

Abstract
Nontargeted effects include radiation-induced genomic instability (RIGI) which is observed in the progeny of cells exposed to ionizing radiation and can be manifested in different ways, including chromosomal instability and micronucleus (MN) formation. Since genomic instability is commonly observed in tumors and has a role in tumor progression, RIGI has the potential of being an important mechanism for radiation-induced cancer. The work presented explores the role of dose and dose rate on RIGI, determined using a MN assay, in normal primary human fibroblast (HF19) cells exposed to either 0.1 Gy or 1 Gy of X-rays delivered either as an acute (0.42 Gy/min) or protracted (0.0031 Gy/min) exposure. While the expected increase in MN was observed following the first mitosis of the irradiated cells compared to unirradiated controls, the results also demonstrate a significant increase in MN yields in the progeny of these cells at 10 and 20 population doublings following irradiation. Minimal difference was observed between the two doses used (0.1 and 1 Gy) and the dose rates (acute and protracted). Therefore, these nontargeted effects have the potential to be important for the low-dose and dose-rate exposure. The results also show an enhancement of the cellular levels of reactive oxygen species after 20 population doublings, which suggests that ionising radiation (IR) could potentially perturb the homeostasis of oxidative stress and so modify the background rate of endogenous DNA damage induction. In conclusion, the investigations have demonstrated that normal primary human fibroblast (HF19) cells are susceptible to the induction of early DNA damage and RIGI, not only after a high dose and high dose rate exposure to low linear energy transfer, but also following low dose, low dose rate exposures. The results suggest that the mechanism of radiation induced RIGI in HF19 cells can be correlated with the induction of reactive oxygen species levels following exposure to 0.1 and 1 Gy low-dose rate and high-dose rate x-ray irradiation.
Keywords: Chromosome instability, dose rate, micronucleus formation
Introduction
The classical paradigm in the radiobiology field states that the biological effects of radiation are due to the deposition of energy and associated DNA damage in the cell nucleus.[1] However, in the early 1990s, significant evidence emerged to challenge classical target theory and highlighted the potential role of nontargeted effects.[1,2] These nontargeted effects, including radiation-induced genomic instability (RIGI) which is observed in the progeny of cells exposed to ionizing radiation, can be manifested in different ways such as gene mutations, chromosomal instability, micronucleus (MN) formation, and an enhanced death rate.[2,3] In addition, there is considerable overlap between RIGI and the genomic instability commonly observed in tumors, which is a well-known enabling characteristic assisting the transition of cells from normal to oncogenic by the acquisition of the various hallmarks of cancer.[4] RIGI is a high-frequency event that occurs at a higher frequency and then can be explained by specific gene mutations. For example, MN formation was used to measure the DNA damage after 7 days in both high-dose rate (HDR) and low-dose rate (LDR)-irradiated mouse lymphocytes cells. The frequency of MN induction in the HDR population cells was higher compared to the equivalent LDR population cells following in vivo exposure to 0–4.45-Gy X-rays.[5] In addition, the findings of Al-Mayah et al. showed that exosomes from the irradiated cell-conditioned media of 2-Gy directly irradiated MCF7 cells (breast epithelial cancer cells) induced early and late (20/24 population doublings following medium/exosome transfer) DNA damage within the unirradiated MCF7 cells. The progeny of irradiated and bystander cells continue to produce and release exosomes, which then affect their own progeny at later times, a response that is observed as Genomic instability (GI).[6] Furthermore, a significant induction of chromosome aberrations (CAs) in the hemopoietic system of CBA/H mice was observed at 24 months following a whole-body exposure to high (neutron) and low (X-ray) linear energy transfer (LET) irradiation.[7] Watson et al. reported the first evidence of in vivo chromosomal instability induced by a bystander mechanism. A mixture of irradiated and labeled unirradiated bone marrow cells of CBA/H mouse transplanted into female recipients displayed a significant induction of CAs in the labeled unirradiated cells.[8] RIGI arises nonclonally within the clonal descendants' multiple generations following the original exposure and can also arise in the descendants of cells that have not been irradiated but as a result of the perturbation of intercellular communication resulting from the irradiation of subpopulations of cells. Unlike conventional observed effects, RIGI has a tendency to plateau with increasing dose.[9,10]
The risk associated with exposure to ionizing radiation has been determined from epidemiological data, most notably on the Japanese atomic bomb survivors.[11] However, there are only limited epidemiological data on cancer risk at very low doses. The UNSCEAR (2017) Committee assessed epidemiological studies analyzing cancer risk based on individual doses due to environmental exposures. The overall findings of these studies do not deliver evidence of a risk of cancer per unit dose higher than that derived from studies of high radiation doses. There is considerable uncertainty in the estimates owing to both limited statistical power and limitations in other aspects such as residual confounding and inaccuracies in exposure assessment. Therefore, the bounds of uncertainty do not cancel a lower risk per unit dose than that observed in studies of higher doses.[12] Thus, the risk associated with the low-dose and LDR radiation exposure typically associated with human exposure is determined by extrapolation from the high-dose and HDR exposure data. This extrapolation relies on a number of assumptions; these include the assumption that DNA is a critical target with the yield of damage increasing linearly with dose, with a corresponding linear increase in the probability of mutations at low doses (and a more rapid increase at higher doses and dose rates) and the relative efficiencies of DNA repair and misrepair. These assumptions form the basis of the linear no-threshold hypothesis. In addition to direct DNA damage, nontargeted effects such as RIGI may also modify cancer risk at low doses, but the overall impact is unclear, although the response is likely to be nonlinear with dose.[11]
The goal of the current study was to explore the role of dose (0.1 and 1 Gy) and dose rate (0.0031 Gy/min compared to 0.42 Gy/min) for X-ray irradiation on the induction of RIGI in normal human diploid lung fibroblast cells. MN assay was used as a marker of RIGI with oxidative stress explored as a potential mechanism. Assays were performed either immediately after irradiation or after approximately 10 and 20 population doublings.
