

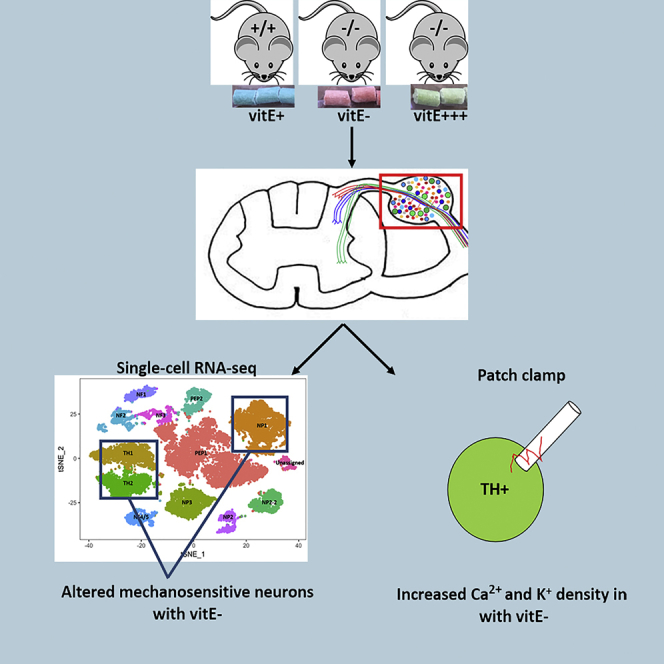
Summary
Ninety percent of Americans consume less than the estimated average requirements of dietary vitamin E (vitE). Severe vitE deficiency due to genetic mutations in the tocopherol transfer protein (TTPA) in humans results in ataxia with vitE deficiency (AVED), with proprioceptive deficits and somatosensory degeneration arising from dorsal root ganglia neurons (DRGNs). Single-cell RNA-sequencing of DRGNs was performed in Ttpa−/− mice, an established model of AVED. In stark contrast to expected changes in proprioceptive neurons, Ttpa−/− DRGNs showed marked upregulation of voltage-gated Ca2+ and K+ channels in mechanosensitive, tyrosine-hydroxylase positive (TH+) DRGNs. The ensuing significant conductance changes resulted in reduced excitability in mechanosensitive Ttpa−/− DRGNs. A highly supplemented vitE diet (600 mg dl-α-tocopheryl acetate/kg diet) prevented the cellular and molecular alterations and improved mechanosensation. VitE deficiency profoundly alters the molecular signature and functional properties of mechanosensitive TH+ DRGN, representing an intriguing shift of the prevailing paradigm from proprioception to mechanical sensation.
Subject Areas: Neuroscience, Molecular Neuroscience, Transcriptomics
Graphical Abstract
Highlights
-
•
vitE deficiency alters gene expression in DRGs
-
•
Mechanosensitive TH+ DRG neurons are most affected
-
•
K+ and Ca2+ current densities are increased in vitE-deficient TH+ DRG neurons
-
•
High-dose vitE supplementation prevents the molecular phenotype
Neuroscience; Molecular Neuroscience; Transcriptomics
Introduction
A mere 10% of the adult American population consume the estimated average requirements (EARs) of vitamin E (vitE) (∼15 mg/day α-tocopherol [α-TOH]) (Fulgoni et al., 2011, Institute of Medicine, 2000, Maras et al., 2004). Severe vitE deficiency stemming from liver diseases, extensive intestinal resections, and the inherited disease of vitE deficiency termed “ataxia with vitE deficiency” (AVED) result in profound ataxia (Muller, 2010). Prevailing functions of vitE have been ascribed to the maintenance of typical neurologic structure and function. Despite identification of vitE as an essential food nutrient (Brigelius-Flohe and Traber, 1999), the molecular, cellular, and functional mechanisms of vitE remain debated. There is extensive evidence supporting the role of vitE, specifically α-tocopherol (α-TOH), as a lipid-soluble antioxidant in vitro (Niki, 2014) and in vivo (Choi et al., 2015, McDougall et al., 2017), and this has been suggested to be the primary functional mechanism of biologic activity (Traber and Atkinson, 2007). Besides this well-established role as an inhibitor of lipid peroxidation, other non-antioxidant properties of α-TOH have been identified, including transcriptional regulation and cell signaling (Azzi, 2018).
AVED results from mutations in the α-tocopherol transfer protein gene (TTPA) (Gotoda et al., 1995, Yokota et al., 1996). Phenotypic variability is due the location of the genetic mutation within TTPA, the amount of vitE in the daily diet, and the time of initiation and dosage of vitE supplementation (Bellayou et al., 2009, Di Donato et al., 2010). Most patients demonstrate symptoms of AVED between 4 and 18 years of age (Cavalier et al., 1998, Gotoda et al., 1995, Schuelke, 1993). Patients with AVED exhibit ataxia, areflexia, decreased fine touch, and vibration discrimination (Gotoda et al., 1995, Schuelke, 1993). Histologic hallmarks of AVED include axonal swellings within the dorsal column medial lemniscus pathway, the sensory pathway that conveys sensations of fine touch vibration, two-point discrimination, and proprioception from the skin and joints. Of note, lesions in the gracile system of the medulla oblongata are also features of physiologic neuroaxonal aging across many species (Bridge et al., 2009). Chronic vitE, specifically α-TOH, deficiency slowly accelerates brain lipid peroxidation and results in cognitive impairment in mice (Fukui et al., 2015) and zebrafish (McDougall et al., 2017). These deficits can be partially restored by vitE supplementation, in the form of α-TOH. Indeed, early α-TOH supplementation in patients with AVED may suppress severe disease symptoms by unknown mechanisms (Aparicio et al., 2001).
Ttpa−/− mice represent an accepted model that recapitulates the phenotype of AVED, with ataxia and histologic lesions, including reduction of myelinated fibers in the gracile fasciculus and chromatolysis of neurons within the nucleus gracilis, as early as 6 months of age (Finno et al., 2018). By 17 months of age, diminished dendritic branching of Purkinje neurons within the cerebellum is evident (Ulatowski et al., 2014). The affected somatosensory tracts originate in the dorsal root ganglia (DRG), with apoptosis of DRG neurons (DRGNs) evident by 12 months of age (Finno et al., 2018). Therefore, it has been suggested that the most pronounced neurologic deficit in Ttpa−/− mice is loss of proprioception. This proprioceptive loss and associated pathology in the spinal cord and cerebellum can be prevented by supplementation of Ttpa−/− mice at weaning with 17x the amount of dl-α-tocopheryl acetate (600 mg/kg feed) (Finno et al., 2018). These studies demonstrate that the interaction of genotype, dietary vitE concentration, and time point in postnatal development are crucial to the development of the neurologic phenotype.
To investigate the underlying mechanisms of functional alterations with vitE deficiency, single-cell RNA-sequencing (scRNA-seq) was performed within the DRG of Ttpa−/− mice. We hypothesized that the most profound transcriptional dysregulation with vitE deficiency would occur in large-diameter myelinated proprioceptive DRGNs. Therefore, we sought to (1) define the transcriptional dysregulation within DRGN subpopulations in Ttpa−/− mice and (2) identify alterations in membrane electrical properties within the targeted DRGN subpopulations of Ttpa−/− mice.
Results
Single-Cell RNA-Sequencing Defines Specific DRGN Subpopulations
To determine the molecular mechanisms leading to sensory deficits in vitE-deficient DRGNs, scRNA-seq was performed on neuronal cells collected from the DRG (between cervical [C1] and lumbar [L6] vertebrae) of mice in three experimental groups: (1) Ttpa+/+ on a basal vitE diet (vitE+; WT), (2) Ttpa−/− on vitE-deficient diet (DEF), and (3) Ttpa−/− on vitE-supplemented diet (SUPP) at 6 months of age (Table S1; see Methods for dietary vitE levels). We have previously identified clinicohistologic evidence of the ataxic phenotype by 6 months of age and completed whole tissue transcriptomic profiling of spinal cord and cerebellum at this time point (Finno et al., 2018). In that study, we identified only minor differences between Ttpa−/− mice maintained on a basal diet (i.e. 35 mg of dl-α-tocopheryl acetate/kg feed) and Ttpa−/− mice on a vitE-deficient diet (DEF; <10 mg of dl-α-tocopheryl acetate/kg feed). Instead, a highly supplemented vitE diet (SUPP; 600 mg of dl-α-tocopheryl acetate/kg feed) was required to prevent the AVED phenotype. Therefore, in the current study, we focused our comparisons on DEF vs. WT and DEF vs. SUPP mice. Contrasts included (1) WT vs. DEF, to determine gene expression changes associated with severe vitE deficiency in DRGNs; (2) SUPP vs. DEF to determine the biologic mechanisms whereby the clinicopathologic AVED phenotype is prevented; and (3) SUPP vs. WT to identify any remaining dysregulated pathways following high-dose vitE supplementation in Ttpa−/− mice. As high-dose vitE supplementation does not restore brain α-TOH to WT concentrations (Finno et al., 2018, Yokota et al., 2001), we postulated that any gene dysregulation within the SUPP vs. WT group may provide insight into this phenomenon.
