

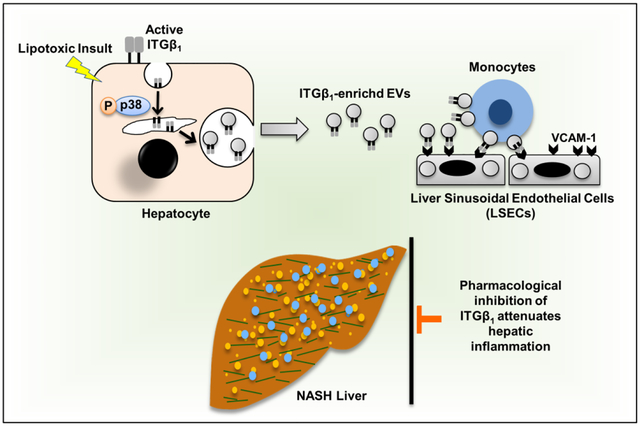
Abstract
Background and aims:
Hepatic recruitment of monocyte-derived macrophages (MoMF) contributes to the inflammatory response in nonalcoholic steatohepatitis (NASH). However, how hepatocyte lipotoxicity promotes MoMF inflammation is unclear. Here we demonstrate that lipotoxic hepatocyte-derived extracellular vesicles (EVs) are enriched with active integrin β1 (ITGβ1), which promotes monocyte adhesion and liver inflammation in murine NASH.
Methods:
Hepatocytes were treated with either vehicle or the toxic lipid mediator lysophosphatidylcholine (LPC); EVs were isolated from the conditioned media and subjected to proteomic analysis. C57BL/6J mice were fed a diet rich in fat, fructose, and cholesterol (FFC) to induce NASH. Mice were treated with anti-ITGβ1 neutralizing antibody (ITGβ1Ab) or control IgG isotype.
Results:
Ingenuity Pathway Analysis of the lipotoxic hepatocyte-derived EV (LPC-EVs) proteome indicated that integrin signaling is an overrepresented canonical pathway. Immunogold electron microscopy and nanoscale flow cytometry confirmed enrichment of LPC-EVs with active ITGβ1. Furthermore, we showed that LPC treatment in hepatocytes activates ITGβ1 and mediates its endocytic trafficking and sorting into EVs. LPC-EVs-enhanced monocytes adhesion to liver sinusoidal endothelial cells (LSECs) was observed by shear stress adhesion assay, and was attenuated in the presence of ITGβ1Ab. FFC-fed, ITGβ1Ab-treated mice displayed reduced inflammation defined by decreased proinflammatory MoMF hepatic infiltration and activation as assessed by immunohistochemistry, mRNA expression, and flow cytometry. Likewise, mass cytometry by time-of-flight (CyTOF) on intrahepatic leukocytes (IHL) displayed reduced infiltrating proinflammatory monocytes. Furthermore, ITGβ1Ab treatment significantly ameliorated liver injury and fibrosis.
Conclusions:
Lipotoxic EVs mediate monocyte adhesion to LSECs mainly by an ITGβ1-dependent mechanism. ITGβ1Ab ameliorates diet-induced NASH in mice by reducing MoMF-driven inflammation, suggesting that blocking ITGβ1 is a potential anti-inflammatory therapeutic strategy in human NASH.
Keywords: extracellular vesicles, integrin β1, integrin α9, adhesion, inflammation, NASH, monocytes, liver sinusoidal endothelial cells, mass cytometry, fibrosis
LAY SUMMARY
Herein, we report that a cell adhesion molecule termed integrin β1 (ITGβ1) plays a key role in the progression of nonalcoholic steatohepatitis (NASH). ITGβ1 is released from hepatocytes under lipotoxic stress as a cargo of extracellular vesicles, and mediates monocyte adhesion to liver sinusoidal endothelial cells, which is an essential step in hepatic inflammation. In a mouse model of NASH, blocking ITGβ1 reduces liver inflammation, injury and fibrosis. Hence, ITGβ1 inhibition may serve as a new therapeutic strategy for NASH.
Graphical Abstract
INTRODUCTION
With the worldwide increase in obesity, nonalcoholic fatty liver disease (NAFLD) is currently the most common chronic liver disease.[1] A subset of patients with NAFLD develops a more severe inflammatory form termed nonalcoholic steatohepatitis (NASH) which can progress to end-stage liver disease. NASH is currently the leading cause of liver-related mortality in many western countries.[2] Therefore, there is an unmet need for mechanism-based therapeutic strategies that reverse established NASH and control the progression of the disease.
Current concepts suggest that excess circulating free fatty acids mediate hepatocyte lipotoxicity in NASH.[3] Moreover, NASH patients are at risk of end-stage liver disease, mainly secondary to the unrelenting sterile inflammatory response triggered by hepatocyte lipotoxicity. This inflammatory response is mediated, in part, by the recruited monocytes that differentiate into macrophages, so-called monocyte-derived macrophages (MoMFs).[4] Although targeting monocyte infiltration in NASH via the dual CC chemokine receptor types 2 and 5 (CCR2/5) antagonist improved fibrosis, it was insufficient to resolve human steatohepatitis. [5] Therefore, key additional signals regulating the trafficking and retention of circulating monocytes in the NASH liver remain undefined.
Hepatocytes release diverse types of membrane-bound, nanometer-sized extracellular vesicles (EVs) into the extracellular milieu under physiological conditions. EVs are efficient messengers, with superior stability and bioavailability of their signature cargos implicated in inflammatory responses.[6–8] Interestingly, many of the EV cargos are selectively transferred through intracellular trafficking pathways and packaged into EVs, reflecting the pathophysiological context of the parent cell.[9] EVs released from lipotoxic hepatocytes are involved in MoMF chemotaxis and the hepatic inflammatory response; [6–8] however, the role of lipotoxic hepatocyte-derived EVs (lipotoxic EVs) in promoting circulating monocyte liver-specific homing through regulating their adhesion to liver sinusoidal endothelial cells (LSECs) in the NASH liver microenvironment has not been explored.
