

Abstract
Environmental microbes frequently live in multispecies biofilms where mutualistic relationships and co-evolution may occur, defining spatial organization for member species and overall community functions. In this context, intrinsic properties emerging from microbial interactions, such as efficient organization optimizing growth and activities in multispecies biofilms, may become the object of fitness selection. However, little is known on the nature of underlying interspecies interactions during establishment of a predictable spatial organization within multispecies biofilms. We present a comparative metatranscriptomic analysis of bacterial strains residing in triple-species and four-species biofilms, aiming at deciphering molecular mechanisms underpinning bacterial interactions responsible of the remarkably enhanced biomass production and associated typical spatial organization they display. Metatranscriptomic profiles concurred with changes in micro-site occupation in response to the addition/removal of a single species, being driven by both cooperation, competition, and facilitation processes. We conclude that the enhanced biomass production of the four-species biofilm is an intrinsic community property emerging from finely tuned space optimization achieved through concerted antagonistic and mutualistic interactions, where each species occupies a defined micro-site favoring its own growth. Our results further illustrate how molecular mechanisms can be better interpreted when supported by visual imaging of actual microscopic spatial organization, and we propose phenotypic adaptation selected by social interactions as molecular mechanisms stabilizing microbial communities.
Subject terms: Biofilms, Transcriptomics, Microbial ecology
Introduction
Environmental microorganisms typically live in dense, surface-associated multispecies communities harboring distinct spatial organization, known as biofilms [1, 2]. Species residing in biofilms live in close vicinity, affecting their fitness through cooperative or competitive activities triggered by interspecies interactions [3–5]. As opposed to both free-living cells and mono-species biofilms, cooperative advantages acquired when living within a mixed community may frequently result in “emerging community properties”, such as enhanced biomass production, access to complex nutrient sources, stress resistance or pathogenicity acquisition [6–9]. In contrast, cumulating evidences from experimental and metabolic model-based studies showed that microorganisms are not living together in pure harmony, as competition for limited resources, such as space and nutrients, is pervasive [10–13].
While fundamental knowledge was gathered from studies of monospecific biofilm cultivation, the focus has gradually shifted toward understanding molecular mechanisms underlying interspecies interactions in complex multispecies biofilms [14]. Advances in RNA sequencing (RNA-seq) technologies have facilitated the study of communal gene expression profiles, revealing the functioning of complex environmental microbial communities [15–18]. Recently, spatial organization within multispecies biofilms gained momentum due to its evidenced critical role in shaping the community composition and functions in vivo [19, 20]. However, unraveling molecular mechanisms underpinning social interactions at the micro-site spatial level is not trivial, as it remains extremely challenging to 3D-visualize environmental microbial communities where sometimes hundreds of species may coexist. Therefore, model biofilms including a limited number of key species stand as an ideal alternative to pave the way in this promising research area.
A previously characterized four-species biofilm composed of the biofilm-forming isolates Stenotrophomonas rhizophila and Xanthomonas retroflexus, and the poor biofilm formers Microbacterium oxydans and Paenibacillus amylolyticus, was previously reported to display significant increase in biomass production compared to co-cultures composed of three species [21]. It has been verified that cooperative interactions mediated locally by the spatial intermixing of X. retroflexus and M. oxydans were crucial in stabilizing the four-species community [22]. Intriguingly, since spatial intermixing of these two species was not observed when co-cultured in either dual-species and triple-species biofilms [21, 22], we concluded that this relationship was sustained by the unique microbial interactions occurring in the four-species community. Gathering additional knowledge on social interactions occurring in this four-species community will further improve our understanding of underlying mechanisms driving the peculiar and highly reproducible micro-site organization and intrinsic properties emerging from microbial interactions, leading to enhanced biomass production.
In this study, we performed RNA-seq on the four-species and two selected triple-species biofilm combinations collected in a continuous drip-flow biofilm reactor (DFR) system, aiming at deciphering the genetic basis of social interactions responsible of the distinct spatial structure observed. We hypothesized that the highly reproducible spatial organization observed in the four-species community is driven by specific bacterial interactions, which will be reflected by changes in transcriptome profiles of individual species at community level when compared to those in triple-species biofilms. To test this, sufficient information was gathered to draft an overview of actual social interactions occurring in the four-species biofilm by purposefully selecting two triple-species biofilms with visually contrasting and distinct spatial structures. While supporting our previous findings regarding the role of cooperation in establishment of higher biofilm volumes, our mRNA-seq results revealed that competition is also an important factor involved in this process. Moreover, phenotypic adaptation (known as phenotypic character displacement) between the two phylogenetic closely related species, S. rhizophila and X. retroflexus, was proposed as molecular mechanisms stabilizing the four-species community. The mRNA-seq results achieved here could help pave the way verifying presumed molecular mechanisms underpinning interspecies interactions in association with experimentally mechanistic follow-up studies.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains S. rhizophila (2), X. retroflexus (3), M. oxydans (5), and P. amylolyticus (7) were isolated from a soil micro-aggregate [23]. For clarity sake, strain numbers (2, 3, 5, and 7) were systematically used throughout the main text. Strains were activated from −80 °C glycerol stocks on tryptic soy agar plates (TSA) (Sigma, St. Louis, USA) for 48 h at 24 °C. Isolated colonies for each strain were inoculated into 5 ml of sterile tryptic soy broth (TSB) (Sigma, St. Louis, USA) and incubated with shaking (250 rpm/min) at room temperature overnight.
Biofilm cultivation
Triple (235, 257) and the four-species biofilms (2357) were grown at room temperature in a continuous DFR system [24]. Briefly, overnight cultures of each strain were sub-cultured to exponential phase and adjusted to an optical density at 600 nm (OD600) of 0.15 in 1/2-strength TSB, mixed with equal amounts to a total volumes of 20 ml, and inoculated into DFR channels. Settings for the bacterial attachment (4 h) and DFR initiation were descried by Liu et al. [21]. Biofilms were collected 24 h after the attachment phase.
Crystal violet assay and viable cell enumeration
Biomasses of triple-species and four-species biofilms on glass slides were quantified by a modified CV assay after 24 h cultivation in DFR [25, 26]. The slides with biomass were incubated for 15 min with 40 ml of 99% methanol for cell fixation, then air-dried for several minutes. After drying, samples were submerged 20 min in 1% (w/v) CV solution for staining. Slides were rinsed at least three times until complete unbound CV removal. Slides were then placed 30 min in 40 ml of 33% acetic acid to release the bound CV. The absorbance was measured at 590 nm by using an EL 340 BioKinetics reader (BioTek Instruments, Winooski, Vt.). The CV-acetic acid suspension was diluted with 33% acetic acid when the OD590 was above 1.1.
Viable cell enumeration was adapted from Jackson et al. [27]. All visible biomass was transferred from glass slides into centrifuge tubes containing 700 µl sterilized phosphate-buffered saline (PBS, pH = 7.4) and glass beads (diameter, 5 mm) using aseptic NuncTM cell scrapers (Thermo Fisher Scientific), after detaching cells with 300 µl sterilized PBS. Samples were homogenized via vortexing (IKA® MS 3 basic) for 30 s at 2500 rpm, and serially diluted down to 1 × 10–7. A 100 µl aliquot of each serial dilution was spread, in triplicate, onto Congo red agar plates (TSA with 40 µg/ml Congo Red and 20 µg/ml Coomassie). Strains 5 and 7 could be visually distinguished based on colony morphologies. The two similar Gammaproteobacteria strains (2 and 3) were discriminated using selective media supplemented with Kanamycin (50 µg/ml), as S. rhizophila is sensitive whereas X. retroflexus is resistant. Agar plates were incubated at 24 °C and colonies for each species were counted after 48 h. Three biological replicates of triple-species and four-species biofilms were collected for CV assay and viable cell numbering, respectively. Results were statistically analyzed using the mucltcomp package [28] in Rgui [29] using ANOVA with a Tukey’s HSD posthoc multiple comparison test (P < 0.05).