ScRNA-seq was performed on an average of 3,614 ± 470 DRGN from two replicate mice per group, with a total of 382 million (M) reads generated (17,862 ± 2,625 reads/cell). Approximately 66.5% of reads mapped to the murine transcriptome, similar to previous reports (Usoskin et al., 2015), and an average of 1,788 ± 283 genes detected per cell, with no difference between experimental groups (Figure S1).
Unsupervised single-cell transcriptome profiling identified 14 initial subpopulations in all mice, using gene profiles as previously reported (Figures 1A and S2A) (Usoskin et al., 2015, Li et al., 2016). Based on further evaluation of subpopulations 0 and 2, both clusters appeared to contain peptidergic neurons and were, therefore, merged into cluster 0.2 (Figure S2B). The top transcripts in each subpopulation are provided in Table S2. A t-distributed stochastic neighbor embedding (t-SNE) plot identified a small number of neuronal cells that could not be classified based on previously reported gene profiles (Figure S2A). This cluster was labeled as “Unassigned.” Each DRG subpopulation set contained differentially expressed genes characteristic for that cluster. These clusters were classified as PEP1, PEP2, NP1, NP2, NP2-2, NP3, NF1, NF2, NF3, NF4-5, TH1, and TH2 as previously described (Usoskin et al., 2015) and “Unassigned” for the unassigned cluster (Table S2). There were no significant differences in cell count per neuronal cluster across experimental groups (p> 0.05 in each subpopulation) (Table S1). A distinct population of microglial cells based on previously reported markers was not identified (Figure 1A). Although previous studies have identified outliers or cells with unresolved identity, we further evaluated the “Unassigned” DRGN subpopulation using the top 15 transcripts defining this cluster (Figure S2A, Table S2). Two of the most specific transcripts representing this cluster, forkhead box protein transcription factor 2 (Foxp2) and olfactomedin 3 (Olfm3), have been previously identified in DRG, primarily in postnatal mice through P14 (Jia et al., 2018, Nakaya et al., 2012). However, these transcripts are also present in adult brain (Ferland et al., 2003, Nakaya et al., 2012, Saunders et al., 2018). Therefore, this subset of neurons most likely represents the small number of remaining developmental neurons undergoing axonal growth.
Figure 1.

An Overall Increase in the Number of Differentially Expressed Transcripts across All DRGN Subpopulations with α-TOH Deficiency
(A) A t-distributed stochastic neighbor embedding (t-SNE) plot of merged datasets of the six experimental mice to define neuronal subpopulations clusters. Thirteen clusters were identified based on previously reported gene expression profiles (Li et al., 2016, Usoskin et al., 2015). The “NP2-2” subgroup contained some genes representative of the NP2 cluster but was distinct from the NP2 cluster and has been previously characterized (Usoskin et al., 2015, Li et al., 2016).
(B) An increasing number of significantly (PFDR<0.05) differentially expressed transcripts (DETs) was identified in each DRGN subpopulation with increasing contrasts of vitE deficiency (i.e. SUPP vs. DEF > WT vs. DEF > SUPP vs. WT).
(C and D) (C) The two most commonly dysregulated transcripts across DRGN subpopulations were carbonic anhydrase 8 (Car8), which was significantly downregulated in 10/13 DRG clusters with vitE deficiency (SUPP vs. DEF), and (D) the lineage-specific transcription factor Runx3, which was significantly upregulated in 10/13 DRG clusters with vitE deficiency (SUPP vs. DEF). p values were adjusted by a false discovery rate of 0.05 and log-transformed. Significance was set at PFDR<0.05, corresponding to a –log Padjusted>1.3 (red line). NF = neurofilament, NP = non-peptidergic, PEP = peptidergic, TH = tyrosine hydroxylase, UNKNOWN = unknown cluster, n = 2 mice per group with ~3,600 cells/mouse profiled.
An Increasing Number of Dysregulated Transcripts with Increasing vitE Deficiency
Within each neuronal cluster, dysregulated transcripts were identified between WT vs. DEF, SUPP vs. WT, and SUPP vs. DEF. For each cell subpopulation, at least two-fold dysregulated (i.e. up- or downregulated at PFDR<0.05) transcripts were identified between vitE treatment groups (i.e. SUPP vs. DEF ≫ WT vs. DEF > SUPP vs WT) (Figure 1B, Tables S3 andS4). The greatest degree of dysregulation in the SUPP vs. DEF group was apparent in the NP1 and TH2 subpopulations (Figure 1B).
Top Dysregulated Transcripts across the Majority of DRG Subpopulations: Car8 and Runx3
When WT and SUPP were compared with the DEF groups, carbonic anhydrase 8 (Car8), encoding for a protein that inhibits inositol trisphosphate receptor 1 (IP3R1) (Zhuang et al., 2015), was significantly (PFDR<0.05) downregulated in 10/13 DRG clusters (Figure 1C). In particular, Car8 was the top downregulated transcript with vitE deficiency in the four neurofilament subgroups (NF1, NF2, NF3, and NF4/5). Car8 was downregulated in these subgroups even in the SUPP vs. WT contrast (Figure 1C). The lineage-specific transcription factor Runx3 was significantly (PFDR<0.05) upregulated in 10/13 DRG clusters when comparing WT and SUPP vs. DEF groups, especially within NP3 and TH2 (Figure 1D). Even within the SUPP vs. WT contrast, Runx3 remained significantly upregulated in 8/13 DRG subpopulations, including NP3 and TH2 (Figure 1D).
As Car8 is the primary inhibitor of IP3R1 (Hirasawa et al., 2007, Hirota et al., 2003) and transcription of Car8 was significantly downregulated with vitE deficiency, we postulated that IP3R1 signaling might be affected by the level α-TOH supplementation. In DEF mice, a pronounced decrease in IP3R1 was apparent in TH+ DRGNs, whereas protein levels appeared increased in SUPP mice (Figure S3A). Similarly, Car8 was increased in TH+ DRGNs in the SUPP group (Figure S3B).
Profound Upregulation of Voltage-Gated Ca2+ and K+ Channels in TH+ DRGNs
Modulation of Ca2+ channel activities mediate changes in neuronal plasticity and, when upregulated, promote neurodegeneration (Chilton, 2006). Transcripts associated with both ligand-gated and voltage-gated Ca2+ and K+ channels were evaluated across genotype and vitE diet groups in our scRNA-seq dataset (Table S5). When comparing SUPP vs. DEF groups, of the voltage-activated Ca2+ channels expressed in DRGNs, intermediate-voltage Cav2.3 (Cacna1e; TH1 PFDR = 1.51 x 10−8, TH2 PFDR = 1.29 x 10−11) channel was most notably upregulated in TH+ DRGNs (Figure 2A). High-voltage-activated P-type Ca2+ channel, Cacna1a, was not significantly altered in any DRGN subpopulation, whereas N-type Ca2+ channel, Cacna1b, was only upregulated in the NP1 DRGN subpopulation of the SUPP vs. DEF groups (PFDR = 0.001) (Table S5). When evaluating K+ channel transcripts significantly (PFDR< 0.05) upregulated in the SUPP vs. DEF groups, the most commonly affected DRGN subpopulations for K+ voltage-gated channel transcript upregulation were TH+ (Table S5). In particular, regulatory subunits of the large-conductance Ca2+-activated channel subfamily M (Kcnmb1; TH1 PFDR = 0.009, TH2 PFDR = 1.68 x 10−4 and Kcnmb2; TH1 PFDR = 0.017, TH2 PFDR = 5.20 x 10−5) were upregulated in TH+ DRGNs (Figure 2B). Although a few of the transcripts encoding voltage-gated K+ channels were upregulated in TH+ DRGNs, most transcripts in this family were upregulated in the non-peptidergic DRGNs (Table S5).
Figure 2.

Upregulation Intermediate Voltage-Gated Ca2+ and K+ Channels in TH+ DRGNs with vitE Deficiency
Heatmaps, plotted by –logPadjusted, comparing the degree of upregulation for R-type intermediate voltage-gated Ca2+, Cacna1e, and Ca2+-activated K+ channel beta subunits channels, Kcnmb1 and Kcnmb2, in DRGN subpopulations with vitE deficiency. Contrast A = SUPP vs. WT, contrast B = WT vs. DEF, contrast C = SUPP vs DEF. Cacna1e= Cav2.3 intermediate voltage-activated Ca2+ channel, Kcnmb = Potassium large conductance calcium-activated channel, subfamily M, beta.
Increase in Cacna1e and Kcnmb2 with vitE Deficiency in TH+ DRG
To confirm that Cav2.3 (Cacna1e) and regulatory subunits Kcnmb1 and Kcnmb2 were indeed upregulated, we further evaluated mRNA and protein levels of Cacna1e and Kcnmb2. In DEF mice, there was a pronounced increase in Cacna1e and Kcnmb2 in TH+ DRGNs at the level of both mRNA (Figures 3A, 3C, and 3E) and respective proteins (Figures 3B and 3D).
