

Abstract
Pre-mRNA splicing is regulated through multiple trans-acting splicing factors. These regulators interact with the pre-mRNA at intronic and exonic positions. Given that most exons are protein coding, the evolution of exons must be modulated by a combination of selective coding and splicing pressures. It has previously been demonstrated that selective splicing pressures are more easily deconvoluted when phylogenetic comparisons are made for exons of identical size, suggesting that exon size–filtered sequence alignments may improve identification of nucleotides evolved to mediate efficient exon ligation. To test this hypothesis, an exon size database was created, filtering 76 vertebrate sequence alignments based on exon size conservation. In addition to other genomic parameters, such as splice-site strength, gene position, or flanking intron length, this database permits the identification of exons that are size- and/or sequence-conserved. Highly size-conserved exons are always sequence-conserved. However, sequence conservation does not necessitate exon size conservation. Our analysis identified evolutionarily young exons and demonstrated that length conservation is a strong predictor of alternative splicing. A published data set of approximately 5000 exonic SNPs associated with disease was analyzed to test the hypothesis that exon size–filtered sequence comparisons increase detection of splice-altering nucleotides. Improved splice predictions could be achieved when mutations occur at the third codon position, especially when a mutation decreases exon inclusion efficiency. The results demonstrate that coding pressures dominate nucleotide composition at invariable codon positions and that exon size–filtered sequence alignments permit identification of splice-altering nucleotides at wobble positions.
Keywords: exon conservation, phylogenetics, splicing, alternative splicing, SNPs
INTRODUCTION
Splicing is a vital step in gene expression that relies on the correct recognition of exons and removal of introns in a process coordinated by the spliceosome. There are many factors that contribute to the identification of an exon and how efficiently it is spliced. Regulation of splicing and alternative splicing requires many trans-acting factors, such as SR and hnRNP proteins, as well as cis-acting elements, such as regulatory sequences within the intron or exon necessary for correct splice-site recognition (Nilsen and Graveley 2010). Some of these cis-acting factors include splice-site (ss) strength at the 5′ or 3′ end of an exon as well as splicing regulatory elements (SREs) to which trans-acting factors bind (Lin and Fu 2007; Venables 2007). Previous work demonstrated that the transition between exon inclusion and exclusion occurs within a very narrow window (Shepard et al. 2011; Busch and Hertel 2015). Mutations within the pre-mRNA sequence that alter how cis- and trans-acting factors cooperate to mediate efficient splicing can result in drastic changes in exon inclusion levels. Therefore, such positions within the pre-mRNA must be under evolutionary pressures to maintain optimal splicing signals needed for intron removal. In addition to ensuring correct splicing of the pre-mRNA, the sequence and identity of encoded amino acids are essential for a functional product. Thus, two sets of evolutionary pressures coexist that impact the generation of functional protein products: pressures to splice correctly and pressures to code for the correct amino acids.
Because of the degenerate nature of the genetic code (Sonneborn 1965), the third position of a codon (wobble position) permits variability within the human genome. It is possible that important splicing pressures could be overrepresented within the wobble position, the most frequent location of synonymous mutations. Synonymous mutations are silent mutations that do not alter the protein sequence but are not always silent with respect to how an exon is spliced or, by extension, how a protein is folded (Gingold and Pilpel 2011). Synonymous mutations have been implicated in the modulation of splicing, translation (Drummond and Wilke 2008; Gingold and Pilpel 2011), and mRNA stability (Duan et al. 2003). Previous studies have systematically characterized the contribution of splicing evolutionary pressures at wobble positions through the identification of synonymous mutations that alter exon inclusion efficiencies (Mueller et al. 2015). The results from these studies demonstrated that splicing changes could not be reliably predicted by simply correlating exon inclusion efficiencies with base-wise conservation of nucleotides across 46 vertebrates (phyloP). However, filtering species alignments based on exon length conservation significantly improved the predictions of splicing outcomes, thus providing a potential approach to deconvoluting splicing from coding pressures (Mueller et al. 2015). Based on these findings, we hypothesized that exon size–filtered sequence alignments may improve the identification of nucleotides that have evolved to mediate efficient exon ligation. To address this hypothesis, an exon size conservation database was generated to report on exon features such as splice-site strength, exon/intron lengths, exon size variation, and the length and sequence conservation across 76 species.
The exon size conservation database permitted the identification of exons that are not only sequence-conserved, but also size-conserved. In general, highly size-conserved exons are always sequence-conserved. Furthermore, the database permitted the identification of exons that are unique to humans/primates and that may be evolutionarily young. Using a published data set of approximately 5000 disease-associated exonic SNPs (Soemedi et al. 2017) allowed for improved splice predictions when nucleotide variations are located at the third codon position. These results demonstrate that coding pressures dictate the nucleotide composition at inflexible codon positions and that the conserved third codon positions frequently upholds splicing pressures.
RESULTS
Architecture of the human genome within the conservation database
Recent studies have highlighted the importance of exon size–filtered alignments in identifying exon conservation between human and other vertebrate species (Mueller et al. 2015). Based on this observation, an exon size conservation database was generated using multiple sequence alignment (multiz100way) of 99 species compared to human exons. However, some of the assemblies in the multiz100way were older and the associated gene annotations were not reliable. Therefore, of the initial 99 species, only 76 vertebrate species were used for aligning length-filtered exons.
A human-centric model to exon length conservation was used to allow for downstream analysis of human disease SNPs. Therefore, for every annotated human exon, our database reports the identity and number of species that conserve the length of the human exon. In addition, the database also reports on the phyloP sequence conservation scores based on multiz100way alignments (Pollard et al. 2010), the exon position within the human gene, exon length, the type of exon (first, internal, last, or single), flanking intron lengths, and 5′ss and 3′ss scores.
Exon length and splice-site score analyses were carried out for 184,796 exons (18,225 genes), representing the exon categories first, internal, last, and single exons (intronless genes). A large proportion of single exons are ∼1000 nt in length (Fig. 1A). First exons have on average shorter exon lengths (Fig. 1B) than last exons (Fig. 1G) and exhibit strong 5′ss scores (MES > 8) (Fig. 1C). Last exons have a larger distribution of exon lengths (Fig. 1G) and generally harbor strong 3′ss scores (Fig. 1H). As expected, internal exons (Fig. 1D), which make up the largest class of the exon types, show a very tight size distribution around 50–250 nt, with an average exon size of 120 nt. This internal exon length distribution is consistent with previous findings that demonstrated that the optimal exon length for efficient splicing is between 50 and 250 nt (Berget 1995; Sterner et al. 1996; Sakharkar et al. 2004;). In addition, internal exons have a tight distribution of 5′ss (Fig. 1E) and 3′ss scores (Fig. 1F), with average scores around 8 MES, consistent with previous findings (Shepard et al. 2011; Busch and Hertel 2015).
