosphates. J. Chromatogr. A. 1973;79:139–146. doi: 10.1016/s0021-9673(01)85282-1. [DOI] [PubMed] [Google Scholar]
- 29.Freier S.M., Kierzek R., Jaeger J.A., Sugimoto N., Caruthers M.H., Neilson T., Turner D.H. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedel M., Nikolajewa S., Sühnel J., Wilhelm T. DiProDB: a database for dinucleotide properties. Nucleic Acids Res. 2009;37:D37–D40. doi: 10.1093/nar/gkn597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goñi J.R., Pérez A., Torrents D., Orozco M. Determining promoter location based on DNA structure first-principles calculations. Genome Biol. 2007;8:R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez A., Noy A., Lankas F., Luque F.J., Orozco M. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 2004;32:6144–6151. doi: 10.1093/nar/gkh954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponnuswamy P.K., Gromiha M.M. On the conformational stability of oligonucleotide duplexes and tRNA molecules. J. Theor. Biol. 1994;169:419–432. doi: 10.1006/jtbi.1994.1163. [DOI] [PubMed] [Google Scholar]
- 34.Cortes C., Vapnik V. Support-vector networks. Machine Learning. 1995;20:273–297. [Google Scholar]
- 35.Wang X., Pardalos P.M. A survey of support vector machines with uncertainties. Ann. Data Sci. 2014;1:293–309. [Google Scholar]
- 36.Qiao Y., Xiong Y., Gao H., Zhu X., Chen P. Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinformatics. 2018;19:14. doi: 10.1186/s12859-018-2009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu X., Ericksen S.S., Mitchell J.C. DBSI: DNA-binding site identifier. Nucleic Acids Res. 2013;41:e160. doi: 10.1093/nar/gkt617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou K.C., Zhang C.T. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 39.Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 40.Fawcett T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006;27:861–874. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.