Figure 3.

Increase in Cacna1e and Kcnmb2 with vitE Deficiency in TH+ DRG
(A–D) Green: Th, Blue: DAPI nuclei, Red: Cacna1e [(A) mRNA, (B) protein] and Kcnmb2 [(C) mRNA, (D) protein]. White box inset magnified in the last column. Fluorescent immunohistochemistry from 4-month WT, DEF, and SUPP mice, n = 1–2 per group. Scale bars represent 10 μm (TH, Cacna1e, Kcnbm2, and merge) and 3 μm (enlarged).
(E) Quantification of mRNA using RNAscope for Cacna1e and Kcnmb2. Mean ± SD, N = 8 counts per experimental group, one-way ANOVA, or Kruskal-Wallis. Scale bars represent 10 μm (TH, Cacna1e, Kcnbm2, and merge) and 3 μm (enlarged).
(F) Von Frey assay, demonstrating a significant increase in sensitivity (i.e. lower Dixon's score) in the SUPP vs. DEF mice. Mean ± SD, N = 8–22 per group, one-way ANOVA, ****p< 0.0001, **p< 0.01, *p< 0.05.
Supplementation with High-Dose α-TOH Increased Mechanical Sensitivity in Ttpa−/− Mice
The von Frey filament assay was performed as previously described (Martinov et al., 2013). SUPP mice had significantly increased mechanical sensitivity compared with DEF diet (Padjusted = 0.007, Figure 3F).
Altered Excitability of DEF Small-Diameter DRGNs
To understand the etiology and mechanisms of DRG neuronal responses, sensory deficits, and degeneration in AVED as shown in Ttpa−/− mice (Finno et al., 2018, Yokota et al., 2001), we focused on the changes in membrane electrical properties. First, we focused on identifying small-diameter mechanosensitive DRGNs. We applied displacement-clamp at DRGN cell-bodies. Using a holding potential of −70 mV, mechanical displacement of the small-diameter DRGN cell body evoked inward currents (Figure 4A, inset shows traces from WT DRGN). The mechanically activated (MA) current (IMA) had amplitudes ranging from 75 to 500 pA (n = 11; Figure 4A). The displacement-response relationships from data from WT DRGNs were fitted with a single Boltzmann function with half-maximal activation displacement (X1/2) and slope factor of 1.1 ± 0.1 μm and 0.3 ± 0.1 μm (n = 11; Figure 4A). Similar data from DEF DRGNs for X1/2 and slope factor were 1.5 ± 0.1 μm and 0.4 ± 0.1 μm (n = 7). Unpaired ttest comparison shows significant differences in the X1/2 (p = 0.016) but not the slope factor (p = 0.51) between the WT and DEF DRGNs. The rightward shift in the displacement-response relations in the DEF DRGNs suggest a decrease in mechanical sensitivity.
Figure 4.

Membrane Properties of Small-Diameter Dorsal Root Ganglion Neurons (DRGNs) from WT and DEF Mice
Current-clamp recordings were performed on DRGNs 6-month-old mice. Membrane input resistance (Ri) was determined by evaluating membrane voltage changes in response to negative and positive current injection. The ohmic relations were fitted with linear regression and the Ri derived from the slope.
(A) Representative traces of displacement-clamp currents recorded using CsCl/NMG-based pipette solution in response to ~250-ms mechanical displacement steps of ~0.42 μm to WT small-diameter DRGN (shown as inset). DRGNs were held at −70 mV. Summary data of displacement-response relationship of mechanically activated (MA) currents (IMA) represented as the I/Imax or open channel probability (Po) against displacement (X) fitted with single Boltzmann function. Data from WT DRGNs (shown in black symbols and fitted with sigmoidal curve in black) and the one-half maximum displacements (X1/2) are 1.1 ± 0.1 μm and 0.3 ± 0.1 μm (n = 11). Data from DEF DRGNs (shown in blue symbols and fitted with sigmoidal curve in blue) and the one-half maximum displacements (X1/2) are 1.5 ± 0.1 μm and 0.4 ± 0.1 μm (n = 7).
(B) Among the small-diameter neurons, there were three distinct classes: fast, medium, and slow adapting. Exemplary plots from fast-adapting DRGNs from WT mice (shown in black, mean Ri in MΩ; 100 ± 6; n = 15), and in DEF (shown in blue, mean Ri in MΩ; 45 ± 5; n = 17: p< 0.0001). The inset is an example of data used to generate the plots. For medium-adapting neurons the Ri (in MW) were as follows: WT (306 ± 23; n = 9) and DEF (162 ± 29; n = 11: p = 0.0014). For slow-adapting neurons the Ri were WT (626 ± 47; n = 13) and DEF (395 ± 38; n = 11: p = 0.0012).
(C) Brief (~5 ms) stepwise positive current was injected to elicit subthreshold (shown in different color codes) and threshold depolarization (WT in black and DEF in blue). The threshold currents are indicated. The threshold voltage was determined, using a dV/dt loop plot (inset, right).
(D and E) Typical voltage response from slow-adapting DRGNs recorded from WT and DEF mice.
(F and G) Action potentials generated using varying pulse durations from fast-adapting DRGNs in WT (F) and DEF (G) mice.
(H) Plots of the relations between threshold potential and pulse duration in WT (in black) and DEF (in blue) mice. The insets show the dV/dt versus membrane potential (V) loops used to determine the thresholds.
Three functional classes of neurons were then assessed based on their evoked spike frequency adaptation kinetics, defined as fast (eliciting 1–2 APs for ∼0.5-s suprathreshold current injection), medium (4–6 APs), and slow (>10 APs) adapting neurons. We first compared WT with DEF DRGNs. On average, the input resistances of the three classes of neurons in WT were ∼2-fold greater than the DEF neurons. Figures 4B and 4C summarize exemplary steady-state input resistances (Ri, in MΩ), of the WT (black; 100 ± 6; n = 15), with ∼0.33 nA required to elicit an AP and DEF (blue; 45 ± 5; n =17), with ∼0.73 nA being the AP threshold for fast-adapting DRGNs. For medium-adapting neurons, the Ri were WT (306 ± 23; n = 9) and DEF (162 ± 29, = 11), and for slow-adapting neurons the Ri were WT (626 ± 47; n = 13) and DEF (395 ± 38; n = 11). The intrinsic membrane properties of the small-diameter DRGNs suggested that membrane excitability was reduced in DEF mice. In agreement with this assertion, DEF slow-adapting DRGNs showed profound attenuation in spike activity in response to current injection compared with WT DRGNs (Figures 4D and4E). AP thresholds for fast-adapting DRGNs were determined by calculating dV/dt, as illustrated in Figure 4C (right panel), and the sensitivity of neurons were tested using the indicated pulse duration. For WT DRGNs, with increasing pulse duration, the threshold voltage amplitude declined in a monotonic fashion. However, for DEF DRGNs, the threshold voltage had a biphasic response relative to pulse duration (Figures 4F–4H).
Increased K+ and Ca2+ Density in DEF Small-Diameter DRGNs
The underlying conductances responsible for the reduced Ri and membrane excitability in the small-diameter Ttpa−/− DRGNs were examined in the voltage-clamp configuration. Ca2+ and K+ currents were isolated under conditions where other ion channel conductances were suppressed. The rationale for focusing on these Ca2+ and K+ conductances stemmed from the scRNAseq analyses (Figures 2 and 3). Whole-cell voltage-clamp of the small-diameter mechanosensitive DRGNs showed outward K+ currents with transient and sustained components in both WT (Figure 5A, upper panel, black) and DEF (Figure 5A, lower panel, blue) mice in response to varying voltage steps (−110 to 40 mV) from a holding potential of −90 mV in 10-mV increments. The difference-current traces plotted in dashed lines (inset) provide the profile of the enhanced outward K+ current in the DEF DRGNs. This difference in currents consisted of mainly a sustained outward current. The total outward K+ current density plotted as a function of voltage showed significant differences in current densities elicited at −30 to 40 mV step voltages comparing data between WT and DEF mice (n = 17; p< 0.05, Figure 5B). To examine Ca2+ currents, we suppressed outward K+ currents, by substituting pipette and bath K+ with NMDG+ and Cs+ (see Methods). Inward Na+ current contamination of Ca2+ currents was suppressed using TTX (1 μM) and by partial substitution of bath Na+ with NMDG. Ca2+ currents were activated using −90 and −40 mV holding voltages, and varying step voltages were applied from −120 to 40 mV. Ca2+ currents in DRGN consist of multiple components: low- and high-voltage-activated currents (Boland and Dingledine, 1990, Wu and Pan, 2004). The difference-current derived by subtracting current traces generated from −40 mV and that at −90 mV holding potentials yielded mainly the transient and low-voltage activated components (Figures 5C and 5D). Summary data from current-voltage relations show that the high-voltage-activated Ca2+ currents were enhanced in DEF DRGNs (Figure 5E). As shown in the inset and illustrated in the current-voltage, rSNX-482 (500 nM), a Cav2.3 (R-type) current-specific blocker (Xie et al., 2016), suppressed a component of the Ca2+ current. Additionally, in three recordings from DEF DRGNs, in which rSNX-482 was applied in the bath solution, the enhanced current was attenuated, suggesting the current was conducted by Cav2.3 channels (Catterall et al., 2005, Xie et al., 2016).
