

Abstract
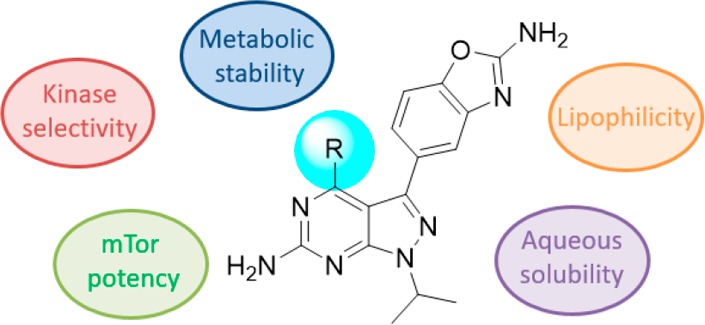
Minor structural modifications—sometimes single atom changes—can have a dramatic impact on the properties of compounds. This is illustrated here on structures related to known mTOR inhibitor Sapanisertib. Subtle changes in the hinge binder lead to strikingly different overall profiles with changes in physical properties, metabolism, and kinase selectivity.
Keywords: mTOR, kinase, selectivity, metabolism, topical application
mTOR (mammalian target of Rapamycin) inhibitors have been extensively studied in oncology, leading to the approval of rapalogs (Rapamycin analogs, binding allosterically to mTOR and preventing the formation of the mTORC1 complex) and the current clinical evaluation of multiple TORKinhibs (ATP-competitive inhibitors of the kinase function of mTOR).1,2 Recently, mTOR inhibition has also entered the dermatology field with reports of topical treatment of psoriasis3−7 and strong links of mTORC1 function with acne.8,9
In this context, identification of mTOR inhibitors for the topical treatment of dermatological diseases started at Nestlé Skin Health. Given the nature of some of these diseases, the compounds were required to display an exquisite safety margin as well as physicochemical properties compatible with topical application. Recent publications have highlighted key parameters to optimize for topical administration, including low molecular weight (<400) and good aqueous solubility (>50 μM) alongside well-balanced lipophilicity (2 < logD < 4). High systemic metabolism in combination with high protein binding are also desired to secure limited systemic effects.10,11
Out of the wealth of potent mTOR inhibitors described in the literature, Sapanisertib (1, Figure 1) was seen as a promising starting point for a topical mTOR inhibitor program. Its potency (<5 nM in a cellular setting on both mTORC1 and mTORC2 complexes) combined with its small size (MW = 309) and logD (SFlogD = 2.3; ChromlogD = 0.4)12,13 were considerable assets on the way to identifying a topical drug. However, its metabolic stability was a clear issue as it could lead to significant systemic exposure even after topical administration. Moreover, although fairly selective vs other kinases, its selectivity profile was deemed suboptimal to avoid potential local toxicities.14
Figure 1.

Structure of known inhibitors Sapanisertib (compound 1) and PF-04691502 (compound 2).
In their published accounts of the discovery of PF-04691502 (2, Figure 1), a dual PI3K/mTOR inhibitor, Pfizer scientists have highlighted the impact of the methyl group at the 4-position of the pyrimidopyridone scaffold on kinase selectivity.15,16 The selectivity arises from the presence of a small lipophilic pocket in the PIKK family of kinases between the hinge region and the conserved Tyrosine residue (Tyr2225 in mTOR, Tyr836 in PI3Kα).
Hybridizing the hinge binder of both Sapanisertib and PF-04691502 suggested synthesis of compounds following the general design paradigm exposed in Figure 2.
Figure 2.
Design paradigm for lead generation.
Strong binding to the hinge region would be ensured by the aminopyrimidine acceptor–donor pair, while careful exploration of the “R” substituent could help increase selectivity vs other kinases. Inspired by other similar campaigns,17−19 an exhaustive approach was thus undertaken to document the impact of different groups at the 4-position of the 2-aminopyrazolopyrimidine scaffold.
Scheme 1 depicts the synthesis of representative compounds evaluated during this exploration.20 A robust synthesis to intermediate 4-chloro-3-iodo-1-(propan-2-yl)-1H -pyrazolo[3,4-d]pyrimidin-6-amine was obtained in three straightforward steps from the commercially available 2-amino-4,6-dichloropyrimidine-5-carbaldehyde. This versatile intermediate allowed the introduction of variations at the 4-position while providing a handle to introduce the aminobenzoxazole unit.