Spatial localization and 3D pairwise cross-correlation (PCC) analyses
Results for spatial localization and pairwise cross-correlation (PCC) analyses of the four-species biofilm were adapted from our previously published work [21, 22]. Specifically, spatial localization analysis was performed by quantifying relative pixels of individual species in three defined layers (top, middle, and bottom) of biofilm images, as described in details by Liu et al. [21]. Image analyses for two types (scant: S and typical: T) of bacterial distribution of the four-species biofilm, defined by Liu et al. [22], were included in this study, in order to avoid bias interpretation with metatranscriptomics achieved from globally collected biofilm samples. Raw z-stack images of triple-species biofilms (three biological replicates) were obtained from Liu et al. [21], and reanalyzed here using custom PCC analysis. In this study, z-stack images of triple-species biofilms were pre-processed and further treated for spatial localization and PCC analyses using previously described methods [21, 22]. R scripts used for image analyses are public available at GitHub (www.github.com/Russel88/RCon3D version 0.9).
Sample collection and total RNA isolation
Triple-species and four-species biofilms were grown up to 24 h, scraped and submerged into 1 ml of Ambion RNAlater® (Thermo Fisher Scientific, Waltham, MA, USA) with sterilized NuncTM cell scrapers (Thermo Fisher Scientific). The RNAlater-preserved samples were kept at 4 °C overnight, after which the supernatant was removed by centrifuging at 8000 × g for 5 min at 4 °C. The pellet was stored at −80 °C before total RNA isolation. This assay was repeated three times at different days to obtain triplicates.
Samples were transferred into 2.0 ml Lysing Matrix E tubes (MP Biomedicals Inc.), and processed with the FastPrep® Instrument (MP Biomedicals Inc.) for 30 s at 6.0 speed. Total RNA was purified from each biofilm sample using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Each sample was suspended in 50 μl of RNA storage solution and RNA quality was determined using Agilent 2100 BioanalyzerTM (Agilent Technologies, Santa Clara, CA, USA).
Purification, enrichment, and sequencing of mRNA
DNA was removed with the DNAfree™ Kit (Ambion) according to the manufacturer’s instructions. Ribosomal RNA (rRNA) was removed using a Ribo-Zero rRNA Removal Kit (Bacteria) (Illumina) according to the manufacturer’s instructions, and mRNA quality was measured using Agilent 2100 BioanalyzerTM (Agilent Technologies, Santa Clara, CA, USA). Bacterial mRNA was fragmented using a RNA fragmentation kit (Ambion), with a size-range of 200–250 bp. Double-stranded cDNA was generated using the SuperScript DoubleStranded cDNA Synthesis Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. An Illumina Paired End Sample Prep kit was used to prepare RNA-seq library according to the manufacturer’s instructions. Samples were sequenced with an Illumina MiSeq using 150 bp paired end libraries (Illumina, San Diego, CA, USA).
Bioinformatic and biostatistic analysis of mRNA profiles
The full genomes of the four strains (Supplementary Table 1) used in this study have been previously sequenced, deposited and annotated in the Rapid Annotation System Technology (RAST) using the SEED hierarchical classification [30]. Peptide sequences obtained from predicted open-reading frames (ORFs) in each genome were exported and combined into three datasets representatives of each tested biofilm conditions. Each peptide datasets were dereplicated using Usearch (Version 8.1.1861) [31].
Raw reads were trimmed for adapters and quality scores using Trimmomatic 0.36 with the supplied adapters list [32]. Since Miseq sequencing often yields poor quality at sequence extremities, all bases in both ends with quality less than three were trimmed. Additionally, a four base window were scanned over the reads and reads with windows with an average quality of <15 were truncated. Core identical protein sequences shared between any two of the four genomes were identified with Usearch (Version 8.1.1861) [31], and removed from the analysis, since they do not contribute in differentiating the strains. Paired and orphan reads were mapped to annotated genes using blastx best hits in Diamond 0.8.22. Blast results were accepted if the e-value was <1 × 10–3, and then combined for downstream processing in Rgui software [29]. mRNA read counts obtained for each protein were corrected according to biomass data with the mean of plate CFU counts (n = 3) to avoid expression biases related to cell number variation of individual species in triple-species and four-species biofilms. Differentially expressed genes were identified using EdgeR package in Rgui [33], relying on internal calibration procedures to compensate and adjust for varying sequencing depths as represented by differing library sizes [34]. While reads per kilobase per million mapped reads (RPKM) stands as the most popular normalization method [35], it was not deemed appropriate for our study. Indeed, this method was initially developed to compare gene