LSECs are highly specialized endothelial cells that serve as a platform for various immune cells, including monocytes, to lodge in the liver.[10] As monocyte receptor-LSEC ligand interactions are not unique to the liver, the question remains whether hepatocyte-specific recruitment processes via EVs exists in NASH. A single prior study reported that adoptive transfer of EVs isolated from the serum of high fat diet-fed mice into chow-fed mice resulted in myeloid cell activation and accumulation in the liver.[11] However, the mechanism by which EVs mediate the hepatic accumulation of myeloid cells was not explored.
Integrins (ITGs) provide the central mechanism for cells in multicellular organisms to interact with and sense their extracellular environment.[12] Integrins are heterodimeric cell surface transmembrane proteins consisting of 24 non-covalently associated α and β subunits which mediate cell-cell and cell-matrix interaction.[13] ITGα9 and β1 exist as heterodimers, and our data indicate that they are particularly enriched in EVs derived from lipotoxic hepatocytes; hence we will use ITGβ1 for simplicity in this manuscript to refer to ITGα9β1. Vascular cell adhesion molecule 1 (VCAM-1) is expressed on the surface of LSECs and is a known ligand for ITGα9β1.[14] Interestingly, ITG can adopt a closed conformation that has a low affinity for ligand (inactive) or an extended open conformation that has a high affinity for ligand (active).[15] Binding of intracellular proteins such as Talin to the dephosphorylated cytoplasmic tail of ITGβ1 regulates its activation and promotes ligand binding.[15] Kinase p38 has been implicated in ITGβ activation via an inside-out, ligand-independent signaling in different disease models.[16, 17] We have previously demonstrated that lipotoxic treatment in hepatocytes induces a mitogen-activated protein kinase (MAPK) signaling cascade leading to the activated phosphorylation of p38.[6, 18, 19] Moreover, ITGs undergo constant endocytic trafficking and recycling that regulate ITG-mediated cell adhesion and migration.[20, 21] This process of ITG trafficking suggests that in lipotoxic hepatocytes, ITGβ1 trafficks through the endocytic-multivesicular body (MVB) pathway to be released in EVs.
Herein we report that ITGβ1, a highly expressed ITG in hepatocytes,[22] is enriched and in an active status in lipotoxic EVs. ITGβ1-enriched EVs enhance monocyte adhesion to LSECs. Most importantly, we demonstrate that ITGβ1 neutralizing antibody attenuates diet-induced NASH in mice, mainly through reducing proinflammatory monocyte hepatic infiltration.
MATERIALS & METHODS:
Please see supplementary material
RESULTS
Lipotoxic hepatocyte-derived EVs are enriched with integrins.
We adopted a non-biased approach to identify and characterize the key proteins on lipotoxic hepatocyte-derived EVs. To this end, we performed proteomics analysis by mass spectrometry (MS) on the EVs derived from primary mouse hepatocytes (PMH) treated with vehicle (Veh) and the toxic lipid mediator lysophosphatidylcholine (LPC). We employed LPC since the toxicity of the saturated free fatty acid palmitate is dependent upon its metabolism to LPC.[23, 24] Unbiased Ingenuity pathway analysis (IPA) of the proteomics data identified ITG signaling among the top represented canonical pathways, particularly in EVs from LPC-treated hepatocytes when compared to EVs from vehicle-treated hepatocytes (Figure 1A). Next, we performed immunoblot analysis for different ITG in hepatocytes treated with vehicle and LPC, and their derived EVs. Western blot identified selective enrichment of ITGβ1, ITGα5, ITGα9, and ITGαv in EVs released from lipotoxic PMH, without changes at the cellular levels (Figure 1B). Similar results were obtained with the human hepatoma cell line Huh7 (Figure 1C). Since ITGβ1 is the most abundant integrin on hepatocytes [22] and the only integrin β expressed on EVs based on our mass spectrometry data, we focused on ITGβ1 as the key functional integrin family member on lipotoxic EVs. Interestingly, the protein level of Talin-1 (a versatile ITGβ1 affinity regulator implicated in adhesion) [25] was also increased in lipotoxic EVs, suggesting that the ITGβ1 on lipotoxic EVs is in active conformation status. To confirm this observation, we employed immunogold electron microscopy, and demonstrated using the active conformation sensitive ITGβ1 antibody (9EG7) enrichment of ITGβ1 in EVs released from lipotoxic PMH (Figure 1D). This observation was further confirmed by nanoscale flow cytometry, which allows the quantification of active ITGβ1-bearing EVs. LPC-treated PMH released more abundant active ITGβ1-positive EVs as compared to Veh-treated PMH (Figure 1F). These findings were also confirmed using Huh7 cells (Figure 1G). Interestingly ITGβ1 expression was increased in the serum EVs of patients with NASH (Figure 1H). Collectively, these data indicate that ITGβ1 in an active conformation is selectively sorted into EVs released from lipotoxic hepatocytes.
Fig. 1. Lipotoxic hepatocyte-derived EVs are enriched with active ITGβ1.

(A) Top ranked canonical pathways identified by IPA of proteomic data on EVs derived from vehicle or LPC-treated PMH. Immunoblot analysis showing protein levels of integrin family members and Talin-1 on EVs and whole cell lysate (WCL) from (B) PMH or (C) Huh7 cells treated with vehicle or 20 μM LPC for 4 hours. Beta-actin, and the EV markers TSG101, CD63 and CD81 were used as loading controls for WCL and EVs, respectively. (D) Immunogold electron microscopy images showing immunoreactivity for ITGβ1 in an active conformation on EVs derived from PMH treated with vehicle (Veh-EV) or LPC (LPC-EV). Nanoscale flow cytometry showing expression levels of active ITGβ1 on EVs, (E) various sizes silica nanoparticles used as calibration beads to define EVs based on the particle size (top panel). ITGβ1+ EVs from PMH treated with Veh or LPC (bottom panel), quantification of ITGβ1-positive EVs from (F) PMH and (G) Huh7. Bar columns represent mean ± standard error of the mean (SEM); n=3–5. (H) Quantification of ITGβ1+ EVs in the serum of patients with simple steatosis (n=8), and NASH with stage 1–2 fibrosis (n=17). Graphs represent mean ± SEM; *p<0.05, **p<0.01 (Unpaired t test).