Materials and Methods
Cell culture
Human fibroblast 19 (HF19) cells, a primary nontransformed human lung fibroblast,[13] were cultured in Minimum Essential Medium with Earle's salts without L-glutamine and supplemented with 10% fetal bovine serum (Sigma: F7524), 2-mM L-glutamine (Gibco: 25030), 1% nonessential amino acids (Gibco: 11140), and 1% (w/v) penicillin/streptomycin solution (Sigma: P0781) in a fully humidified 5% CO2 incubator at 37°C. Cells were passaged twice weekly. During irradiations, the solid caps of the flasks were sealed tight in order to maintain the optimal pH of the medium during irradiation while out of the gassed incubator and subsequently opened to allow gas exchange just prior to replacing into the a fully humidified 5 % CO2 incubator at 37°C.
X-ray irradiation
Irradiations were performed at the Oxford Institute for Radiation Oncology, University of Oxford, with 250-kV constant potential X-rays filtered with 1.25-mm Sn, 0.25-mm Cu, and 1.5-mm Al. Cells were seeded at 1.5 × 106 in T75 flasks and incubated for ≈ 24 h prior to irradiation [Figure 1]. HDR irradiations at 0.42 Gy/min were performed with the flask on a Perspex sheet positioned 500 mm above source with the tube operated at 12 mA, whereas LDR irradiations of 0.0031 Gy/min were performed with the flask placed on a Perspex shelf 850 mm above the source with the tube operated at 0.3 mA. The shelf supporting the flask is the center of the Perspex box of a Stuart incubator which was used to maintain the temperature at ≈ 37°C during the irradiation. Dosimetry rate measurements were performed using Gafchromic EBT3 film (Imaging Equipment Ltd., Chilcompton, UK) placed on the base of a T75 flask filled with an identical volume of liquid used for the biological experiments. The exposed film was scanned 24 h latter in transmission mode at 48-bit red, green, and blue (RGB) (16 bits per color) with 300 dots per inch resolution using a flatbed scanner (Epson Expression 10000XL). All the films were scanned centrally using a template and in an identical orientation as recommended by the manufacturer's guidelines. The dose was calculated using the optical density of the red channel corrected using the blue channel to compensate for small nonuniformities in the film. The batch of EBT3 film was calibrated from 0 to 5 Gy following recommendations of the report of American Association of Physicists in Medicine (AAPM) Task Group 61.[14]
Figure 1.
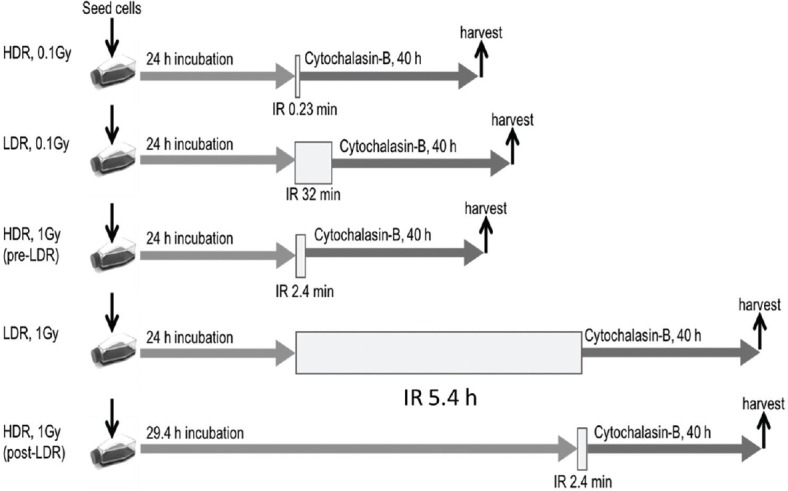
Experimental design for high dose rate (HDR) and low dose rate (LDR) x-ray irradiation.
Micronucleus assay
The frequency of MN induction in HF19 cells was measured using a cytokinesis-block protocol technique adapted from Erexson and Kligerman, and Erexson et al.[15,16] Immediately following irradiation, cells were treated with 6-μg/ml cytochalasin-B and subsequently incubated for 40 h. Cells were then harvested and centrifuged (at 200 × g at room temperature for 10 min), the supernatant was discarded, and the remaining pellets were resuspended. Once resuspended, 1 ml of hypotonic solution (warmed KCl; 0.55-g potassium chloride (Sigma, P3911) and 100-ml ultrapure water kept in a 37°C water bath) was subsequently added in a drop-wise manner followed by a further 10-ml KCl. The tubes were then incubated at 37°C for 5 min prior to adding 3 drops of 25% glacial acetic acid in methanol (3:1 fixative); all tubes were inverted once and centrifuged. Following removal of the supernatant, the pellets were resuspended in 10 ml of 3:1 fixative (added drop-wise) and left at room temperature for 10 min. Cells were further centrifuged, the supernatant was discarded, and pellets were resuspended in 0.5–1 ml of 3:1 fixative depending on the pellet size. The resulting fixed cell suspension was dropped onto individually labeled, clean/degreased microscope slides, and these were left to dry at room temperature before analysis. In addition, MN assay was used to investigate the induction of MNs at 5 and 10 passages, corresponding to approximately 10 and 20 population doublings (15 and 20 days, respectively), following irradiation.
