

Abstract
Esophageal squamous cell carcinoma (ESCC) is a prevalent aggressive malignant tumor with poor prognosis. Investigations into the molecular changes that occur as a result of the disease, as well as identification of novel biomarkers for its diagnosis and prognosis, are urgently required. Long non-coding RNAs (lncRNAs) have been reported to play a critical role in tumor progression. The present study performed data mining analyses for ESCC via an integrated study of accumulated datasets and identification of the differentially expressed lncRNAs from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases. The identified intersection of differentially expressed genes (lncRNAs, miRNAs and mRNAs) in ESCC tissues between the GEO and TCGA datasets was investigated. Based on these intersected lncRNAs, the present study constructed a competitive endogenous RNA (ceRNA) network of lncRNAs in ESCC. A total of 81 intersection lncRNAs were identified; 67 of these were included in the ceRNA network. Functional analyses revealed that these 67 key lncRNAs primarily dominated cellular biological processes. The present study then analyzed the associations between the expression levels of these 67 key lncRNAs and the clinicopathological characteristics of the ESCC patients, as well as their survival time using TCGA. The results revealed that 31 of these lncRNAs were associated with tumor grade, tumor-node-metastasis (TNM) stage and lymphatic metastasis status (P<0.05). In addition, 15 key lncRNAs were demonstrated to be associated with survival time (P<0.05). Finally, 5 key lncRNAs were selected for validation of their expression levels in 30 patients newly diagnosed with ESCC via reverse transcription-quantitative PCR (RT-qPCR). The results suggested that the fold changes in the trends of up- and downregulation between GEO, TCGA and RT-qPCR were consistent. In addition, it was also demonstrated that a select few of these 5 key lncRNAs were significantly associated with TNM stage and lymph node metastasis (P<0.05). The results of the clinically relevant analysis and the aforementioned bioinformatics were similar, hence proving that the bioinformatics analysis used in the present study is credible. Overall, the results from the present study may provide further insight into the functional characteristics of lncRNAs in ESCC through bioinformatics integrative analysis of the GEO and TCGA datasets, and reveal potential diagnostic and prognostic biomarkers for ESCC.
Keywords: esophageal cancer, long non-coding RNA, clinical characteristics, overall survival
Introduction
According to statistics from the World Health Organization (WHO) in 2015, esophageal cancer ranks seventh in terms of global cancer incidence (572,000 new cases per year) and sixth overall in global cancer-associated mortality (509,000 deaths) (1). Approximately 85% of all esophageal cancer cases are esophageal squamous cell carcinoma (ESCC), and the most common cause of death is metastasis (2). Tumor staging and grading systems are important in the clinical diagnosis of cancer, but are currently not adequate for prognosis and prediction of the disease (3). Frequently, when patients with ESCC are diagnosed in clinical practice, the majority have already progressed to advanced stage and/or lymphatic metastasis. Furthermore, the survival rate in patients with ESCC is very poor, and there is a lack of specific biomarkers for early diagnosis and prognosis (4). Therefore, more efficient and accurate ESCC diagnostic and prognostic biomarkers are urgently required in order to improve these areas, including screening for the early stages of ESCC, as well as new effective treatment methods.
Accumulating evidence has revealed that dysregulated long non-coding RNAs (lncRNAs) play a number of key biological roles in the progression of various types of cancer (5,6). An increasing number of studies have suggested that aberrant expression of lncRNAs in ESCC is closely associated with histological type, tumor-node-metastasis (TNM) stage, lymph node metastasis and prognosis (7,8).
ESCC is a multistep disease, which involves multiple interactions between genetic and environmental factors. Furthermore, lncRNAs play an important regulatory role in epigenetics. Investigating the ESCC-associated alterations of lncRNAs may aid the identification of valuable biomarkers for ESCC diagnosis and prognosis. Previous studies have primarily focused on the diagnostic and prognostic performance of a small portion of lncRNAs in ESCC, but a larger number of lncRNAs remain unexplored. Therefore, elucidating the functions of dysregulated lncRNAs, particularly in ESCC, is currently an important research topic.
Currently, high-throughput RNA sequencing technologies are being widely used for the detection of lncRNA alterations in carcinogenesis and screening for potential biomarkers of numerous diseases (9). However, the small sample sizes used for microarray detection often present a bias toward the identification of ESCC-associated lncRNAs due to lack of RNA sequencing data and, therefore, often generate errors (10). By using large sample sizes that integrate multiple RNA sequencing datasets, sufficient information regarding patients with ESCC can be obtained, thereby providing more convincing results. Thus far, with the advent of high-throughput RNA sequencing technologies, The Cancer Genome Atlas (TCGA; http://portal.gdc.cancer.gov/) and Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database platforms have allowed the collected RNA sequencing data from microarray chips to be uploaded and standardized for quality control (11). Therefore, identification of the ESCC-associated lncRNAs would be more reliable with the use of large sample sizes integrating multiple analyses from different RNA sequencing databases.
The RNA sequencing databases obtained from the tissues of patients with ESCC are expected to provide a novel analysis strategy to identify diagnostic and prognostic biomarkers for this disease. In the present study, data mining analyses for ESCC were performed by integrating the significant differences in RNA obtained from the GEO and TCGA databases. Through these efforts, co-differentially expressed lncRNAs in ESCC may be identified. Based on these lncRNAs, subsequent analyses were performed, including gene functional enrichment analyses, competing endogenous RNA network construction, assessment of the association between differentially expressed lncRNAs and the clinicopathological characteristics of patients with ESCC, and survival analyses. Finally, reverse transcription-quantitative PCR (RT-qPCR) was used to validate the bioinformatics analysis results in the tumor and adjacent non-tumor tissues of 30 patients with newly diagnosed ESCC. This new approach may contribute to an improved method for identifying potential lncRNA biomarkers for the diagnosis of ESCC, as well as its classification and prediction of prognosis.
Materials and methods
Microarray dataset collection
The present study collected RNA sequencing expression datasets (lncRNAs, miRNAs and mRNAs) comprising tissue samples from 802 patients with ESCC, which included cancer tissues and adjacent normal esophageal tissues from the GEO genomics database. The RNA sequencing datasets of patients with ESCC were downloaded from the GSE23400 (12) (208 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23400), GSE26886 (13) (69 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26886), GSE45670 (14) (38 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45670), GSE97049 (14 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97049), GSE6188 (15) (257 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6188) and GSE55856 (16) (216 tissue samples: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE55856) datasets. All the aforementioned GEO datasets were obtained from Affymetrix Human Genome Array platforms. A total of 312 patients with ESCC were obtained from the TCGA database (up to November 1, 2018). Annotation information of the RNA sequencing datasets was obtained using the Affymetrix Human Genome Array platforms. The present study was fully compliant with the publication guidelines provided by the GEO and TCGA databases. The details of GEO and TCGA database ESCC patient tissue RNA sequencing datasets, sample descriptions and clinicopathological characteristics are provided in Tables I and II. A flow diagram of the integrated bioinformatics analysis from the GEO and TCGA databases is provided in Fig. 1.
Table I.
Details of ESCC studies and RNA sequencing microarray datasets from the GEO database.
| GSE | Publication | RNA sequencing styles | Sample size for each group |
|---|---|---|---|
| GSE23400 | Clinical cancer research | lncRNA | Tumor, 104 |
| mRNA | Adjacent normal tissues, 104 | ||
| GSE26886 | BMC Cancer | lncRNA | Tumor, 35 |
| mRNA | Adjacent normal tissues, 34 | ||
| GSE45670 | Annals of oncology | lncRNA | Tumor, 28 |
| mRNA | Adjacent normal tissues, 10 | ||
| GSE97049 | Unrecorded | miRNA | Tumor, 7 |
| Adjacent normal tissues, 7 | |||
| GSE6188 | Cancer research | miRNA | Tumor, 153 |
| Adjacent normal tissues, 104 | |||
| GSE55856 | Gut | miRNA | Tumor, 108 |
| Adjacent normal tissues, 108 |
ESCC, esophageal squamous cell carcinoma; GEO, Gene Expression Omnibus.
Table II.