Hepatocyte lipotoxic treatment induces ITGβ1 activation and endocytic trafficking.
We further examined the activation and endocytic trafficking of ITGβ1 in hepatocytes under lipotoxic stress (Figure 2A). Conformation-specific antibodies against ITGβ1 cannot detect the SDS-denatured target protein, and thus are not suitable for immunoblot assay. Therefore, to determine if hepatocyte ITGβ1 is activated by lipotoxic treatment, lysates from Veh or LPC-treated Huh7 cells were immunoprecipitated with the inactive (MAB13) or the active (9EG7) conformation-specific ITGβ1 antibodies, and immunoblotted with an antibody for total ITGβ1 (Figure 2B). Based on our prior report of p38 activation with lipotoxic treatment in hepatocytes, [6, 19] and the established role of p38 in ITG activation in different disease models, [16, 17] we examined whether p38 mediates LPC-induced ITGβ1 activation in hepatocyte. LPC treatment causes a significant decrease in inactive (tyrosine phosphorylated ITGβ1 tail, higher molecular weight), and increase in active (tyrosine dephosphorylated tail, lower molecular weight) ITGβ1, which is reduced in the presence of the p38 inhibitor SB203580, indicating that lipotoxic stress-induced hepatocyte ITGβ1 activation occurs via a p38-mediated pathway. Similar results were obtained with the mouse hepatocyte cell line AML12 (Figure 2C), (the inactive conformation sensitive antibody MAB13 reacts only with human species hence it was not used with the mouse AML12 cells). To confirm that the ITGβ1 9EG7 antibody immunoprecipitates active ITGβ1, we analyzed the ITGβ1 immunoprecipitates from vehicle and LPC-treated PMH by MS as previously described by us.[6] The MS data was searched allowing for phosphostyrosine as a variable modification and showed absence of phosphorylation on the first NPxY motif on the ITGβ1 C terminal, confirming that the pulled ITGβ1 is in an active conformation (Supplementary table 1). Furthermore, we employed immunofluorescence (IF) microscopy and confirmed that LPC treatment induced activation of ITGβ1, which was diminished with SB203580 (Figure 2D). Moreover, we noted that in lipotoxic hepatocytes, the active ITGβ1 accumulated in cytoplasmic structures consistent with intracellular vesicles (Figure 2D, second panel). To further explore the intracellular trafficking of active ITGβ1, we examined the co-localization of active ITGβ1 with the early endosome marker early endosome antigen (EEA) 1, the MVB marker CD63, and the late endosome marker Rab7. ITGβ1 co-localization with EEA1, CD63 or Rab7 (Figure 2E) was increased with LPC treatment when quantified using the Pearson’s correlation coefficient. To examine if lipotoxicity regulates active ITGβ1 lysosomal degradation, we assessed the co-localization of active ITGβ1 with the lysosome marker LAMP1; however, there was no obvious co-localization of ITGβ1 with LAMP1 (Supplementary Figure 1). Collectively, these results suggest that hepatocyte lipotoxic treatment induces ITGβ1 activation and endocytic trafficking, resulting in active ITGβ1 release in EVs.
Fig. 2. Hepatocyte lipotoxic treatment induces ITGβ1 activation and endocytic trafficking.

(A) Schematic representation of activation and endocytic trafficking of ITGβ1. (B) Huh7 cells and (C) AML12 cells were treated with either vehicle or 20 μM LPC for 15–30 min ±10 μM p38 inhibitor SB203580 (SB). Cell lysates were immunoprecipitated with active conformation-sensitive ITGβ1 antibody (9EG7), inactive conformation-sensitive ITGβ1 antibody (Mab13) or isotype IgG. Beta-actin was used as a loading control. Huh7 cells were treated with either vehicle or 5 μM LPC for 20 min ± 10 μM SB203580, (D) active ITGβ1 was labeled with 9EG7. (E) Co-localization of active ITGβ1 with early endosomes, late endosomes, or MVBs was assessed using anti-EEA1, anti-Rab7, and anti-CD63 antibodies, respectively. Scale bar: 5μm, n=3, quantification of co-localization between two fluorophores was done by Pearson’s correlation coefficient; ***p<0.001, ****p<0.0001 (Unpaired t test).
Lipotoxic Hepatocytes Release EVs in the Circulation.
We have previously demonstrated that LPC treatment increases hepatocytes EVs release in vitro.[6, 8] To assure that this observation is not an artifact of the in vitro system, we developed a mouse model to track circulating EVs of hepatocyte origin (Supplementary Figure 2A). We then quantified the hepatocyte-derived EVs in the plasma by nanoscale flowcyometry, and identified a 5-fold increase with the NASH-inducing diet (Supplementary Figure 2B). These data conclusively demonstrate for the first time that lipotoxic hepatocytes release large number of EVs in the circulation in vivo.
Lipotoxic hepatocyte-derived EVs promote monocytes adhesion to LSECs via an ITGβ1-dependent mechanism.
We have previously reported that EVs from lipotoxic hepatocytes induce MoMF chemotaxis.[6] To understand the biological functions exerted by lipotoxic hepatocyte-derived EVs on monocytes, we performed RNA sequencing (RNAseq) on primary mouse monocytes incubated with EVs from LPC (LPC-EVs) or vehicle (Veh-EVs)-treated PMH. IPA of RNAseq data showed leukocyte adhesion and diapedesis-related signaling among the top overrepresented canonical pathways in monocytes stimulated with LPC-EVs, suggesting the involvement of LPC-EVs in monocyte adhesion to LSECs (Figure 3A).
Fig. 3. toxic hepatocyte-derived EVs promote monocyte adhesion to LSECs via an ITGβ1-dependent mechanism.