Figure 5.

Increased K+ and Ca2+ Current Density in DEF versus WT DRGNs
(A)Whole-cell onward K+ currents were elicited using depolarizing steps from −110 to 40 mV (ΔV = 10 mV). The tail currents were at −40 mV. Current traces recorded from WT and DEF DRGNs are shown in black and blue, respectively. To obtain a profile of currents that are enhanced in DEF DRGNs, we determined the “difference currents” between DEF and WT neurons at −70 and 40 mV step voltages (traces are plotted with dashed lines in inset).
(B) Summary of steady-state currents was normalized to individual membrane capacitance (Cm), from 6-month-old WT mice (shown with black line and symbol) and DEF (shown with gray traces). Data were generated from 14 DRGNs from each experimental group. The mean current densities (in pA/pF) in WT and DEF DRGNs at 0 mV step voltage were 26.1 ± 2.6 and 39.9 ± 2.8; n = 14, p = 0.0015.
(C and D) Inward Ca2+ currents recorded from a 12-pF DRGNs in WT (in black) and DEF (in blue) mice from −90 and −40 mV holding potentials. Currents were generated using voltage steps ranging from −110 to 40 mV. The difference-current traces (−90 mV) - (−40 mV) are plotted in dashed lines as an inset.
(E) Peak Ca+ current density (I)-voltage (V) relation from data amassed from 12 DRGNs in each group. The current densities generated from a holding voltage of −40 mV are plotted with WT in black and DEF in blue. The high-voltage activated component of the Ca2+ current was enhanced in the DEF DRGNs. The peak current density (in pA/pF) for currents elicited from a holding potential of −40 mV for WT DRGNs was 20.1 ± 1.5 (n = 9) and for DEF DRGNs was 30.9 ± 2.5 (n = 9, p = 0.002). After application of 500 nM rSNX-482 to the DEF DRGNs the peak current density plummeted to 12.6 ± 1.2 (n = 6, p< 0.0001).
Since high-dose vitE supplementation on DEF DRGN appeared to restore the molecular and cellular hallmarks of vitE deficiency, we examined the membrane properties of small-diameter DRGNs in SUPP mice. We examined sensitivity of SUPP DRGNs to mechanical displacements as described in Figure 4A. The displacement-response relationships from data from SUPP DRGNs were fitted with a single Boltzmann function (shown in green) with half-maximal activation displacement (X1/2) and slope factor of 1.09 ± 0.1 μm and 0.3 ± 0.1 μm (n = 6; Figure 6A). We superimposed data from WT (in black) and DEF (in blue) DRGNs for comparison. A one-way ANOVA was performed to compare the X1/2 and the slope factor in WT, DEF, and SUPP DRGNs displacement-response curves. There were significant differences at the p< 0.05 level for the three conditions (X1/2 F(2,21) = 6.088, p = 0.008). Post-hoc comparisons using the Tukey HSD test indicated that data from WT and DEF (p = 0.011) as well as DEF and SUPP (p = 0.024) are significantly different. There was no significant difference between WT and SUPP. Additionally, there were no significant differences in the slope factor in the three experimental conditions.
Figure 6.

Membrane Properties of Small-Diameter Dorsal Root Ganglion Neurons (DRGNs) from SUPP Mice
(A) Current-clamp recordings were performed on DRGNs 6-month-old mice supplemented with 600 mg dL-alpha-tocopheryl/kg feed. Data were assessed from DRGNs with capacitance <15 pF. Membrane input resistance (Ri) was determined by evaluating membrane voltage changes in response to negative and positive current injection.
(B) The ohmic relations were fitted with linear regression and the Ri derived from the slope. SUPP mice fast-adapting DRGNs had a mean Ri of 115 ± 9 MW (n = 11).
(C) Similar to WT DRGNs, as the pulse duration was prolonged, the threshold voltage declined.
(D) Typical voltage response from slow-adapting DRGNs recorded from SUPP mice. Compared with WT DRGNs, the slow-adapting neurons in SUPP did not recover fully despite vitE supplementation.
(E)Whole-cell onward K+ currents were elicited using depolarizing steps from −130 to 30 mV (ΔV = 10 mV), from a holding potential of −90 mV. The tail currents were at −60 mV. Current traces recorded from SUPP DRGNs are shown.
(F) Summary of steady-state currents was normalized to individual membrane capacitance (Cm), from 6-month-old SUPP mice. Data were generated from 15 DRGNs.
(G and H) Inward Ca2+ currents recorded from a 10-pF DRGNs in SUPP mice from −90 and −40 mV holding potentials. Currents were generated using voltage steps ranging from −120 to 40 mV.
(I) The current-voltage relations generated at −90 mV and −40 mV holding potential is plotted. The I-V relations of Ca2+ currents from DEF DRGNs from Figure 5E is re-plotted in blue for comparison.
Excitability of SUPP Small-Diameter DRGNs
For SUPP mice, fast-adapting DRGNs had a mean Ri 115 ± 9 MΩ (n = 11) (Figure 6B, shown with green symbols and line). The Ri between WT (in black line), DEF (in blue line), and DRGNs (Figure 4B) is replotted for comparison. There were significant differences at the p< 0.05 level for the three conditions (F(2,40) = 174.7, p = 0.001). Post-hoc comparisons using the Tukey HSD test indicated that Ri from WT vs. DEF (p = 0.001), WT vs. SUPP (p = 0.001), and DEF vs. SUPP (p = 0.001) are significantly different. Similar to WT DRGNs, as the pulse duration was prolonged, the threshold voltage amplitude declined (Figure 6C). However, the apparent preventative effects of vitE supplementation were not visibly seen in the slow-adapting small-diameter DRGNs. Compared with WT DRGNs, the slow-adapting neurons in SUPP did not recover fully despite vitE supplementation (Figure 6D). We examined whole-cell onward K+ currents by applying depolarizing voltage steps from −130 to 30 mV (ΔV = 10 mV), from a holding potential of −90 mV in SUPP DRGNs (Figure 6E). The steady-state outward K+ currents normalized to individual membrane capacitance (Cm) (Figure 6F) showed that vitE supplementation is sufficient to restore the outward K+ current (plotted in green and compared with the mean data from WT [in black] and DEF [in blue]) (see Figure 5B). For example, the total outward current density elicited at 0 mV in SUPP DRGNs was 24.7 ± 2.9 pA/pF (n = 11). There were significant differences at the p< 0.05 level for the three conditions; steady-state K+ current elicited at 0 mV F(2,36)= 47.6, p = 0.001. Post-hoc comparisons using the Tukey HSD test indicated that WT vs. DEF (p = 0.001) and DEF vs. SUPP (p = 0.001) are significantly different. There is no significant difference between WT vs. SUPP (p = 0.58). We recorded inward Ca2+ currents from ∼10-pF DRGNs in SUPP mice using −90 and −40 mV holding voltages. Currents were generated using voltage steps ranging from −120 to 40 mV (Figures 6G and 6H). The current-voltage relations generated at −90 mV and −40 mV holding voltages are plotted, and the data suggest that vitE supplementation seemingly reversed the enhanced high-voltage-activated Ca2+ current observed in DEF DRGNs (Figure 6I). Summary data from DEF DRGNs in Figure 5E (in blue) is replotted for comparison.
Upregulation of Pro-apoptotic Transcripts with vitE Deficiency
Based on previous studies (Finno et al., 2018), evidence exists for increased apoptosis in the DRGNs with vitE deficiency by 1 year of age in DEF mice. In both the TH2 (PFDR = 1.13 x 10−4) and NP1 (PFDR = 0.01) DRGN subpopulations, apoptosis-inducing factor, mitochondrion-associated 3 (Aifm3), was upregulated with vitE deficiency (WT vs. DEF and SUPP vs. DEF; Figure S4A). Additionally, tumor necrosis factor receptor superfamily, member 21 (Tnfrsf21), which promotes apoptosis by release of cytochrome c from the mitochondria into the cytoplasm, was significantly upregulated in the NP2 (PFDR = 2 x 10−4) and TH2 (PFDR = 0.008) DRGN subpopulations (SUPP vs. DEF; Figure S4A). These results suggest that molecular signatures of apoptosis, most pronounced in TH+ DRGNs, occur by 6 months of age in DEF mice.