Clinical information and samples size for TCGA ESCC datasets.
| Variables | Total cases, n=312 (%) | Alive, n=138 (%) | Deceased, n=174 (%) |
|---|---|---|---|
| Sex | |||
| Male | 211 (67.63) | 83 (60.14) | 128 (73.56) |
| Female | 101 (32.37) | 55 (39.86) | 46 (26.44) |
| Race | |||
| White | 162 (51.92) | 5 (39.86) | 107 (61.49) |
| Asia | 127 (40.71) | 68 (49.28) | 59 (33.91) |
| Black | 23 (7.37) | 15 (10.87) | 8 (4.60) |
| Age, years | |||
| ≤50 | 86 (27.56) | 53 (38.41) | 33 (18.97) |
| >50 | 226 (72.44) | 85 (61.59) | 141 (81.03) |
| Tumor grade | |||
| GI | 49 (15.71) | 31 (22.46) | 18 (10.34) |
| GII | 146 (46.79) | 48 (34.78) | 98 (56.32) |
| GIII–IV | 117 (37.50) | 59 (42.75) | 58 (33.33) |
| TNM stage | |||
| I/II | 99 (31.73) | 30 (21.74) | 69 (39.66) |
| III/IV | 213 (68.27) | 108 (78.26) | 105 (60.34) |
| Lymph node status | |||
| No metastasis | 35 (11.22) | 32 (23.19) | 3 (1.72) |
| Metastasis | 277 (88.78) | 106 (76.81) | 171 (98.28) |
TCGA, The Cancer Genome Atlas; ESCC, esophageal squamous cell carcinoma.
Figure 1.

Flowchart for integrated bioinformatics analysis of ESCC publicly available RNA sequencing datasets from the GEO and TCGA databases. ESCC, esophageal squamous cell carcinoma; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas.
Annotation of microarray probes
In order to enhance the comparability of the GEO database RNA sequencing data, the signature values of the GSE23400, GSE26886, GSE45670, GSE97049, GSE6188 and GSE55856 RNA sequencing datasets were downloaded and converted to new comparable transcripts. Probe reannotation methods were used to obtain a standard measurement of the RNA sequencing expression profiles. Once the aforementioned ESCC RNA sequencing probe signature values and probe IDs were downloaded from the GEO database, they underwent a mapped detection analysis in a database of human genomes, and those that were mismatched were excluded. The remaining probe positions on the chromosomes were identified using the GENCODE tool (https://www.gencodegenes.org/). Probes matched to both protein-coding genes and lncRNAs were also excluded. If multiple probes matched the same gene, the median values were instead used as the expression level of that gene. In addition, prior to any further analyses, RNA sequencing data normalization was performed to ensure the data were fully compliant with the publication guidelines of TCGA.
Integration of microarray data and differential expression analysis
All the ESCC tissue samples from the GEO database were downloaded from three or more datasets in order to enlarge the sample number and avoid generating less reliable results. A differential analysis was then separately performed for each dataset, comparing ESCC tumor tissues to adjacent normal tissues using the limma R 3.4.4 software (https://www.r-project.org/) [false discovery rate (FDR) <0.05, fold change >2, P<0.05]. Subsequently, overlapping subclass analyses were used to identify the separate co-differentially expressed genes in each dataset, including lncRNAs, miRNAs and mRNAs, with the Venn 2.1 tool (http://bioinfogp.cnb.csic.es/tools/venny/index.html). The integrated ESCC tissue dysregulated lncRNA, miRNA and mRNA lists were saved for further analysis.
TCGA database provided the normalized RNA sequencing data, which included lncRNAs and mRNAs from patients with ESCC using the RNASeqV2 system. Furthermore, ESCC level 3 normalized miRNA sequencing data (Illumina HiSeq 2000 microRNA sequencing platforms) were also downloaded from TCGA. The significantly differentially expressed lncRNAs, mRNAs and miRNAs in the 312 tumor tissues and 47 adjacent non-tumor esophageal epithelial tissues in patients with ESCC were then analyzed (FDR<0.05, fold change >2, P<0.05). The significantly different lncRNAs, mRNAs and miRNAs were selected for further analysis.
Finally, according to the fold changes of differentially expressed lncRNAs, mRNAs and miRNAs in ESCC tissues from the GEO and TCGA databases, the common genes were selected for subsequent analysis.
Construction of the competing endogenous (ce) RNA network
In the present study, an lncRNA, miRNA and mRNA ceRNA network was built based on the theory that lncRNAs can regulate miRNA abundance by sequestration binding, acting as ‘miRNA sponges’ through miRNA binding to the mRNAs and negatively regulating gene expression. Common ESCC tissues with significantly differentially expressed lncRNAs, mRNAs and miRNAs (FDR<0.05, fold change >2, P<0.05) were selected to build the ceRNA network, in which the fold changes of genes were rooted in the TCGA database, in order to investigate whether these intersection lncRNAs, mRNAs and miRNAs were involved in ceRNA regulation. MiRcode (https://omictools.com/mircode-tool), miRanda (http://www.microrna.org/microrna/home.do) and Targetscan (http://www.targetscan.org/) were used to predict the miRNA target lncRNAs and miRNA-mRNA interactions in the different databases. Finally, the predicted miRNA target genes and the significantly differentially expressed intersection genes in the GEO and TCGA databases were used to build the ceRNA network. A flow chart for the lncRNA, miRNA and mRNA ceRNA network construction is presented in Fig. 2.
Figure 2.

Flowchart for lncRNA, miRNA and mRNA ceRNA network construction. lncRNA, long non-coding RNA.
During this process, the lncRNA, miRNA and mRNA ceRNA network was constructed using the fold change data of the significantly expressed RNA of patients with ESCC from TCGA database using the R package. Pearson's correlation matrix models were used to identify the potential relevance of all pair-wise genes. Finally, based on the weighted adjacency matrix, the network connectivity of genes with other genes was investigated, and the ceRNA network was built using Cytoscape software (version 3.0.).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis
In order to investigate the potential gene functional enrichment and signaling pathway regulation of the consensus mRNAs that were involved in the ceRNA network, GO (http://www.geneontology.org) and KEGG pathway analyses were performed. The mRNAs that were involved in the ceRNA network were uploaded to the GO database to investigate the enriched molecular functions of these mRNAs. Upregulated and downregulated mRNAs from the ceRNA network were analyzed. Furthermore, the KEGG tool (http://www.kegg.jp/) was used to identify the potential regulated signaling pathways of these genes. R software was used to visualize the GO and KEGG results.
Association between ceRNA network key lncRNAs and ESCC clinical status from TCGA
Based on the co-expression of the lncRNAs, miRNAs and mRNAs in the ceRNA network, the key lncRNAs that were involved in the network were selected as target lncRNAs potentially associated with ESCC progression. Subsequently, the potential association between ceRNA network key lncRNAs and TCGA ESCC patients' clinicopathological characteristics were investigated, which included sex, TNM stage, tumor grade, lymphatic metastasis status and pathological stage using multiple linear regression analysis.
Kaplan-Meier survival analysis
In order to investigate whether the key lncRNAs that were involved in the ceRNA network were associated with the TCGA database status, a Kaplan-Meier survival analysis of the ESCC patients from TCGA was performed. Based on the TCGA ESCC patient datasets, the fold changes of the aforementioned selected key lncRNAs in the cancer tissues and overall survival rates of patients with ESCC were determined using the GEPIA tool (http://gepia.cancer-pku.cn/). Kaplan-Meier survival analysis parameters were calculated using the publicly available TCGA ESCC patient datasets and GEPIA tools. The survival distributions of different TCGA with ESCC, and the expression changes in key lncRNAs, were examined using Kaplan-Meier analysis, log-rank test and hazard ratio (HR).
Preparation of human ESCC samples and RT-qPCR validation of bioinformatics analysis results
Next, 30 tumor and paired non-tumor esophageal tissues from patients with ESCC were collected from the Gansu Wuwei Tumor Hospital (Wuwei, China); the patients were aged 40–75 years. The samples were collected and stored at −80°C. All patients were diagnosed with ESCC according to their pathological examination results. Clinical basic information and informed consent forms were obtained from all ESCC patients (Table III). The collection of the tumor samples from patients with ESCC was approved by the Ethics Committee of the Gansu Wuwei Tumor Hospital.
Table III.