Lipo (A) Top represented canonical pathways in monocytes stimulated with LPC-EVs vs Veh-EVs. (B) Equal number of Huh7 cells were treated with either veh or LPC. EVs were collected from the conditioned media and labelled with DiO. THP1 cells were co-cultured with human LSECs in the presence of labelled EVs. Scale bar: 20 μm for the top panel, and 5 μm for the bottom panel. (C) Z-stack confocal microscopy of THP1 incubated with DiO-labelled EVs from LPC-treated Huh7 cells (white arrows). (D) Primary mouse monocytes were stimulated with Veh-EV or LPC-EV from PMH ± ITGβ1Ab, and infused in microfluidic chambers coated with a monolayer of primary mouse LSECs. Adherent cells were quantified. (E) Immunoblot analysis showing ITGβ1 knockdown in shITGβ1 cell line. Beta-actin was used as a loading control. (F) THP1 cells were stimulated with either Veh-EV or LPC-EV from wild-type (WT) Huh7 cells, or shITGβ1 Huh7 cells, and infused in microfluidic chambers coated with a monolayer of primary human LSECs ± VCAM-1 Ab. Adherent THP1 cells were quantified similar to D. VCAM-1 is expressed on human LSECs under basal condition as shown by flow cytometry; bar graphs represent mean±SEM; n=6, ***p<0.001, ****p<0.0001 (One-way ANOVA with Bonferroni’s multiple comparison).
To examine the interaction between lipotoxic EVs-stimulated monocytes and LSECs, we co-cultured the human monocyte cell line, THP1, with EVs derived from equal number of hepatocytes treated with either vehicle or LPC. EVs were labelled with a fluorescent lipophilic dye DiO. Confocal microscopy revealed that monocytes incubated with LPC-EVs were more likely to adhere to LSECs (Figure 3B). We then subjected monocytes to live cell imaging with Z stack microscopy following incubation with EVs. EVs were observed both on the surface and on deeper focal plane (intracellular) of the THP1 cells (Figure 3C), suggesting that ITGβ1-enriched EVs interact with monocytes in a topography that allows them to potentially tether monocytes to LSECs. Interestingly, many of these EVs are also internalized by monocytes (Figure 3C), which has implications for ITGβ1 recycling to the cell surface. To further explore if LPC-EVs enriched with ITGβ1 mediates monocyte adhesion to LSECs, a key stage in liver inflammation, we employed a flow-based adhesion assay using microfluidic chambers (Supplementary Figure 3A–B) coated with a monolayer of mouse primary LSECs. Monocytes stimulated with LPC-EVs have enhanced adhesion to LSECs (Figure 3D). Interestingly this enhanced adhesion was diminished when monocytes were incubated with anti-ITGβ1 neutralizing antibody (ITGβ1Ab), suggesting that ITGβ1 on lipotoxic EVs may be responsible for EVs-induced monocytes adhesion to LSECs. We further confirmed this finding using ITGβ1-knockdown Huh7 by shRNA technology (shITGβ1) (Figure 3E–F, Supplementary Figure 3C). Likewise the adhesion of lipotoxic EVs-stimulated monocytes to endothelial cells was reduced in the presence of ITGα9 neutralizing antibody (Supplementary Figure 3D). Moreover, pretreatment of LSECs with a neutralizing antibody against VCAM-1 (an ITGα9β1 ligand expressed on LSECs under basal conditions as examined by flowcytometry), significantly diminished the adhesion of LPC-EVs-stimulated monocytes to LSECs (Figure 3F). Taken together, these results support a role for LPC-EV ITGα9β1 in monocyte adhesion to LSEC via an ITGα9β1-VCAM-1 binding interactions. Interestingly, monocyte inflammatory activation markers expression was also enhanced with lipotoxic EVs stimulation (Supplementary Figure 4), supporting the proinflammatory role of lipotoxic EVs.
Anti-ITGβ1 antibody treatment does not alter the metabolic phenotype or the steatosis in FFC diet-fed mice.
Based on the in vitro findings supporting a key role of lipotoxic EV ITGβ1 in monocyte adhesion to LSECs and potentially in liver inflammation; we examined the potential beneficial effect of ITGβ1 neutralizing antibody in our mouse model of diet-induced NASH. Eight-week-old C57BL/6J wild-type mice were fed either chow or a diet high in saturated fat, fructose, and cholesterol (FFC) for 24 weeks. At 20 weeks of the diet mice were treated with either anti-ITGβ1 neutralizing antibody (ITGβ1Ab) or control IgG isotype antibody (IgG) twice per week for 4 weeks. Firstly, we assessed the metabolic status of each group of mice. Comprehensive Laboratory Animal Monitoring System (CLAMS) study showed that total daily caloric intake (Supplementary Figure 5A), physical activity, energy expenditure, and respiratory quotient (Figure 4A) were similar between FFC-fed ITGβ1Ab-treated versus control IgG-treated mice. Body weight during the whole study period (Figure 4B, Supplementary Figure 5B), liver weight (Supplementary Figure 5C), and liver to body weight ratio (Figure 4C) at the time of sacrifice were significantly increased with the FFC diet, but similar between ITGβ1Ab-treated and control IgG-treated groups. Likewise, homeostasis model assessment of insulin resistance (HOMA-IR) (Figure 4D), and triglyceride content in liver tissue (Figure 4E) were increased with the FFC diet, but were not different between the 2 treatment groups on the FFC diet; although HOMA-IR has some limitations in assessing insulin sensitivity in vivo. Moreover, histological examination of the liver by hematoxylin and eosin (H&E) stain displayed similar extent of steatosis in the FFC-fed mice from the different treatment groups (Figure 4F). Interestingly, ITGβ1Ab-treated mice had less inflammatory infiltrates compared to IgG-treated, FFC-fed mice (Figure 4F). Consistent with the in vitro data, active ITGβ1 expression was increased with FFC diet when assessed by immunohistochemistry and reduced with the ITGβ1 neutralizing Ab (Supplementary Figure 6A). Collectively, ITGβ1Ab treatment in FFC-fed mice was well tolerated, and did not affect the metabolic phenotype or the hepatic steatosis.