expression within one sample or for one organism by rescaling counts according to gene and library size [36]. Nevertheless, this normalization fails to integrate the important biomass changes observed in our multispecies biofilm data. Furthermore, gene size and expression were very weakly and negatively correlated together in our experiments (Supplementary Fig. 1), and RPKM is also known to strongly influence the mean-to-variance relationship within the data, which stands as a crucial step for accurate modeling of contingency data and meaningful downstream statistics [37]. Differential expression levels between mRNA profiles obtained from each biofilm consortia were extracted by means of generalized linear model under negative binomial distribution (nbGLM) using a likelihood ratio test with a post hoc false discovery rate correction test (LRT, FDR-corrected P < 0.05, log2-fold change > 2).
Results
Distinct spatial organization in triple-species and four-species biofilms
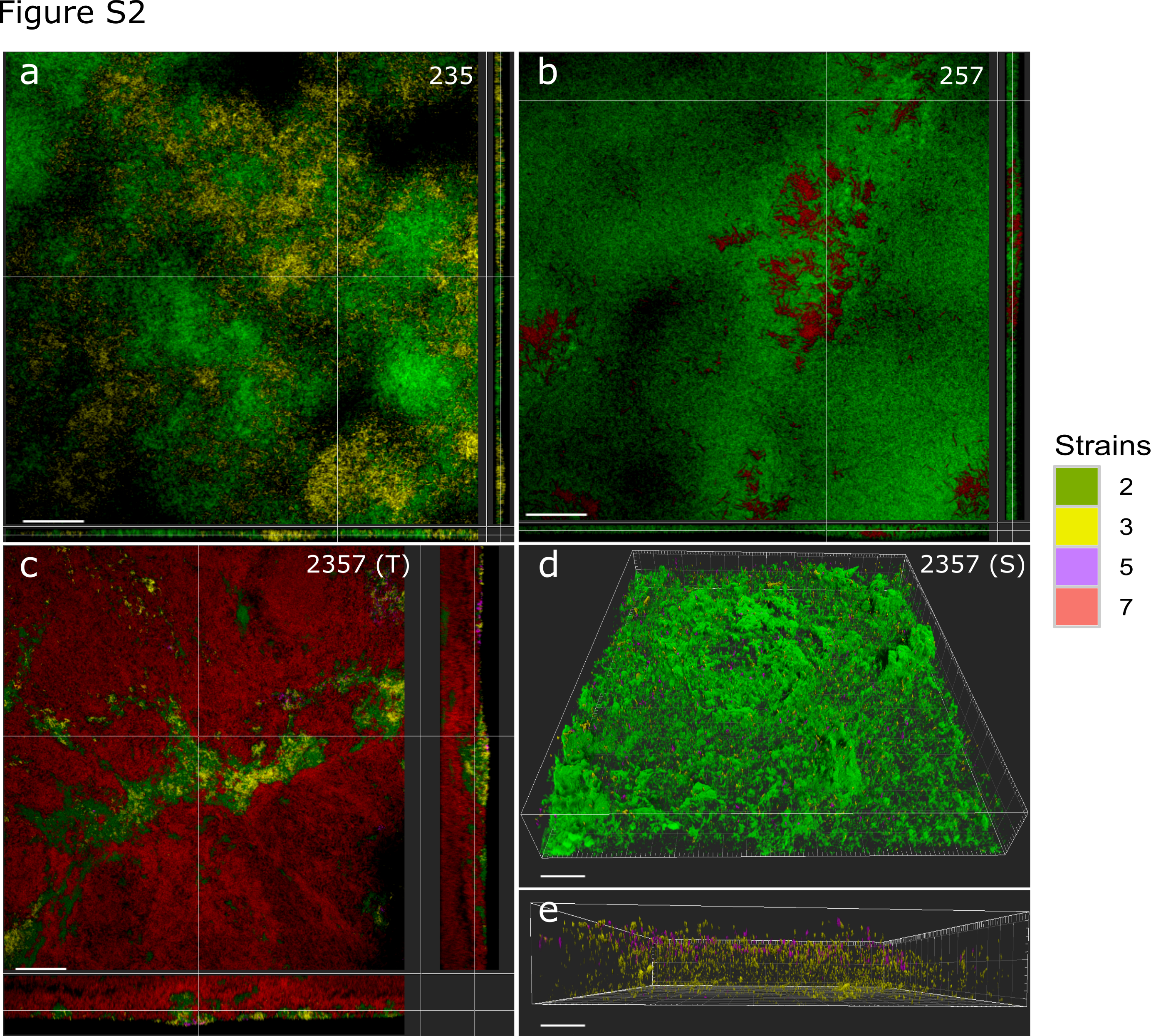
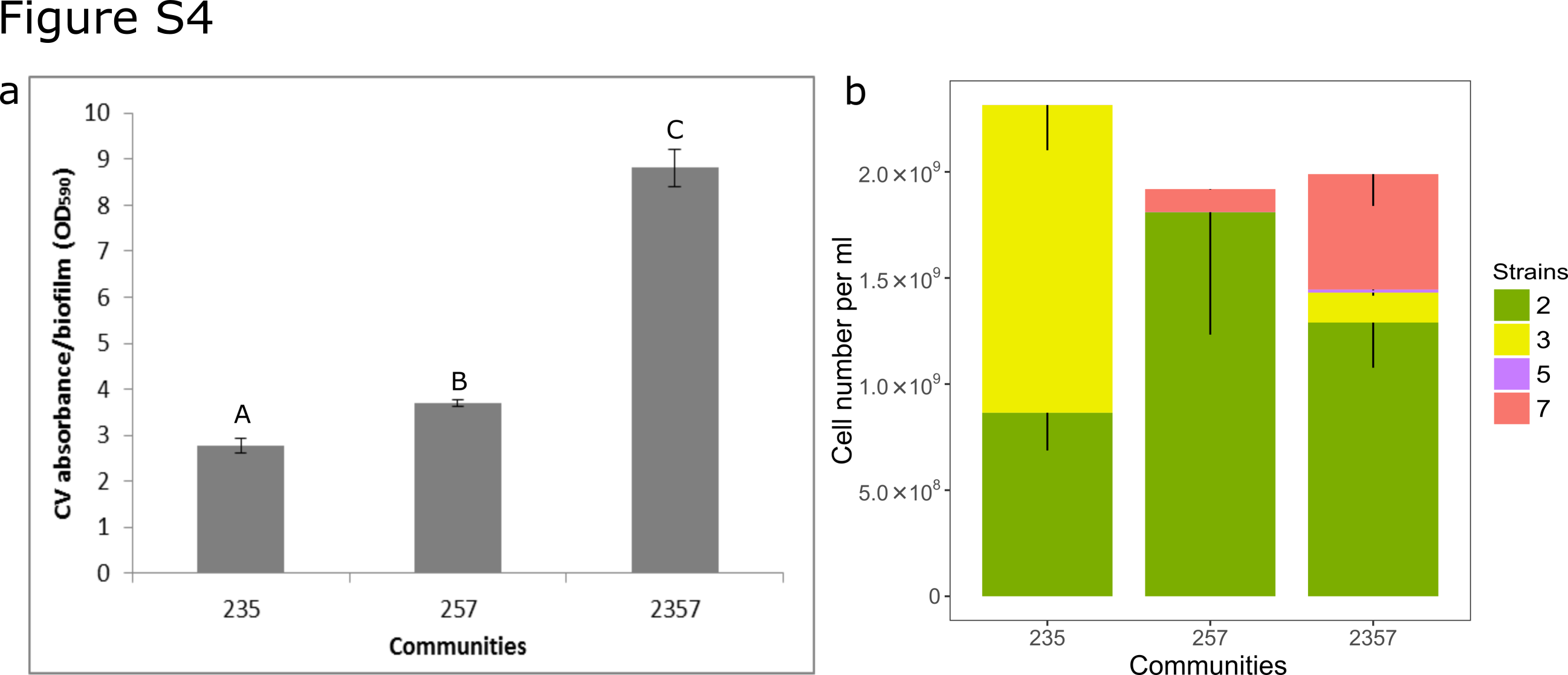
In our previous studies [21, 22], two triple-species biofilms (235, 257) displayed peculiar spatial structure compared to the four-species biofilm (2357) (Fig. 1; Supplementary Fig. 2), where two types of organizations could be defined based on the distribution pattern of X. retroflexus (strain 3) and M. oxydans (strain 5) (Fig. 1c, d). As reported by Liu et al. [21], P. amylolyticus (strain 7) displayed distinct scattered growth as micro-colonies inside biomass of S. rhizophila (strain 2) in the triple-species biofilm (257) after 24 h cultivation in DFR, similar to the spatial pattern achieved through monospecific culture (Supplementary Fig. 3). However, the spatial organization was markedly changed in the four-species biofilm consortia, as the presence of X. retroflexus facilitated the colonization of strain 7 (Fig. 2a; One-way ANOVA, P < 0.01), leading to its predominance in terms of bio-volumes, concomitant to the observed enhanced total biomass formation via CV assay performed in this study (Supplementary Fig. 4a).
Fig. 1.

Spatial structure of investigated biofilms and overview of experimental design. Confocal microscopy images showing the spatial organization of triple-species a, b and four-species c, d biofilms composed of S. rhizophila (2, green), X. retroflexus (3, yellow), M. oxydans (5, purple), and P. amylolyticus (7, red). Samples with distinct spatial patterns showed in c, d were collected at different locations in the same biofilm. Magnification: ×40. Scale bar: 50 µm. Modified with permission from Liu et al. [21]. 2357 (S) and 2357 (T) represent local regions with scant (S) and typical (T) presence of M. oxydans (5) in the four-species biofilm, respectively. e Triple-species and four-species biofilms were cultivated in a DFR system for 24 h and scraped off the glass slide. mRNA was extracted and purified, followed by sequening on an Illumina MiSeq platform. Comparative meta-transcriptomics analyses were performed between triple-species and four-species communities
Fig. 2.

Cell numbers and distribution of individual species in triple-species and four-species biofilms cultivated for 24 h. a Individual species show distinct cell densities in different communities, however total cell numbers are not significantly different. b Top, middle, and bottom biofilms layers are defined as follows: Top layer is defined from the very top to the section where the bio-volume is occupied up to 60%; bottom layer is defined as the last 20 sections of stacks if any; middle layer is defined as the rest of the sections in stacks if any. c The relative bio-volume of pixels from each species is plotted in three defined layers (top, middle, and bottom) of biofilms. 2; S. rhizophila, 3; X. retroflexus, 5; M. oxydans and 7; P. amylolyticus. 2357 (S) and 2357 (T) represent local regions with scant (S) and typical (T) presence of M. oxydans (5) in the four-species biofilm, respectively. Strain 5 is not visible in consortia 235 and 257, because it is present in extremely low density (Supplementary Fig. 5; Supplementary Table 1). Error bars represent standard error of the mean of three biological replicates (n = 3)
Spatial localization analysis obtained in the current study further confirmed previous observations. Species (2/3 and 2/7) maintained stably relative bio-volumes in the three defined layers of triple-species biofilms (235 and 257) (Fig. 2b, c; One-way ANOVA, P > 0.05). Strain 5 was mainly occupying the bottom layer, but in extremely low bio-volumes throughout triple-species biofilms (235 and 257) (Supplementary Fig. 5; One-way ANOVA, Tukey’s HSD test: P < 0.05). However, the colonization of strain 5, mainly occupying the top layer, was strongly promoted in both types of the four-species biofilm (Fig. 2c; Supplementary Fig. 2; One-way ANOVA, Tukey’s HSD test: P < 0.05). The abundance/density of strain 3, mainly occupying top layers of the four-species biofilm (Fig. 2c; One-way ANOVA, Tukey’s HSD test: P < 0.01), was considerably reduced by the presence of strain 7 (Fig. 2a; One-way ANOVA, P < 0.001).