Scheme 1. Synthesis of Representative Compounds from Table 1.

Reagents and conditions: (i) NH2–NH2, Et3N, THF/H2O, 60 °C, 57%; (ii) NIS, DMF, 80 °C, 98%; (iii) 2-iodopropane, Cs2CO3, DMF, r.t. 76%; (iv) 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[d]oxazol-2-amine, Pd(dppf)Cl2·CH2Cl2, K2CO3, dioxane, 100 °C, microwave, 34–41%; (v) ZnMe2 in heptane, Pd(PtBu3)2, THF, 0 °C, 97%; (vi) ZnEt2, Pd(PtBu3)2, THF, 0 °C, 21%; (vii) H2, Pd/C, MeOH, r.t. 29%; (viii) NaOMe, MeOH, r.t. 95%; (ix) NH3 in H2O (32% w/w), dioxane, 100 °C, 97%; (x) BBr3 in CH2Cl2, CH2Cl2, 0 °C, 5%.
Multiparametric evaluation of the compounds was conducted with potency being evaluated on mTOR enzyme and both mTOR complexes (mTORC1 and mTORC2) in a cellular setting.20 Selectivity against PI3Kα was monitored as a surrogate for lipid kinase selectivity. General kinome selectivity was generated at 10 μM on selected compounds and is expressed as S35, the ratio of the number of kinases with less than 35% residual activity over the total number of kinases.21 Lipophilicity and kinetic aqueous solubility were monitored, alongside metabolic stability in human microsomes and hepatocytes (Tables 1 and 2).20
Table 1. R Exploration.

| Entry | R | mTOR enz Ki nMa | mTORC1/2 IC50 nMa | PI3Kα Ki nMa | S35 @ 10 μMb | ChromlogD6.5d | Solubility μMd |
|---|---|---|---|---|---|---|---|
| 1 | n/a | 1 | 1/5 | 28 | 0.20 | 0.4 | 82 |
| 3a | -Me | 62 | 40/200 | 990 | -c | 0.8 | 74 |
| 4a | -Me | 33 | 30/90 | 2200 | 0.02 | 1.4 | 5.2 |
| 3b | -Et | 50 | 60/140 | 85 | 0.02 | 1.2 | 62 |
| 3c | -CH2OH | 4000 | -c/-c | >10000 | -c | 0.2 | 82 |
| 3d | -OMe | 370 | 420/680 | 250 | -c | 1.6 | <1 |
| 3e | -Cl | 10 | 8/20 | 920 | 0.02 | 1.7 | 2 |
| 4b | -Cl | 11 | 7/20 | 4600 | 0.02 | 2.3 | <1 |
| 3f | -F | 13 | 40/80 | 2600 | -c | 1.8 | <1 |
| 3g | -Br | 21 | 35/60 | 1000 | -c | 1.8 | 4 |
| 3h | -CF3 | 2300 | -c/-c | >10000 | -c | 2.4 | <1 |
| 3i | -CN | 2200 | -c/-c | >10000 | -c | 1.8 | <1 |
| 3j | -NH2 | 4 | 4/11 | 150 | 0.10 | –0.1 | 88 |
| 3k | -H | 6 | 7/15 | 780 | 0.26 | 1.1 | 1.6 |
Geomean of at least 2 determinations. See Supporting Information for assay details.
S35 is defined as the ratio of the number kinases with less than 35% residual activity over the total number of kinases. The test was performed at DiscoverX.21
Not tested.
See Supporting Information for assay details.
Table 2. Microsome and Hepatocyte Data for R Exploration.
| Entry | Human Mics %parent compound remaining @15 mina | Human heps Clint μL/min/1e6 cellsa |
|---|---|---|
| 1 | >95 | <5 |
| 3a | >95 | <5 |
| 3b | 93 | <5 |
| 3c | >95 | <5 |
| 3d | >95 | -b |
| 3e | 92 | 11 |
| 3f | 83 | 57 |
| 3g | 94 | 55 |
| 3h | >95 | <5 |
| 3i | >95 | <5 |
| 3j | >95 | <5 |
| 3k | 94 | 150 |
See Supporting Information for assay details.