Demographic and clinical characteristics of 30 patients with ESCC.
| Variables | Total cases, n=30 (%) |
|---|---|
| Age, years | 59±9.72 |
| (mean ± standard deviation) | |
| Sex | |
| Male | 20 (66.67) |
| Female | 10 (33.33) |
| TNM stage | |
| I/II | 8 (26.67) |
| III/IV | 22 (73.33) |
| Lymph node status | |
| No metastasis | 9 (30.00) |
| Metastasis | 21 (70.00) |
ESCC, esophageal squamous cell carcinoma.
Total RNA was isolated from tissue samples using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Reverse Transcription Kit (Promega Corporation) and GoTaq® qPCR Master Mix of Power SYBR® Green (Promega Corporation) were used to synthesize cDNA and perform RT-qPCR analysis. The reaction was performed at 95°C for 2 min, followed by 40 cycles at 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. A dissociation curve was analyzed from 60–95°C. Finally, RT-qPCR was performed using the Step One Plus™ PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.). RT-qPCR relative fold change results were calculated using the 2−ΔΔCq method (17).
Statistical analysis
All ESCC tissue RNA sequencing datasets from the GEO database were obtained from at least three independent datasets. R software was used to normalize the RNA sequencing data and compare significantly differentially expressed genes. Cytoscape 3.0 and GEPIA software tools were used to construct the ceRNA network and perform the survival analysis. Two-tailed Student's t-test was used to assess the differences between subgroups. P<0.05 was considered to indicate a statistically significant difference.
Results
Convergence of gene expression signatures across the different datasets of patients with ESCC from GEO
The ESCC RNA sequencing data and other information were obtained from the GEO database. In order to increase the veracity and reliability of the signal values, and also decrease the possibility of false-positives, three independent datasets were downloaded, including ESCC tissue lncRNA, miRNA and mRNA sequencing results. Differentially expressed genes were identified using the limma package according to the threshold of FDR<0.05, fold change >2 and P<0.05. Volcano plots and Venn analysis revealed the number of differentially expressed genes identified from each dataset (Fig. 3). The GSE23400 dataset contained 5,310 differentially expressed genes, including 792 differentially expressed lncRNAs and 4,518 mRNAs. The GSE26886 dataset contained 8,747 differentially expressed genes, including 1,340 differentially expressed lncRNAs and 7,407 mRNAs. The GSE45670 dataset contained 6,336 differentially expressed genes, including 914 differentially expressed lncRNAs and 5,422 mRNAs. The GSE97049 dataset contained 178 differentially expressed miRNAs. The GSE6188 dataset contained 198 differentially expressed miRNAs. Finally, the GSE55856 dataset contained 198 differentially expressed miRNAs. The detailed information of these six datasets and the number of differentially expressed genes identified from each dataset are presented in Table IV.
Figure 3.

(A) Volcano plots of differentially expressed genes in GEO datasets; (B) Venn diagram demonstrates the differentially expressed intersection genes (lncRNAs, miRNAs and mRNAs) of GEO datasets. GEO, Gene Expression Omnibus; lncRNA, long non-coding RNA.
Table IV.
Details information of differentially expressed genes in GEO database.
| GSE | Differentially expressed genes | Upregulated | Downregulated | Intersection differentially expressed genes | |
|---|---|---|---|---|---|
| GSE23400 | lncRNA | 792 | 378 | 414 | 108 |
| GSE26886 | lncRNA | 1340 | 599 | 741 | |
| GSE45670 | lncRNA | 914 | 508 | 406 | |
| GSE23400 | mRNA | 4518 | 2219 | 2299 | 1234 |
| GSE26886 | mRNA | 7407 | 3933 | 3474 | |
| GSE45670 | mRNA | 5422 | 2599 | 2823 | |
| GSE97049 | miRNA | 178 | 68 | 110 | 45 |
| GSE6188 | miRNA | 198 | 48 | 150 | |
| GSE55856 | miRNA | 219 | 109 | 110 | |
GEO, Gene Expression Omnibus.
Following the Venn tool intersection, differentially expressed genes were analyzed, and it was demonstrated that there were 108 common lncRNAs, 1,234 mRNAs and 45 miRNAs with significantly changed levels in ESCC tissues from the GEO database (Fig. 3; Table IV). Among those, 59 lncRNAs, 779 mRNAs and 30 miRNAs were upregulated, whereas 49 lncRNAs, 455 mRNAs and 15 miRNAs were downregulated. Detailed information on these common significantly differentially expressed genes in ESCC tissues from GEO database can be found in Tables SI–SII.
ESCC tissues with co-differentially expressed genes identified in the GEO and TCGA databases
The ESCC patient tissues RNA sequencing results and their clinicopathological information were collected from TCGA database. Compared with the tumor and non-tumor tissue RNA sequencing results of patients with ESCC from TCGA, we identified a total of 889 differentially expressed lncRNAs (597 upregulated and 292 downregulated, 4,796 differentially expressed mRNAs (3,170 upregulated and 1,626 downregulated), and 438 differentially expressed mRNAs (299 upregulated and 139 downregulated) (Table IV). The Venn diagram in Fig. 4 presents the intersections of genes between the GEO and TCGA databases. A total of 81 co-differentially expressed lncRNAs, 39 miRNAs and 357 mRNAs were identified (Fig. 4 and Table V).
Figure 4.

Venn diagram demonstrating the intersections of genes between GEO and TCGA data. (A) The intersection of lncRNAs; (B) The intersection of miRNAs; (C) The intersection of mRNAs. GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; lncRNA, long non-coding RNA.
Table V.
ESCC-related intersection co-differentially expressed lncRNAs, miRNAs and mRNAs in GEO and TCGA.
| Genes | Co-differentially expressed lncRNAs, miRNAs and mRNAs |
|---|---|
| lncRNAs | ALOX12P2, AOC4P, BTN2A3P, C20orf166-AS1, C21orf62-AS1, CMAHP, CYP2D7, CYP4Z2P, DDX12P, DIRC3, DUSP5P1, ENST00000570167.1, ENST00000584492.5, FAM66A, FAM86HP, FAM86JP, FAR2P1, FIRRE, FOXD2-AS1, GUCY1B2, HAND2-AS1, HAVCR1P1, HCP5, HNRNPA3P1, IPW, LINC00176, LINC00341, LINC00346, LINC00472, LINC00476, LINC00663, LINC00689, LINC00887, LINC00889, LINC00950, LINC00982, LINC01001, LINC01105, LINC01588, LOC100128164, LOC100499484-C9ORF174, LOC101928316, LOC148696, LOC202181, LOC283856, LOC399815, LOC728743, MBL1P, MEG3, MIR4435-2HG, MIR503HG, MIR600HG, NONHSAT068116.2, NONHSAT075748.2, NONHSAT179718.1, NONHSAT198787.1, PART1, PCAT18, PSMG3-AS1, PTGES2-AS1, PVT1, PWAR5, PWARSN, RAMP2-AS1, RPLP0P2, SBF1P1, SLC26A4-AS1, SLC8A1-AS1, SMIM10L2A, SMIM10L2B, SNHG4, TCAM1P, TP73-AS1, UCA1, UG0898H09, XR_253656.2, XR_946740.1, ZFAS1, ZFP91-CNTF, ZNF300P1, ZNF542P |