Pairwise cross-correlation analyses (PCC) were performed on the pre-processed stacked images generated by CLSM and the output indicates whether two species tend to intermix (PCC > 1), are randomly distributed (PCC = 1), or segregate (PCC < 1) from each other. Specifically, the presence of strain 7 considerably enhanced intermixing patterns of species pairs 2/3 and 3/5, but it did not affect the relative localization of the species pair 2/5 in short between-species distances (<10 µm) in the four-species biofilm (2357) compared to those in the triple-species biofilm (235) (Fig. 3). Respectively, species pairs 5/7 and 2/5 were more segregated and intermixing (in <10 µm between-species distances) in the four-species biofilm (2357) compared to those in the triple-species biofilm (257), while the intermixing pattern of the species pair 2/7 was not remarkably impacted (Fig. 3). In addition, species pairs 2/5, 3/5, and 5/7, derived from the 24 h four-species biofilm (2357), showed distinct spatial localization in regions with typical and scant presence of strain 5, while species pairs 2/3 and 2/7 exhibited similar intermixing pattern in these two regions (Supplementary Fig. 6).
Fig. 3.

Pairwise cross-correlation analyses of triple-species and four-species biofilms. The mean PCC value (continuous line) and the standard error of the mean (shaded areas) are plotted against distances spaced at intervals of 1 µm. The dashed horizontal line (PCC value of 1) corresponds to random positioning. The PCC value indicates whether two species tend to intermix (PCC > 1), are randomly distributed (PCC = 1), or segregate (PCC < 1) from each other at specific between-species distances. 2; S. rhizophila, 3; X. retroflexus, 5; M. oxydans, and 7; P. amylolyticus. 2357 (T) represents local regions with typical presence of M. oxydans in the four-species biofilm. PCC values of species pairs were calculated from three biological replicates (n = 3). Note different scales on y-axes in top and bottom row plots
Metatranscriptomes and expression patterns
Metatranscriptome libraries were created from four-species and triple-species biofilms (Fig. 1e; all in three biological replicates). In average, 1.2 million of the transcripts were mapped to coding sequences of the genomes with sample sizes ranging between ~3 and ~8 million of reads after core identical protein sequences were removed (Supplementary Table 2). Species (2/3 and 2/7) showed distinctively relative protein coding transcripts in triple-species and four-species consortia before CFU normalization (Fig. 4a).
Fig. 4.

Metatranscriptomic analysis of triple-species and four-species biofilms. a The percentages of protein-coding transcripts of individual species in 24 h triple-species and four-species biofilms (three biological replicates for each sample). b Redundancy analysis (RDA) on mRNA profiles from tested consortia. Metatranscriptomes were transformed into relative abundance and the constrained model was built using bio-volumes (3-D imaging), CFU counts, and presence/absence of strain 3, X. retroflexus, and strain 7, P. amylolyticus, (capscale function, R package vegan, Bray–Curtis dissimilarity, 10,000 perm.). Axes are displaying the percentage of explained variance by the model
Redundancy analysis (RDA) based on Bray–Curtis dissimilarity obtained from the mRNA profiles prior to CFU normalization was performed after read counts were normalized by library sizes (Fig. 4b). The result revealed a significant, non-random distribution of samples according to their respective biofilm consortia (P = 4.1E−3), making them prone for pairwise statistical comparison. The first component was separating biofilm consortia 235 from both 257 and 2357 (RDA1, 79.7%), indicating the overruling effect of strain 7 presence/absence, correlating with higher bio-volumes. On the other hand, the second component was clearly showing a separation between the biofilm consortia 257 with the others (RDA2, 9.4%), indicating that the presence/absence of strain 3 was accounting for the second most important source of variance in this analysis.
Quantitative analysis of differentially expressed functions in the three communities
The pairwise differential gene expression analysis after biomass normalization allowed the detection of 378 RAST genetic functions with significantly altered expression levels between four-species versus triple-species communities (Supplementary Table 3, LRT, FDR-corrected P < 0.05; >2 log2-fold differential expression change). Strains 2 and 3 shared 81% expressed genetic functions well classified in the SEED database in the triple-species biofilm (235), which was expected due to their close phylogenetic relatedness (Supplementary Fig. 7; Supplementary Table 4). However, the percentage was sharply decreased to 43% when co-existing in the four-species biofilm (Supplementary Fig. 7; Supplementary Table 5), revealing that these two species showed distinct differences in gene expression profiles in response to strain 7 (Supplementary Table 3). In addition, strains 2 and 7, in the four-species biofilm consortia (2357) did not significantly respond to strain 3 in terms of differential gene expression compared to the triple-species biofilm (257) when biomass normalization was applied.
Respectively, 64 and 314 genetic functions from strains 2 and 3 were differentially expressed in the four-species biofilm (2357) in response to strain 7 compared to control expression levels in the triple-species biofilm consortia (235) (Supplementary Table 3). Amongst those functions, 312 were up-regulated in strain 3 (only two down-regulated), while 62 were down-regulated in strain 2 (two up-regulated), indicating a pronounced but converse response of these closely related Gammaproteobacterial strains to strain 7. Due to low biomass and subsequent low mRNA sequencing resolution for strain 5, no significant differences in expression of genetic functions could be attributed to this strain. However, differences could be seen at higher SEED classification levels of “RNA metabolism” and “Virulence/Disease/Defense” (Table 1; One-way ANOVA, Tukey’s HSD test: P < 0.05).
Table 1.
Differentially expressed functional categories of M. oxydans between three biofilm context
| Functional category for M. oxydans | 235 (%) | 2357 (%) | 257 (%) |
|---|---|---|---|
| RNA metabolism | 2.53 ± 0.42a | 8.76 ± 1.95c | 4.7 ± 1.0b |
| Virulence, disease and defense | 13.55 ± 1.66a | 30.19 ± 4.67b | 28.84 ± 2.38b |
a, b and c represent significant differences in pair-wise comparisons (One-way ANOVA, Tukey’s HSD test: P < 0.05). Pair-wise comparisons were taken place between any two of three biofilm communities (235, 257 and 2357)
The active response of X. retroflexus to P. amylolyticus in gene expression (2357 vs. 235)
To provide further understanding of the functions altered in strains 2 and 3 for the four-species vs. three-species biofilms (2357 vs. 235), differentially expressed genes were classified into SEED functions [38] and distributions within the categories were illustrated in Fig. 5 (genetic functions annotated to ‘None’ category are not shown) and Supplementary Table 3. Notably, the sets of genetic functions up-regulated or down-regulated were mainly annotated to ‘Amino Acids and Derivatives’, ‘Carbohydrates’, ‘Membrane Transport’, ‘Respiration’, ‘Protein Metabolism’, and ‘Stress Response’.
Fig. 5.