Fig. 4. Anti-ITGβ1 antibody treatment did not alter neither the metabolic phenotype nor the steatosis in FFC diet-fed mice.

Wild-type C57BL/6J mice were fed either chow or FFC diet, and treated with either ITGβ1Ab or control IgG isotype. (A) Physical activity, energy expenditure, and respiratory quotient were assessed by CLAMS chambers. (B) Body weight curves. (C) Liver to body weight ratio at the time of sacrifice. (D) HOMA-IR at 23 weeks. (E) Hepatic triglyceride content. (F) Representative images of H&E staining of liver tissues (scale bar, 100 μm). Arrows indicate inflammatory cells infiltrate; bar graphs represent mean±SEM; ***p<0.001, ****p<0.0001, ns, nonsignificant; n=5–6 per group (One-way ANOVA with Bonferroni’s multiple comparison).
Anti-ITGβ1 antibody treatment in FFC-fed mice attenuates hepatic inflammation.
Given the key role of ITGβ1 in monocyte adhesion to LSECs (Figure 3D–F), we examined whether ITGβ1Ab reduces hepatic proinflammatory monocyte recruitment and macrophage-mediated liver inflammation in our dietary mouse model of NASH. Immunostaining of liver tissues revealed that ITGβ1Ab-treated mice had reduced positive area for Mac-2, a marker of phagocytically active macrophages (Figure 5A–B). This finding was supported by the decrease in hepatic mRNA expressions of the macrophage marker Cd68, the infiltrating proinflammatory monocyte marker Ccr2, proinflammatory cytokines Tnf-α, Il12b (Figure 5C, Supplementary Figure 6B) in FFC-fed ITGβ1Ab-treated mice. Furthermore, flow cytometric analysis of the IHL population identified an increase in CD45+ cells in the FFC-fed mice, without significant alteration with ITGβ1Ab treatment (Figure 5D). In contrast, ITGβ1Ab-treated FFC-fed mice did display a significant decrease in the infiltrating proinflammatory monocytes (M1 polarized) defined as CD45+CD11bhiF4/80intCCR2+ cells. Collectively, these findings suggest that blockade of ITGβ1 reduces hepatic proinflammatory monocyte infiltration and MoMF-mediated liver inflammation.
Fig. 5. Anti-ITGβ1 antibody treatment in FFC-fed mice attenuates hepatic inflammation.

(A) Representative images of macrophage galactose-specific lectin (Mac-2) staining of liver sections. (B) Mac-2 positive areas were quantified in 10 random 20x microscopic fields and averaged for each animal. (C) Hepatic mRNA expression levels of Cd68, Ccr2 and Tnf-α were assessed by real-time PCR. Fold change was determined after normalization to 18s expression and expressed relative to Chow-IgG mice. (D) Flow cytometric analysis of the IHL population: top panels show the gating strategy; infiltrating monocytes were defined as CD45+ CD11bhi F4/80int CCR2+cells. Bottom panels show quantification of each population. Bar graphs represent mean±SEM; n=3–5 per group; *p<0.05, **p<0.01, ***p < 0.001, ****p<0.0001 (One-way ANOVA with Bonferroni’s multiple comparison, unpaired t test for panel D.).
Anti-ITGβ1 antibody treatment in FFC-fed mice reduces the pro-inflammatory monocyte hepatic infiltration.
Macrophages are characterized using a variety of criteria, including ontogeny (yolk sac- vs. bone marrow-derived) and function (pro-inflammatory vs. restorative).[4] Liver macrophages are also frequently classified as resident macrophages (Kupffer cells) or recruited macrophages (i.e., circulating bone marrow-derived monocytes differentiating into macrophages). Functionally, macrophages exist as a continuum, with tissue damaging or pro-inflammatory at one end of the spectrum (M1-like), and restorative macrophages involved in tissue repair and healing at the other end (M2-like). While MoMFs play a crucial role in NASH pathogenesis and progression,[26] various other immune cells including neutrophils, dendritic cells, and lymphocytes are involved in NASH pathogenesis.[27, 28] Moreover, ITGβ1 neutralizing antibodies are known to inhibit T lymphocyte trafficking, [29] and might also affect neutrophil trafficking to the liver. Therefore, to determine the contribution of the different subset of macrophages, and other immune cells in ITGβ1Ab protective effect in NASH, we profiled B lymphocytes, T lymphocytes, natural killer cells, NKT cells, dendritic cells and neutrophils in addition to monocytes and macrophages using the state of the art technology mass cytometry by time-of-flight (CyTOF). Twenty eight clusters were obtained (Figure 6A) based on the intensities of 24 different cell surface markers (Figure 6B). Each group of mice displayed a characteristic pattern of clusters abundance (Figure 6C and D). Out of 28 clusters obtained by CyTOF, 13 clusters were differentially expressed between the study groups and categorized into distinct leukocyte subpopulations based on the intensities of individual cell surface markers (Supplementary Table 2). In particular, clusters 5 and 9 had typical expression markers of infiltrating proinflammatory MoMFs, the abundance of these clusters was increased with the FFC-diet, but significantly reduced with ITGβ1Ab treatment (Figure 7A), confirming the flow cytometry data. Likewise, clusters 7 and 17 (Figure 7B) defined as infiltrating MoMF, were reduced in the FFC-fed ITGβ1Ab-treated mice. In contrast, clusters 1 and 2 had typical marker expression patterns of alternative, M2 polarized, or restorative macrophages defined by increased expression of the anti-inflammatory surface marker CD206, as well as the hepatic macrophage markers Lgals, MERTK, and F4/80. The abundance of clusters 1 and 2 was decreased in the FFC-fed mice, but significantly increased with ITGβ1 blockade (Figure 7C). We next assessed other clusters defined as B cell-like cluster 8 (Supplementary Figure 7A), neutrophil-like cluster 15 (Supplementary Figure 7B), dendritic cell-like cluster 19 (Supplementary Figure 7C), and T lymphocyte cluster 16 and 21 (Supplementary Figure 7D). These clusters showed no statistically significant difference between the FFC-fed experimental groups, indicating that the protective effect of ITGβ1 blockade in the FFC-diet induced NASH is mainly through reduced proinflammatory monocyte trafficking and retention in the liver without significant effect on other immune cells.