Staining of slides
In brief, two Coplin jars were each filled with a 50-ml prepared phosphate buffer solution (pH 6.8). A 0.0031-g acridine orange (Sigma: A6014) was added to the first jar. Slides were stained for 25 s in the acridine orange/buffer solution and then quickly dipped for a few seconds in the buffer-only jar.[10,11] Finally, they were left to dry at room temperature before analysis on a fluorescent microscope.
Scoring micronuclei
Slides were coded and analyzed in a blind and random fashion. MNs were scored only in binucleated (BN) cells, and at least 500 BN cells were scored per group for four biological replicates. MN induction was quantified as the percentages of BN cells within MNs (%MN/BN).[17]
Oxidative stress
The Muse oxidative stress reagent has been widely used to detect reactive oxygen species (ROS) in cellular populations and is based on dihydroethidium (DHE).[18] This reagent penetrates the cell where DHE is oxidized by superoxide anions and forms the DNA-binding fluorophore ethidium bromide or a structurally similar product. This reagent intercalates with DNA giving rise to a red fluorescence-enabling ROS+ cells (cells exhibiting ROS) to be distinguishable from ROS− cells. Cells were seeded at 1.5 × 106 cells per T75 for 24 h prior to irradiation. Samples were collected ≈ 1.5 h following irradiation and again after 30 days corresponding to approximately 20 population doublings. The media were collected and saved in a labeled sterile universal tube. The flasks were washed twice with 10-ml phosphate-buffered saline (PBS): the first PBS was added to the universal tube but the second wash of 10-ml PBS was discarded. The cells were detached from each flask by adding 1.5-ml (0.025%) trypsin-ethylenediaminetetraacetic acid (EDTA) solution for 30–60 s and incubated at 37°C for 3–5 min to allow cells to dissociate from the flask base. The trypsin-EDTA solution was also collected and added to the universal contents. Dissociated cells were collected with the universal tube contents (saved media, PBS, and trypsin) and added back to the universal tube. All cells were centrifuged, the supernatants were discarded, and the remaining pellets were resuspended. Once resuspended, 200 μl of sterileDulbecco's phosphate-buffered saline (DPBS) was subsequently added to each tube. Ten microliters of the resulting cell suspension was added to 190 μl of the Muse oxidative stress reagent working solution. The cell suspension was then mixed thoroughly by vortexing at medium speed for 3–5 s. Samples were incubated for 30 min at 37°C in the dark. Finally, samples were run on the Muse cell analyzer (Muse™ Oxidative Stress Kit, Millipore) according to the manufacturer's protocol.
Statistical analysis
Data for the distribution of MN among BN cells in a pooled dataset were examined for normality. The MN data were shown not to have normal distribution; it was therefore further subjected to the Fisher's exact test to calculate the P values. P ≤ 0.05 was considered statistically significant. While for oxidative stress measurements, the combined data from three independent experiments were compared by the Student's t-test, with two-tailed P < 0.05 indicating statistically significant differences. The error bars represent the standard error of the mean of replicate experiments (standard error of the mean).
Results
Micronucleus assay analysis
The variation in the percentage of BN cells with MNs (%MN/BN) produced with the assay performed immediately, after ≈ 10 population doublings, and after ≈ 20 population doublings is shown in Figures 2-4, respectively.
Figure 2.
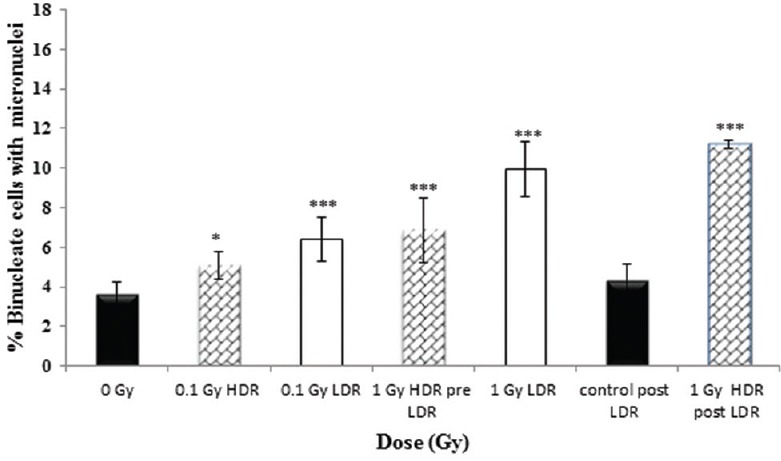
The percentage of binucleate cells (BN) with micronuclei (MN) (%MN/BN) for high (0.42 Gy/min) and low (0.0031 Gy/min) dose rate at 0, 0.1 and 1 Gy immediately following irradiation, with addition of cytochalasin B immediately following irradiation (<5 minutes). Data represent the combined data of four independent but parallel experiments performed on different days. Error bars represent the standard error of the mean (SEM)
Figure 4.