Not tested.
Although less potent than Sapanisertib, the methyl compounds (3a and 4a) provided proof of concept that compounds with the general structure highlighted in Figure 2 could be active against mTor. The selectivity vs PI3Kα of the isopropyl analog (3a) was unchanged, while the n-butyl analog (4a) displayed slightly increased selectivity. The overall S35 value in the kinase panel was improved, but no conclusions could be drawn regarding selectivity given the observed loss of potency on mTor. The overall properties of these two methyl-substituted compounds (3a and 4a) were in line with their lipophilicity, with the more lipophilic n-butyl group leading to poor aqueous solubility. Moving to the ethyl group in the 4-position, analog 3b gave a very similar profile with the exception of an increased activity against PI3Kα. The hydroxymethyl derivative 3c gave rise to a sharp activity loss, highlighting the need for lipophilic substituents in this region. Similarly, the methoxy group (3d) was not well tolerated. The added lipophilicity resulted once again in a significantly reduced aqueous solubility.
The incorporation of halogen atoms at the 4-position of the pyrazolopyrimidine yielded promising compounds with improved potency vs alkyl groups. The fluoro (3f), the chloro (3e and 4b), and the bromo (3g) analogs all displayed robust mTOR inhibition with improved selectivity vs PI3Kα and, for chloro derivatives 3e and 4b, a reduced S35 value. As discussed below when describing the cocrystal structures, potential halogen bond or halogen−π interactions could explain the potency gain in the case of the chloro or bromo analogs. It is, however, harder to rationalize the fluoro analog’s potency. These three compounds had relatively high lipophilicity with consequentially low aqueous solubility.
Moving to electron withdrawing groups such as trifluoromethyl (3h) or nitrile (3i) resulted in a significant loss in potency. The bis-amino analog (3j) gave quite promising results with potency on par with Sapanisertib and improved selectivity vs PI3Kα and more importantly vs the general kinome. The low lipophilicity imparted good aqueous solubility, and the scaffold was seen as a potential starting point for optimization.
Finally, the nonsubstituted analog (3k) generated particularly unexpected results. The potency on mTOR remained quite high with improved selectivity vs PI3Kα. As demonstrated by Pfizer scientists, removing the substituent facing Tyrosine Tyr2225 had a negative impact on general kinome selectivity.16 Moving the amino group from the 2- to the 4-position of the pyrazolopyrimidine had a considerable impact on lipophilicity (ΔChromlogD = +0.7) with the expected negative impact on aqueous solubility.
Crystal structures of Sapanisertib and three of the compounds described above were obtained in closely related PI3Kα (Figure 3).22 One major difference between the interaction of these compounds in the PI3Kα and the mTOR hinge region is the position of the key Tyrosine residue (Tyr2225 in mTOR and Tyr836 in PI3Kα), as this residue has been shown to move significantly in mTOR when bound to closely related analog PP-242.23 In PI3Kα, this shift of the tyrosine residue was not detected with the compounds presented here, and a hydrogen bond between the sp2 nitrogen of benzoxazole was detected in all of our crystal structures. All of the compounds make the expected interaction with hinge residue Val851 (Val2240 in mTOR). Sapanisertib makes an additional hydrogen bond with Glu849 (Gly2238 in mTOR; Figure 3A), whereas the 2-amino substituted analogs make a hydrogen bond with Val851. The chlorine atom in analog 3e is too far away to interact both with the backbone carbonyl of Glu849 through a halogen bond24 and with Tyr836 through a halogen−π interaction25,26 (Figure 3C). However, either or both these interactions could potentially take place in mTOR and explain the increased affinity. Lastly, the bis-amino analog 3j makes three strong hydrogen bonds with the hinge residues (Figure 3D).
Figure 3.

X-ray cocrystal structure of inhibitors bound to PI3Kα. (A) Sapanasertib compound 1 (PDB code: 6GVF); (B) compound 3a (PDB code: 6GVG); (C) compound 3e (PDB code: 6GVH); and (D) compound 3j (PDB code: 6GVI). Hydrogen bonds are represented by green dashed lines.