To further investigate the pathways leading to apoptosis of DRGNs with vitE deficiency by 1 year of age (Finno et al., 2018), and the potential preference for TH+ DRGNs, protein expression of total and cleaved caspase 3, caspase 8, and caspase 9 was evaluated with both immunohistochemistry and Western blot analyses in 4-month-old mice. There was no evidence of total caspase 3, caspase 8, or caspase 9 activation (data not shown). However, a significant (p< 0.01) increase in cleaved caspase 3 and caspase 9, localized to TH+ DRGNs, was apparent in DEF mice (Figures S4B and S4C). There was no evidence of cleaved caspase 8 activation (Figure S4D).
Additional Transcript and Pathway Dysregulated with vitE Deficiency
In order to determine if the altered gene expression in the DEF DRGN was modulated by the vitE binding proteins Ttpa (Kono et al., 2013) and tocopherol binding protein (Tap or Sec14l2) (Zingg, 2015), we interrogated these transcripts in our dataset. Ttpa was not expressed in the DRGN dataset. Sec14l2 was expressed in DRGNs but only differentially expressed between SUPP vs. DEF in the NP1 subpopulation (upregulated in DEF group; PFDR = 0.004). Additionally, the scRNA-seq dataset was evaluated for genes encoding enzymes that have been previously found to directly bind to vitE, including protein kinase C (Prkca); PH domain and leucine-rich repeat protein phosphatase 2 (Phlpp2); cyclooxygenase-2 (Ptgs2); and lipoxygenases 5, 12, and 15 (Alox5, Alox12, and Alox15) (Domijan et al., 2014, Zingg, 2015). Ptgs2, Alox5, Alox12, and Alox15 were not expressed in DRGN and, although Phlpp2 was expressed, there were no differences between experimental groups in any DRGN subpopulation. Prkca was significantly downregulated in non-peptidergic DEF DRGN subpopulations (NP1, PFDR = 0.01; NP2, PFDR = 0.001; and NP3, PFDR = 0.03) and upregulated in TH+ DRGN subpopulations (TH1, PFDR = 0.03 and TH2, PFDR = 0.002) when comparing SUPP vs. DEF.
With severe vitE deficiency, degenerative axons are identified within the caudal medulla oblongata and spinal cord with AVED (Yokota et al., 2000). Therefore, we interrogated our dataset for dysregulation of transcripts associated with axonal guidance, synaptic plasticity, and myelination. When SUPP were compared with the DEF groups, upregulation of associated transcripts was identified across DRGN subpopulations, with most transcripts upregulated in TH2 DRGNs (Table S6). Myelin basic protein (Mbp) was upregulated in 4/13 SUPP vs. DEF DRGNs (Table S4) but was only downregulated in the TH2 subpopulation (PFDR = 0.004).
Because Ca2+ signaling triggers growth cone development during neurodevelopment (Chilton, 2006), transcripts associated with Ca2+ binding were interrogated in our scRNA-seq dataset. Most Ca2+ binding transcripts were most upregulated in TH+ DRGNs with increasing vitE deficiency (Figure S5A, contrast “C”).
To further investigate the transcripts expressed among DRGN subtypes and across vitE diet groups, pathway analyses were performed using Panther Pathway overrepresentation analysis (http://pantherdb.org/). When WT and SUPP were compared with the DEF groups, the most commonly dysregulated pathways were upregulation of both the heterotrimeric G-protein signaling pathways Giα and Gsα (P00026) and Gqα and Goα (P00027) (Figures S5B andS5C). Significant (PFDR< 0.05) upregulation of these two G-protein signaling pathways was identified in 9/13 DRG clusters (Table S7, Figure S6). For many of these analyses, G-protein coupled receptor (GPCR) pathways were also significantly (PFDR< 0.05) overrepresented (Table S7).
To further investigate the potential role of enhanced G-protein signaling with vitE deficiency in specific DRGN subpopulations, specific somatosensory genes were evaluated, including G-coupled receptors (GPCRs) and the transient receptor potential vanilloid 1 (Trpv1) channel, as recently reviewed (Yudin and Rohacs, 2018). Associated transcripts were most significantly upregulated in TH+ expressing neurons in the SUPP vs. WT contrast (Figure S5D). These results indicate a positive association between G-protein-coupled receptor transcripts and vitE deficiency, especially in TH+ DRGNs.
Transcripts for GO-Slim biologic processes known to regulate mitochondrial functions were significantly (PFDR< 0.05) downregulated in TH+ and NP1 DRG subpopulations (Table S7). This discovery prompted further evaluation of transcripts associated with oxidative phosphorylation. Between 30% and 50% of the transcript of mitochondrial complex I, NADH:ubiquinone oxidoreductase supernumerary subunits (NDUF) were significantly downregulated (PFDR< 0.05) in NP1 and TH2 DRG subpopulations with vitE deficiency (Figure S5E, Table S8).
Discussion
Although the antioxidant role of vitE is well established, with evidence to support protection of critical fatty acids during development of the nervous system (Miller et al., 2012, Lebold et al., 2013), the proposed non-antioxidant roles for this vitamin remain unclear (Gohil et al., 2010, Azzi, 2007). Additionally, it remains unclear why, although all central nervous system tissues are α-TOH deficient in the Ttpa−/− mouse model, the primary clinicopathologic lesions are localized to sensory tracts. Based on the phenotype associated with AVED (Gotoda et al., 1995, Yokota et al., 1996) and recapitulated in the Ttpa−/− mouse model (Yokota et al., 2001, Finno et al., 2018), we expected a profound shift in the transcriptomic profile of the proprioceptive DRGN subpopulation. Unexpectedly, we instead identified the most pronounced message alterations in NP1 and TH+ DRGNs, responsible for mechanosensation. Both NP1 and TH+ DRGNs are small to medium in diameter and unmyelinated, whereas proprioceptive DRGNs are large-diameter myelinated neurons. Intriguingly, both NP1 and TH+ subpopulations of DRGNs arise from the same developmental lineage for C-fibers, separating into distinct lineages by E16.5–17.5 in the mouse (reviewed in Olson et al., 2016).
Upregulation of intermediate voltage-gated Ca2+ and K+ channels in SUPP vs. DEF groups (Figure 2) prioritized the TH+ DRGN subpopulation for further evaluation. Within TH+ DRGNs, Ca2+ dysfunction with vitE deficiency was implicated by upregulation of the Cav2.3 (Cacna1e) and the Ca2+-activated K+ channel subunit Kcnmb2 (Figure 3). These findings, in combination with decreased IP3R1 (Figure S3A) and upregulation of Gq-coupled receptor pathways responsible for the formation of inositol 1,4,5 triphosphate (IP3) (Figures S5B and S5C) in DEF mice, indicate that abnormal Ca2+ dynamics in TH+ DRGNs may be central to the etiology of AVED. Additionally, downregulation of Car8 (Figures 1C and S3B), a major inhibitor of IP3R1 (Hirasawa et al., 2007, Hirota et al., 2003), indicated coordinated transcriptional changes that could contribute to overactive Ca2+ signaling in TH+ DRGNs. Consistent with this interpretation was the observation that acute vitE deficiency (4–6 months) reduced membrane excitability (Figures 4 and 5) and caspase activation (Figure S4), whereas long-term deficiency resulted in apoptosis of DRGNs (Finno et al., 2018). Supplementation with high-dose α-TOH in DEF mice at weaning prevented the transcriptomic and biochemical profiles described, partially prevented the electrophysiological abnormalities of TH+ DRGNs, and significantly improved mechanical sensitivity as assessed from the displacement-response curves (Figure 6A) and via the von Frey filament assay (Figure 3F). As peripheral neuropathy is a clinical feature of AVED (Gotoda et al., 1995, Fogel and Perlman, 2007), altered Ca2+ signaling within mechanosensitive DRGNs could provide a mechanism for loss of peripheral sensation.
Collectively, our findings are the first to link TH+ DRGNs with vitE deficiency and AVED. TH+ DRGNs constitute approximately 10%–15% of all mouse lumbar DRGNs (Brumovsky et al., 2006); are located in haired skin; and are responsive to brush, pressure, and pinch but not temperature (Li et al., 2016). In patients with AVED, loss of vibration sense and sensitivity to light touch are the frequently reported symptoms (Gotoda et al., 1995), as the dorsal column medial lemniscal neuroanatomic tract is targeted in vitE deficiency (Finno et al., 2018, Yokota et al., 1996). Therefore, we postulate that these symptoms may be due to impaired excitability and abnormal Ca2+ signaling. Specifically, reduced membrane excitability of mechanosensitive DRGNs could account for the peripheral neuropathy associated with AVED. Supplemental α-TOH has been demonstrated to improve peripheral neuropathy in AVED patients (Martinello et al., 1998).