FIGURE 1.

Architecture of the human genome. Exons categorized into four distinct types (single, first, internal, and last exons) contain varying exon length distributions but maintain strong splice-site scores. (A) Exon length distribution of single exons. (B) Exon length distribution of first exons. (C) 5′ss score of first exons. (D) Exon length distribution of internal exons. (E) 5′ss score of internal exons. (F) 3′ss score of internal exons. (G) Exon length distribution of last exons. (H) 3′ss score of last exons.
Overall, these findings demonstrate that the majority of first, internal, and last exons can be classified as containing strong 3′ss and 5′ss scores. Furthermore, first exons generally maintain an average distribution of exon lengths (Fig. 1B) comparable to internal exons (Fig. 1D), whereas last exon size distributions are much broader and are characteristically longer in length (Fig. 1G).
Distribution of length-conserved exons across 76 species
The exon size or length conservation score defined here as the Ultra-In score (see Materials and Methods), was obtained by comparing 76 vertebrate species to the human reference genome and determining for each exon the number of species that maintain the human exon size. This score ranges from 0, representing a unique exon size in human, to 76, which represents exon size conservation across all species evaluated. For the purpose of this analysis, low length conservation is defined as an Ultra-In score of <10 (10 or less species with exon length conservation), moderate length conservation is defined by a score between 10 and 40, and high length conservation as a score >40. An analysis of size conservation frequencies for all exons highlights the emergence of two general populations (Fig. 2A). These two populations are those with low exon length conservation, observed only in a small number of species, mainly primates (data not shown), and those with high exon length conservation across a larger number of vertebrate species.
FIGURE 2.

Distribution of exons across 76 length-conserved species. The length conservation score is defined as the number of species with length-conserved exons when compared to human. The number of exons in each length conservation bin is plotted for (A) all types of exons, (B) all exons between 1 and 49 nt in length, (C) exons between 50 and 250 nt in length, and (D) exons longer than 250 nt in length.
When categorizing exons by length, striking differences are observed for short exons of <50 nt (Fig. 2B), average-sized exons between 50 and 250 nt in length (Fig. 2C), or long exons >250 nt (Fig. 2D). The exon length cutoffs used are based on what is known to be an efficient nucleotide length for the recognition of exons by the spliceosome (Berget 1995). The distribution of length-conserved exons seen in Figure 2A is mainly reproduced by the 50- to 250-nt exon size group (Fig. 2C). However, a large proportion of the low length-conserved exon population is no longer present in the 50–250 nt exon size bin. Rather, this low length-conserved population is overrepresented in the exon size group >250 nt in length (Fig. 2D). The fact that the majority of exons between 50 and 250 nt are highly length-conserved suggests that optimal exon size is an important evolutionary feature.
Length-conserved exons are sequence-conserved
It is unknown to what degree exon length conservation and sequence conservation covary or evolve independently. To evaluate the association between sequence and architectural features, the average exon phyloP score (representing sequence conservation) and average exon length conservation score were correlated for all internal exons within each gene. This correlation demonstrates that for internal exons, there is strong positive linear relationship between length and sequence conservation (Fig. 3). The majority of genes fall within a population characterized by high exon length and high exon sequence conservation (length conservation score > 40, phyloP score > 3), whereas fewer genes are either not exon length– and/or exon sequence–conserved. Therefore, genes that consist of internal exons with high sequence conservation also demonstrate high exon length conservation, but not all highly size-conserved exons are sequence-conserved. This strong correlation most likely represents a convergence between size and sequence over the evolutionary lineage of an internal exon in defining optimal length and sequence elements for efficient exon recognition. In contrast to internal exons, first and last exons display a weak correlation between sequence and length conservation (Supplemental Fig. 1A,C). This is consistent with what would be expected for terminal exons, given that their variability may likely be dictated by their noncoding nature, allowing for targeted regulation by the capping and polyadenylation machineries, as well as trans-acting factors.
FIGURE 3.
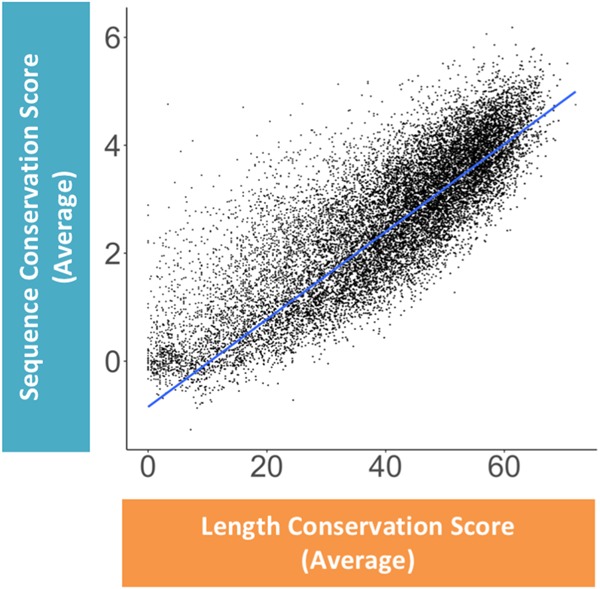
Correlation between average length and sequence conservation of internal exons. The correlation between the average sequence conservation score and the average length conservation score of all internal exons within a single gene is shown. Each dot denotes a single gene represented by the average sequence and length conservation score for all its internal exons. The blue line represents a regression fit to the data, R = 0.84.
Distribution of length conservation across grouped species
To analyze the correlation between exon size conservation and exon recognition features, internal exons were binned based on their Ultra-In length conservation score. The average exon length is longer for exons that have poor length conservation (Supplemental Fig. 2A). In addition, the sum of splice-site scores was observed to be lower for the low length-conserved group 0–10 (Supplemental Fig. 2B). Exon length and sequence conservation are highly correlated with minimal sequence conservation overlap between the two extreme categories 0–10 and 70–76 (Supplemental Fig. 2C). These observations support the conclusion that length and sequence conservation are highly correlated evolutionary features.