Metabolism in human microsomes and human hepatocytes was measured for all relevant analogs (Table 2). Screening in both microsomes and hepatocytes in parallel was used not only to identify compounds with metabolism liabilities but also to highlight ones more susceptible to phase II metabolism. Indeed, the conjugation event often associated with phase II metabolism would most likely generate inactive metabolites, which are highly desirable to reduce any systemic pharmacology. In general, the compounds were stable in both assays with four notable exceptions.
All halogen substituted analogs (compounds 3e, 3f, and 3g) displayed a high turnover in hepatocytes with limited turnover in microsomes. This discrepancy suggested a possible phase II metabolism and prompted more in-depth metabolism studies. A Met-ID study on the halogen containing compounds in human hepatocytes revealed that all three formed a glutathione adduct (compound 5, Figure 4).27,35 This result clearly highlighted the 4-position of these compounds as reactive to nucleophiles. Given the potential risk of irritation with the high doses found in the upper layers of the skin, 4-halogeno derivatives were thus deprioritized.28
Figure 4.

Main metabolite of halogen containing compounds 3e, 3f, and 3g and main metabolite of 2-aminopyrazolopyrimidine analog 3k.
Nonsubstituted analog 3k was also quickly metabolized in human hepatocytes while being essentially stable in microsomes, prompting once again a more in-depth analysis. The main metabolite was identified as pyrimidone derivative 6a (Figure 4),29,30 which could potentially come from aldehyde-oxidase mediated metabolism.31−33 Pyrimidone 6a was synthesized and evaluated in the hope of identifying an inactive molecule (Table 3). Pyrimidone 6a turned out to be quite active on mTOR with an increased selectivity vsPI3Kα and low S35 value. If clearly not an inactive metabolite, it served as a new platform to identify potent and selective mTOR inhibitors.
Table 3. Pyrazolopyrimidones Exploration.

| Entry | R1 | mTOR enz Ki nMa | mTORC1/2 IC50 nMa | PI3Kα Ki nMa | S35b | ChromlogD6.5d | Solubility μMd | Human Mics %PP @15 mind | Human heps Clint μL/min/1e6 cellsd |
|---|---|---|---|---|---|---|---|---|---|
| 1 | n/a | 1 | 1/5 | 28 | 0.20 | 0.4 | 82 | >95 | <5 |
| 3k | n/a | 6 | 7/15 | 780 | 0.26 | 1.1 | 1.6 | 94 | 150 |
| 6a | -NH2 | 20 | 40/200 | 3200 | 0.05 | 0.4 | 9.2 | >95 | <5 |
| 6b | -NHMe | 190 | 240/360 | >10000 | -c | 1.5 | <1 | >95 | <5 |
| 6c | -NMe2 | 1700 | -c/-c | >10000 | -c | 2.1 | 2 | 90 | <5 |
| 6d | -Me | 40 | 45/90 | >10000 | -c | 1.2 | 3 | >95 | <5 |
| 6e | -H | 28 | 30/130 | >10000 | 0.02 | 0.9 | 3 | >95 | <5 |
Geomean of at least two determinations. See Supporting Information for assay details.
S35 is defined as the ratio of the number of kinases with less than 35% residual activity over the total number of kinases. The test was performed at DiscoverX.
Not tested.
See Supporting Information for assay details.
At this stage, the parallel was made with published work by Merck scientists on a series of IRAK4 inhibitors.34,35 In their case, chloro, methyl, and hydrogen substituents were similarly tolerated in the hinge region of the IRAK4’s ATP binding pocket (compounds 7a, 7b, and 7c, respectively, in Figure 5).36,37
Figure 5.

The pyrimidone derivative (7e) also turned out to be quite promising and even more potent on IRAK4 than the corresponding nonsubstituted compound (7d). This was all the more surprising as X-ray crystallography proved the pyrimidone tautomer altered the binding mode and shifted the hydrogen bonding pattern to the hinge region.