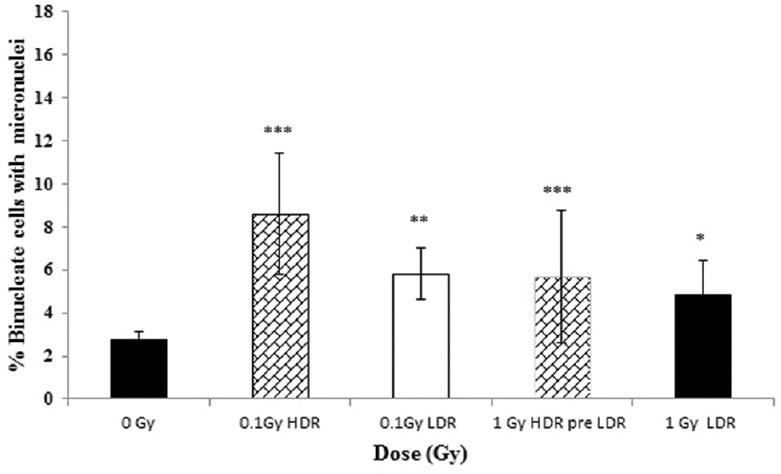
The percentage of binucleate cells (BN) with micronuclei (MN) (%MN/BN) at high (0.42 Gy/min (and low (0.0031 Gy/min) dose rate at 0, 0.1 and 1 Gy after 20 population doublings. Data from four independent experiments, error bars represent the standard error of the mean (SEM)
Under all radiation conditions studied, the data demonstrate a significant induction of BN cells with MNs produced during the first division following irradiation when compared to unirradiated controls [Figure 2]. A similar response was observed for 0.1- and 1-Gy HDR acute exposures when the assay was performed 24 h after seeding the flasks (pre-LDR). While no significant difference was observed between MN inductions following a 0.1-Gy acute exposure and a 0.1-Gy LDR protracted exposure, the data showed an enhanced MN induction following a 1-Gy protracted exposure (over a 5.4 h) compared to a 1-Gy acute exposure (P ≤ 0.001). However, no significant difference was observed between this protracted exposure and the delayed 1-Gy acute exposure performed directly following the 1-Gy protracted exposure (post-LDR: ≈29.4 h postseeding the flask), although the control assay performed at this time (0 Gy post-LDR) showed no difference to the main 0-Gy control (0 Gy pre-LDR). This suggests that the timing of the assay and subsequent harvest postseeding may potentially modulate the measured yield of radiation-induced MNs.
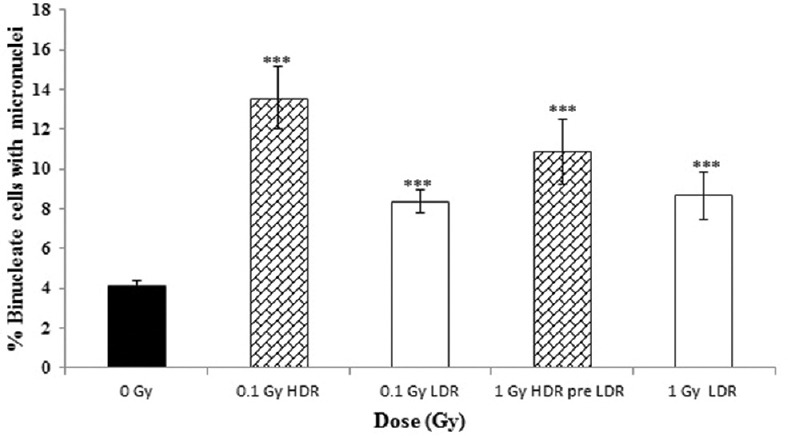
Likewise, the results of the delayed cellular response following 10 population doublings [Figure 3] and 20 population doublings [Figure 4] following irradiation all show a significant increase in MN induction across all irradiated groups when compared to unirradiated controls. Neither HDR nor LDR exposure showed that a significant difference was observed between the 0.1- and 1-Gy responses, although there is a suggestion that acute exposures may have a slightly enhanced yield when compared to protracted exposures.
Figure 3.
The percentage of binucleate cells (BN) with micronuclei (MN) (%MN/BN) at high (0.42 Gy/min) and low (0.0031 Gy/min) dose rate at 0, 0.1 and 1 Gy at 10 population doublings post irradiation. Error bars represent the standard error of the mean (SEM)
Oxidative stress
The variation in the percentage HF19 cells exhibiting enhanced levels of ROS following irradiation 1.5 h postexposure and 20 population doublings later are displayed in the Figure 5 and Figure 6 respectively. For both time points, there was a significant enhancement in the response for both acute and protracted 1-Gy exposures. The degree of enhancement observed was similar for acute and protracted exposures, although there is an indication that the fraction of cells responding was slightly lower at the later time point. For the lower 0.1-Gy exposures, only the protracted 0.1-Gy exposure produced a statistically significant enhancement over controls in the fraction of cells responding 1.5 h postexposure, but no enhancement was observed at the later time point.
Figure 5.
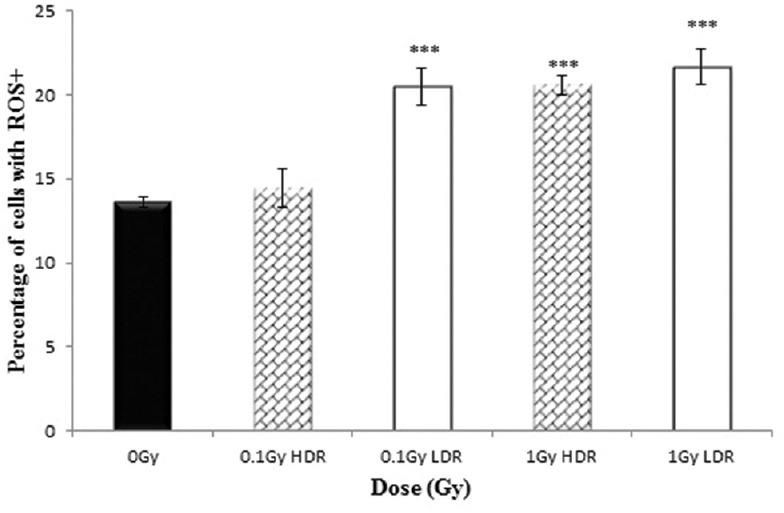
Percentage of HF19 cells with ROS+ at 1.5 hours following x-ray irradiation at 0.1 and 1 Gy HDR and LDR. Combined data from three independent experiments. Error bars represent the standard error of the mean (SEM)
Figure 6.