| miRNAs | let-7c-5p, let-7g-3p, miR-101-3p, miR-101-5p, miR-106b-5p, miR-125a-5p, miR-130b-3p, miR-133a-3p, miR-135b-5p, miR-141-3p, miR-143-3p, miR-145-5p, miR-15b-3p, miR-15b-5p, miR-16-5p, miR-182-5p, miR-183-5p, miR-185-5p, miR-18a-5p, miR-195-5p, miR-200a-3p, miR-200b-3p, miR-200c-3p, miR-200c-5p, miR-205-5p, miR-20b-5p, miR-21-3p, miR-224-5p, miR-28-5p, miR-31-5p, miR-320a, miR-32-5p, miR-328-3p, miR-330-5p, miR-33a-5p, miR-425-5p, miR-484, miR-497-5p, miR-93-5p |
| mRNAs | ABCA8, ABCC8, ACACB, ACADL, ACADSB, ACTG2, ACVR2A, ADAMTSL1, ADCY2, ADCY5, ADCY6, ADGRD1, ADH1B, ADHFE1, AFF3, AGPS, ALAD, ALDH6A1, ALDH7A1, ANGPTL1, ANK2, AOX1, APLP1, AQP4, AR, ARHGDIG, ARHGEF6, ARRB1, ASPA, ASXL3, ATP1A2, ATP4A, ATP4B, AZI2, B3GAT1, B4GALNT2, BID, BIRC5, BMP3, BMP8B, BMPER, BMS1, C16orf89, C2orf40, C6, C7, CA4, CAB39L, CACNA2D2, CADM2, CADM3, CALM1, CASQ2, CCBE1, CCKAR, CCKBR, CD1E, CD44, CDC6, CDH19, CDH2, CDK6, CELF4, CFLAR, CGNL1, CHGA, CHGB, CHMP2B, CHRDL1, CHST11, CKB, CKM, CKMT2, CLCNKA, CLDN1, CLDN16, CNKSR2, CNN1, CNTFR, CNTN2, CNTN3, COL2A1, COL4A3, CPA2, CPEB1, CPEB3, CPLX2, CTNND2, CTSC, CUX2, CYBRD1, CYFIP2, CYP2U1, CYP4B1, DES, DHX36, DIRAS1, DLG2, DLG3, DNER, DPP10, DPP6, DPT, E2F2, E2F3, EDA, EDNRB, EFNA5, EIF4EBP2, ELOVL6, EME1, ENAM, ENPP5, EPHA5, EPHB1, ERBB4, ESPL1, ESRRB, ESRRG, ETNPPL, EXO1, FAM107A, FAR1, FAXDC2, FGA, FGF2, FGFR1, FGG, FNDC5, FRMD1, FXYD1, FZD4, GAB1, GAB2, GALNT2, GALNT6, GATA5, GC, GFRA1, GHRL, GIF, GKN1, GKN2, GNAQ, GPD1L, GPER1, GPM6A, GPR155, GREM2, GRIA1, GRIA3, GRIA4, GRIK3, GRIK5, GRIN2A, GSTM5, H2AFJ, H2AFX, HCFC2, HDC, HIPK2, HMGA2, HMGCS2, HMP19, HOXA10, HPN, HPSE2, HS6ST3, HSPB6, HSPB7, ICOS, ID4, IGF1R, IGF2BP1, IKBKE, IL1RAP, IL6ST, INPP5A, IQSEC3, IRS1, ITGA8, ITGB8, ITPR2, KAT2B, KCNB1, KCNE2, KCNJ10, KCNJ11, KCNJ16, KCNK2, KCNMA1, KCNMB2, KCTD8, KIAA0408, KIAA2022, KIF5A, KLF15, KSR1, LAMC2, LAMTOR3, LDB3, LIFR, LIPF, LMOD1, LONRF2, MAGI1, MAGI3, MAMDC2, MAOA, MAP3K13, MAP4K4, MAPK4, MAPT, MARVELD3, MASP1, MFAP5, MFSD4A, MME, MMP14, MOCS1, MT1M, MYH11, MYLK, MYO18B, MYOC, MYOCD, MYRIP, NBEA, NCAM1, NEGR1, NRXN1, NTN4, OAS2, OGN, OMD, P2RX2, PANX1, PCDH9, PCSK2, PDCD4, PDCD6IP, PDE1A, PDE2A, PDE7B, PDZRN4, PEBP4, PGA3, PGA4, PGA5, PGM5, PGR, PI16, PKHD1L1, PLAU, PLCXD3, PLN, PLP1, PML, PPP1R12B, PPP1R1A, PPP1R9A, PPP2R3A, PRICKLE2, PRIMA1, PRKAA2, PRKACB, PRKAR2B, PRKCB, PRSS1, PSAPL1, PSMB2, PSME4, PTGER3, PTGIS, PTGS1, PTPN2, PTPRN, RAB11A, RAB11FIP2, RAB2A, RAD51, RAG1, RANBP3L, RAP1A, RAPGEF2, RBL1, RBPMS2, RELN, RGN, RIC3, RIMS4, RNF125, RORC, RPRM, RPS6KA2, RPS6KA6, RSPO2, RYR2, S1PR1, SCARA5, SCG3, SCIN, SCN7A, SCUBE2, SEMA3E, SERPINA5, SESN3, SFRP1, SGCA, SH3GL2, SH3GLB1, SIGLEC11, SIX4, SLC1A2, SLC26A7, SLC2A1, SLC2A4, SLC5A7, SLC9A4, SLIT2, SLK, SORCS1, SORT1, SOX10, SOX4, SST, STMN1, STMN2, STUM, SYNPO2, SYT4, TACR1, TCEAL2, TCF3, TFDP2, TGFBR2, THRB, TMEM132C, TNFAIP3, TNFRSF10B, TNXB, TP53INP2, TRA2B, TRIM50, VAMP2, VIP, VIPR2, WASF3, WDR17, WISP2, XKR4, YWHAZ, ZBTB16, ZFP36, ZFP36L2, ZNF385B, ZNF471 |
ESCC, esophageal squamous cell carcinoma; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; lncRNA, long non-coding RNA.
Construction of ceRNA network
In order to determine whether the intersection of co-differentially expressed lncRNAs, miRNAs and mRNAs in GEO and TCGA (Table V) exist in competing endogenous regulating relationships, the intersected 81 lncRNAs, 39 miRNAs and 357 mRNAs were applied to construct the ceRNA network. The aforementioned 39 miRNAs target lncRNAs and mRNAs were then predicted based on MiRcode, miRanda and Targetscan. Subsequently, the intersection genes of the abovementioned miRNAs target predicted the lncRNAs and mRNAs to build the ceRNA network (Table V). The ceRNA network was visualized using Cytoscape software (version 3.0). The results revealed that there were 67 lncRNAs, 37 miRNAs and 80 mRNAs involved in the ceRNA network (Fig. 5). The detailed information of ceRNA network is presented in Table SIII.
Figure 5.

The lncRNA-miRNA-mRNA ceRNA network. Red, upregulated genes; green, downregulated genes; triangles, lncRNAs; squares, miRNAs; balls, mRNAs. lncRNA, long non-coding RNA.
Functional analysis of mRNAs in the ceRNA network
The present study identified 67 lncRNAs, 37 miRNAs and 80 mRNAs involved in the ceRNA network. Therefore, it may be suggested that the co-differentially expressed genes in the ESCC tissues that were included in the ceRNA network may play key roles in ESCC progression. Based on these suggestions, the present study investigated the potential biological regulatory functions of these 80 mRNAs that were involved in the ceRNA network via GO enrichment of functions and KEGG pathway analyses. The upregulated and downregulated intersected mRNAs were further analyzed. The results suggested that the most enriched GO function by upregulated mRNAs was ‘Regulation of biological process (GO:0050789)’. The most enriched GO function by downregulated mRNAs was ‘Single-organism cellular process (GO:0044763)’ (Fig. 6). The KEGG pathway analysis indicated 25 signaling pathways involved in regulating upregulated mRNAs, and the most enriched pathway was ‘MicroRNAs in cancer (hsa05206)’. In addition, 95 signaling pathways were involved in regulating downregulated mRNAs, and the most enriched pathway was ‘Insulin secretion (hsa04911)’. Some of these signaling pathways, such as the ‘Wnt signaling pathway’, were involved in the progression of ESCC (18), the ‘MAPK signaling pathway’ was revealed as a key pathway affecting esophageal carcinoma cell proliferation and apoptosis (19), and the ‘mTOR signaling pathway’ was involved in the progression of ESCC (20). Furthermore, ‘Bladder cancer, Non-small-cell lung cancer, Pathways in cancer and PI3K-AKT signaling pathway’ were also reported as cancer-associated signaling pathways (21–24) (Fig. 7).
Figure 6.

Top 20 enrichment of GO terms for mRNAs in the ceRNA network (the bar plot shows the enrichment scores of the significant enrichment GO terms). GO, Gene Ontology.
Figure 7.

Top 20 enrichment of pathways for mRNAs in ceRNA network (the bar plot shows the enrichment scores of the significant enrichment pathways).