Our findings identify altered Ca2+ signaling in TH+ DRGNs with vitE deficiency, in particular, IP3R1 pathways. Upregulation of Ca2+-binding transcripts, the voltage-gated Cav2.3 (Cacna1e) channel, and Ca2+-activated K+ channel subunit Kcnmb2 are likely mechanistically interrelated alterations that further contribute to the neuropathology in TH+ DRGNs. First, electrophysiological recordings showed enhanced K+ and Ca2+ current densities in small-diameter DRGNs isolated from DEF mice. Second, upregulation of GPCRs and downregulation of Car8 support a role of altered GPCR/phospholipase C (PLC)/inositol triphosphate (IP3) signaling in vitE-deficient TH+ DRGNs. Reactive oxygen species can activate Gq-coupled receptors, catalyzing the conversion of plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) by PLC to the intracellular secondary molecules IP3 and diacylglycerol (DAG) (Servitja et al., 2000, Vaarmann et al., 2010). As vitE remains the most potent inhibitor of lipid peroxidation (Choi et al., 2015, McDougall et al., 2017, Niki, 2014), the protection afforded at the plasma membrane may affect GPCR signaling. The decrease in small-diameter DRGN membrane excitability of DEF mice may, therefore, result from altered Ca2+ signaling in TH+ DRGNs. In keeping with reduced excitability of DEF DRGNs, the rate of change of membrane voltage at the upstroke phase of APs (dV/dt) was shallow compared with WT neurons (Figure 4C), which may implicate alterations in Na+ current magnitude and kinetics (Han et al., 2012). Although there were no significantly dysregulated Na+ channel transcripts in the TH+ DRGNs (Table S4), posttranslational modifications of Na+ channels may have occurred. Additionally, we cannot determine from this study if the observed effects on membrane excitability are the result of the observed gene expression changes following vitE deficiency or the insufficient vitE concentrations in the DRGNs. Experiments requiring the addition of exogenous α-TOH to DRGNs from DEF mice would be required to determine if membrane excitability can be restored with the addition of vitE. Despite this inability to definitively determine causality, the clinical relevance of these findings indicate that gene expression and membrane excitability changes within the DRGN may underlie the peripheral neuropathy observed with AVED.
By 12 months of age, apoptosis within most large- and small-diameter DRGNs is evident in DEF mice (Finno et al., 2018). Increased TUNEL staining of DRGNs in DEF mice is not observed by 6 months of age (unpublished results). In this study, scRNA-sequencing was performed only on viable cells (see Cell count/viability and RNA quality assurance). Therefore, within the 6-month DEF mice examined in this study, increased activity of cleaved caspase-3 and -9 in DEF TH+ DRGNs is likely a proximal mechanism of promoting apoptosis.
Using whole tissue spinal cord homogenate, we previously identified altered nuclear receptor activation in DEF mice as they aged from weaning to 6 months (Finno et al., 2018). With sufficient vitE, retinoid orphan-related receptor alpha (RORA)-targeted transcripts were activated in the spinal cord, whereas insufficient vitE led to the activation of liver X receptor (LXR)-targeted transcripts. This effect was not observed in whole-tissue spinal cord when simply comparing groups at 6 months of age (WT vs. DEF vs. SUPP). Similarly, we did not identify upregulation of most previously investigated (Finno et al., 2018) RORA-targeted transcripts in the DRGNs between SUPP and DEF groups at 6 months of age in this current study. Comparison of DRGNs subpopulations in WT, DEF, and SUPP experimental groups between weaning and 6 months of age requires further investigation to fully elucidate the role for RORA with vitE deficiency.
Elevations in 7-oxygenated cholesterol products have been previously described in both the Ttpa−/− (Finno et al., 2018) and atherosclerotic apolipoprotein vitE-deficient mouse models (Rosenblat and Aviram, 2002). Both 7α-hydroxycholesterol and 7-ketocholesterol are inverse agonists for the constitutively active RORA (Wang et al., 2010). Therefore, we surmise that increased oxysterols suppress constitutive RORA signaling during vitE deficiency, a hypothesis that has been supported by other studies in both the Ttpa−/− mouse (Gohil et al., 2003, Gohil et al., 2004) and the vitE-deficient horse (Finno et al., 2016). RORA activation is required for synaptic maintenance (Landis and Sidman, 1978, Sotelo and Changeux, 1974) and the neuroprotective effect is mediated by the antioxidant proteins glutathione peroxidase 1 and peroxiredoxin 6 (Boukhtouche et al., 2006). Of note, RORA controls the expression of IP3R1 (Gold et al., 2003, Sarachana and Hu, 2013). Therefore, the underlying molecular mechanism whereby vitE deficiency leads to AVED may involve differential nuclear receptor activation with age, leading to altered IP3R1 signaling in TH+ DRGNs, in addition to alterations in redox status at the plasma membrane (Figure 7). Alternatively, specific oxidized polyunsaturated fatty-acid-derived lipid mediators, which are protected by vitE from oxidation and destruction (Choi et al., 2015, Lebold et al., 2013, Lebold and Traber, 2014, Ulatowski and Manor, 2013), may be involved in this protective mechanism.
Figure 7.

Proposed Mechanism of Action for α-TOH in TH+ DRGN
With adequate α-TOH (left), constitutive activity of the RAR-related orphan receptor alpha (RORA) transcription factor is maintained, increasing IP3R1 transcription (Gold et al., 2003, Sarachana and Hu, 2013). Although there is evidence that vitE can affect the plasma membrane structure and bind to signaling enzymes to affect their activity (Zingg, 2015, Habermehl et al., 2005), we propose in this model that signaling through the PLC/IP3/IP3R1 axis maintains Ca2+ homeostasis. VitE can suppress PLC activity (Domijan et al., 2014) and, by stimulating DAGK (Koya et al., 1997), DAG is removed and PKC inhibited, providing a protective effect. With α-TOH deficiency (right), cholesterol is oxidized and resulting oxysterols repress constitutive RORA activity (Wang et al., 2010), leading to decreased IP3R1 transcription. ROS activate the PLC/IP3/IP3R1 axis (Servitja et al., 2000, Vaarmann et al., 2010); however, without sufficient IP3R1, [Ca2+]i cannot increase. Additionally, loss of DAGK stimulation increases DAG and PKC. We propose this leads to the identified alterations in membrane excitability and activation of apoptotic pathways in DRGNs. BK= big potassium channel; DAG = diaglycerol; DAGK= diaglycerol kinase; IP3 = inositol triphosphate; IP3R1 = inositol 1,4,5 triphosphate receptor 1; PIP2 = phosphatidylinositol 4,5-bisphosphate; PKC = protein kinase C; PLC = phospholipase C; RORA= RAR-related orphan receptor alpha; ROS = reactive oxygen species.
Single-cell RNA-seq generated a total of 382 million reads in our study, with an average of 3,614 DRGNs profiled per mouse (i.e. 7,228 neurons per experimental group). In the pioneer scRNA-seq study of DRG, pooled lumbar DRG were used to generate 2.76 billion reads across 622 single mouse neurons (Usoskin et al., 2015). A total of 3,574 ± 2,010 distinct genes were identified in each cell (Usoskin et al., 2015), as compared with our study of 1,788 ± 283 genes per cell. A subsequent publication used neuron-size-based hierarchical clustering and high-coverage scRNA-seq of 203 neurons collected from lumbar DRG of five pooled male mice (10,950 ± 1,218 genes per neuron) (Li et al., 2016). Despite the overall lower depth of sequencing across genes in our study, we were able to identify the previously characterized (Usoskin et al., 2015) neuronal subpopulations within the DRG. Previous reports (Usoskin et al., 2015, Li et al., 2016) clustered the unmyelinated TH+ neuronal population into one population. In our dataset, two distinct TH+ subgroups (TH1 and TH2) were identified across all samples. Despite highly overlapping transcriptional profiles, these subtypes were not closely enough related to merge, unlike the PEP1 subpopulation (i.e. Clusters 0 and 2). The distinct sub-clustering of TH+ subpopulations in our study warrants further investigation, as the most profound alterations with vitE deficiency in our model occurred primarily in the TH2 subpopulation. Our mice were 6 months of age, older than those used in previously reported scRNA-seq profiling studies in DRG (Li et al., 2016, Usoskin et al., 2015).
The von Frey filament assay is used on glabrous skin, which is innervated by both alpha and beta low-threshold mechanoreceptors, both not TH+ C-low threshold mechanoreceptors (Li et al., 2011). However, it has been observed that von Frey filament can activate sensitized non-peptidergic type C nociceptive fibers (i.e. NP1 subpopulations) under certain conditions (Pinto et al., 2019). In our study, the NP1 subpopulation, responsible for neuropathic pain, was also notably implicated with vitE deficiency. VitE has been demonstrated to act as an analgesic in rodent models of neuropathic pain (Kim et al., 2006), although a more recent study suggests that vitC is required concurrently (Lu et al., 2011). Foot withdrawal thresholds in response to mechanical stimuli were used to assess neuropathic pain followed by spinal cord ligation, with increased thresholds for 6 h after vitE injection (Kim et al., 2006). As neurobehavioral assays through tactile stimuli cannot reliably differentiate different DRGN activities (Li et al., 2011), we elected to document electrophysiological dysfunction in TH+-specific mechanosensitive DRGNs with vitE deficiency using whole-cell membrane recordings specific to small-diameter mechanosensitive DRGNs. However, further investigation into the role of vitE in NP1 nociceptive DRGNs is warranted.