Primates display the greatest similarity in exon architecture with humans
Using exon length conservation as a measure of reassessing species closeness, species with similar exon lengths when compared to human were identified. The number of times an exon had the same length in each of the 76 species located within the 0–10 group relative to human was plotted. The assumption made in this analysis is that species that are more closely related to humans should exhibit the highest representation of exon length conservation in the 0–10 category in which minimal overall length conservation is seen. Highest exon size similarities were observed for primates (Fig. 4) with a striking drop-off from marmoset to bush baby. Marmosets are small monkeys, considered to be part of the New World monkeys, appearing ∼30 million years ago. Bush babies are considered to be prosimians, primate-like mammals that appeared much earlier, ∼60 million years ago (Siepel 2009). The difference between the evolutionary appearance between these two mammals could explain the reduction in exon size variation that is seen, given that New World monkeys most likely evolved from prosimians (Siepel 2009). Interestingly, this exon size conservation analysis is highly consistent with what is known about the phylogeny of primates. Chimps are the most closely related primate to humans, followed by gorillas and orangutans and lesser apes such as the gibbon, the Old World monkeys, the baboons, and New World monkeys such as the squirrel monkey and marmoset (Siepel 2009). In summary, this analysis demonstrates that exon size conservation is a genomic feature that significantly contributes to evolutionary trends in mammals. Exon size conservation could therefore participate in evolutionary fitness of the species overall and can be used as a genomic feature to retrace the lineage of species creation.
FIGURE 4.

Exon size comparison between human and other species. The number of size-conserved internal exons within the 0–10 Ultra-In group of exons are reported for each of the 76 species used in the exon size database. Species names are listed on the x-axis and count of exons per each species is depicted on the y-axis.
Further investigation into the length-conserved exons within primates (10,491 exons) demonstrated that the majority of these exons are coding genes. Only a small fraction (59 exons) appear to be ALU-derived, whereas the majority of these exons are of an unknown origin. Interestingly, the genes from which these 59 ALU exons were derived appear to be involved in the regulation of leukocyte-mediated immunity.
Distribution of intron lengths flanking internal exons of different size conservation
It is known that the length of introns flanking internal exons defines how an exon will be recognized and spliced (Fox-Walsh et al. 2005), either through exon or intron definition. To understand the variation of flanking intron lengths around exons with high length and sequence conservation, human upstream and downstream intron lengths were recorded and analyzed. Four main intron length categories were designated based on the length transition between intron and exon definition. This length of an intron was defined by the transition from intron definition to an exon definition model of splice-site recognition, based on previous findings (Fox-Walsh et al. 2005). An internal exon can be flanked on either side by introns that are both short in length (<250 nt), designated as SS (“short short”) exons, or a short upstream intron and a long downstream intron (>250 nt) designated as SL (“short long”) exons. Internal exons can also be flanked by long introns designated as LL (“long long”) exons or a long upstream and short downstream intron designated as LS (“long short”) exons (Fig. 5A). A correlation of internal exons and their flanking intron size clearly highlights the exon definition population of internal exons (LL) and the population of exons that are at least partially intron-defined (LS, SL, SS). Interestingly, this genome view demonstrates an underrepresentation of flanking intron sizes that are at the transition point between exon and intron definition, and illustrates a remarkable demarcation for minimal intron size (∼75 nt) (Fig. 5B). Using these four exon architectural groups, the sum of the 5′ss and 3′ss scores was correlated. On average, significantly stronger splice-site scores are observed when flanking introns are long compared to splice-site scores of internal exons flanked by SS introns (Fig. 5C). This observation suggests that exons undergoing exon definition require stronger splice sites on average than those that undergo intron definition. In addition, internal exons flanked by LL introns were on average significantly longer in length by ∼24 nt than those flanked by LS, SL, and SS introns (Fig. 5D). The average length of these exons, however, was still within the optimal exon length window of 50–250 nt. Regardless, this exon size difference could represent the need for LL exons to provide additional trans-acting factor binding sites to ensure efficient spliceosomal recognition.
FIGURE 5.

Distribution of intron lengths flanking internal exons. (A) Cartoon depiction of the four intron length-defined exon groups. Exons that are flanked by (SS) short short, (SL) short long, (LL) long long, or (LS) long short introns. Gray boxes represent flanking exons, red boxes represent internal exons, and black lines represent introns of various lengths. The numbers above the introns indicate length definitions. (B) Correlation between upstream and downstream intron lengths flanking internal exons. Each quadrant is defined by the length of the upstream and downstream intron length, respectively. The red dotted lines depict the intron length of 250 nt, the established transition from intron to exon definition. Each black dot represents a single internal exon. The four exon groups were correlated with (C) the sum of the 5′ss and 3′ss scores, (D) the internal exon length, (E) the length conservation score, and (F) the sequence conservation score. All intron length comparisons are statistically significant (P < 0.05) unless marked by “ns” (not significant).
When exon length conservation was evaluated for the four intron groups, exons flanked by LL introns demonstrated significantly higher size conservation than exons flanked by SS introns (Fig. 5E). Similarly, exons flanked by LL introns are characterized by significantly higher sequence conservation when compared to exons flanked by SS introns (Fig. 5F). Interestingly, the distribution of intron lengths downstream from first exons is noticeably broader than that observed for internal exons (Supplemental Fig. 3A), whereas last intron size did not reveal differences in size distribution (Supplemental Fig. 3B). These results demonstrate that first introns, consistent with previous work (Park et al. 2014), are in general longer than subsequent introns.
Distribution of splice-site scores across internal exons
Splice-site scores mediate efficient exon recognition. As previously mentioned, the shift between inclusion and exclusion of an exon is demarcated by a distinct switch in splice-site strength designation (MES of 7 to 8) (Shepard et al. 2011), in which, based on these findings, a MES of 8 was used as a cutoff for determining splice-site groups. Therefore, exons were defined as those that either have a strong 3′ss and a strong 5′ss “strong strong” (SS), “strong weak” (SW), “weak strong” (WS), or “weak weak” (WW) splice-site scores (Supplemental Fig. 4A). Significantly longer exon lengths were identified when flanking splice-site scores were weak (WW) (Supplemental Fig. 4B). Furthermore, high splice scores correlate with increased exon length conservation (Supplemental Fig. 4C) and nucleotide sequence conservation (Supplemental Fig. 4D). These observations suggest that the exon/intron architecture modulates the required pattern of splice-site strengths across an exon. On average, exons with WW splice sites require longer lengths to aid in efficient recognition, potentially through increasing trans-acting factor binding. The positive correlation between length and sequence conservation is likely to reflect the importance of conserving splice-site sequences for effective recognition and splicing of internal exons.