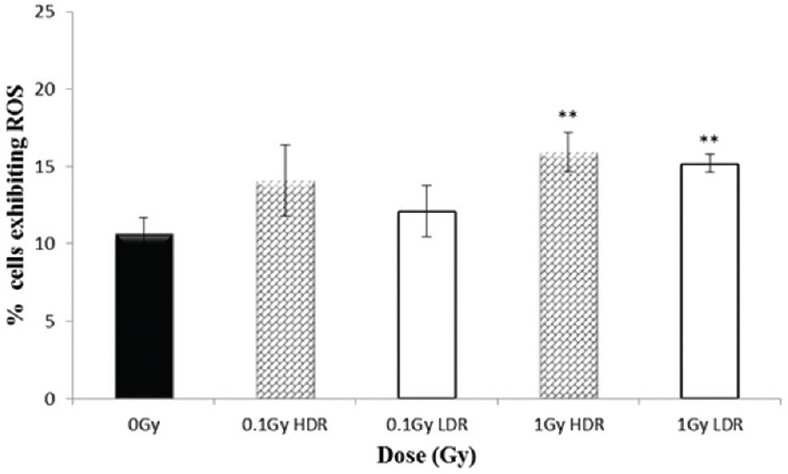
Percentage of HF19 cells with ROS+ at 20 population doublings following x-ray irradiation at 0.1 and 1 Gy HDR and LDR. Combined data from three independent experiments. Error bars represent the standard error of the mean (SEM)
Discussion
CAs present during the first mitosis and associated MN following division are predominantly produced as a result of the initial DNA damage produced by the interaction of IR within the cell and associated repair and misrepair. While the formation of these sites of initial DNA damage such as double-strand breaks (DSBs) increases linearly with the dose,[19] the probability of misrepair between sites and the formation of aberrations and MNs is dependent on the relative separation of these sites of damage. Therefore, they typically follow a linear-quadratic dose–response for low-LET radiation due to the increasing chance of interaction between sites due to their increasing density with increasing dose. The MN containing acentric chromosome or chromatid fragments are a result of unrepaired or misrepaired DNA breaks. This occurs when the repair capacity of the cells is insufficient to repair an excessive level of DSB production. This is due to either inappropriate function of enzymes involved in the nonhomologous end-joining pathway[20] or misrepair of DSBs caused by the dysfunctional homologous recombination.[21] Recently, Pantelias et al. have demonstrated the hypothesis that the premature chromosome condensation dynamics in asynchronous micronucleated cells rigger chromosome shattering in a single catastrophic event. This is a hallmark of chromothripsis which underlies the disruption of tumor suppressor genes and activation of oncogenes.[22] In peripheral blood lymphocytes, this occurs in a reproducible fashion for a given radiation quality, and the induction of CAs (such as dicentric chromosomes) and MNs is routinely used for biodosimetry[23] It is therefore not unexpected that the data presented here indicate that the yield of MN is greater for 1 Gy than 0.1 Gy. DNA DSBs are typically repaired with a half time of ≈ 20 min,[24] and so for protracted exposures (LDR), where total irradiation times are comparable to or exceed the repair half-time, there is a significant reduction in biological effectiveness for a range of endpoints, due to the decrease in lesion density (and therefore the probability of misrepair) resulting from lesion repair. It was not unexpected that the immediate yield of MN formation following a 0.1-Gy LDR was similar to that of a 0.1-Gy HDR, due to the limited number and therefore low density of DNA lesions and the total LDR irradiation time only slightly longer than the DSB repair time. Although it was a little surprising that the pre-LDR 1-Gy acute exposure was lower than the 1-Gy protracted exposure, still the timing of the addition of cytochalasin-B and subsequent harvest is significantly different and potentially results in different MN detection efficiencies. The difference between the HDR and LDR groups (under the same conditions of collection) in terms of the timing of adding cytochalasin-B and the timing of the subsequent harvesting of cells can lead to different collection efficiencies as like is not being compared with like. It was therefore not too surprising to see different results, and this was one of the reasons pre- and post-acute experiments were performed [Figures 1, row 2 and 3]. When the acute irradiation was performed 29.4 h, rather than 24 h, after plating (corresponding to the end of the protracted exposure), then the MN yield was found to be similar to that for the protracted exposure. The limited difference between the HDRs compared to LDR yields is potentially a result of the DSB yield following a 1-Gy exposure at a low enough density that the probability of interaction between acute exposures is relatively small and a higher dose may be required to detect a significant difference between the acute and protracted exposures. Typical dose–response curves obtained for MN[25] are often relatively linear up to 1–2 Gy, with the dose-square component becoming significant at higher doses. Due to loss of the replicative capacity of many of these cells containing IR-induced CA and MN, the number of these cells quickly falls with time and subsequent cell division,[26] with those found at later times experiencing RIGI. Shimura and Kojima noted that the lowest dose of photons required to observe a statistically significant increase in the formation of MN and chromosomal aberrations in peripheral blood lymphocytes irradiatedin vitro was of the order of 0.1 Gy.[27]
The initial pioneering experiments of Kadhim et al[28] showed a significant induction of RIGI in hemopoietic stem cells only following exposure to high-LET alpha-particles but not following 3Gy low-LET X-ray exposure. Subsequent studies did, however, show a significant induction of RIGI following lower doses of 0.1 Gy and 1 Gy.[29] RIGI was also shown to be induced and persist in hemopoietic stem cells for the remaining lifetime after whole-body irradiation of CBA/H mice to 3 Gy of 250-kV X-rays. Kadhim et al. also observed RIGI in the progeny of HF19 cells exposed to 3 Gy of X-rays but not those of HF12 cells.[30] Likewise, experiments in other systems have demonstrated that low-LET radiation is capable of inducing RIGI.[31,32] While some studies have shown an increasing response with increasing dose, numerous other studies have indicated that RIGI (measured using CA or MNs) lacks a radiation dose–response which is dose independent or has a threshold dose above which no additional RIGI is induced.[33]