Association between lncRNA signature and the clinical characteristics of patients with ESCC
The aforementioned 67 lncRNAs that were involved in the ceRNA network were selected and further investigated in order to identify the association between these key lncRNAs and TCGA database with the clinicopathological characteristics of the 312 patients with ESCC. The ESCC patients' clinicopathological characteristics included age, sex, race, tumor grade, TNM stage and lymphatic metastasis, and were obtained from TCGA database. The relevance analysis results suggested that 31 lncRNAs were significantly differentially expressed in ESCC patients with different clinicopathological characteristics (P<0.05). It was revealed that PTGES2-AS1, CMAHP, LINC00472, LINC01105, BTN2A3P, LOC728743, LINC00346, PSMG3-AS1, SBF1P1, LINC01588, DIRC3, RPLPOP2, SMIM10L2A, ZNF300P1 and TP73-AS1 were associated with tumor grade; LOC148696, HCP5, LOC148696, LINC00472, RPLP0P2, DDX12P, SMIM10L2A, PVT1, ZNF300P1, SMIM10L2B and HAND2-AS1 were associated with TNM stage; and PTGES2-AS1, FOXD2-AS1, LOC148696, TCAM1P, LINC00982, LINC00176, DIRC3, ALOX12P2 and SBF1P1 were associated with lymphatic metastasis in patients with ESCC. Furthermore, it was indicated that PART1 and LINC00341 may be associated with sex and race, respectively (Table VI).
Table VI.
Associations between lncRNA signature and ESCC patients' clinicopathological characteristics.
| Comparisons | Upregulated | Downregulated |
|---|---|---|
| Sex (male vs. female) | C20orf166-AS1, UG0898H09, PART1 | |
| Race (Caucasian vs. Asian) | ALOX12P2, DUSP5P1 | PART1, LINC00341, LINC00982, DIRC3 |
| Tumor grade | PTGES2-AS1, LINC01588, BTN2A3P, | PSMG3-AS1, CMAHP, LINC00472, LINC01105, |
| (GIII–IV vs. GI–II) | LOC728743, RPLPOP2, LINC00346 | SBF1P1, DIRC3, SMIM10L2A, ZNF300P1, TP73-AS1 |
| TNM stage (T3 + T4 vs. T1 + T2) | RPLP0P2, DDX12P, PVT1, HCP5 | LINC00472, LOC148696, SMIM10L2A, ZNF300P1, SMIM10L2B, HAND2-AS1 |
| Lymphatic metastasis (yes vs. no) | PTGES2-AS1, FOXD2-AS1, TCAM1P, LINC00176 | DIRC3, LINC00982, LOC148696, SBF1P1 |
ESCC, esophageal squamous cell carcinoma; lncRNA, long non-coding RNA.
Prognostic analysis of lncRNA expression levels and survival of patients with ESCC
Based on the RNA sequencing data and clinical information of the patients with ESCC from TCGA, a Kaplan-Meier survival analysis was used to identify the associations between the ceRNA network 67 key lncRNAs and overall survival time of the ESCC patients. The expression levels of these 67 key lncRNAs and ESCC prognosis data in TCGA database were synthetically calculated using a Cox proportional hazard regression model and it was revealed that there were 15 lncRNAs statistically associated with the overall survival rate (log-rank P<0.05). Among these 15 lncRNAs, 7 (ZNF300P1, TP73-AS1, UG0898H09, CMAHP, DIRC3, LOC148696 and SMIM10L2A) were negatively associated with the prognosis of patients with ESCC (P<0.05), and 8 (RPLPOP2, HCP5, PVT1, LINC01588, PTGES2-AS1, DDX12P, CYP2D7 and LOC399815) were positively associated with the prognosis of patients with ESCC (P<0.05) (Fig. 8).
Figure 8.

Kaplan-Meier survival curves for 15 lncRNAs associated with ESCC patients' overall survival time [horizontal axis, overall survival time (days); vertical axis, survival function]. ESCC, esophageal squamous cell carcinoma; lcRNA, long non-coding RNA.
RT-qPCR validation
Through comprehensive analysis of the aforementioned results, it was inferred that LINC00982, TP73-AS1, SMIM10L2A, PVT1 and FOXD2-AS1 may play important roles in ESCC progression. Therefore, these five lncRNAs were selected and their actual expression levels were detected in 30 patients with newly diagnosed ESCC and paired non-tumor esophageal epithelial tissue samples via RT-qPCR to assess the reliability and validity of the bioinformatics analysis results (Tables III and VII). The results revealed that LINC00982, TP73-AS1 and SMIM10L2A were downregulated, whereas PVT1 and FOXD2-AS1 were upregulated in ESCC tumor tissues (Fig. 9). The results suggested that the RT-qPCR validation in 30 newly diagnosed patients with ESCC and the aforementioned bioinformatics analysis results (Table VII) exhibited the same trends (Fig. 5).
Table VII.
Randomly selected lncRNAs with absolute FC>2, P<0.05.
| Name (lncRNAs) | Gene ID | Regulation | TCGA (mean FC) | GEO (mean FC) |
|---|---|---|---|---|
| LINC00982 | 440556 | Down | −17.539 | −3.991 |
| TP73-AS1 | 57212 | Down | −3.845 | −5.579 |
| SMIM10L2A | 399668 | Down | −7.850 | −4.852 |
| PVT1 | 5820 | Up | 7.193 | 9.997 |
| FOXD2-AS1 | 84793 | Up | 6.470 | 14.211 |
FC, fold change; TCGA, The Cancer Genome Atlas; GEO, Gene Expression Omnibus; lncRNA, long non-coding RNA.
Figure 9.
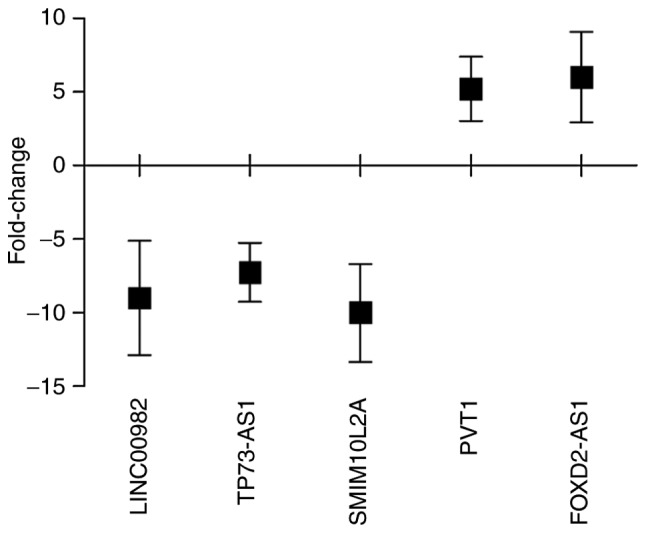
Box plot showing the median and quartiles of specific lncRNAs in donor samples. lncRNA, long non-coding RNA.
Subsequently, the association between the real expression levels of the aforementioned 5 lncRNAs and the clinical characteristics of the 30 newly diagnosed patients with ESCC was assessed. The results revealed that TP73-AS1, SMIM10L2A and PVT1 were significantly associated with TNM stage (P<0.05), whereas LINC00982, SMIM10L2A and FOXD2-AS1 were significantly associated with lymph node metastasis status (P<0.05). In addition, it was also revealed that TP73-AS1, SMIM10L2A and FOXD2-AS1 were correlated with the age and sex of the patients with ESCC (P<0.05; Fig. 10). The expression levels of the 5 lncRNAs were detected, and the results from the clinically relevant analysis and the aforementioned bioinformatics analysis (Tables V and VII) were similar, suggesting that the bioinformatics analysis used in the present study was credible.
Figure 10.

Box plot showing the association of the fold change in LINC00982, TP73-AS1, SMIM10L2A, PVT1 and FOXD2-AS1 expression with clinicopathological characteristics in 30 ESCC patients. ESCC, esophageal squamous cell carcinoma; y, years. *P<0.05.
Discussion
ESCC is a common malignant tumor originating from the mucosa of the esophagus at vulnerable sites, including the pharyngoesophageal junction, the part of the esophagus crossing with the posterior surface of the left bronchus, and the part of the esophagus passing through the diaphragmatic esophageal hiatus (25). Gastrointestinal endoscopy detection cannot identify all precancerous conditions or the early stages of ESCC. The incidence and mortality rate of ESCC in the general population are high, and a large number of patients are usually diagnosed late and have a poor prognosis (26). In addition, ESCC lymph node metastasis may be regional, bidirectional, continuous or jumping, and effective complete eradication methods are currently lacking (27). In order to improve survival rate and the detection and treatment of early-stage ESCC, more effective therapeutic tools are required, including the identification of novel diagnostic and prognostic biomarkers. Recent studies that have focused on the abnormal expression of lncRNAs have revealed a new investigative approach to the pathological changes observed in ESCC, and have also indicated that the search for novel biomarkers may hold promise for ESCC diagnosis and prognosis (28–30). The main concern is that small amounts of RNA sequencing samples may not accurately reflect the abnormal changes in lncRNAs when used as biomarkers that are associated with the diagnosis and prognosis of ESCC (31,32). Therefore, the ESCC-associated dysregulated lncRNA expression profiles should be identified based on large sample sizes in order to improve their accuracy and reliability as diagnostic and prognostic markers.