Length and sequence conservation across alternatively spliced exons
To understand to what degree alternative splicing correlates with exon length and sequence conservation, the exon conservation database was correlated with the EST HEXEvent database, a resource that reports exon inclusion levels and alternative 3′ss or 5′ss selection preferences for all annotated human exons (Busch and Hertel 2013). Interestingly, the correlation between exon size conservation and exon skipping frequencies (Fig. 6A) is slightly more predictive than the correlation between sequence conservation and exon skipping frequencies (Fig. 6B). No significant correlations were observed between length or sequence conservation across alternative 3′ss and 5′ss usage events (data not shown). These results demonstrate that low levels of exon inclusion can be predicted by low exon length and sequence conservation score. We conclude that exon length conservation is an equivalent or slightly improved predictor of alternative exon inclusion than exon sequence conservation.
FIGURE 6.

Correlation between the frequency of alternative exon skipping and exon length or sequence conservation. (A) Correlation between exon length conservation (Ultra-In score) and the frequency of exon skipping (Pearson correlation coefficient: −0.484, P-value: 1.5 × 10−97). (B) Correlation between exon sequence conservation (phyloP score) and the frequency of exon skipping (Pearson correlation coefficient: −0.327, P-value: 1.5 × 10−42). Each black dot represents a skipped exon. X-axis scale: 0 = no exon skipping, 1 = 100% exon skipping.
To investigate whether individual exons are under selection, independent from other exons within the same gene, the average exon length conservation score for an entire gene was compared between a group of genes that harbor at least one alternatively spliced exon and a group of genes that do not harbor an alternatively spliced exon. Interestingly, genes with alternative exons are characterized by higher average exon size conservation (Supplemental Fig. 5), suggesting that alternatively spliced exons are embedded within genes that contain exons of high size conservation. Considering the correlation between alternative exon inclusion and exon size conservation (Fig. 6A), these results support the notion that at minimum alternatively spliced exons are under independent selection pressures. For example, exon four of the TARS (Threonyl-tRNA Synthetase) gene is a skipped exon with poor size and sequence conservation (scores of 2 and −0.05, respectively). However, the average exon length conservation score for the entire gene is 61, and the average sequence conservation score is 3.9. Therefore, the TARS gene is highly size- and sequence-conserved, with a skipped exon that is poorly size- and sequence-conserved. Although this trend of divergent alternative exon size conservation is evident for alternative exons that are characterized by low inclusion levels (<10% inclusion), high inclusion alternative exons (>40% inclusion) display different size conservation features. For example, exon 4 of the DBT (Dihydrolipoamide Branched Chain Transacylase E2) gene is an alternatively spliced exon with high size and sequence conservation (scores of 71 and 4.8, respectively). The average exon length conservation score for the entire gene is 51 and the average sequence conservation score is 3.3. In this case, the alternative exon displays a greater size and sequence conservation when compared with the remaining DBT exons, suggesting that the alternative exons experienced increased selective pressures when compared with the other exons of the gene. These data and examples support the idea that alternative exons appear to evolve as independent units within a gene.
Using the exon conservation database to predict splice-altering SNPs
Previous studies have suggested that ∼20% of disease-associated alleles alter splicing (Lim et al. 2011). Recently, the splicing outcome of 5132 disease-associated SNPs was reported using an exon trap model (Soemedi et al. 2017). The authors generated wild-type (WT) and SNP versions of each analyzed exon and tested their effects on exon inclusion using a reporter assay. The published data set provided the opportunity to test whether the exon size–filtered conservation approach described above could identify SNPs that induce splicing changes. Given the discussion about the coevolution of coding and splicing features, such predictions would be expected to be most accurate for SNPs located at the wobble position. Thus, high nucleotide conservation at wobble positions of size-conserved exons could be predictive of splice-mediating positions.
Using the splicing SNP data set (Soemedi et al. 2017) as a starting point allowed for testing whether exon size–filtered sequence alignments could be used as a method for splicing prediction. The distribution of the data within the SNP database can be divided into two groups: SNPs that occur at the exon/intron boundary (or junction) and those that occur within the exon (nonjunction). The junction and nonjunction data can be further subdivided by the location of the SNP within the context of a codon to differentiate between SNPs that lead to protein-coding defects or SNPs that occur within the wobble position. Most of the 5132 disease-associated SNPs analyzed represent nucleotide changes that lead to amino acid changes, given that the majority of SNPs evaluated occur within position one and two of the codon (Supplemental Fig. 6A). Most of the 5′ss SNPs (those located at the exon/intron junction of the 5′ss) alter exon inclusions levels consistent with predicted changes in splice-site score (Supplemental Fig. 6B), confirming that nucleotide alterations at splice sites impact splicing efficiency through changes in the complementarity between the pre-mRNA and spliceosomal factors. More importantly, this junction analysis demonstrates that the SNP splicing data generated through exon trap experimentation conforms to expectations that a mutation leading to altered splice-site strengths results in altered exon inclusion levels.
To predict the impact a nonjunction SNP has on exon splicing, the sequence conservation of each SNP was evaluated within an upstream and downstream 5-nt window. Each SNP was then categorized depending on its reading frame location (first, second, or wobble position) and whether the SNP was ever observed in other species with size-conserved exons. A SNP was defined as a “mutation not important” if that SNP was present alone in other exon size-conserved species, but no other nucleotide changes were seen within the flanking 5 nt. These SNPs are considered not important for splicing because they are seen within size-conserved exons in other species. The second category of SNPs was defined as “mutation not observed,” in which a SNP is never seen across all the exon size–conserved species. Such nucleotide invariability suggests that the nucleotide identity at the SNP position is evolutionarily important and could be essential for pre-mRNA splicing.
The third category of SNPs are those that have “SNPs with covariance.” For this category, a SNP was only seen in exon size–conserved species when other nucleotide variations were present within 5 nt upstream of or downstream from that SNP. These additional nucleotide changes could be important for splicing, as they may act to compensate for defects caused by a single mutation, as previously described (Mueller et al. 2015). Close to 17% of nonjunction SNPs changed exon inclusion by >10%, the chosen splice difference significance cutoff. Of these splice-altering SNPs, 60% reduced exon inclusion (as calculated by negative delta percent spliced in [dpsi] values). The remaining 40% had positive dpsi values, indicating that the SNP increased exon inclusion. As a control, SNPs that resulted in exon inclusion changes of <0.2% were used as a “no change” control group. For each of these splice effect groups (exclusion, inclusion, no change/control) the codon position of the queried SNP was determined (first, second, or wobble position) before each SNP was categorized at the evolutionary level as defined above. The major evolutionary category of SNPs at nonjunction sites, regardless of codon position, was the “mutation not observed” category, suggesting that these SNP positions harbor overlapping splicing and coding pressures (Fig. 7A; Supplemental Fig. 7). The wobble position, however, revealed the greatest variability between the three evolutionary SNP categories, with the expected higher percentage of “mutation not important” and “SNPs with covariance” categories when compared to their observed frequency at codon positions one and two (Supplemental Fig. 7A,B).