No cocrystal structures of compound 6a (or closely related analogs) bound to PI3Kα could be obtained, and no concrete proof could be secured, showing the pyrimidone derivatives described in this article bound to mTOR using their carbonyl tautomer. Inspired by the Merck IRAK4 reports (e.g. optimized compound 7f)34 analogs with modified amino groups at the 2-position were profiled. Substitution led to a decrease in potency (compounds 6b and 6c), suggesting either the importance of the hydrogen on the amino group or a steric clash in this region. However, replacing the amino group by a methyl (compound 6d) maintained potency, suggesting a nonessential role of the 2-amino group. Finally, deleting the amino group altogether (compound 6e) displayed similar potency with exquisite selectivity vs PI3Kα. No sign of inhibition was detected on other PI3K isoforms at 10 μM.21
Although quite appealing from a potency/selectivity point of view, the pyrimidones were deemed less promising starting points. Indeed, securing good aqueous solubility was seen as a significant challenge for this series, even in low logD space (Chart 1), potentially highlighting the high crystallinity of these derivatives. One reasonable explanation could come from the tendency of pyridones/pyrimidones to form strong H-bond interactions in their crystal packing.38,39
Chart 1. Aqueous Solubility vs ChromlogD6.5 for All of the Compounds Presented in This Articlea.
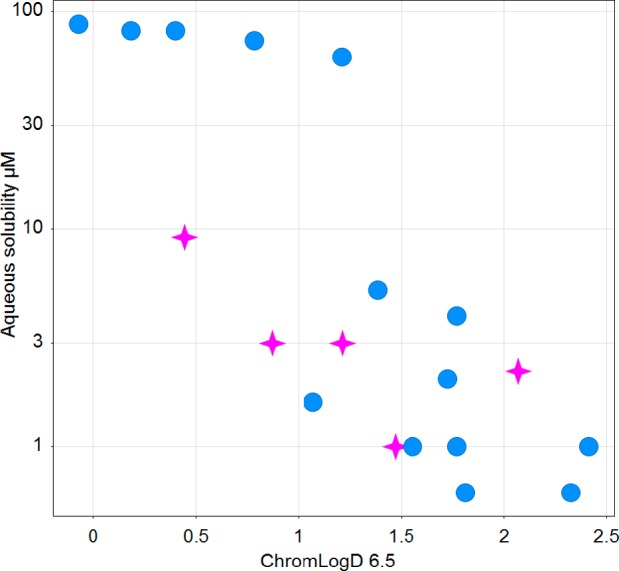
a Pink stars represent pyrimidone compounds; blue circles, pyrimidine compounds.
In conclusion, the body of work presented here highlights that simple changes to the hinge binding core of Sapanisertib could have drastic impact on the compound’s kinase selectivity, physical properties, and metabolism. The move of an amino group from the 4-position to the 2-position of the pyrazolopyrimidine core (1 vs 3k) is particularly striking, as the new analog displays significantly higher lipophilicity (+0.7) and a completely different metabolic fate. Another example is the replacement of the amino group by a phenol (1 vs 6e) with an impressive impact on the selectivity vs PI3K isoforms. Optimization of potency, metabolic instability, and solubility in formulation leading to the identification of a clinical topical mTOR inhibitor will be the subject of an upcoming publication.
Acknowledgments
Researchers at Evotec (Toulouse, France) are acknowledged for their help in the synthesis of key intermediates for the synthesis of the compounds disclosed in this article.
Glossary
Abbreviations
- mTOR
mammalian target of Rapamycin
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PIKK
PI3K-related kinase
- IRAK4
(interleukin-1 receptor-associated kinase 4
- Met-ID
metabolite identification
- THF
tetrahydrofuran
- NIS
N-iodosuccinimide
- DMF
dimethylformamide
- r.t.
room temperature.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00401.
Assay descriptions, synthetic schemes, synthetic methods, and full kinase selectivity profiles (for compounds 1, 3b, 3e, 3j, 3k, 4a, 4b, 6a, and 6e) (PDF)
Author Present Address
§ G. Ouvry: Evotec, 114 Innovation Drive, Milton Park, Abingdon, United Kingdom.
Author Present Address
∥ L. Clary: Nuvisan SARL, 2400 route des Colles, 06410 Biot, France.
Author Present Address
⊥ L. L. Tomas, C. Bouix-Peter, C. S. Harris, E. Vial, L. F. Hennequin: Galderma S.A., Rx Strategy & Innovation Group, Rue d-Entre-Deux-Villes 10, 1814 La Tour de Peilz, Switzerland.