The development of high throughput RNA sequencing technologies has allowed thousands of dysregulated RNAs to be observed in a number of diseases (33–36). The present study identified abnormally expressed lncRNAs implicated in the pathogenesis of ESCC by synthetically analyzing RNA sequencing datasets obtained from the GEO and TCGA databases. A number of studies have proved that integration of multiple RNA sequencing datasets is a better method for enhancing the accuracy and reliability of results compared with using individual small samples (37). RNA sequencing allows reannotation, identification of differentially expressed RNAs in the GEO and TCGA databases, ceRNA network construction based on gene discovery, association between key lncRNAs and clinical characteristics of ESCC patients and survival analysis, in order to investigate the potential biomarkers for the diagnosis and prognosis of ESCC.
A multiple subset analysis and integrated bioinformatics approach was applied in order to identify dysregulated RNAs in ESCC in the present study. In order to increase the gene information accuracy, the RNA sequencing signals from the GEO database were matched to their chromosomal position using the GENCODE tool. All dataset samples were used in order to increase the sample numbers so as to avoid errors in the results and narrow the gene objects range. In addition, to avoid errors occurring from different ESCC individual tissues, only paired RNA sequencing sample datasets were included in the GEO database.
According to the integrated bioinformatics approach, it was revealed that there were 81 lncRNAs, 39 miRNAs and 357 mRNAs co-differentially expressed ESCC-associated intersection genes between the GEO and TCGA database. Among these 81 dysregulated lncRNAs, some have been indicated to exhibit significantly different expression levels in ESCC, including downregulated lncRNA HAND2-AS1 which may act as an anti-oncogene that can inhibit ESCC cell proliferation, migration and invasion (38). Dong et al (39) and Huang et al (40) have reported that downregulated MEG3 in ESCC tissues can inhibit ESCC cell growth and induce apoptosis. In addition, upregulated lncRNAs PVT1, TP73-AS1, UCA1 and ZFAS1 were also reported to be significantly differentially expressed in esophageal cancer, and also to be involved in regulating the progression of this disease (41–44). To the best of our knowledge, the functions of the remaining dysregulated lncRNAs in the development and progression of ESCC have not yet been reported.
Based on the theory that lncRNAs can regulate miRNA abundance by sequestration binding, acting as ‘miRNA sponges’, and the fact that miRNAs can bind to mRNAs and negatively regulate gene expression (45), the present study built an lncRNA-miRNA-mRNA ceRNA network according to their negative regulatory associations. Among 81 ESCC-associated intersection lncRNAs, a total of 67 key lncRNAs were included in the lncRNA-miRNA-mRNA ceRNA network. The ceRNA network construction can reveal the potential regulatory associations between lncRNAs, miRNAs and mRNAs in ESCC. According to the ceRNA network created in the present study, some of these 67 lncRNAs, such as FIRRE, FOXD2-AS1, IPW and LINC00472, were also reported as potential diagnostic and prognostic biomarkers in numerous human diseases, including ESCC (46–49). In order to investigate the potential regulatory functions of these 67 key lncRNAs, the present study further analyzed the 80 negatively regulated mRNAs in the ceRNA network, and revealed that some of those also play important roles in the progression of ESCC, including CYBRD1, EDA, ERBB4, FGF2 and IKBKE (50–52). Subsequently, the present study analyzed the 67 key lncRNAs in the ceRNA network and identified enrichment of the functions of 80 mRNAs that were indirectly involved in signaling pathways. The GO analysis results revealed that the enriched GOs primarily targeted the metabolism and cell biological process. The KEGG analysis revealed that certain pathways were also associated with cancer, such as the Wnt signaling pathway, the MAPK signaling pathway, non-small cell lung cancer, pathways in cancer and the PI3K-AKT signaling pathway (53). Therefore, the present bioinformatics analysis results revealed that these 67 key lncRNAs in the ceRNA network may be implicated in the progression of ESCC.
The associations between the expression levels of the 67 aforementioned key lncRNAs and the clinicopathological characteristics from TCGA database were analyzed, and it was revealed that 32 lncRNAs were associated with the characteristics of the 312 patients with ESCC. These lncRNAs were primarily associated with tumor grade, TNM stage and lymphatic metastasis status. Among these 31 lncRNAs, PVT1, FOXD2-AS1 and PART1 have been reported to be the key genes involved in lymphatic metastasis and invasion and as diagnostic biomarkers for ESCC (54–56). However, the functions of other lncRNAs have not yet been reported in association with ESCC progression. Therefore, the present study also analyzed the association between the 67 key lncRNAs in the ceRNA network and overall survival of patients with ESCC in TCGA database. It was revealed that 15 lncRNAs were associated with overall survival time. Among these 15 lncRNAs, only TP73-AS1 and PVT1 have been reported to be associated with survival in ESCC (42,57), whereas other lncRNAs have not been reported to date. The bioinformatics analysis results in the present study revealed potential lncRNAs biomarkers for the diagnosis, classification and prognosis of ESCC.
Subsequently, RT-qPCR validation of 5 randomly selected lncRNAs in 30 ESCC tissue samples was performed and the accuracy and credibility of the bioinformatics results were assessed. The results of the RT-qPCR validation were almost the same as the expression data in the GEO and TCGA databases. The present study then analyzed the association between these 5 randomly selected lncRNAs and the collected clinicopathological data of the patients with ESCC. The results also suggested that the results with these 5 lncRNAs were similar to the aforementioned bioinformatics analysis results. Therefore, the synthetic bioinformatics analysis results of the present study are reliable.
In summary, the present study has successfully identified specific ESCC-associated lncRNAs from large-scale samples through integrated analysis of RNA expression profile datasets of patients with ESCC from the GEO and TCGA databases. Differentially expressed lncRNAs and their potential functions in ESCC were investigated in the present study, along with specific ESCC-associated lncRNAs by different clinicopathological characteristics and overall survival time of patients with ESCC. Overall, these 67 lncRNAs are worth investigating further in terms of their applicability as biomarkers in the diagnosis, clinicopathological classification and prognosis of patients with ESCC.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The present study was supported by the Fundamental Research Funds for the Central Universities (grant no. lzujbky-2018-13), the National Natural Science Foundation of China (grant no. 81803188), and the Gansu Province Science and Technology Project (grant no. 1606RJYA270).
Availability of data and materials
The authors declare that all relevant raw data will be made freely available to any researchers who wish to use them for non-commercial purposes, whilst preserving any necessary confidentiality and anonymity. All data generated or analyzed during this study are included in this published article.