FIGURE 7.

Splice-altering SNPs at the wobble position are selected against. The evolutionary conservation of exonic SNPs at wobble positions are compared between the delta psi groups that lead to increased exon exclusion, increased exon inclusion, or the control group that do not change exon inclusion levels. The relative representation of “Mutation-Not-Important” (red bars), “Mutation-Not-Observed” (blue bars), or “SNPs-With-Covariance” (green bars) are shown for (A) nonjunction exonic positions and (B) junction exonic positions.
To test whether splice-altering SNP positions are enriched for any of the three evolutionary categories, their relative representation was compared between those SNPs that lead to exon exclusion and the control group or those SNPs that increase exon inclusion and the control group. SNP positions at the nonjunction wobble site that reduce exon inclusion were enriched for the “mutation not observed” category and selected against the “mutation not important” group (Fig. 7A). These observations are consistent with the notion that splice-altering nucleotide changes are selected against, especially when the nucleotide change results in exon skipping. Interestingly, the conservation features for SNP positions that result in increased exon inclusion are quite different. The most prominent enrichment is seen for the “SNPs with covariance” category (Fig. 7A). It is possible that this category represents events of local compensatory nucleotide changes that reestablish efficient splicing. Qualitatively identical, but quantitatively more striking selection trends are observed for junction SNPs at wobble positions (Fig. 7B), reinforcing the interpretation that splice-altering SNPs are identifiable using the exon size–filtered phylogenetic conservation approach.
Using the published exon trap data set (Soemedi et al. 2017), it was possible to determine whether interrogated SNPs change the amino acid sequence, whether they create premature stop codons (PTCs), or whether they are synonymous nucleotide alterations. The first unanticipated observation regarding the disease-associated SNP data set was the fact that wobble position entries are underrepresented (Supplemental Fig. 8A), even for those SNPs that induce splicing changes of >10% (Supplemental Fig. 8B–D). Thus, it appears that disease-associated SNPs at wobble positions that alter splicing are selected against in the data set.
Furthermore, many of the “mutation not observed” SNPs either induce a PTC or they change the encoded amino acid (Supplemental Fig. 8B). Interestingly, SNPs that reduce exon inclusion display a greater proportion of nucleotide changes that create stop codons, suggesting that the loss of splicing could represent a molecular mechanism to avoid the incorporation of a PTC-containing exon. This trend is also observed in the “SNPs with covariance” group (Supplemental Fig. 8C). Remarkably, SNPs that increase exon inclusion in that group display an inverse relationship, strengthening the notion that splicing assists in the avoidance of PTC containing exons. In summary, the use of exon size–filtered sequence alignments allows for improved identification of splice-altering SNPs and, moreover, assists in predicting the direction of splicing changes.
DISCUSSION
The hypothesis that exon size–filtered species alignments improve the identification of nucleotides evolved to mediate efficient exon ligation was tested. The generation of the exon size conservation database allowed for the identification of fundamentally important architectural parameters of the human genome. The majority of internal exons are between 50 and 250 nt long and most exons harbor strong 5′ss and 3′ss scores.
Displaying exon size conservation across 76 species allowed for the identification of two diverse exon populations (Fig. 2A). One population of high length-conserved exons was mainly represented by internal exons of length 50–250 nt (Fig. 2C; Supplemental Fig. 9B) and a second population of low length-conserved exons that were largely represented by first and last exons longer in length (exons >250 nt) (Supplemental Fig. 9A,C). These results demonstrate that optimal exon size (50–250 nt) is an evolutionarily conserved feature. Terminal exons, because of their nature of being mostly noncoding, would be expected to represent the greatest variability in length conservation, as necessitated by transcription, translation, and other regulatory controls.
Single exons also reveal unique size characteristics (Fig. 1A). The majority of single exons demonstrated low length conservation (<10) (Supplemental Fig. 9D). Interestingly, 30% of single exons are olfactory genes. These genes are characterized by low sequence conservation and low length conservation (60% in 0–10 category, 40% in 10–40 category). Of the remaining single-exon genes, ∼70% percent are involved in signal transduction pathways and metabolic processes, representing their importance for species survival. Previous studies have suggested that intronless genes have appeared relatively recently (Shabalina et al. 2010), and their emergence could be due to gene duplication or retroposition of mRNA (Marques et al. 2005). The overall low length-conserved nature of these intronless genes could be explained by the burst of retroposition during the sudden emergence of primate evolution (Marques et al. 2005). It is also possible that these intronless genes could have formed through the “intron late” model or through exon fusion, with their lack of introns allowing for rapid gene processing to achieve a certain level of expression (Chen et al. 2002). Intronless genes have been shown to accumulate in the cytoplasm (Lei et al. 2011), yet their lack of engagement with the splicing machinery suggests their nuclear export is not mediated by splicing-dependent export pathways (Lei et al. 2011). This independence could explain how intronless genes achieve appropriate levels of expression, unaided by the association between splicing and nuclear export.
A strong correlation between exon length and exon sequence conservation was identified (Fig. 3; Supplemental Fig. 1B), suggesting that exons have evolved to optimize sequence and length, presumably satisfying coding pressures and splicing pressures alike. For every given exon within a gene, there are varying degrees of sequence and length conservation. As illustrated for alternatively spliced exons, one exon in a gene may have very high sequence and size conservation, whereas the flanking downstream exon may have very low sequence and size conservation. However, these differences in exon size and sequence conservation are not only unique to alternatively spliced exons. For example, CPXM1 is a gene that potentially encodes for a protein associated with members of the membrane-bound carboxypeptidase family, which may be important for cell–cell interactions. Exon two of CPXM1 has a low phyloP score of 0.53 and a very low length conservation score of 1. However, within the same gene, exon 12 has a high phyloP score of 4.39 and a high length conservation score of 66. Another example is the gene CPSF, an important protein component of the polyadenylation machinery. Exon two has a high phyloP score of 4.3 but a low length conservation score of 10, whereas exon three has a low phyloP score of 0.62 and a moderately high length conservation score of 39. In combination with the observations made for alternatively spliced exons, these examples demonstrate great variation between sequence and length conservation of exons within the same gene. This supports the notion that exons, rather than entire genes, are units of evolutionary selection, consistent with the idea that split genes increase the diversification potential of species.