Bar chart of SEED functions that are differentially expressed in strain 2 (S. rhizophila, green) and strain 3 (X. retroflexus, yellow) in the four-species biofilm (2357) compared with those in the triple-species biofilm (235). Left and right panels represent the counts of down-regulated and up-regulated functions in the four-species biofilm (2357), respectively
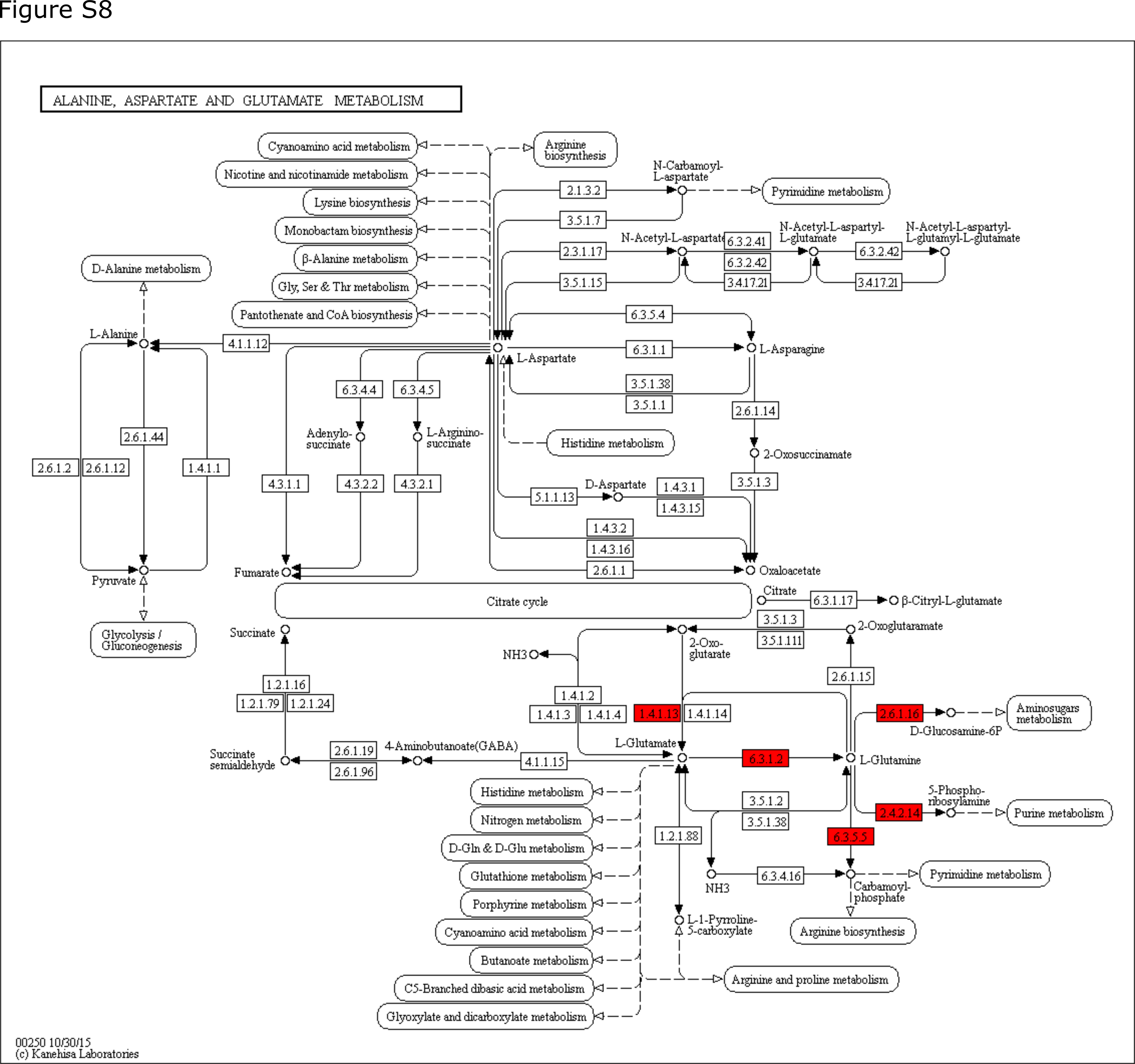
Strain 3 up-regulated 15 genetic functions annotated to ‘Amino Acids and Derivatives’ in response to the presence of strain 7 in the four-species consortia. Proteins with these genetic functions were mapped back to Kyoto Encyclopedia of Genes and Genomes (KEGG) database [39], providing an overview of their involvement in specific metabolic pathways. Specifically, two genetic functions (EC 2.1.2.1 and EC 1.4.4.2) were involved in glycine metabolism and three were related to branched-chain amino acids (BCAA) degradation (EC 4.2.1.9, EC 1.2.4.4 and EC 2.3.1.168). Ammonia generated by glycine and BCAA degradation could be used for the active glutamine synthesis, which was reflected by the up-regulation of glnA (EC 6.3.1.2) and gltA (EC 1.4.1.13). Glutamine was more likely to be further processed for aminosugars, purine, and pyrimidine metabolisms, as the essential enzymes (EC 2.4.2.14, EC 2.6.1.16 and EC 6.3.5.5) catalyzing glutamine into substrates participating in these pathways were significantly up-regulated (Supplementary Fig. 8). Twelve genetic functions involved in cell division or cell wall synthesis, and nine involved in purine or pyrimidine metabolism were up-regulated, suggesting ongoing cell replication/activities of strain 3 in the four-species consortia.
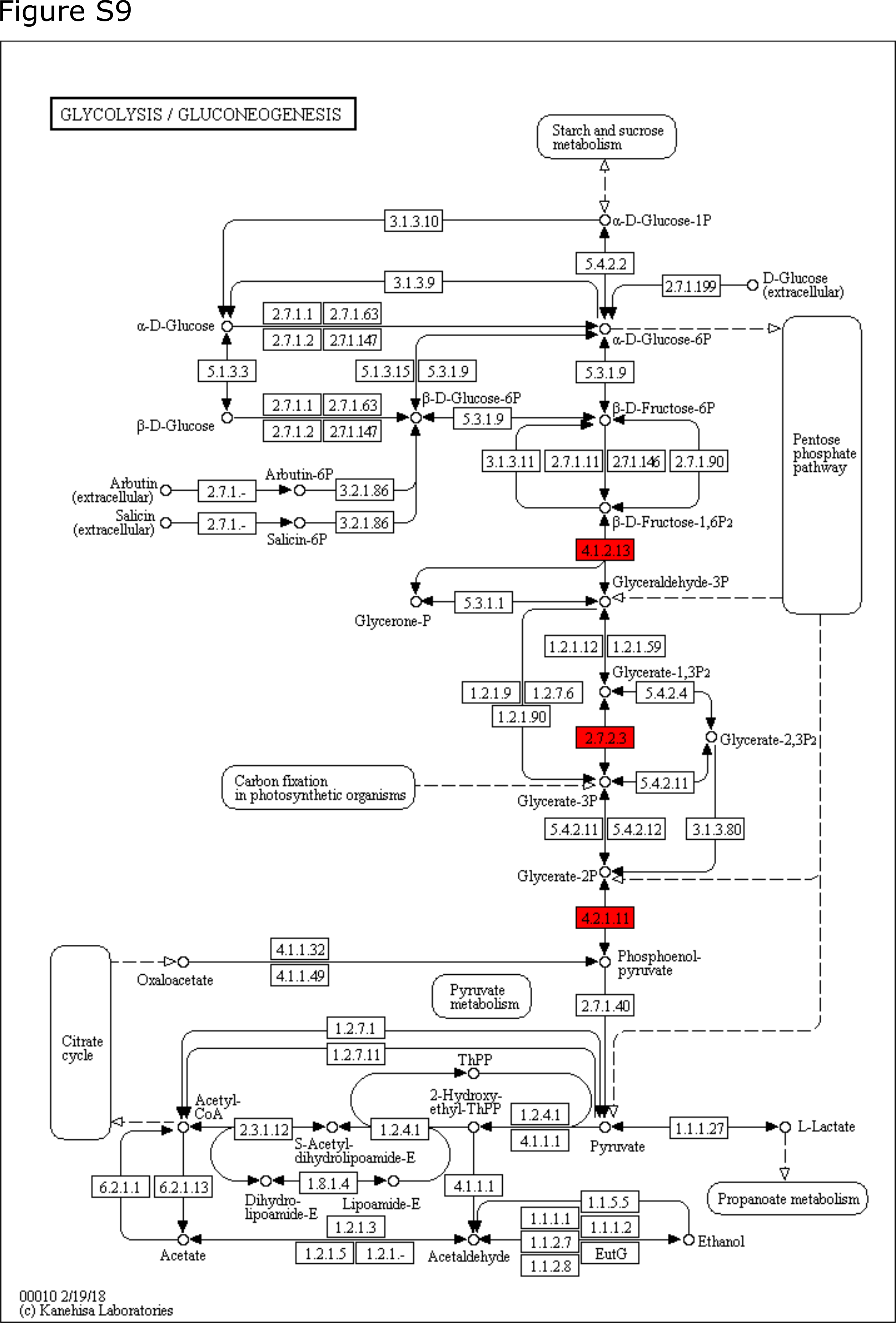
In addition, of the 15 up-regulated functions from strain 3 annotated to ‘Carbohydrates’, three (EC 4.1.2.13, EC 2.7.2.3, and EC 4.2.1.11) were involved in glycolysis (Supplementary Fig. 9), and four (EC 1.2.4.1, EC 1.8.1.4, EC 4.2.1.3 and EC 2.3.1.61) were associated with citrate cycle (TCA cycle) (Supplementary Fig. 10). Furthermore, 14 functions associated with oxidative phosphorylation (‘respiration’) were up-regulated, which in combination indicate that strain 3 was actively utilizing glucose as carbon source for aerobic respiration. Furthermore, one genetic function involved in cyclic-di-GMP phosphodiesterase synthesis, and seven involved in flagellar synthesis and regulation were up-regulated, suggesting enhanced motility potential of strain 3 in the 24 h four-species biofilm compared to the triple-species biofilm excluding strain 7. Strain 2 did not show significantly different expression in all these genetic functions differentially expressed by strain 3.