One of the most notable findings identified in the DEF mouse model is the complete prevention of the clinical and histologic phenotype with high-dose α-TOH supplementation at weaning (Finno et al., 2018, Yokota et al., 2001). To further support these clinical findings, we have now demonstrated complete prevention of the transcriptomic and partial rescue of the electrophysiologic dysfunction within the DRGNs at the single-cell level. VitE supplementation in Ttpa−/− mice suffices to partially restore K+ and Ca2+ current properties in small-diameter mechanosensitive DRGNs, but it remains unclear why slow-adapting DRGNs are impervious to vitE supplementation. Of note, α-TOH concentrations in the brain of these SUPP mice are still significantly lower than those of WT mice (Finno et al., 2018, Yokota et al., 2001), indicating that α-TTP in the brain functions in maintaining local concentration of α-TOH. Despite the lower brain α-TOH concentrations in SUPP mice compared with WT mice, the AVED clinicohistologic phenotype is completely prevented. Therefore, a minimum concentration of vitE is likely required at this critical time in postnatal development. Even more important is the fact that the phenotype is rescuable when vitE is provided at P21, a time point that is considered the end of postnatal development in the mouse. Therefore, it may be possible that synaptogenesis and axonal elongation continue into adulthood, thereby requiring sufficient vitE from 1–6 months of age in mice.
Although this study focused on DRGNs, vitE has been demonstrated to have effects in other regions of the nervous system. Within the hippocampus, α-TOH induced long-term potentiation (Xie and Sastry, 1993), which is involved in learning and memory (Thompson, 1986). The action of vitE in hippocampal CA1 neurons occurred without a significant alteration in the membrane potential and the input resistance. Rats that are fed a vitE-deficient diet for three months had impaired long-term potential induction, with a reduction in post-tetanic potentiation, suggesting that either neurotransmitter release or postsynaptic mechanisms are involved (Xie and Sastry, 1995). It remains to be determined if the postulated mechanism for the neuroprotective effect of vitE in the DRG (Figure 7) is applicable to other regions of the nervous system.
In conclusion, we have identified the most profound transcriptomic, biochemical, and electrophysiologic changes in small-diameter TH+ mechanosensitive DRGNs, rather than the proprioceptive subpopulation, with vitE deficiency. Increased upregulation of voltage-gated Ca2+ and K+ channels led to AP abbreviation and reduced membrane excitability in TH+ DRGNs. Concurrently, alterations in IP3R1 expression were identified, in addition to evidence of apoptosis via caspase-3- and caspase-9-mediated pathways. A highly supplemented α-TOH diet rescues the cellular and molecular alterations and represses the loss of mechanosensation found in DEF mice. The peripheral neuropathy associated with AVED likely encompasses defects in membrane properties and Ca2+ signaling in mechanosensitive TH+ DRGNs, providing targets for therapeutic intervention.
Limitations of the Study
We cannot determine from this study if the observed effects on membrane excitability are the result of the observed gene expression changes following vitE deficiency or the insufficient vitE concentrations in the DRGNs. Experiments requiring the addition of exogenous α-TOH to DRGNs from DEF mice would be required to determine if membrane excitability can be restored with the addition of vitE.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Support for this work was provided by the National Institutes of Health (NIH) to C.J.F. (K01OD015134 and L40 TR001136). ENY was supported by (NIH: AG051443, DC015135, AG060504, and DC016099). Support for sequencing was provided by the University of California Davis Genome Center. We thank Dr. Isaac Pessah for his careful review of this manuscript.
Author Contributions
CJF and ENY were responsible for the conceptualization, funding acquisition, methodology, investigation, formal analysis, resources, and writing of the manuscript. J.P., M.K., S.P., M.H.B., M.P.F., and J.H.L. contributed to the study and analysis of results, and B.D.J. and M.S. contributed to the informatics methodology and analysis of results. All authors have reviewed the final manuscript.
Declaration of Interests
The authors declare no competing financial interests.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.064.
Supplemental Information
References
- Aparicio J.M., Belanger-Quintana A., Suarez L., Mayo D., Benitez J., Diaz M., Escobar H. Ataxia with isolated vitamin E deficiency: case report and review of the literature. J. Pediatr. Gastroenterol. Nutr. 2001;33:206–210. doi: 10.1097/00005176-200108000-00022. [DOI] [PubMed] [Google Scholar]
- Azzi A. Molecular mechanism of alpha-tocopherol action. Free Radic. Biol. Med. 2007;43:16–21. doi: 10.1016/j.freeradbiomed.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Azzi A. Many tocopherols, one vitamin E. Mol. Aspects Med. 2018;61:92–103. doi: 10.1016/j.mam.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Bellayou H., Dehbi H., Bourezgui M., Slassi I., Nadifi S. Ataxia with vitamin E deficiency (AVED); an example of the contribution of research in molecular genetic to counselling in Morocco. Pathol. Biol. (Paris) 2009;57:425–426. doi: 10.1016/j.patbio.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Boland L.M., Dingledine R. Multiple components of both transient and sustained barium currents in a rat dorsal root ganglion cell line. J. Physiol. 1990;420:223–245. doi: 10.1113/jphysiol.1990.sp017909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhtouche F., Vodjdani G., Jarvis C.I., Bakouche J., Staels B., Mallet J., Mariani J., Lemaigre-Dubreuil Y., Brugg B. Human retinoic acid receptor-related orphan receptor alpha1 overexpression protects neurones against oxidative stress-induced apoptosis. J. Neurochem. 2006;96:1778–1789. doi: 10.1111/j.1471-4159.2006.03708.x. [DOI] [PubMed] [Google Scholar]
- Bridge K.E., Berg N., Adalbert R., Babetto E., Dias T., Spillantini M.G., Ribchester R.R., Coleman M.P. Late onset distal axonal swelling in YFP-H transgenic mice. Neurobiol. Aging. 2009;30:309–321. doi: 10.1016/j.neurobiolaging.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohe R., Traber M.G. Vitamin E: function and metabolism. FASEB J. 1999;13:1145–1155. [PubMed] [Google Scholar]
- Brumovsky P., Villar M.J., Hokfelt T. Tyrosine hydroxylase is expressed in a subpopulation of small dorsal root ganglion neurons in the adult mouse. Exp. Neurol. 2006;200:153–165. doi: 10.1016/j.expneurol.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Catterall W.A., Perez-Reyes E., Snutch T.P., Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Cavalier L., Ouahchi K., Kayden H.J., Di Donato S., Reutenauer L., Mandel J.L., Koenig M. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am. J. Hum. Genet. 1998;62:301–310. doi: 10.1086/301699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilton J.K. Molecular mechanisms of axon guidance. Dev. Biol. 2006;292:13–24. doi: 10.1016/j.ydbio.2005.12.048. [DOI] [PubMed] [Google Scholar]
- Choi J., Leonard S.W., Kasper K., McDougall M., Stevens J.F., Tanguay R.L., Traber M.G. Novel function of vitamin E in regulation of zebrafish (Danio rerio) brain lysophospholipids discovered using lipidomics. J. Lipid Res. 2015;56:1182–1190. doi: 10.1194/jlr.M058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato I., Bianchi S., Federico A. Ataxia with vitamin E deficiency: update of molecular diagnosis. Neurol. Sci. 2010;31:511–515. doi: 10.1007/s10072-010-0261-1. [DOI] [PubMed] [Google Scholar]
- Domijan A.M., Kovac S., Abramov A.Y. Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca(2)(+) signal. J.Cell Sci. 2014;127:21–26. doi: 10.1242/jcs.138370. [DOI] [PubMed] [Google Scholar]
- Ferland R.J., Cherry T.J., Preware P.O., Morrisey E.E., Walsh C.A. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J. Comp. Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Finno C.J., Bordbari M.H., Gianino G., Ming-Whitfield B., Burns E., Merkel J., Britton M., Durbin-Johnson B., Sloma E.A., McMackin M. An innate immune response and altered nuclear receptor activation defines the spinal cord transcriptome during alpha-tocopherol deficiency in Ttpa-null mice. Free Radic. Biol. Med. 2018;120:289–302. doi: 10.1016/j.freeradbiomed.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finno C.J., Bordbari M.H., Valberg S.J., Lee D., Herron J., Hines K., Monsour T., Scott E., Bannasch D.L., Mickelson J., Xu L. Transcriptome profiling of equine vitamin E deficient neuroaxonal dystrophy identifies upregulation of liver X receptor target genes. Free Radic. Biol. Med. 2016;101:261–271. doi: 10.1016/j.freeradbiomed.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel B.L., Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6:245–257. doi: 10.1016/S1474-4422(07)70054-6. [DOI] [PubMed] [Google Scholar]