Author Present Address
∇ E. Borde: Genochem, 34 Traverse de la Paoute, 06130 Grasse, France.
Author Present Address
○ L. Chantalat: Nestlé Skin Health S.A., Avenue Gratta-Paille 2, 1018 Lausanne, Switzerland.
Author Present Address
□ C. Defoin-Platel, M. Forissier, L. Lamy, A.-P. Luzy, A. Taddei: Syneos Health Les Templiers, 2400 route des Colles, 06410 Biot, France.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare the following competing financial interest(s): All authors were Nestle Skin Health R&D or Synchrotron Soleil full-time employees at the time this work was carried out.
Supplementary Material
References
- Zask A.; Verheijen J. C.; Richard D. J. Recent advances in the discovery of small-molecule ATP competitive mTOR inhibitors: a patent review. Expert Opin. Ther. Pat. 2011, 21, 1109–1127. 10.1517/13543776.2011.584871. [DOI] [PubMed] [Google Scholar]
- Roohi A.; Hojjat-Farsangi M. Recent advances in targeting mTOR signaling pathway using small molecule inhibitors. J. Drug. Target. 2017, 25, 189–201. 10.1080/1061186X.2016.1236112. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S. K.; Raychaudhuri S. P. mTOR Signaling Cascade in Psoriatic Disease: Double Kinase mTOR Inhibitor a Novel Therapeutic Target. Indian J. Dermatol. 2014, 59, 67–70. 10.4103/0019-5154.123499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.; Lin X.; Meng X.; Lin M. Phosphoinositide-3 kinase/protein kinase-B/mammalian target of rapamycin pathway in psoriasis pathogenesis. A potential therapeutic target?. Acta Derm. Venereol. 2014, 94, 371–379. 10.2340/00015555-1737. [DOI] [PubMed] [Google Scholar]
- Wei K. C.; Lai P. C. Combination of everolimus and tacrolimus: a potentially effective regimen for recalcitrant psoriasis. Dermatol. Ther. 2015, 28, 25–27. 10.1111/dth.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo M. S.; Sivamani R. K. Phytochemical modulation of the Akt/mTOR pathway and its potential use in cutaneous disease. Arch. Dermatol. Res. 2014, 306, 861–871. 10.1007/s00403-014-1480-8. [DOI] [PubMed] [Google Scholar]
- Ormerod A. D.; Shah S. A.; Copeland P.; Omar G.; Winfield A. Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double-blind trial. Br. J. Dermatol. 2005, 152, 758–764. 10.1111/j.1365-2133.2005.06438.x. [DOI] [PubMed] [Google Scholar]
- Agamia N. F.; Abdallah D. M.; Sorour O.; Mourad B.; Younan D. N. Skin expression of mammalian target of rapamycin and forkhead box transcription factor O1, and serum insulin-like growth factor-1 in patients with acne vulgaris and their relationship with diet. Br. J. Dermatol. 2016, 174, 1299–1307. 10.1111/bjd.14409. [DOI] [PubMed] [Google Scholar]
- Melnik B. C.; Zouboulis C. C. Potential role of FoxO1 and mTORC1 in the pathogenesis of Western diet-induced acne. Exp. Dermatol 2013, 22, 311–315. 10.1111/exd.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an exhaustive and essential review on topical design see: Dermal Drug Candidate Selection and Development: An Industrial Perspective; Trottet L., Maibach H., Eds.; Springer, 2017. [Google Scholar]
- For a recent study highlighting key features to optimize flux through skin see:Ullrich T.; Sasmal S.; Boorgu V.; Pasagadi S.; Cheera S.; Rajagopalan S.; Bhumireddy A.; Shashikumar D.; Chelur S.; Belliappa C.; Pandit C.; Krishnamurthy N.; Mukherjee S.; Ramanathan A.; Ghadiyaram C.; Ramachandra M.; Santos P. G.; Lagu B.; Bock M. G.; Perrone M. H.; Weiler S.; Keller H. 3-alkoxy-pyrrolo[1,2-b]pyrazolines as selective androgen receptor modulators with ideal physicochemical properties for transdermal administration. J. Med. Chem. 2014, 57, 7396–7411. 10.1021/jm5009049. [DOI] [PubMed] [Google Scholar]
- ChromLogD6.5 was calculated from an HPLC measured Chromatographic Hydrophobicity Index at pH 6.5 using the following equation: ChromLogD6.5 = 0.086*CHI6.5 −3.5. pH 6.5 is used to mimick the more acidic nature of skin. For additional references on CHI, see:Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- For this series of compounds, there was always a significant but reproducible shift between shake-flask logD7.4 and ChromlogD6.5 that could not be explained by the pH difference given that the molecules described in this article are all neutral.