Moreover, strain 3 up-regulated seven genetic functions involved in the regulation of redox state (‘oxidative stress’) in response to the presence of strain 7, indicating that strain 3 was challenged when co-cultured in the four-species consortia. This was consistent with the observation that viable cell numbers of strain 3 were reduced 10-fold in the four-species consortia compared to those of the triple-species consortia (235) (Supplementary Table 1; One-way ANOVA, p < 0.001). Concomitantly, six genetic functions associated with DNA repair, 13 for protein biosynthesis and degradation, six for phosphorus metabolism and 11 for iron acquisition (‘membrane transport’) were significantly up-regulated in strain 3. Conversely, strain 2 remarkably down-regulated eight genetic functions involved in iron acquisition, and did not show differential expression of genetic functions associated with stress response.
Discussion
Microbes residing in multispecies biofilms occupy distinct, non-randomly distributed micro-sites, which are changing over time as the biofilm develops due to local heterogeneity driven by interspecies interactions [40, 41]. As such, biofilm represents a perfect model proving understanding of how emerging properties may become the object of selection [42–44]. It has been proposed that the fitness gain achieved through community-emerging properties resulting from social interactions is a major driving force in multispecies biofilm ecology and evolution, where distinct spatial organization is crucial [45, 46]. Recently, specialized computational and experimental models have been increasingly used to establish links between social interactions (cooperation, competition and facilitation) and spatial organization, with direct implications for our conceptual view of biofilms [47, 48]. However, few studies have tried to decipher molecular mechanisms underlying interspecies interactions within model multispecies communities [48], and to our knowledge this study is the first aiming to link gene expression patterns with spatial organization of member species.
In the present study, we show that intermixing patterns of species pairs in the triple-species consortia (235) were remarkably changed in response to the presence of strain 7 in the four-species consortia (2357). We hypothesized that this was tightly linked to changes in gene expression profiles of cells residing in different microbial communities. Indeed, strain 3 changed expression of 314 genetic functions in response to the presence of strain 7 in the four-species biofilm. Specifically, strain 3 significantly up-regulated genetic functions involved in glycolysis, TCA cycle, and oxidative phosphorylation, correlating with its spatial location in top layers of the four-species biofilm with expected high availability of oxygen and glucose [49, 50]. Genetic functions involved in flagella synthesis were also significantly up-regulated in strain 3, which was assumed to be the strategy applied for stably co-existing in top layers. Localization in the top biofilm layers is believed to provide direct growth advantages via continuous supply and access to resources, which is a known strategy used by this species and other fast growers to compete in biofilms [51–53]. Strain 2, although phylogenetic closely related to strain 3, did not show significantly different expression in all these genetic functions significantly altered in strain 3, supporting observations that strain 2 was evenly present in three defined layers of biofilms (235 and 2357) in spite of the presence of strain 7 (Fig. 2c). In addition, metatranscriptome profiles indicated that strain 3 was challenged when co-cultured with strain 7 up to 24 h, showing significant up-regulation of genetic functions involved in “Stress Response” and a 10-fold reduction of strain 3 cell numbers (7.1% of total CFU counts) in the presence of strain 7. Herschend et al. reported that strain 3 accounted for 20.4% of total CFU counts of the same four-species biofilm grown up to 48 h, indicating this species stabilized in the community [54]. Collectively, strain 3 was actively responding to strain 7 in ways facilitating its occupation of the top layers with favorable access to nutrient and oxygen, which therefore seems to be a successful strategy for better co-existence in the community. Our results confirmed that the previously observed spatial segregation of strains 3 and 7 in the four-species biofilm [22] was driven by competing for resources, such as oxygen and available nutrients in the growth media.
It was previously verified experimentally that antagonism is more likely to occur among closely related species sharing similar metabolic pathways [55]. Indeed, strains 2 and 3 displayed decreased growth fitness even when co-cultured in nutrient-rich media as compared to mono-species biofilms [22], suggesting a competitive relationship between these closely related species. Here, the intermixing level between these two species increased in response to strain 7 in the four-species biofilm, as only limited space in middle and bottom layers was available due to the dominance of the latter species. We hypothesized that niche competition between these two species for the limited nutrient resources was strengthened, as strain 7 was found to enhance metabolic activities related to fermentation and nitrate/nitrite ammonification [54]. However, strain 2 was not displaying up-regulation of genetic functions involved in stress response as strain 3 did. Instead, strain 2 remarkably down-regulated genetic functions involved in membrane transport and iron acquisition, being a requisite for successful competition in nutrient-limited environments [56, 57]. Our results suggest that the niche overlap between strains 2 and 3 in the four-species biofilm was minimized by differential expression of similar genes resulting in a phenotypic character displacement. Such a functional niche displacement mechanism could be an important strategy facilitating resource partitioning among sympatric microbes in dense multispecies communities such as biofilms [58].
Iron is a versatile redox-active catalyst and crucial cofactor for many biological functions and processes [59]. Strain 2 remarkably down-regulated genetic functions involved in iron acquisition in response to strain 7, however, its growth fitness was slightly enhanced in the four-species biofilm (Supplementary Table 1; One-way ANOVA, Tukey’s HSD test: P < 0.05). In addition, no significant gene regulation was detected in strain 2 in the presence/absence of strain 3 (257 vs. 2357), indicating that the gene expression profile of strain 2 was mainly affected by strain 7 (235 vs. 2357). Overall, results suggest that the growth of strain 2 was facilitated by sharing costly products for iron scavenging when co-cultured with strain 7. Moreover, we previously observed that the colonization of strain 7 was promoted during the four-species biofilm development as compared to that excluding strain 2 [21], suggesting metabolic dependencies as a biotic factor affecting their population dynamics [60]. In addition, these two species did not show segregation within 30 µm between-species distances neither in the triple- nor four-species biofilms (257 and 2357) (Fig. 3), further supporting the hypothesis that they mutually facilitated their growths via cooperation through weak metabolic interdependencies [41].
Here, no total viable cell count difference was noticed amongst the three investigated consortia (Supplementary Fig. 4b), however, strain 7 colonization was facilitated by the presence of strain 3 (257 vs. 2357, Fig. 2a), thus increasing biomass production of the four-species biofilm due to large cell size of strain 7 [21]. The variation in cell numbers of individual species may lead to different community functions, which could also be reflected by distinct intermixing patterns between species pairs in triple-species and four-species biofilms (Fig. 3). Unexpectedly, we did not detect any significant impact by strain 3 on strain 7 gene expression in the four-species biofilm (2357). This may be due to only very local responses by strain 7 to the presence of strain 3, which are averaged when sampling and analyzing total RNA. Alternatively, the growth facilitating effect on strain 7 by strain 3 is not reflected in an altered gene expression pattern. This highlights the importance of investigating interspecies interactions at subpopulation microscale levels, which could be visually defined by spatial organization of co-occurring species [61, 62]. However, state-of-the-art mRNA sequencing requires large amounts of raw material to achieve decent extraction yields, thus not being compatible with the fine sampling scale required to target subpopulations. In addition, PCC analyses showed distinct intermixing patterns between species pairs in two specific types of local regions in the four-species biofilm (Supplementary Fig. 6), suggesting heterogeneous gene expression of an individual species due to differences in the surrounding micro-environments [63].