Exons flanked by longer introns showed greater sequence and length conservation in our analysis (Fig. 5E,F). It is known that splice-site recognition through exon definition is less efficient (Fox-Walsh et al. 2005). Interestingly, “exon definition” exons have stronger splice sites and more optimal exon sizes, perhaps to maintain efficient exon inclusion (Fig. 5C). Using similar arguments, exons flanked by short introns will be included more efficiently; therefore, stronger splice-site scores and more optimal exon lengths are less important exon features (Fig. 5C,D). In addition, the stronger the average splice-site score, the greater the length and sequence conservation (Supplemental Fig. 4C,D). Previous studies have shown that certain exonic splicing elements (the 5′ss/3′ss and exonic splicing enhancers/silencers) may have coevolved in a manner that maintains exon strength (Xiao et al. 2007). These findings, consistent with our comparisons, further illustrate that there are different strategies to maintain efficient internal exon recognition within the context of variable exon/intron architectures.
It has been shown that there is a higher incidence of alternative splicing in higher eukaryotes than lower eukaryotes (Kim et al. 2007; Keren et al. 2010). These observations highlight the evolutionary lineage of alternative splicing within the phylogenetic tree and suggest that alternative splicing is an evolutionary driving force for the generation of new exons. Furthermore, it has been demonstrated that new exons display a higher frequency of alternative splicing (Alekseyenko et al. 2007; Corvelo and Eyras 2008). Therefore, it is possible that length- and/or sequence-conserved exons correlate with alternative splicing frequencies. In support of this hypothesis, we showed that exons are more likely skipped when length and sequence conservation is poor (Fig. 6). Surprisingly, exon length conservation was observed to be a good predictor of alternative splicing frequencies, indicating that exon architectural features aid in the evolution of newly emerged exons. These results suggest that exon length and sequence conservation play a convergent role in the evolution of an exon.
Exon conservation database improves splice pattern prediction
SNPs are the most abundant type of variation within DNA sequences (Shastry 2002), and many disease-associated SNPs have been suggested to play a role in splicing (Yang et al. 2009; Lalonde et al. 2011; Lim et al. 2011; Motta-Mena et al. 2011; Xiong et al. 2015). Understanding the functional impact of SNPs in human diversity and disease and their influence on splicing may be key to understanding and improving treatments for various diseases. Using an exon trap splicing data set (Soemedi et al. 2017) and the exon conservation database described here, it was possible to test whether exon size–filtered sequence alignments could be used as an approach to identify splice-altering nucleotide changes. Using the SNP categories “mutation not important,” “mutation not observed,” and “SNPs with covariance,” it was possible to differentiate between mutations that are more likely to have an impact on splicing and those that do not.
Codon position played an important role in whether nucleotide changes were tolerable. The wobble position of the codon demonstrated the greatest conservation variability between the splice altering and control SNP categories (Fig. 7). Consistent with this prediction, wobble position SNPs that are conserved across exon size–conserved comparator species are enriched for nucleotide changes that impact splicing, in particular for SNPs that decrease exon inclusion. The comparative analysis also identified “SNPs with covariance” that strongly correlate with increased exon inclusion, demonstrating the important nature of compensatory mutations in rescuing any defects that could be generated by the presence of isolated SNPs.
It is known that single mutations at a regulatory binding site have a high probability of disrupting the binding of regulatory proteins (Liu et al. 2001; Fairbrother et al. 2002; Wang et al. 2004, 2006; Soukarieh et al. 2016). However, additional nucleotide changes within 6 nt upstream of or downstream from that SNP have the potential to rescue splicing, presumably through the creation of alternative binding sites for regulatory proteins (Mueller et al. 2015). Such compensatory nucleotide covariation is highlighted in cases in which a PTC-inducing wobble position SNP is associated with additional nucleotide changes to induce exon skipping (Supplemental Fig. 8), a process referred to as nonsense-associated altered splicing (Wang et al. 2002, 2004; Wilkinson and Shyu 2002). Thus, the enrichment of exon skipping observed for “SNPs with covariance” could be a path that is used to evade the inclusion of PTC-containing exons in the final transcripts.
At its core, splicing is a fundamental process that has led to the discovery of key elements important for maintaining genomic stability. Mutations within the genome have supported diversity and variations within species; however, these changes have also led to a number of human diseases. The potential to predict splicing-associated diseases based on sequence analysis is critical, not only for understanding splicing regulation mechanistically, but also for developing disease-appropriate therapeutic approaches. The exon size–filtered phylogenetic comparison approach described here aids in increasing the prospect of identifying splice-altering SNPs at wobble positions and potentially improves the prognostic power of predicting the direction of splicing changes.
MATERIALS AND METHODS
Generation of exon conservation database using ultra-conserved exons
Orthologous genes in different species vary in sequence, length, and the number of exons per gene. To find corresponding exons of the same gene in two or more species, it is necessary to compare the sequence of all matching exons and not to rely solely on the exon number or its relative position in that gene.
To generate a database of human exons that contains information pertaining to exons that are conserved in both sequence and length, multiple sequence alignment (multiz100way) of 99 species compared to human exons was used. This multiple alignment file was generated by the Multiz tool (Blanchette et al. 2004) downloaded from the UCSC Genome Browser. For each human exon, the genomic location of aligned sequences from other species to the sequence of that exon were extracted from this multiple alignment data. In each species, if that location overlapped with an annotated exon (covering at least 20% of the exon), that exon was listed as a matching exon to the human exon. If an exon matched with a similar sequence in another species but also maintained its structure (length difference of ≤3 nt between the human exon and the matching species exon), that exon was defined as ultra-conserved in that species. This generated a list of matching exons from all species with exon length information stored within the database. Some of the assemblies used in multiz100way were older and the associated gene annotations were not found or trusted, therefore the process was continued with only 76 species, including human. Additionally, only the canonical version of each gene was used from the human annotation (hg19 RefSeq).
Exon conservation database parameters
For each exon in the human genome listed in the exon conservation database, an Ultra-In number was generated by comparing the length of that exon and the matching exons in other species. This number, denoted as a score, represents the number of species in which a particular exon is ultra-conserved for length.