Authors' contributions
CYL conceived and designed the study. CYL and XHW performed the experiments. WWZ, JLW and JL analyzed and interpreted the results. JLX and XHW performed the patient tissue sample collection and quality control. CYL performed analysis and quality control, and was a major contributor to writing the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of the Gansu Wuwei Tumor Hospital. All patients provided written informed consent to participate in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Jiang C, Chen Y, Zhu Y, Xu Y. Systematic review and meta-analysis of the accuracy of 18F-FDG PET/CT for detection of regional lymph node metastasis in esophageal squamous cell carcinoma. J Thorac Dis. 2018;10:6066–6076. doi: 10.21037/jtd.2018.10.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M. Evaluation of a web-based App demonstrating an exclusionary algorithmic approach to TNM cancer staging. JMIR Cancer. 2015;1:e3. doi: 10.2196/cancer.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Wang Y, Zhao L, Sang S, Zhang L. Prognostic value of platelet-to-lymphocyte ratio in oncologic outcomes of esophageal cancer: A systematic review and meta-analysis. Int J Biol Markers. 2018 Apr 1; doi: 10.1177/1724600818766889. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 5.Quan J, Pan X, Zhao L, Li Z, Dai K, Yan F, Liu S, Ma H, Lai Y. LncRNA as a diagnostic and prognostic biomarker in bladder cancer: A systematic review and meta-analysis. Onco Targets Ther. 2018;11:6415–6424. doi: 10.2147/OTT.S167853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian T, Wang M, Lin S, Guo Y, Dai Z, Liu K, Yang P, Dai C, Zhu Y, Zheng Y, et al. The impact of lncRNA dysregulation on clinicopathology and survival of breast cancer: A systematic review and meta-analysis. Mol Ther Nucleic Acids. 2018;12:359–369. doi: 10.1016/j.omtn.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Zhang Y, Chen M, Shi L, Xu L, Zou X. A genome-wide analysis of long noncoding RNA profile identifies differentially expressed lncRNAs associated with Esophageal cancer. Cancer Med. 2018;7:4181–4189. doi: 10.1002/cam4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khadirnaikar S, Narayanan SP, Shukla SK. Decoding the LncRNA transcriptome of esophageal cancer: Identification of clinically relevant LncRNAs. Biomark Med. 2018;12:1083–1093. doi: 10.2217/bmm-2018-0032. [DOI] [PubMed] [Google Scholar]
- 9.Ji S, Yang W, Liu J, Zhao J, Chen L, Ni Q, Long J, Yu X. High throughput gene sequencing reveals altered landscape in DNA damage responses and chromatin remodeling in sporadic pancreatic neuroendocrine tumors. Pancreatology. 2018;18:318–327. doi: 10.1016/j.pan.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Krishnamurthy A, Ferl RJ, Paul AL. Comparing RNA-Seq and microarray gene expression data in two zones of the Arabidopsis root apex relevant to spaceflight. Appl Plant Sci. 2018;6:e01197. doi: 10.1002/aps3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Zhou L, Song Z, Xiong M, Zhang Y, Yang Y, Chen K, Chen Z. The identification of new biomarkers for bladder cancer: A study based on TCGA and GEO datasets. J Cell Physiol. 2019 Feb 18; doi: 10.1002/jcp.28208. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 12.Su H, Hu N, Yang HH, Wang C, Takikita M, Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ, et al. Global gene expression profiling and validation in esophageal squamous cell carcinoma and its association with clinical phenotypes. Clin Cancer Res. 2011;17:2955–2966. doi: 10.1158/1078-0432.CCR-10-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Ma C, Kemmner W. Wdr66 is a novel marker for risk stratification and involved in epithelial-mesenchymal transition of esophageal squamous cell carcinoma. BMC Cancer. 2013;13:137. doi: 10.1186/1471-2407-13-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen J, Yang H, Liu MZ, Luo KJ, Liu H, Hu Y, Zhang X, Lai RC, Lin T, Wang HY, Fu JH. Gene expression analysis of pretreatment biopsies predicts the pathological response of esophageal squamous cell carcinomas to neo-chemoradiotherapy. Ann Oncol. 2014;25:1769–1774. doi: 10.1093/annonc/mdu201. [DOI] [PubMed] [Google Scholar]
- 15.Guo Y, Chen Z, Zhang L, Zhou F, Shi S, Feng X, Li B, Meng X, Ma X, Luo M, et al. Distinctive microRNA profiles relating to patient survival in esophageal squamous cell carcinoma. Cancer Res. 2008;68:26–33. doi: 10.1158/0008-5472.CAN-06-4418. [DOI] [PubMed] [Google Scholar]
- 16.Jang HJ, Lee HS, Burt BM, Lee GK, Yoon KA, Park YY, Sohn BH, Kim SB, Kim MS, Lee JM, et al. Integrated genomic analysis of recurrence-associated small non-coding RNAs in oesophageal cancer. Gut. 2017;66:215–225. doi: 10.1136/gutjnl-2015-311238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Cao B, Yang W, Jin Y, Zhang M, He T, Zhan Q, Herman JG, Zhong G, Guo M. Silencing NKD2 by promoter region hypermethylation promotes esophageal cancer progression by activating Wnt signaling. J Thorac Oncol. 2016;11:1912–1926. doi: 10.1016/j.jtho.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Zhu Y, Zhu L, Chen X, Xu Y, Zhao Y, Shao Y, Li F, Jiang Y, Lu J, et al. Eupatilin inhibits the proliferation of human esophageal cancer TE1 cells by targeting the Akt-GSK3β and MAPK/ERK signaling cascades. Oncol Rep. 2018;39:2942–2950. doi: 10.3892/or.2018.6390. [DOI] [PubMed] [Google Scholar]
- 20.Jiang JH, Pi J, Jin H, Cai JY. Oridonin-induced mitochondria-dependent apoptosis in esophageal cancer cells by inhibiting PI3K/AKT/mTOR and Ras/Raf pathways. J Cell Biochem. 2019;120:3736–3746. doi: 10.1002/jcb.27654. [DOI] [PubMed] [Google Scholar]
- 21.He J, Zhao J, Zhu W, Qi D, Wang L, Sun J, Wang B, Ma X, Dai Q, Yu X. MicroRNA biogenesis pathway genes polymorphisms and cancer risk: A systematic review and meta-analysis. PeerJ. 2016;4:e2706. doi: 10.7717/peerj.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu M, Zhu S, Xiong S, Xue X, Zhou X. MicroRNAs and the PTEN/PI3K/Akt pathway in gastric cancer (Review) Oncol Rep. 2019;41:1439–1454. doi: 10.3892/or.2019.6962. [DOI] [PubMed] [Google Scholar]
- 23.Xiao YF, Yong X, Tang B, Qin Y, Zhang JW, Zhang D, Xie R, Yang SM. Notch and Wnt signaling pathway in cancer: Crucial role and potential therapeutic targets (Review) Int J Oncol. 2016;48:437–449. doi: 10.3892/ijo.2015.3280. [DOI] [PubMed] [Google Scholar]
- 24.Toren P, Zoubeidi A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (Review) Int J Oncol. 2014;45:1793–1801. doi: 10.3892/ijo.2014.2601. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi Y, Nishida T, Tsujii M, Tsutsui S, Yamamoto K, Isohashi F, Yamasaki M, Miyata H, Kato M, Yamada T, et al. Lymph node enlargement after definitive chemoradiotherapy for clinical stage I esophageal squamous cell carcinoma. BMC Cancer. 2014;14:706. doi: 10.1186/1471-2407-14-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng YF, Chen HS, Wu SC, Chen HC, Hung WH, Lin CH, Wang BY. Esophageal squamous cell carcinoma and prognosis in Taiwan. Cancer Med. 2018;7:4193–4201. doi: 10.1002/cam4.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mine S, Watanabe M, Kumagai K, Okamura A, Yuda M, Hayami M, Yamashita K, Imamura Y, Ishizuka N. Comparison of mediastinal lymph node metastases from adenocarcinoma of the esophagogastric junction versus lower esophageal squamous cell carcinoma with involvement of the esophagogastric junction. Dis Esophagus. 2019 Feb 22; doi: 10.1093/dote/doz002. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 28.Huang GW, Xue YJ, Wu ZY, Xu XE, Wu JY, Cao HH, Zhu Y, He JZ, Li CQ, Li EM, Xu LY. A three-lncRNA signature predicts overall survival and disease-free survival in patients with esophageal squamous cell carcinoma. BMC Cancer. 2018;18:147. doi: 10.1186/s12885-018-4058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Sun D, Wu K, Liu J, Zhao M, Li X, Xu Y, Li B. Genome-wide analysis of long non-coding RNAs in esophageal squamous cell carcinoma reveals their potential role in invasion and metastasis. Thorac Cancer. 2019;10:78–89. doi: 10.1111/1759-7714.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alaei S, Sadeghi B, Najafi A, Masoudi-Nejad A. LncRNA and mRNA integration network reconstruction reveals novel key regulators in esophageal squamous-cell carcinoma. Genomics. 2019;111:76–89. doi: 10.1016/j.ygeno.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Campbell JD, Liu G, Luo L, Xiao J, Gerrein J, Juan-Guardela B, Tedrow J, Alekseyev YO, Yang IV, Correll M, et al. Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA. 2015;21:164–171. doi: 10.1261/rna.046060.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins JE, Wali N, Sealy IM, Morris JA, White RJ, Leonard SR, Jackson DK, Jones MC, Smerdon NC, Zamora J, et al. High-throughput and quantitative genome-wide messenger RNA sequencing for molecular phenotyping. BMC Genomics. 2015;16:578. doi: 10.1186/s12864-015-1788-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozsolak F. Attomole-level genomics with single-molecule direct DNA, cDNA and RNA sequencing technologies. Curr Issues Mol Biol. 2016;18:43–48. [PubMed] [Google Scholar]