- Fraser C.; Carragher N. O.; Unciti-Broceta A. eCF309: a potent, selective and cell-permeable mTOR inhibitor. MedChemComm 2016, 7, 471–477. 10.1039/C5MD00493D. [DOI] [Google Scholar]
- Liu K. K.-C.; Bagrodia S.; Bailey S.; Cheng H.; Chen H.; Gao L.; Greasley S.; Hoffman J. E.; Hu Q.; Johnson T. O.; Knighton D.; Liu Z.; Marx M. A.; Nambu M. D.; Ninkovic S.; Pascual B.; Rafidi K.; Rodgers C. M.-L.; Smith G. L.; Sun S.; Wang H.; Yang A.; Yuan J.; Zou A. 4-Methylpteridinones as orally active and selective PI3K/mTOR dual inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 6096–6099. 10.1016/j.bmcl.2010.08.045. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Bagrodia S.; Bailey S.; Edwards M.; Hoffman J.; Hu Q.; Kania R.; Knighton D. R.; Marx M. A.; Ninkovic S.; Sun S.; Zhang E. Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04691502 through structure based drug design. MedChemComm 2010, 1, 139–144. 10.1039/c0md00072h. [DOI] [Google Scholar]
- Patel L.; Chandrasekhar J.; Evarts J.; Haran A. C.; Ip C.; Kaplan J. A.; Kim M.; Koditek D.; Lad L.; Lepist E.-I.; McGrath M. E.; Novikov N.; Perreault S.; Puri K. D.; Somoza J. R.; Steiner B. H.; Stevens K. L.; Therrien J.; Treiberg J.; Villaseñor A. G.; Yeung A.; Phillips G. 2,4,6-Triaminopyrimidine as a novel hinge binder in a series of PI3Kδ selective inhibitors. J. Med. Chem. 2016, 59, 3532–3548. 10.1021/acs.jmedchem.6b00213. [DOI] [PubMed] [Google Scholar]
- Barlaam B.; Cosulich S.; Fitzek M.; Germain H.; Green S.; Hanson L. L.; Harris C. S.; Hancox U.; Hudson K.; Lambert-van der Brempt C.; Lamorlette M.; Magnien F.; Ouvry G.; Page K.; Ruston L.; Ward L.; Delouvrie B. Discovery of a novel aminopyrazine series as selective PI3Kα inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3030–3035. 10.1016/j.bmcl.2017.05.028. [DOI] [PubMed] [Google Scholar]
- Perreault S.; Chandrasekhar J.; Cui Z.-H.; Evarts J.; Hao J.; Kaplan J. A.; Kashishian A.; Keegan K. S.; Kenney T.; Koditek D.; Lad L.; Lepist E.-I.; McGrath M. E.; Patel L.; Phillips B.; Therrien J.; Treiberg J.; Yahiaoui A.; Phillips G. Discovery of a phosphoinositide 3-kinase (PI3K) β/δ inhibitor for the treatment of phosphatase and tensin homolog (PTEN) deficient tumors: building PI3Kβ potency in a PI3Kδ-selective template by targeting nonconserved Asp856. J. Med. Chem. 2017, 60, 1555–1567. 10.1021/acs.jmedchem.6b01821. [DOI] [PubMed] [Google Scholar]
- Assay details, complete synthesis schemes and detailed syntheses of all of the compounds can be found in the Supporting Information.
- see Supporting Information for complete data.
- PDB codes: compound 1, 6GVF; compound 3a, 6GVG; compound 3e, 6GVH; compound 3j, 6GVJ.