Fig. 6.

Links between bacterial interactions and spatial organization in multispecies biofilms. a Overview of bacterial interactions occurring in the four-species community. Arrow-ending and bar-ending represent positive (facilitation) and negative (inhibition) interactions in terms of growth, respectively. All species pair interactions were predicted by comparing growth fitness of individual species when grown alone to that when co-cultured with other member species in dual-species, three-species, and four-species biofilms [21, 22]. Red lines indicate that bacterial interactions were further supported by comparative metatranscriptomics profiles presented in this study. b Strategies proposed for stabilizing the microbial community enhanced biomass production of the four-species biofilm. (1) Bacterial spatial segregation (e.g. X. retroflexus and P. amylolyticus); (2) bacterial spatial intermixing (X. retroflexus and M. oxydans) strengthened by the competitive force from P. amylolyticus; (3) phenotypic character displacement (occurring between S. rhizophila and X. retroflexus), where each species utilizes different nutrient resources in order to avoid strong competition caused by niche overlap; (4) Weak cooperation (occurring between S. rhizophila and P. amylolyticus). Dotted line denotes that predictable information is required from mRNA-seq to support whether S. rhizophila promotes the colonization of P. amylolyticus through secreting biological molecules benefiting the latter species. S. rhizophila (green circle), X. retroflexus (yellow circle), M. oxydans (purple circle) and P. amylolyticus (red circle); Square and triangle represent nutrients originating from media or secreted by bacterial species
In all three tested biofilms, no differences in genetic functions from strain 5 were detected due to its relatively low abundance. However, up-regulation of “RNA metabolism” and “Virulence/Disease/Defense” could be identified at higher SEED classification levels. RNA metabolism refers to any event in the life cycle of ribonucleic acid (RNA) molecules, reflecting the metabolic activity of samples investigated [64]. Strain 5 increased RNA metabolism expression and also exhibited a 14-fold increase in the number of viable cells in the four-species consortia (Supplementary Table 1), thus confirming the growth advantage obtained in the four-species community. Frequently, microorganisms up-regulate genetic functions involved in “Virulence/Disease/Defense” to maintain their competitiveness when stressed by local conditions [65, 66]. Strain 5 up-regulated this category only in response to strain 7 in the four-species biofilm (Table 1), thus indicating benefits gained from co-existence with strain 3. Substantially, the total community gained the most biomass production when strains 3 and 5 collaboratively made it to top layers. This supports the hypothesis that spatial intermixing in top layers between these two species was strongly driven by synergistic interactions [22]. As such, these two species could be seen as keystone species when co-cultured in the four-species biofilm (2357), but not in the triple-species biofilm (235). Indeed, their interaction turn them into so-called “ecosystem engineers” in ecology, defined as organisms able to significantly affect ecological properties of a given ecosystem resulting in modification, maintaining and/or creation of habitats [67], in this case as a biofilm. Pinpointing such crucial keystones species in complex environmental microbial communities is challenging, and multispecies biofilm models are perfectly suited to address this issue. Moreover, cooperative interactions between these two species seemed to be strengthened by the presence of competitive force from strain 7. This suggests that both cooperative and competitive interactions played important roles in fulfilling the enhanced biomass production. It should however be emphasized that these exploratory findings need to be verified and further investigated in detailed mechanistic studies and tested in mathematical models.
Overall, we conclude that the intrinsic community property of enhanced biomass production was closely connected with fine-tuned spatial organization of the four-species consortia, where each species occupied specific micro-localization optimizing growth fitness, in combination with a phenotypic character displacement. Hence, co-existence in the four-species biofilm seems to be stabilized by a combination of optimized spatial organization and phenotypic modifications both minimizing competition (Fig. 6). Our results illustrate how metatranscriptomic data can be better interpreted when combined with visual imaging of actual spatial organization, providing a new framework and perspectives on how phenotypic character displacement may stabilize social interactions of co-existing species in biofilms. Moreover, our results highlight the necessity and importance of investigating molecular mechanisms of complex multispecies communities at subpopulation levels in consideration of the non-random spatial organization driven by local interspecies interactions.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was partly funded by grants from The Danish Council for Independent Research; ref no. DFF-1323-00235 (SIMICOM), from the Villum Foundation (ref nos. 10098 and 11735).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Wenzheng Liu, Samuel Jacquiod
Supplementary information
The online version of this article (10.1038/s41396-019-0494-9) contains supplementary material, which is available to authorized users.
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Nadell CD, Drescher K, Foster KR. Spatial structure, cooperation and competition in biofilms. Nat Rev Microbiol. 2016;14:589–600. doi: 10.1038/nrmicro.2016.84. [DOI] [PubMed] [Google Scholar]
- 3.Hansen SK, Rainey PB, Haagensen JAJ, Molin S. Evolution of species interactions in a biofilm community. Nature. 2007;445:533–6. doi: 10.1038/nature05514. [DOI] [PubMed] [Google Scholar]
- 4.Mitri S, Foster KR. The genotypic view of social interactions in microbial communities. Annu Rev Genet. 2013;47:247–73. doi: 10.1146/annurev-genet-111212-133307. [DOI] [PubMed] [Google Scholar]
- 5.Burmølle M, Ren D, Bjarnsholt T, Sørensen SJ. Interactions in multispecies biofilms: do they actually matter? Trends Microbiol. 2014;22:84–91. doi: 10.1016/j.tim.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drescher K, Nadell CD, Stone HA, Wingreen NS, Bassler BL. Solutions to the public goods dilemma in bacterial biofilms. Curr Biol. 2014;24:50–5. doi: 10.1016/j.cub.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KWK, Periasamy S, Mukherjee M, Xie C, Kjelleberg S, Rice SA. Biofilm development and enhanced stress resistance of a model, mixed-species community biofilm. ISME J. 2014;8:894–907. doi: 10.1038/ismej.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren D, Madsen JS, Sørensen SJ, Burmølle M. High prevalence of biofilm synergy among bacterial soil isolates in cocultures indicates bacterial interspecific cooperation. ISME J. 2015;9:81–9. doi: 10.1038/ismej.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, et al. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun. 2011. 10.1038/ncomms1597. [DOI] [PubMed]
- 12.Foster KR, Bell T. Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol. 2012;22:1845–50. doi: 10.1016/j.cub.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien EJ, Monk JM, Palsson BO. Using genome-scale models to predict biological capabilities. Cell. 2015;161:971–87. doi: 10.1016/j.cell.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Røder HL, Sørensen SJ, Burmølle M. Studying bacterial multispecies biofilms: where to start? Trends Microbiol. 2016;24:503–13. doi: 10.1016/j.tim.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 15.Ghoul M, Mitri S. The ecology and evolution of microbial competition. Trends Microbiol. 2016;10:833–45. doi: 10.1016/j.tim.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kukurba KR, Montgomery SB. RNA sequencing and analysis. Cold Spring Harb Protoc. 2015;2015:951–69. doi: 10.1101/pdb.top084970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacquiod S, Nunes I, Brejnrod A, Hansen MA, Holm PE, Hohansen A, et al. Long-term soil metal exposure impaired temporal variation in microbial metatranscriptomes and enriched active phages. Microbiome. 2018. 10.1186/s40168-018-0606-1. [DOI] [PMC free article] [PubMed]
- 19.Jemielita M, Taormina MJ, Burns AR, Hampton JS, Rolig AS, Guillemin K. Spatial and temporal features of the growth of a bacterial species colonizing the zebrafish gut. mBio. 2014. 10.1128/mBio.01751-14. [DOI] [PMC free article] [PubMed]