Total number of exons analyzed were 184,796 exons (18,225 genes), representing single exons (1116 exons), first exons (10,933 exons), last exons (16,364 exons), and internal exons (156,383 exons). In addition to relative exonic position in the transcript, basic characteristics of each exon were also listed in the exon conservation database including genomic coordinates, exon length, and upstream and downstream intron lengths (as extracted from the hg19 gene annotation). It is important to note that there is a discrepancy between first and last exons owing to mismatched annotation files. The multiple alignment data used was not from the same canonical genome annotation as the reference for exon annotations. These mismatched annotation files lead to a larger number of first exons than last exons.
Additionally, sequence conservation scores and 3′ss and 5′ss scores were also included. The Maximum Entropy Score (MES) is a computationally derived value assigned to splice-site sequences based on the modeling of short sequence motifs around the 5′ss or 3′ss using the maximum-entropy principle (Yeo and Burge 2004). This numerical scoring permits the designation of strong splice sites as those with a MES > 8 or weak splice sites as MES < 6–7 (Shepard et al. 2011), with an average 5′ss score for constitutive exons of MES = 8.3 and MES = 8.8 for the 3′ss (Busch and Hertel 2015). The sequence conservation score was obtained through phyloP (Pollard et al. 2010), generated by averaging the nucleotide-based values already available for multiz100way alignment, as phylop100way tracks on the UCSC Genome Browser. Splice-site scores were also obtained from MaxEnt scores (Yeo and Burge 2004) for both the 3′ss and 5′ss. The corresponding genomic sequence was extracted for each end of the exon and the scores were calculated using the web interface of MaxEntScan. In addition, 233 exons were filtered out because of poor annotations within the human genome itself. It is important to note that using phyloP as a measure of sequence conservation has its limitations. PhyloP currently looks at base-wise conservation across 46 species only, whereas our size conservation score used in the exon conservation database is generated across 76 species. Future developments in phyloP will aid in better comparisons, leading to a larger pool of species from which to compare and a potentially tighter correlation between length and sequence conservation
Alternative splicing events
The exon conservation database, using bedtools intersect (v2.25.0) (Quinlan and Hall 2010) was merged with the EST HEXEvent database (hg19 build), which contained counts of canonical and alternative splicing events for each exon (Busch and Hertel 2013). The resulting merged database yielded conservation scores and splicing event counts for each exon. The resulting “.bed” file was filtered using the Pandas Python library (v0.18.1) to remove exons with fewer than 75 events recorded and exons with a sum of splice-site strengths of <0. Skipped exons were obtained by eliminating exons that were included in the final transcript >95% of the time and by further eliminating exons that cannot participate in exon skipping (first and last exons). Alternative 3′ and 5′ splicing events were obtained using the same >75 events and >0 sum of splice-site score filters. Exons with a canonical 3′ss or 5′ss usage frequency of 1 were removed for each splicing event, respectively; first exons were removed to obtain alternative 3′ss exons and terminal exons were removed to obtain alternative 5′ss exons.
Inclusion or canonical 3′ss and 5′ss usage frequencies were plotted against conservation scores using the Matplotlib Python library (v1.5.3), and Pearson correlation scores were obtained using the Scipy.stats Python library (v1.0.0).
Exonic SNP conservation database
Using the exon conservation database, those exons that included one or more mutations as catalogued (Soemedi et al. 2017) were separated and further analyzed to derive a second database: a SNP conservation database. For each one of the SNP mutations, parameters within the database included the following: position of the mutation within the human exon as well as 5 nt upstream of and downstream from that position, Ultra-In score (from the multiple alignment data), the associated amino acid sequence for the WT and mutant SNP, and the position of that SNP within the context of a codon. Additionally, if the mutation was located at a junction, defined as a position located within 3 nt of splice sites, the splice-site scores (MaxEnt scores) were generated for both the WT and mutated versions of 3′ss and 5′ss.
SNP conservation database mutation categories
Mutations within the SNP database were categorized into three groups based on their occurrences in the Ultra-In species and any variations observed in the neighboring nucleotides. The first category was defined as SNPs that are “mutation not observed.” These SNPs include mutations listed in (Soemedi et al. 2017) that did not occur in any Ultra-In species. In the second category, SNPs were defined as “mutation not important” when a SNP was observed in a subset of Ultra-In species but the neighboring nucleotides did not show any variation between species. This mutation was present in the Ultra-In species; however, no other mutations were nearby and the SNP containing exon still had the same length as the human exon. Hence the mutation did not influence splicing, and this mutation was not important for splicing. The third group, “SNPs with covariance,” represents cases in which the annotated mutation was observed in some species, but at least one other variation was also seen in the 10 nt around that mutation in a subset of those species. The combination of this mutation and these nearby variations may not affect the splicing pattern; therefore, it is possible that these variations had a compensatory effect on the main mutation.
If the aligned sequence from other species compared to the 11 nt (mutation and surrounding 10 nt) in human contained three or more indels or “N,” they were not considered in the final mutation categorization. Although the exons were already matched by sequence similarity, the difference that was not significant when comparing the whole exon could cover a noticeable portion of these 11 nt. If the mutation was located close to a splicing junction, a sequence of “Z” was used to show the border of the exon in the codon containing the mutation and “X” was used in the associated amino acid column.
The delta MaxEnt score was calculated for either the 5′ss or 3′ss as mutant (MT) MaxEnt score – wild-type (WT) MaxEnt score. The percent spliced in (psi) value was calculated as the ratio of Spliced/(Unspliced + Spliced). The delta psi (dpsi) value was calculated as: MT psi – WT psi. For all analysis, a 10% dpsi cutoff was used to generate the “dpsi−” (exclusion) and “dpsi+” (inclusion) categories. For the control group (no change group or dpsiC), a 0.2% cutoff was used. This cutoff variable was necessary because a cutoff of zero did not have sufficient data points.
Bedtools v2.25.0 was used to find the overlap between genomic regions when necessary in finding matching exons. Binary files, wigToBigwig and bigWigSummary, downloaded from the UCSC Genome Browser, were used in conservation score calculation. All the other scripts were written in Python 2.7, and all analysis and graphs were generated using R v3.5.1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the members of our laboratory for helpful discussions and comments on this manuscript as well as funding support from the National Institutes of Health (NIH) (R01 GM062287 and R01 GM110244 to K.J.H.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.070987.119.