- Yang H.; Rudge D. G.; Koos J. D.; Vaidialingam B.; Yang H. J.; Pavletich N. P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–224. 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y. N.; Inoue Y.; Nakanishi I.; Kitaura K. Cl-π interactions in protein-ligand complexes. Protein Sci. 2008, 17, 1129–1137. 10.1110/ps.033910.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Similar halogen−π interactions have been well characterized in the Factor Xa inhibitor literature, as exemplified in:Perzborn E.; Roehrig S.; Straub A.; Kubitza D.; Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discovery 2011, 10, 61–75. 10.1038/nrd3185. [DOI] [PubMed] [Google Scholar]
- This halogen displacement has already been documented on similar heterocycles in:Inoue K.; Ohe T.; Mori K.; Sagara T.; Ishii Y.; Chiba M. Aromatic Substitution Reaction of 2-Chloropyridines Catalyzed by Microsomal Glutathione S-Transferase 1. Drug Metab. Dispos. 2009, 37, 1797–1800. 10.1124/dmd.109.027698. [DOI] [PubMed] [Google Scholar]
- One routine test for assessing skin irritancy is the DPRA assay (https://www.oecd.org/chemicalsafety/testing/Draft_DPRA_TG_final_15May2014.pdf) which essentially quantifies the reaction of a compound with cysteine and lysine containing peptides.
- There is significant literature suggesting that the pyrimidone should be the preferred tautomer in this case:Galvão T. L.; Rocha I. M.; Ribeiro da Silva M. D.; Ribeiro da Silva M. A. From 2-hydroxypyridine to 4(3H)-pyrimidinone: computational study on the control of the tautomeric equilibrium. J. Phys. Chem. A 2013, 117, 12668–12674. 10.1021/jp410004x. [DOI] [PubMed] [Google Scholar]
- Taylor P. J.; van des Zwan G.; Antonov L.. Tautomerism: Introduction, History, and Recent Developments in Experimental and Theoretical Methods in Tautomerism: Methods and Theories; Antonov L.; Ed.; Wiley-VCH, 2014; pp 1–24. [Google Scholar]
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde Oxidase: An Enzyme of Emerging Importance in Drug Discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Manevski N.; King L.; Pitt W. R.; Fabien Lecomte F.; Toselli F.. Metabolism by Aldehyde Oxidase: Drug Design and Complementary Approaches to Challenges in Drug Discovery. J. Med. Chem. 2019, 10.1021/acs.jmedchem.9b00875. [DOI] [PubMed] [Google Scholar]
- Oxidation by aldehyde oxidase was reported on the related heterocyclic core of Famciclovir:Rashidi M. R.; Smith J. A.; Clarke S. E.; Beedham C. In vitro oxidation of famciclovir and 6-deoxypenciclovir by aldehyde oxidase from human, guinea pig, rabbit, and rat liver. Drug Metab. Dispos. 1997, 25, 805–813. [PubMed] [Google Scholar]
- Seganish W. M.; Fischmann T. O.; Sherborne B.; Matasi B.; Lavey B.; McElroy W. T.; Tulshian D.; Sondey C.; Garlisi C. G.; Devito K.; Fossetta J.; Lundell D.; Niu X. Discovery and Structure Enabled Synthesis of 2,6-Diaminopyrimidin-4-one IRAK4 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 942–947. 10.1021/acsmedchemlett.5b00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy W. T.; Seganish W. M.; Herr R. J.; Harding J.; Yang J.; Yet L.; Komanduri V.; Prakash K. C.; Lavey B.; Tulshian D.; Greenlee W. J.; Sondey C.; Fischmann T. O.; Niu X. Discovery and hit-to-lead optimization of 2,6-diaminopyrimidine inhibitors of interleukin-1 receptor-associated kinase 4. Bioorg. Med. Chem. Lett. 2015, 25, 1836–1841. 10.1016/j.bmcl.2015.03.043. [DOI] [PubMed] [Google Scholar]
- To our knowledge, there is no reported strong homology between IRAK4 and mTOR.
- Of note, Merck scientists also noted a displacement of the chlorine atom by glutathione in their series.
- As an example, the melting point of 2-pyridone (107–108 °C) is significantly higher than that of 2-aminopyridine (59–60 °C).
- As a side note, and of no influence for topical administration, 6a was found to be extensively effluxed in Caco2 when compared to close analog bis amino derivative 3j (efflux ratio 5 and 100 for 3j and 6a, respectively).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.