Figure 6. Intrahepatic leukocyte profiling by mass cytometry by time-of-flight (CyTOF).

CyTOF was performed on IHL of chow-fed mice, and FFC-fed mice treated with either ITGβ1Ab or control IgG isotype. IHL from IgG-treated chow-fed mice were used as control. (A) Twenty-eight unique clusters of IHL were defined by a 24 cell surface marker panel using the Rphenograph clustering algorhithm and were visualized on a t-distributed stochastic neighbor embedding (tSNE) plot. (B) Heat map demonstrating the distribution and relative intensity of the cell surface markers used in the clustering analysis. (C) Heat map showing the relative abundance of each cluster for each mouse. (D) Representative tSNE plots of each group. Red indicates high frequency categorization of cells to a cluster; blue indicates low frequency; n=3 per group.
Fig. 7. nti-ITGβ1 antibody reduces the pro-inflammatory monocyte hepatic infiltration in the FFC-fed mice.

A Differentially expressed clusters between the groups (top graphs); clusters categorized into distinct leukocyte subpopulations based on intensities of individual cell surface markers (bottom graphs). (A) Cluster 5 and 9 represent infiltrating pro-inflammatory MoMF, (B) clusters 7, and 17 represent infiltrating MoMF, (C) cluster 1, 2 and 28 represent restorative macrophage, and (D) cluster 10 represents hepatic macrophage (n=3 per group); bar graphs represent mean±SEM (top panel); *p < 0.05, **p<0.01***, p<0.001, ****p< 0.0001 (One-way ANOVA with Bonferroni’s multiple comparison).
Anti-ITGβ1 antibody treatment reduces FFC diet-induced liver injury and fibrosis in murine NASH.
To determine if reduced hepatic inflammation through ITGβ1 blockade may protect against NASH progression and liver fibrosis, we examined liver injury and fibrosis. FFC-fed, ITGβ1Ab-treated mice were relatively protected against hepatocyte apoptosis compared to control IgG-treated mice, as demonstrated by reduced TUNEL-positive cells (Figure 8A) and serum alanine aminotransferase (ALT) levels (Figure 8B) as well as reduced NAFLD activity score (NAS) (Figure 8C) when compared to IgG-treated mice on the same diet. Next, we examined the expressions of fibrosis-related genes, mRNA levels of both Collagen 1a1 and Osteopontin were elevated in the FFC-fed mice, and significantly decreased with ITGβ1Ab treatment (Figure 8D), indicating the possible anti-fibrotic effect of ITGβ1Ab. This finding was further confirmed by Sirius red staining (Figure 8E) as well as α-smooth muscle actin (α-SMA) immunohistochemistry (Figure 8F). Taken together, these findings indicate a protective effect of ITGβ1Ab against NASH-associated liver injury and fibrosis in diet-induced NASH.
Fig. 8. Anti-ITGβ1 antibody treatment reduces FFC diet-induced liver injury and fibrosis in murine NASH.

(A) Representative images of TUNEL staining of liver sections, quantification of TUNEL-positive cells. (B) Serum ALT levels. (C) NAS scores. (D) Hepatic mRNA expression of Collagen1a1 and Osteopontin. (E) Representative images of Sirius red staining, quantification of Sirius red-positive areas. (F) Representative images of α-SMA staining of liver sections, quantification of α-SMA-positive areas. Scale bars: 100 μm; n=5–6 per group; bar graphs represent mean±SEM; *p< 0.05, **p<0.01, ***p<0.001, ****p<0.0001(One-way ANOVA with Bonferroni’s multiple comparison).
DISCUSSION
The current study provides insights regarding the mechanism by which lipotoxic hepatocyte-derived EVs may regulate peripheral blood monocyte adhesion to LSECs and hepatic recruitment and retention during NASH. The current data indicate that: i) lipotoxic insult in hepatocyte activates ITGβ1 and facilitates its endocytic trafficking and release in EVs; ii) lipotoxic hepatocyte-derived EVs enhance monocyte adhesion to LSECs mainly via their ITGα9β1 cargo binding interaction with LSEC VCAM-1; in FFC diet-induced NASH; iii) ITGβ1 neutralizing antibody reduces proinflammatory monocyte hepatic infiltration; iv) blocking ITGβ1 attenuates liver injury, inflammation and fibrosis. To our knowledge, our report is the first comprehensive profiling of IHL in murine NASH using CyTOF, and the first study of a therapeutic effect of ITGβ1 inhibition in diet-induced NASH. Our findings are discussed in greater details below.
Herein, we build on our prior observation implicating EVs released from lipotoxic hepatocytes in the sterile pro-inflammatory response in NASH via their chemotactic cargo CXCL10 [6] and examine the signaling molecules responsible for monocyte homing to the liver and adhesion to LSECs, a key step for the initiation of inflammation in NASH. We demonstrate for the first time that toxic lipid treatment in hepatocytes induces an active conformation switch of ITGβ1 via a p38 signaling pathway.