The results presented here clearly show a significant induction of MN in HF19 fibroblast cells compared to unirradiated controls at 10 and 20 population doublings following irradiation with 0.1 Gy and 1 Gy delivered either as an acute (HDR) or protracted (LDR) exposure. The MN formation was higher in 0.1 Gy which could be due to the bystander effects as the change in gene expression occurs as early as 1 h postradiation in doses as low as 0.05 Gy in bystander cells.[34] Our findings of induction of GI instability at low-dose (0.1 Gy) low-LET X-ray irradiation in HF19 cells agree with the results of Kadhim who has observed that the delayed chromosomal aberrations were increased over controls in bone marrow cells from both CBA/H and C57BL/6 in bred strains following exposure to 0.1 (low)- and 1 (high)-Gy dose X-ray irradiation.[35] There is a minimal difference between the observed levels of MN produced between the two doses used, and the data suggest that LDR exposure may be slightly more effective than the corresponding HDR. These data are consistent with the data of Belyakov et al., which demonstrated RIGI in the progeny of irradiated primary human AG01522B fibroblast cells which were subsequently subcultured for up to 30 days postirradiation with X-ray doses from 1 to 8 Gy (240 kV at 0.78 Gy/min) and observed using MN assay.[32] The fraction of MNs from 8 days onward was observed to be significantly smaller than the initial response which peaked at around 3 days. Likewise, Trott et al. observed delayed MN in the progeny of surviving V-79 Chinese hamster cells up to 4 weeks following irradiation with 250-kV X-rays (dose rate: 0.8–0.9 Gy/min), with the frequency increasing with dose up to 3 Gy, above which the level was constant.[9] Werner, Wang, and Doetsch (2017) also showed persistent MN induction in the progeny of mouse epithelial cells 7 days postirradiation with 320-kV X-rays (for 2, 4, and 6 Gy).[36] Turner et al. showed an increase in persistent MN formation in mouse lymphocytes 7 days postirradiation of C57BL/6 mice with 320-kV X-rays for both protracted (0.31 cGy/min) and acute exposures (1.03 Gy/min).[5] There was no statistically significant difference in the response between the two-dose rates and the response increase up to 2.2 Gy with no further increase at 4.45 Gy.[5] In addition, Graupner et al. have observed a significant increase in MN formation in the blood samples taken from CBAB6 F1 mice 2 days following low-LET irradiation with a total body absorbed dose of ∼3 Gy (3.2-Gy chronic LDR γ-ray [2.2 mGy/h] and 2.6-Gy acute HDR X-ray [1.3 Gy/min]). Both HDR and LDR groups displayed more induction of MN/BN, tenfold and threefold higher respectively, compared to the blood from unirradiated mice.[37]
It has previously been shown that RIGI does not require genetic alterations for the effect to be initiated or perpetuated,[38,39] and this along with the high frequency of instability responses has led to the speculation that alterations in gene expression modulating cellular homeostasis may underlie the instability response.[40] Gene expression is regulated through epigenetic alterations such as chromatin remodeling, DNA methylation, and microRNA expression.[41] A number of studies have demonstrated that IR can result in changes in methylation[42,43] and microRNA expression.[44] Inflammatory processes have also been proposed as a potential mechanism[45] involving proinflammatory cytokines linked with oxidative stress and ROS which can modulate the background level of DNA damage.[38,46,47] This would be consistent with the results presented above, which show an increase in the level of cellular ROS following 1-Gy high and LDR X-ray exposure, not only 1.5 h following exposure but also after 20 population doublings. Although there is a slight suggestion of enhancement of the level of ROS at 20 population doublings later for the 0.1-Gy exposures, this is not significantly different to the 0-Gy control.
Conclusion
The work presented explores the role of dose and dose rate on RIGI, determined using a MN assay, in normal primary HF19 cells following exposure to either 0.1 Gy or 1 Gy of X-rays, delivered either as an acute (0.42 Gy/min) or protracted (0.0031 Gy/min) exposure. While the expected increase in MN was observed following the first mitosis of the irradiated cells compared to unirradiated controls, the results also demonstrate a significant enhancement in MN yields in the progeny of these cells at 10 and 20 population doublings following irradiation. Minimal difference was observed between the two doses used (0.1 and 1 Gy) and the dose rate (acute and protracted). The results also show an enhancement of the cellular levels of ROS after 20 population doublings, which suggests that IR could potentially perturb the homeostasis of oxidative stress and so modify the background rate of endogenous DNA damage induction. Therefore, these nontargeted effects have the potential to be important for the low-dose and dose-rate exposures. There are two main messages of this manuscript: (a) moderate radiation dose can be harmful not more than low dose regarding the induction of late (or delayed) genotoxic effect and (b) MNs are not the best endpoint to quantify RIGI in cultured fibroblasts.