- 34.Lacaze É, Gendron AD, Miller JL, Colson TL, Sherry JP, Giraudo M, Marcogliese DJ, Houde M. Cumulative effects of municipal effluent and parasite infection in yellow perch: A field study using high-throughput RNA-sequencing. Sci Total Environ. 2019;665:797–809. doi: 10.1016/j.scitotenv.2019.02.155. [DOI] [PubMed] [Google Scholar]
- 35.Ge L, Liu S, Xie L, Sang L, Ma C, Li H. Differential mRNA expression profiling of oral squamous cell carcinoma by high-throughput RNA sequencing. J Biomed Res. 2015;29 doi: 10.7555/JBR.29.20140088. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao M, Zhong A, Patel N, Alur C, Vyas D. High throughput RNA sequencing utility for diagnosis and prognosis in colon diseases. World J Gastroenterol. 2017;23:2819–2825. doi: 10.3748/wjg.v23.i16.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma T, Liang F, Oesterreich S, Tseng GC. A joint bayesian model for integrating microarray and RNA sequencing transcriptomic data. J Comput Biol. 2017;24:647–662. doi: 10.1089/cmb.2017.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Y, Li S, Wang S, Rubegni P, Tognetti L, Zhang J, Yan L. Long noncoding RNA HAND2-AS1 inhibits cancer cell proliferation, migration, and invasion in esophagus squamous cell carcinoma by regulating microRNA-21. J Cell Biochem. 2019;120:9564–9571. doi: 10.1002/jcb.28233. [DOI] [PubMed] [Google Scholar]
- 39.Dong Z, Zhang A, Liu S, Lu F, Guo Y, Zhang G, Xu F, Shi Y, Shen S, Liang J, Guo W. Aberrant methylation-mediated silencing of lncRNA MEG3 functions as a ceRNA in esophageal cancer. Mol Cancer Res. 2017;15:800–810. doi: 10.1158/1541-7786.MCR-16-0385. [DOI] [PubMed] [Google Scholar]
- 40.Huang ZL, Chen RP, Zhou XT, Zhan HL, Hu MM, Liu B, Wu GD, Wu LF. Long non-coding RNA MEG3 induces cell apoptosis in esophageal cancer through endoplasmic reticulum stress. Oncol Rep. 2017;37:3093–3099. doi: 10.3892/or.2017.5568. [DOI] [PubMed] [Google Scholar]
- 41.Yang S, Ning Q, Zhang G, Sun H, Wang Z, Li Y. Construction of differential mRNA-lncRNA crosstalk networks based on ceRNA hypothesis uncover key roles of lncRNAs implicated in esophageal squamous cell carcinoma. Oncotarget. 2016;7:85728–85740. doi: 10.18632/oncotarget.13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zang W, Wang T, Wang Y, Chen X, Du Y, Sun Q, Li M, Dong Z, Zhao G. Knockdown of long non-coding RNA TP73-AS1 inhibits cell proliferation and induces apoptosis in esophageal squamous cell carcinoma. Oncotarget. 2016;7:19960–19974. doi: 10.18632/oncotarget.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou XL, Wang WW, Zhu WG, Yu CH, Tao GZ, Wu QQ, Song YQ, Pan P, Tong YS. High expression of long non-coding RNA AFAP1-AS1 predicts chemoradioresistance and poor prognosis in patients with esophageal squamous cell carcinoma treated with definitive chemoradiotherapy. Mol Carcinog. 2016;55:2095–2105. doi: 10.1002/mc.22454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi H, Liu Z, Pei D, Jiang Y, Zhu H, Chen B. Development and validation of nomogram based on lncRNA ZFAS1 for predicting survival in lymph node-negative esophageal squamous cell carcinoma patients. Oncotarget. 2017;8:59048–59057. doi: 10.18632/oncotarget.19937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang P, Guo Q, Gao Y, Zhi H, Zhang Y, Liu Y, Zhang J, Yue M, Guo M, Ning S, et al. Improved method for prioritization of disease associated lncRNAs based on ceRNA theory and functional genomics data. Oncotarget. 2017;8:4642–4655. doi: 10.18632/oncotarget.13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Q, Tai S, Wang J. Oncogenicity of lncRNA FOXD2-AS1 and its molecular mechanisms in human cancers. Pathol Res Pract. 2019;215:843–848. doi: 10.1016/j.prp.2019.01.033. [DOI] [PubMed] [Google Scholar]
- 47.Stelzer Y, Sagi I, Yanuka O, Eiges R, Benvenisty N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat Genet. 2014;46:551–557. doi: 10.1038/ng.2968. [DOI] [PubMed] [Google Scholar]
- 48.Zang Y, Zhou X, Wang Q, Li X, Huang H. LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem Biophys Res Commun. 2018;501:131–138. doi: 10.1016/j.bbrc.2018.04.194. [DOI] [PubMed] [Google Scholar]
- 49.Su C, Shi K, Cheng X, Han Y, Li Y, Yu D, Liu Z. Long noncoding RNA LINC00472 inhibits proliferation and promotes apoptosis of lung adenocarcinoma cells via regulating miR-24-3p/DEDD. Technol Cancer Res Treat. 2018;17:1533033818790490. doi: 10.1177/1533033818790490. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Lu T, Chen D, Wang Y, Sun X, Li S, Miao S, Wo Y, Dong Y, Leng X, Du W, Jiao W. Identification of DNA methylation-driven genes in esophageal squamous cell carcinoma: A study based on the cancer genome atlas. Cancer Cell Int. 2019;19:52. doi: 10.1186/s12935-019-0770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang W, Qu Y, Tan B, Jia Y, Wang N, Hu P, Wang J. Prognostic significance of preoperative IKBKE expression in esophageal squamous cell carcinoma. Onco Targets Ther. 2018;11:1305–1314. doi: 10.2147/OTT.S156818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maehara O, Suda G, Natsuizaka M, Ohnishi S, Komatsu Y, Sato F, Nakai M, Sho T, Morikawa K, Ogawa K, et al. Fibroblast growth factor-2-mediated FGFR/Erk signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis. 2017;38:1073–1083. doi: 10.1093/carcin/bgx095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Unger JM, Vaidya R, Hershman DL, Minasian LM, Fleury ME. Systematic review and meta-analysis of the magnitude of structural, clinical, and physician and patient barriers to cancer clinical trial participation. J Natl Cancer Inst. 2019;111:245–255. doi: 10.1093/jnci/djy221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li PD, Hu JL, Ma C, Ma H, Yao J, Chen LL, Chen J, Cheng TT, Yang KY, Wu G, et al. Upregulation of the long non-coding RNA PVT1 promotes esophageal squamous cell carcinoma progression by acting as a molecular sponge of miR-203 and LASP1. Oncotarget. 2017;8:34164–34176. doi: 10.18632/oncotarget.15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao J, Zhou C, Zhang J, Mo J, Ye Q, He J, Diao J. Upregulation of the long noncoding RNA FOXD2-AS1 predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Biomark. 2018;21:527–533. doi: 10.3233/CBM-170260. [DOI] [PubMed] [Google Scholar]
- 56.Kang M, Ren M, Li Y, Fu Y, Deng M, Li C. Exosome-mediated transfer of lncRNA PART1 induces gefitinib resistance in esophageal squamous cell carcinoma via functioning as a competing endogenous RNA. J Exp Clin Cancer Res. 2018;37:171. doi: 10.1186/s13046-018-0845-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Chen LP, Wang H, Zhang Y, Chen QX, Lin TS, Liu ZQ, Zhou YY. Robust analysis of novel mRNA-lncRNA cross talk based on ceRNA hypothesis uncovers carcinogenic mechanism and promotes diagnostic accuracy in esophageal cancer. Cancer Manag Res. 2018;11:347–358. doi: 10.2147/CMAR.S183310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant raw data will be made freely available to any researchers who wish to use them for non-commercial purposes, whilst preserving any necessary confidentiality and anonymity. All data generated or analyzed during this study are included in this published article.