The role of ITG-enriched EVs has been established in organotropism, and tumor cell migration.[21, 30] Thus, EV-mediated intercellular integrin signaling is a biologically plausible concept; and our study is the first to identify a non-neoplastic role of ITG on EVs. Moreover, we define the molecular mediators engaged in the adhesion process by employing microfluidic chambers, the optimal technology to study cross talk between two different cell types, in a flow-based paradigm [31]. Here we demonstrate that EVs stimulated monocyte adhesion to LSECs was diminished with Itgβ1 knockdown in the EV donor lipotoxic hepatocytes or with pharmacological inhibition of ITGβ1, ITGα9 or its LSECs ligand VCAM-1. Interestingly, recent in vivo study using zebrafish embryo showed targeting of ITGβ1-enriched EVs to the venous endothelium and macrophages.[32] Inspection of tumor-derived EVs in close proximity to the vessel wall revealed arrest of EVs following a rolling behavior, suggesting that it could be driven by progressive activation of adhesion molecules.[32] A very similar behavior was observed for endogenous EVs in zebrafish.[33] Furthermore, EVs either surfed on the filopodia or were taken up by macrophages leading to reduced macrophage motility, and polarization to the M1-like phenotype. Although the exact molecular mediators of the interaction of EVs with both endothelial cells and macrophages were not examined in these studies, these findings relate to ours and support that ITGβ1-enriched EVs interact with monocytes in a topography that allows binding to LSECs either by fusion/surfing on the cell membrane or endocytosis and recycling back to the surface. Future effort will be concentrated on defining the molecular mediators responsible for hepatocyte-derived EVs uptake by target cells (LSECs, and MoMF). Furthermore our data demonstrate for the first time that the number of circulating EVs of hepatocyte origin is increased in mice with NASH (Supplementary Figure 2). The hepatocyte-derived EV gradient is the highest in the liver microenvironment, mainly the sinusoidal space where EVs likely confer the liver homing signal in response to lipotoxic injury. Our findings are also in line with published proteomics analysis of circulating EVs from NAFLD mice, showing enrichment with cell adhesion-related proteins.[34]
To examine the role of ITGβ1 in monocyte adhesion and liver inflammation in vivo, we utilized a well-established dietary mouse model with high fidelity to human NASH.[35] In the current study, we demonstrate that the FFC diet induces similar changes in both ITGβ1Ab and IgG isotype-treated mice in the metabolic profile and hepatic steatosis. Interestingly, ITGβ1Ab-treated mice on the FFC diet have a relative attenuation of all the injurious features of NASH, when compared with isotype-treated mice. Moreover, our CyTOF data did not show significant alteration in the T lymphocyte, B lymphocyte, NK cells, neutrophil and dendritic cells populations with ITGβ1 antibody treatment in FFC-fed mice, suggesting that the therapeutic benefit of ITGβ1 antibody is mainly through reduced proinflammatory monocyte infiltration.
Inflammation correlates with liver fibrosis and disease progression in NASH patients.[26] Hence improved liver fibrosis in ITGβ1Ab-treated FFC-fed mice might be a consequence of reduced MoMF-mediated hepatic inflammation. However, we cannot exclude the possibility that blockade of endogenously expressed ITGβ1 in other liver cell types such as hepatic stellate cells might have contributed to the reduced fibrosis.
EVs are emerging as key players in cell-to-cell communication. Hence, modulation of EV interaction with target cells by ITGβ1 pharmacological inhibition would offer a specific therapeutic strategy to block the proinflammatory signal originating from lipotoxic hepatocytes. Integrin-based therapeutics have shown clinically significant benefits in patients with chronic inflammatory diseases, [29] and may have an expanded indication for use in patients with NASH. Thus, the current study advances our understanding of the pathogenic mechanisms linking integrin signaling to liver inflammation in NASH, and identify new potential anti-inflammatory therapeutic strategies, that reduces the propensity of LSECs to recruit harmful proinflammatory monocytes.
Supplementary Material
HIGHLIGHTS.
Hepatocytes under lipotoxic stress release active ITGβ1-enriched extracellular vesicles (EVs)
Lipotoxic hepatocyte-derived EVs enhance monocyte adhesion to liver sinusoidal endothelial cells mainly via their ITGβ1 cargo
ITGβ1 neutralizing antibody reduces proinflammatory monocyte hepatic infiltration in murine NASH
Blocking ITGβ1 attenuates liver inflammation, injury and fibrosis in murine NASH
Acknowledgments:
We thank Dr. Gregory J. Gores for his thorough review of the manuscript. We thank Dr. Nathan K. LeBrasseur and his laboratory members, especially Dr. Thomas White, for their assistance in the Comprehensive Laboratory Animal Monitoring System data. We thank Dr. Bing Q. Huang for his help in the studies employing electron microscopy. We also thank Dr. Cristine Charlesworth, and the Mayo Clinic Medical Genome Facility-Proteomics Core and its supporting grant, NCI Cancer Center Support Grant 5P30 CA15083–43C1 for performing the mass spectrometry analysis, as well as the optical microscopy core NIDDK P30DK084567.
Grant Support: this work was supported by NIH grant DK111397 (to SHI), North American Society of Pediatric Gastroenterology Hepatology and Nutrition Young Investigator Award/Nestle Nutrition Award and Gilead Science career development award (to SHI), the Mayo Clinic K2R pipeline and Children Research Center. F.L is funded by a postdoctoral fellowship from the Fonds de Recherche du Quebec-Sante (FRQS).
List of abbreviations:
- AAV
adeno-associated viral vector
- ALT
alanine aminotransferase
- α-SMA
alpha smooth muscle actin
- CCR2
CC chemokine receptor 2
- CD
cluster of differentiation
- CLAMS
comprehensive lab animal monitoring system
- CLEC4
type-C lectin domain family 4
- CyTOF
mass cytometry by time-of-flight
- DAB
diaminobenzidine
- DMEM
Dulbecco’s modified Eagle’s medium
- EEA1
early endosome antigen 1
- EGFP
enhanced green fluorescent protein
- ELISA
enzyme-linked immunosorbent assay
- EV
extracellular vesicle
- FBS
fetal bovine serum
- FFC
fat, fructose and cholesterol
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H&E
hematoxylin and eosin
- HOMA-IR
homeostasis model assessment of insulin resistance
- IACUC
institutional animal care and use committee
- IHL
intrahepatic leukocyte
- IP
immunoprecipitation
- IPA
ingenuity pathway analysis
- ITG
integrin
- LAMP1
lysosomal-associated membrane protein 1
- LPC
lysophosphatidylcholine
- LSEC
liver sinusoidal endothelial cell
- Mac-2
macrophage galactose-specific lectin
- MAPK
mitogen-activated protein kinase
- mRNA
messenger RNA
- MoMF
monocyte-derived macrophage
- MS
mass spectrometry
- MVB
multivesicular body
- MWCO
molecular weight cut off
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- NTA
nanoparticle-tracking analysis
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PFP
platelet-free plasma
- PMH
primary mouse hepatocyte
- PMT
photomultiplier tube
- RNAseq
RNA sequencing
- rRNA
ribosomal RNA
- SDS
sodium dodecyl sulfate
- SEM
standard error of the mean
- shRNA
small hairpin RNA
- TBG
thyroxine binding protein
- TGF-α
transforming growth factor-α
- TNF-α
tumor necrosis factor-α
- TSG101
tumor susceptibility gene 101
- tSNE
t-distributed stochastic neighbor embedding
- TUNEL
terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling
- UF-SEC
ultrafiltration and size-exclusion chromatography
- VCAM-1
vascular cell adhesion molecule 1
- WCL
whole cell lysate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: the authors have no conflict of interest related to the manuscript.