Financial support and sponsorship
Egyptian Government.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effectsin vitro 2003. Radiat Res. 2012;178:AV223–36. doi: 10.1667/rrav19.1. [DOI] [PubMed] [Google Scholar]
- 2.Kadhim M, Salomaa S, Wright E, Hildebrandt G, Belyakov OV, Prise KM, et al. Non-targeted effects of ionising radiation – implications for low dose risk. Mutat Res. 2013;752:84–98. doi: 10.1016/j.mrrev.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller AC, Brooks K, Stewart M, Anderson B, Shi L, McClain D, et al. Genomic instability in human osteoblast cells after exposure to depleted uranium: Delayed lethality and micronuclei formation. J Environ Radioact. 2003;64:247–59. doi: 10.1016/s0265-931x(02)00053-x. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Turner HC, Shuryak I, Taveras M, Bertucci A, Perrier JR, Chen C, et al. Effect of dose rate on residual γ-H2AX levels and frequency of micronuclei in X-irradiated mouse lymphocytes. Radiat Res. 2015;183:315–24. doi: 10.1667/RR13860.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Mayah A, Bright S, Chapman K, Irons S, Luo P, Carter D, et al. The non-targeted effects of radiation are perpetuated by exosomes. Mutat Res. 2015;772:38–45. doi: 10.1016/j.mrfmmm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Watson GE, Pocock DA, Papworth D, Lorimore SA, Wright EG. In vivo chromosomal instability and transmissible aberrations in the progeny of haemopoietic stem cells induced by high- and low-LET radiations. Int J Radiat Biol. 2001;77:409–17. doi: 10.1080/09553000010028476. [DOI] [PubMed] [Google Scholar]
- 8.Watson GE, Lorimore SA, Macdonald DA, Wright EG. Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res. 2000;60:5608–11. [PubMed] [Google Scholar]
- 9.Trott KR, Jamali M, Manti L, Teibe A. Manifestations and mechanisms of radiation-induced genomic instability in V-79 Chinese hamster cells. Int J Radiat Biol. 1998;74:787–91. doi: 10.1080/095530098141078. [DOI] [PubMed] [Google Scholar]
- 10.Prise KM. New advances in radiation biology. Occup Med (Lond) 2006;56:156–61. doi: 10.1093/occmed/kql010. [DOI] [PubMed] [Google Scholar]
- 11.Ozasa K, Shimizu Y, Suyama A, Kasagi F, Soda M, Grant EJ, et al. Studies of the mortality of atomic bomb survivors, report 14, 1950-2003: An overview of cancer and noncancer diseases. Radiat Res. 2012;177:229–43. doi: 10.1667/rr2629.1. [DOI] [PubMed] [Google Scholar]
- 12.UNSCEAR. Sources, Effects and Risks of Ionizing Radiation. UNSCEAR 2017 Report. United Nations Scientific Committee on the Effects of Atomic Radiation. [Last accessed on accessed 2019 Oct 03]. Available from: https://www.unscear.org/docs/publications/2017/UNSCEAR_2017_Report.pdf .
- 13.Cox R, Masson W. X-Ray Survival Curves of Cultured Human Diploid Fibroblasts. 1975. [Last accessed on 2019 Feb 08]. Available from: https://inis.iaea.org/search/searchsinglerecord.aspx?recordsFor=SingleRecord&RN=7240058 .
- 14.Ma CM, Coffey CW, DeWerd LA, Liu C, Nath R, Seltzer SM, et al. AAPM protocol for 40-300 kV X-ray beam dosimetry in radiotherapy and radiobiology. Med Phys. 2001;28:868–93. doi: 10.1118/1.1374247. [DOI] [PubMed] [Google Scholar]
- 15.Erexson GL, Kligerman AD. A modified mouse peripheral blood lymphocyte culture system for cytogenetic analysis. Environ Mol Mutagen. 1987;10:377–86. doi: 10.1002/em.2850100407. [DOI] [PubMed] [Google Scholar]
- 16.Erexson GL, Kligerman AD, Allen JW. Diaziquone-induced micronuclei in cytochalasin B-blocked mouse peripheral blood lymphocytes. Mutat Res. 1987;178:117–22. doi: 10.1016/0027-5107(87)90093-5. [DOI] [PubMed] [Google Scholar]
- 17.Azzam EI, De Toledo SM, Spitz DR, Little JB. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res. 2002;62:5436–42. [PubMed] [Google Scholar]
- 18.Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: Selective imaging with hydroethidine. J Neurosci. 1996;16:1324–36. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc Natl Acad Sci U S A. 2003;100:5057–62. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–68. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31:961–7. doi: 10.1093/carcin/bgq069. [DOI] [PubMed] [Google Scholar]
- 22.Pantelias A, Karachristou I, Georgakilas AG, Terzoudi GI. Interphase cytogenetic analysis of micronucleated and multinucleated cells supports the premature chromosome condensation hypothesis as the mechanistic origin of chromothripsis. Cancers (Basel) 2019;11 doi: 10.3390/cancers11081123. pii: E1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.IAEA. Cytogenetic Dosimetry: Applications in Preparedness for and Response to Radiation Emergencies. IAEA. 2011. [Last accessed on 2018 Dec 15]. Available from: https://www-pub.iaea.org/MTCD/publications/PDF/EPR-Biodosimetry%202011_web.pdf .