- 20.Tropini C, Earle KA, Huang KC, Sonnenburg JL. The gut microbiome: connecting spatial organization to function. Cell Host Microbe. 2017;21:433–42. doi: 10.1016/j.chom.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Russel J, Røder HL, Madsen JS, Burmølle M, Sørensen SJ. Low-abundant species facilitates specific spatial organization that promotes multispecies biofilm formation. Environ Microbiol. 2017;19:2893–905. doi: 10.1111/1462-2920.13816. [DOI] [PubMed] [Google Scholar]
- 22.Liu W, Russel J, Burmølle M, Sørensen SJ, Madsen JS. Micro-scale intermixing: a requisite for stable and synergistic co-establishment in a four-species biofilm. ISME J. 2018. 10.1038/s41396-018-0112-2. [DOI] [PMC free article] [PubMed]
- 23.de la Cruz-Perera CI, Ren D, Blanchet M, Dendooven L, Marsch R, Sørensen SJ, et al. The ability of soil bacteria to receive the conjugative IncP1 plasmid, pKJK10, is different in a mixed community compared to single strains. FEMS Microbiol Lett. 2013;338:95–100. doi: 10.1111/1574-6968.12036. [DOI] [PubMed] [Google Scholar]
- 24.Goeres DM, Hamilton MA, Beck NA, Buckingham-Meyer K, Hilyard JD, Loetterle LR, et al. A method for growing a biofilm under low shear at the air–liquid interface using the drip flow biofilm reactor. Nat Protoc. 2009;4:783–8. doi: 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 25.Ren D, Madsen JS, de la Cruz-Perera CI, Bergmark L, Sørensen SJ, Burmølle M. High-throughput screening of multispecies biofilm formation and quantitative PCR-based assessment of individual species proportions, useful for exploring interspecific bacterial interactions. Micro Ecol. 2014;68:146–54. doi: 10.1007/s00248-013-0315-z. [DOI] [PubMed] [Google Scholar]
- 26.Peeters E, Nelis HJ, Coenye T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J Microbiol Methods. 2008;72:157–65. doi: 10.1016/j.mimet.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Jackson G, Beyenal H, Rees WM, Lewandowski Z. Growing reproducible biofilms with respect to structure and viable cell counts. J Microbiol Methods. 2001;47:1–10. doi: 10.1016/S0167-7012(01)00280-9. [DOI] [PubMed] [Google Scholar]
- 28.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50:346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 29.R development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016. [Google Scholar]
- 30.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42:206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 32.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2010;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Mccarthy D, Robinson M, Smyth GK. edgeR: differential expression analysis of digital gene expression data. User’s guide. 2015. http://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf. [DOI] [PMC free article] [PubMed]
- 35.Li P, Piao Y, Shon HS, Ryu KH Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinform. 2015. 10.1186/s12859-015-0778-7. [DOI] [PMC free article] [PubMed]
- 36.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 37.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–32. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 38.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008 doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Røder HL, Madsen JS, Bjarnsholt T, Sørensen SJ, Burmølle M. Interspecific bacterial interactions are reflected in multispecies biofilm spatial organization. Front Microbiol. 2016 doi: 10.3389/fmicb.2016.01366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 42.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–50. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 43.Blouin M, Karimi B, Mathieu J, Lerch TZ. Levels and limits in artificial selection of communities. Ecol Lett. 2015;18:1040–8. doi: 10.1111/ele.12486. [DOI] [PubMed] [Google Scholar]
- 44.Madsen JS, Sørensen SJ, Burmølle M. Bacterial social interactions and the emergence of community-intrinsic properties. Curr Opin Microbiol. 2018;42:104–9. doi: 10.1016/j.mib.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 45.Hansen LB, Ren D, Burmølle M, Sørensen SJ. Distinct gene expression profile of Xanthomonas retroflexus engaged in synergistic multispecies biofilm formation. ISME J. 2017;11:300–3. doi: 10.1038/ismej.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madsen JS, Lin Y-C, Squyres GR, Price-Whelan A, de Santiago Torio A, Song A, et al. Facultative control of matrix production optimizes competitive fitness in Pseudomonas aeruginosa PA14 biofilm models. Appl Environ Microbiol. 2015;81:8414–26. doi: 10.1128/AEM.02628-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitri S, Xavier JB, Foster KR. Social evolution in multispecies biofilms. Proc Natl Acad Sci USA. 2011;108:10839–46. doi: 10.1073/pnas.1100292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Momeni B, Brileya KA, Fields MW, Shou W. Strong inter-population cooperation leads to partner intermixing in microbial communities. Elife. 2013 doi: 10.7554/eLife.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cronenberg CCH, van den Heuvel JC. Determination of glucose diffusion coefficients in biofilms with micro-electrodes. Biosens Bioelectron. 1991;6:255–62. doi: 10.1016/0956-5663(91)80011-L. [DOI] [PubMed] [Google Scholar]
- 50.Zhou X, Zhang M, Yu T, Liu Y, Shi H. Oxygen profiles in biofilms undergoing endogenous respiration. Chem Eng J. 2013;220:452–8. doi: 10.1016/j.cej.2013.01.004. [DOI] [Google Scholar]
- 51.Jacquiod S, Franqueville L, Cécillon S, Vogel TM, Simonet P. Soil bacterial community shifts after chitin enrichment: an integrative metagenomic approach. PLoS ONE. 2013. 10.1371/journal.pone.0079699. [DOI] [PMC free article] [PubMed]
- 52.Kreft JU. Biofilms promote altruism. Microbiology. 2004;150:2751–60. doi: 10.1099/mic.0.26829-0. [DOI] [PubMed] [Google Scholar]
- 53.Xavier JB, Foster KR. Cooperation and conflict in microbial biofilms. Proc Natl Acad Sci USA. 2007;104:876–81. doi: 10.1073/pnas.0607651104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herschend J, Damho ZBV, Marquard AM, Svensson B, Sørensen SJ, Hägglund P, et al. A meta-proteomics approach to study the interspecies interactions affecting microbial biofilm development in a model community. Sci Rep. 2017. 10.1038/s41598-017-16633-6. [DOI] [PMC free article] [PubMed]
- 55.Russel J, Røder HL, Madsen JS, Burmølle M, Sørensen SJ. Antagonism correlates with metabolic similarity in diverse bacteria. Proc Natl Acad Sci USA. 2017;114:10684–8. doi: 10.1073/pnas.1706016114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skaar EP. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010. 10.1371/journal.ppat.1000949. [DOI] [PMC free article] [PubMed]
- 57.Raines DJ, Moroz OV, Blagova EV, Turkenburg JP, Wilson KS, Duhme-klair A. Bacteria in an intense competition for iron: key component of the Campylobacter jejuni iron uptake system scavenges enterobactin hydrolysis product. Proc Natl Acad Sci USA. 2016;113:5850–5. doi: 10.1073/pnas.1520829113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tannock GW, Wilson CM, Loach D, Cook GM, Eason J, O’Toole PW, et al. Resource partitioning in relation to cohabitation of Lactobacillus species in the mouse forestomach. ISME J. 2012;6:927–38. doi: 10.1038/ismej.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheldon JR, Heinrichs DE. Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol Rev. 2018;39:592–630. doi: 10.1093/femsre/fuv009. [DOI] [PubMed] [Google Scholar]
- 60.Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci USA. 2015;112:6449–54. doi: 10.1073/pnas.1421834112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Werner E, Roe F, Bugnicourt A, Franklin MJ, Heydorn A, Molin S, et al. Stratified growth in Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2004;70:6188–96. doi: 10.1128/AEM.70.10.6188-6196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kearns DB, Losick R. Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev. 2005;19:3083–94. doi: 10.1101/gad.1373905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mielich-süss B, Lopez D. Molecular mechanisms involved in Bacillus subtilis biofilm formation. Environ Microbiol. 2015;17:555–65. doi: 10.1111/1462-2920.12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–64. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gellatly SL, Hancock RE. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. 2018;67:159–73. doi: 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- 66.Eleftherianos I, Boundy S, Joyce SA, Aslam S, Marshall JW, Cox RJ, et al. An antibiotic produced by an insect-pathogenic bacterium suppresses host defenses through phenoloxidase inhibition. Proc Natl Acad Sci USA. 2007;104:2419–24. doi: 10.1073/pnas.0610525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones CG, Lawton JH, Shachak M. Organisms as ecosystem engineers. Oikos. 1994;69:373–86. doi: 10.2307/3545850. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.