REFERENCES
- 1.Bellentani S, The epidemiology of non-alcoholic fatty liver disease. Liver Int, 2017. 37 Suppl 1: p. 81–84. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol, 2019. 70(3): p. 531–544. [DOI] [PubMed] [Google Scholar]
- 3.Neuschwander-Tetri BA, Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology, 2010. 52(2): p. 774–88. [DOI] [PubMed] [Google Scholar]
- 4.Krenkel O and Tacke F, Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol, 2017. 17(5): p. 306–321. [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL, et al. , A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology, 2018. 67(5): p. 1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim SH, et al. , Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology, 2016. 63(3): p. 731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirsova P, et al. , Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology, 2016. 64(6): p. 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsova P, et al. , Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology, 2016. 150(4): p. 956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand S, et al. , Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim Biophys Acta Proteins Proteom, 2019. [DOI] [PubMed] [Google Scholar]
- 10.Knolle PA and Wohlleber D, Immunological functions of liver sinusoidal endothelial cells. Cellular & Molecular Immunology, 2016. 13(3): p. 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng ZB, et al. , Immature Myeloid Cells Induced by a High-Fat Diet Contribute to Liver Inflammation. Hepatology, 2009. 50(5): p. 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massey VL, et al. , The Hepatic “Matrisome” Responds Dynamically to Injury: Characterization of Transitional Changes to the Extracellular Matrix in Mice. Hepatology, 2017. 65(3): p. 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hynes RO, Integrins: bidirectional, allosteric signaling machines. Cell, 2002. 110(6): p. 673–87. [DOI] [PubMed] [Google Scholar]
- 14.Humphries JD, Byron A, and Humphries MJ, Integrin ligands at a glance. Journal of Cell Science, 2006. 119(19): p. 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridgewater RE, Norman JC, and Caswell PT, Integrin trafficking at a glance. Journal of Cell Science, 2012. 125(16): p. 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, et al. , Sequential activation of p38 and ERK pathways by cGMP-dependent protein kinase leading to activation of the platelet integrin alphaIIb beta3. Blood, 2006. 107(3): p. 965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun H, et al. , Distinct chemokine signaling regulates integrin ligand specificity to dictate tissue-specific lymphocyte homing. Dev Cell, 2014. 30(1): p. 61–70. [DOI] [PubMed] [Google Scholar]
- 18.Tomita K, et al. , Mixed-lineage kinase 3 pharmacological inhibition attenuates murine nonalcoholic steatohepatitis. JCI Insight, 2017. 2(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomita K, et al. , Mixed Lineage Kinase 3 Mediates the Induction of CXCL10 by a STAT1-Dependent Mechanism During Hepatocyte Lipotoxicity. J Cell Biochem, 2017. 118(10): p. 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwamoto DV and Calderwood DA, Regulation of integrin-mediated adhesions. Curr Opin Cell Biol, 2015. 36: p. 41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung BH, et al. , Directional cell movement through tissues is controlled by exosome secretion. Nature Communications, 2015. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogorad RL, et al. , Nanoparticle-formulated siRNA targeting integrins inhibits hepatocellular carcinoma progression in mice. Nat Commun, 2014. 5: p. 3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kakisaka K, et al. , Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. American Journal of Physiology-Gastrointestinal and Liver Physiology, 2012. 302(1): p. G77–G84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccolis M, et al. , Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol Cell, 2019. 74(1): p. 32–44 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tadokoro S, et al. , Talin binding to integrin beta tails: a final common step in integrin activation. Science, 2003. 302(5642): p. 103–6. [DOI] [PubMed] [Google Scholar]
- 26.Kazankov K, et al. , The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol, 2019. 16(3): p. 145–159. [DOI] [PubMed] [Google Scholar]
- 27.Rensen SS, et al. , Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS One, 2012. 7(12): p. e52411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Soloski MJ, and Diehl AM, Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology, 2005. 42(4): p. 880–5. [DOI] [PubMed] [Google Scholar]
- 29.Ley K, et al. , Integrin-based therapeutics: biological basis, clinical use and new drugs. Nature Reviews Drug Discovery, 2016. 15(3): p. 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoshino A, et al. , Tumour exosome integrins determine organotropic metastasis. Nature, 2015. 527(7578): p. 329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel D, et al. , Using reconfigurable microfluidics to study the role of HGF in autocrine and paracrine signaling of hepatocytes. Integr Biol (Camb), 2015. 7(7): p. 815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyenne V, et al. , Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Developmental Cell, 2019. 48(4): p. 554–+. [DOI] [PubMed] [Google Scholar]
- 33.Verweij FJ, et al. , Live Tracking of Inter-organ Communication by Endogenous Exosomes In Vivo. Developmental Cell, 2019. 48(4): p. 573–+. [DOI] [PubMed] [Google Scholar]
- 34.Povero D, et al. , Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One, 2014. 9(12): p. e113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan A, et al. , A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol, 2017. 312(6): p. G666–G680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.