- 24.Gulston M, de Lara C, Jenner T, Davis E, O'Neill P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004;32:1602–9. doi: 10.1093/nar/gkh306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vral A, Fenech M, Thierens H. The micronucleus assay as a biological dosimeter of in vivo ionising radiation exposure. Mutagenesis. 2011;26:11–7. doi: 10.1093/mutage/geq078. [DOI] [PubMed] [Google Scholar]
- 26.Anderson RM, Marsden SJ, Paice SJ, Bristow AE, Kadhim MA, Griffin CS, et al. Transmissible and nontransmissible complex chromosome aberrations characterized by three-color and mFISH define a biomarker of exposure to high-LET alpha particles. Radiat Res. 2003;159:40–8. doi: 10.1667/0033-7587(2003)159[0040:tancca]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 27.Shimura N, Kojima S. The lowest radiation dose having molecular changes in the living body. Dose Response. 2018;16:4. doi: 10.1177/1559325818777326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kadhim MA, Macdonald DA, Goodhead DT, Lorimore SA, Marsden SJ, Wright EG. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature. 1992;355:738–40. doi: 10.1038/355738a0. [DOI] [PubMed] [Google Scholar]
- 29.Kadhim MA, Hill MA, Moore SR. Genomic instability and the role of radiation quality. Radiat Prot Dosimetry. 2006;122:221–7. doi: 10.1093/rpd/ncl445. [DOI] [PubMed] [Google Scholar]
- 30.Kadhim MA, Marsden SJ, Wright EG. Radiation-induced chromosomal instability in human fibroblasts: Temporal effects and the influence of radiation quality. Int J Radiat Biol. 1998;73:143–8. doi: 10.1080/095530098142518. [DOI] [PubMed] [Google Scholar]
- 31.Little JB, Nagasawa H, Pfenning T, Vetrovs H. Radiation-induced genomic instability: Delayed mutagenic and cytogenetic effects of X rays and alpha particles. Radiat Res. 1997;148:299–307. [PubMed] [Google Scholar]
- 32.Belyakov OV, Prise KM, Trott KR, Michael BD. Delayed lethality, apoptosis and micronucleus formation in human fibroblasts irradiated with X-rays or alpha-particles. Int J Radiat Biol. 1999;75:985–93. doi: 10.1080/095530099139746. [DOI] [PubMed] [Google Scholar]
- 33.Smith LE, Nagar S, Kim GJ, Morgan WF. Radiation-induced genomic instability: Radiation quality and dose response. Health Phys. 2003;85:23–9. doi: 10.1097/00004032-200307000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Furlong H, Mothersill C, Lyng FM, Howe O. Apoptosis is signalled early by low doses of ionising radiation in a radiation-induced bystander effect. Mutat Res. 2013;741-742:35–43. doi: 10.1016/j.mrfmmm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Kadhim MA. Role of genetic background in induced instability. Oncogene. 2003;22:6994–9. doi: 10.1038/sj.onc.1206883. [DOI] [PubMed] [Google Scholar]
- 36.Werner E, Wang Y, Doetsch PW. A single exposure to low- or high-LET radiation induces persistent genomic damage in mouse epithelial cells in vitro and in lung tissue. Radiat Res. 2017;188:373–80. doi: 10.1667/RR14685.1. [DOI] [PubMed] [Google Scholar]
- 37.Graupner A, Eide DM, Brede DA, Ellender M, Lindbo Hansen E, Oughton DH, et al. Genotoxic effects of high dose rate X-ray and low dose rate gamma radiation in apcMin/+ mice. Environ Mol Mutagen. 2017;58:560–9. doi: 10.1002/em.22121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lorimore SA, Kadhim MA, Pocock DA, Papworth D, Stevens DL, Goodhead DT, et al. Chromosomal instability in the descendants of unirradiated surviving cells after alpha-particle irradiation. Proc Natl Acad Sci U S A. 1998;95:5730–3. doi: 10.1073/pnas.95.10.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snyder AR, Morgan WF. Lack of consensus gene expression changes associated with radiation-induced chromosomal instability. DNA Repair (Amst) 2005;4:958–70. doi: 10.1016/j.dnarep.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Baverstock K. Radiation-induced genomic instability: A paradigm-breaking phenomenon and its relevance to environmentally induced cancer. Mutat Res. 2000;454:89–109. doi: 10.1016/s0027-5107(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 41.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–56. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rugo RE, Mutamba JT, Mohan KN, Yee T, Chaillet JR, Greenberger JS, et al. Methyltransferases mediate cell memory of a genotoxic insult. Oncogene. 2011;30:751–6. doi: 10.1038/onc.2010.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miousse IR, Kutanzi KR, Koturbash I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int J Radiat Biol. 2017;93:457–69. doi: 10.1080/09553002.2017.1287454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Zhu M, Ye P, Chen G, Wang W, Chen M. Ionizing radiation-induced microRNA expression changes in cultured RGC-5 cells. Mol Med Rep. 2015;12:4173–8. doi: 10.3892/mmr.2015.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorimore SA, Wright EG. Radiation-induced genomic instability and bystander effects: Related inflammatory-type responses to radiation-induced stress and injury? A review. Int J Radiat Biol. 2003;79:15–25. [PubMed] [Google Scholar]
- 46.Limoli CL, Giedzinski E. Induction of chromosomal instability by chronic oxidative stress. Neoplasia. 2003;5:339–46. doi: 10.1016/S1476-5586(03)80027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kundu JK, Surh YJ. Inflammation: Gearing the journey to cancer. Mutat Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]