REFERENCES
- Alekseyenko AV, Kim N, Lee CJ. 2007. Global analysis of exon creation versus loss and the role of alternative splicing in 17 vertebrate genomes. RNA 13: 661–670. 10.1261/rna.325107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget SM. 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715. 10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. 2013. HEXEvent: a database of Human EXon splicing Events. Nucleic Acids Res 41: D118–D124. 10.1093/nar/gks969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. 2015. Splicing predictions reliably classify different types of alternative splicing. RNA 21: 813–823. 10.1261/rna.048769.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Gentles AJ, Jurka J, Karlin S. 2002. Genes, pseudogenes, and Alu sequence organization across human chromosomes 21 and 22. Proc Natl Acad Sci 99: 2930–2935. 10.1073/pnas.052692099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvelo A, Eyras E. 2008. Exon creation and establishment in human genes. Genome Biol 9: R141 10.1186/gb-2008-9-9-r141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352. 10.1016/j.cell.2008.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. 2003. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 12: 205–216. 10.1093/hmg/ddg055 [DOI] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh R-F, Sharp PA, Burge CB. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007–1013. 10.1126/science.1073774 [DOI] [PubMed] [Google Scholar]
- Fox-Walsh KL, Dou Y, Lam BJ, Hung S-P, Baldi PF, Hertel KJ. 2005. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci 102: 16176–16181. 10.1073/pnas.0508489102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y. 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481 10.1038/msb.2011.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11: 345–355. 10.1038/nrg2776 [DOI] [PubMed] [Google Scholar]
- Kim E, Magen A, Ast G. 2007. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res 35: 125–131. 10.1093/nar/gkl924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde E, Ha KCH, Wang Z, Bemmo A, Kleinman CL, Kwan T, Pastinen T, Majewski J. 2011. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res 21: 545–554. 10.1101/gr.111211.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Dias AP, Reed R. 2011. Export and stability of naturally intronless mRNAs require specific coding region sequences and the TREX mRNA export complex. Proc Natl Acad Sci 108: 17985–17990. 10.1073/pnas.1113076108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. 2011. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci 108: 11093–11098. 10.1073/pnas.1101135108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Fu X-D. 2007. SR proteins and related factors in alternative splicing. Adv Exp Med Biol 623: 107–122. 10.1007/978-0-387-77374-2_7 [DOI] [PubMed] [Google Scholar]
- Liu HX, Cartegni L, Zhang MQ, Krainer AR. 2001. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet 27: 55–58. [DOI] [PubMed] [Google Scholar]
- Marques AC, Dupanloup I, Vinckenbosch N, Reymond A, Kaessmann H. 2005. Emergence of young human genes after a burst of retroposition in primates. PLoS Biol 3: e357 10.1371/journal.pbio.0030357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta-Mena LB, Smith SA, Mallory MJ, Jackson J, Wang J, Lynch KW. 2011. A disease-associated polymorphism alters splicing of the human CD45 phosphatase gene by disrupting combinatorial repression by heterogeneous nuclear ribonucleoproteins (hnRNPs). J Biol Chem 286: 20043–20053. 10.1074/jbc.M111.218727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller WF, Larsen LSZ, Garibaldi A, Hatfield GW, Hertel KJ. 2015. The silent sway of splicing by synonymous substitutions. J Biol Chem 290: 27700–27711. 10.1074/jbc.M115.684035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Hannenhalli S, Choi SS. 2014. Conservation in first introns is positively associated with the number of exons within genes and the presence of regulatory epigenetic signals. BMC Genomics 15: 526 10.1186/1471-2164-15-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121. 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar MK, Chow VTK, Kangueane P. 2004. Distributions of exons and introns in the human genome. In Silico Biol 4: 387–393. [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Spiridonov AN, Novichkov PS, Spiridonov NA, Koonin EV. 2010. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol Biol Evol 27: 1745–1749. 10.1093/molbev/msq086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry BS. 2002. SNP alleles in human disease and evolution. J Hum Genet 47: 561–566. 10.1007/s100380200086 [DOI] [PubMed] [Google Scholar]
- Shepard PJ, Choi E-A, Busch A, Hertel KJ. 2011. Efficient internal exon recognition depends on near equal contributions from the 3′ and 5′ splice sites. Nucleic Acids Res 39: 8928–8937. 10.1093/nar/gkr481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A. 2009. Phylogenomics of primates and their ancestral populations. Genome Res 19: 1929–1941. 10.1101/gr.084228.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, Bayrak-Toydemir P, McDonald J, Fairbrother WG. 2017. Pathogenic variants that alter protein code often disrupt splicing. Nat Genet 49: 848–855. 10.1038/ng.3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneborn TM. 1965. Degeneracy of the genetic code: extent, nature, and genetic implications. In Evolving genes and proteins (ed. Bryson V, Vogel HJ), pp. 377–397. Academic, New York. [Google Scholar]
- Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frébourg T, Tosi M, Martins A. 2016. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet 12: e1005756 10.1371/journal.pgen.1005756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DA, Carlo T, Berget SM. 1996. Architectural limits on split genes. Proc Natl Acad Sci 93: 15081–15085. 10.1073/pnas.93.26.15081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP. 2007. Downstream intronic splicing enhancers. FEBS Lett 581: 4127–4131. 10.1016/j.febslet.2007.08.012 [DOI] [PubMed] [Google Scholar]
- Wang J, Chang YF, Hamilton JI, Wilkinson MF. 2002. Nonsense-associated altered splicing: a frame-dependent response distinct from nonsense-mediated decay. Mol Cell 10: 951–957. 10.1016/S1097-2765(02)00635-4 [DOI] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. 2004. Systematic identification and analysis of exonic splicing silencers. Cell 119: 831–845. 10.1016/j.cell.2004.11.010 [DOI] [PubMed] [Google Scholar]
- Wang Z, Xiao X, Van Nostrand E, Burge CB. 2006. General and specific functions of exonic splicing silencers in splicing control. Mol Cell 23: 61–70. 10.1016/j.molcel.2006.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MF, Shyu A-B. 2002. RNA surveillance by nuclear scanning? Nat Cell Biol 4: E144–E147. 10.1038/ncb0602-e144 [DOI] [PubMed] [Google Scholar]
- Xiao X, Wang Z, Jang M, Burge CB. 2007. Coevolutionary networks of splicing cis-regulatory elements. Proc Natl Acad Sci 104: 18583–18588. 10.1073/pnas.0707349104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JO, Kim W-Y, Bhak J. 2009. ssSNPTarget: genome-wide splice-site single nucleotide polymorphism database. Hum Mutat 30: E1010–1020. 10.1002/humu.21128 [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.