- Fukui K., Nakamura K., Shirai M., Hirano A., Takatsu H., Urano S. Long-term vitamin E-deficient mice exhibit cognitive dysfunction via elevation of brain oxidation. J. Nutr. Sci. Vitaminol. (Tokyo) 2015;61:362–368. doi: 10.3177/jnsv.61.362. [DOI] [PubMed] [Google Scholar]
- Fulgoni V.L., 3rd, Keast D.R., Bailey R.L., Dwyer J. Foods, fortificants, and supplements: where do Americans get their nutrients? J. Nutr. 2011;141:1847–1854. doi: 10.3945/jn.111.142257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil K., Godzdanker R., O'Roark E., Schock B.C., Kaini R.R., Packer L., Cross C.E., Traber M.G. Alpha-tocopherol transfer protein deficiency in mice causes multi-organ deregulation of gene networks and behavioral deficits with age. Ann. N Y Acad. Sci. 2004;1031:109–126. doi: 10.1196/annals.1331.012. [DOI] [PubMed] [Google Scholar]
- Gohil K., Schock B.C., Chakraborty A.A., Terasawa Y., Raber J., Farese R.V., Jr., Packer L., Cross C.E., Traber M.G. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in alpha-tocopherol transfer protein. Free Radic. Biol. Med. 2003;35:1343–1354. doi: 10.1016/s0891-5849(03)00509-4. [DOI] [PubMed] [Google Scholar]
- Gohil K., Vasu V.T., Cross C.E. Dietary alpha-tocopherol and neuromuscular health: search for optimal dose and molecular mechanisms continues! Mol. Nutr. Food Res. 2010;54:693–709. doi: 10.1002/mnfr.200900575. [DOI] [PubMed] [Google Scholar]
- Gold D.A., Baek S.H., Schork N.J., Rose D.W., Larsen D.D., Sachs B.D., Rosenfeld M.G., Hamilton B.A. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–1131. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoda T., Arita M., Arai H., Inoue K., Yokota T., Fukuo Y., Yazaki Y., Yamada N. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N. Engl. J. Med. 1995;333:1313–1318. doi: 10.1056/NEJM199511163332003. [DOI] [PubMed] [Google Scholar]
- Habermehl D., Kempna P., Azzi A., Zingg J.M. Recombinant SEC14-like proteins (TAP) possess GTPase activity. Biochem. Biophys. Res. Commun. 2005;326:254–259. doi: 10.1016/j.bbrc.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Han C., Hoeijmakers J.G., Liu S., Gerrits M.M., te Morsche R.H., Lauria G., Dib-Hajj S.D., Drenth J.P., Faber C.G., Merkies I.S. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain. 2012;135:2613–2628. doi: 10.1093/brain/aws187. [DOI] [PubMed] [Google Scholar]
- Hirasawa M., Xu X., Trask R.B., Maddatu T.P., Johnson B.A., Naggert J.K., Nishina P.M., Ikeda A. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol.Cell.Neurosci. 2007;35:161–170. doi: 10.1016/j.mcn.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota J., Ando H., Hamada K., Mikoshiba K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem. J. 2003;372:435–441. doi: 10.1042/BJ20030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L.F., Wang L., Chopp M., Li C., Zhang Y., Szalad A., Zhang Z.G. MiR-29c/PRKCI regulates axonal growth of dorsal root ganglia neurons under hyperglycemia. Mol. Neurobiol. 2018;55:851–858. doi: 10.1007/s12035-016-0374-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K., Kim J.H., Gao X., Zhou J.L., Lee I., Chung K., Chung J.M. Analgesic effect of vitamin E is mediated by reducing central sensitization in neuropathic pain. Pain. 2006;122:53–62. doi: 10.1016/j.pain.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Kono N., Ohto U., Hiramatsu T., Urabe M., Uchida Y., Satow Y., Arai H. Impaired alpha-TTP-PIPs interaction underlies familial vitamin E deficiency. Science. 2013;340:1106–1110. doi: 10.1126/science.1233508. [DOI] [PubMed] [Google Scholar]
- Koya D., Lee I.K., Ishii H., Kanoh H., King G.L. Prevention of glomerular dysfunction in diabetic rats by treatment with d-alpha-tocopherol. J. Am. Soc. Nephrol. 1997;8:426–435. doi: 10.1681/ASN.V83426. [DOI] [PubMed] [Google Scholar]
- Landis D.M., Sidman R.L. Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J. Comp. Neurol. 1978;179:831–863. doi: 10.1002/cne.901790408. [DOI] [PubMed] [Google Scholar]
- Lebold K.M., Kirkwood J.S., Taylor A.W., Choi J., Barton C.L., Miller G.W., La Du J., Jump D.B., Stevens J.F., Tanguay R.L., Traber M.G. Novel liquid chromatography-mass spectrometry method shows that vitamin E deficiency depletes arachidonic and docosahexaenoic acids in zebrafish (Danio rerio) embryos. Redox Biol. 2013;2:105–113. doi: 10.1016/j.redox.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebold K.M., Traber M.G. Interactions between alpha-tocopherol, polyunsaturated fatty acids, and lipoxygenases during embryogenesis. Free Radic. Biol. Med. 2014;66:13–19. doi: 10.1016/j.freeradbiomed.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.L., Li K.C., Wu D., Chen Y., Luo H., Zhao J.R., Wang S.S., Sun M.M., Lu Y.J., Zhong Y.Q. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 2016;26:967. doi: 10.1038/cr.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Rutlin M., Abraira V.E., Cassidy C., Kus L., Gong S., Jankowski M.P., Luo W., Heintz N., Koerber H.R. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell. 2011;147:1615–1627. doi: 10.1016/j.cell.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Kallenborn-Gerhardt W., Geisslinger G., Schmidtko A. Additive antinociceptive effects of a combination of vitamin C and vitamin E after peripheral nerve injury. PLoS One. 2011;6:e29240. doi: 10.1371/journal.pone.0029240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras J.E., Bermudez O.I., Qiao N., Bakun P.J., Boody-Alter E.L., Tucker K.L. Intake of alpha-tocopherol is limited among US adults. J. Am.Diet Assoc. 2004;104:567–575. doi: 10.1016/j.jada.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Martinello F., Fardin P., Ottina M., Ricchieri G.L., Koenig M., Cavalier L., Trevisan C.P. Supplemental therapy in isolated vitamin E deficiency improves the peripheral neuropathy and prevents the progression of ataxia. J. Neurol. Sci. 1998;156:177–179. doi: 10.1016/s0022-510x(98)00038-0. [DOI] [PubMed] [Google Scholar]
- Martinov T., Mack M., Sykes A., Chatterjea D. Measuring changes in tactile sensitivity in the hind paw of mice using an electronic von Frey apparatus. J. Vis. Exp. 2013;82:e51212. doi: 10.3791/51212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall M., Choi J., Truong L., Tanguay R., Traber M.G. Vitamin E deficiency during embryogenesis in zebrafish causes lasting metabolic and cognitive impairments despite refeeding adequate diets. Free Radic. Biol. Med. 2017;110:250–260. doi: 10.1016/j.freeradbiomed.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Medicine . The National Academeies Press; Washington, DC: 2000. Dietary Reference Intakes: Vitamin C, Vitamin E, Selenium, and Carotenoids. [PubMed] [Google Scholar]
- Miller G.W., Ulatowski L., Labut E.M., Lebold K.M., Manor D., Atkinson J., Barton C.L., Tanguay R.L., Traber M.G. The alpha-tocopherol transfer protein is essential for vertebrate embryogenesis. PLoS One. 2012;7:e47402. doi: 10.1371/journal.pone.0047402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D.P. Vitamin E and neurological function. Mol. Nutr. Food Res. 2010;54:710–718. doi: 10.1002/mnfr.200900460. [DOI] [PubMed] [Google Scholar]
- Nakaya N., Sultana A., Lee H.S., Tomarev S.I. Olfactomedin 1 interacts with the Nogo a receptor complex to regulate axon growth. J. Biol. Chem. 2012;287:37171–37184. doi: 10.1074/jbc.M112.389916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: in vitro and in vivo evidence. Free Radic. Biol. Med. 2014;66:3–12. doi: 10.1016/j.freeradbiomed.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Olson W., Dong P., Fleming M., Luo W. The specification and wiring of mammalian cutaneous low-threshold mechanoreceptors. Wiley Interdiscip. Rev. Dev. Biol. 2016;5:389–404. doi: 10.1002/wdev.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L.G., Souza G.R., Kusuda R., Lopes A.H., Sant'Anna M.B., Cunha F.Q., Ferreira S.H., Cunha T.M. Non-peptidergic nociceptive neurons are essential for mechanical inflammatory hypersensitivity in mice. Mol. Neurobiol. 2019;56:5715–5728. doi: 10.1007/s12035-019-1494-5. [DOI] [PubMed] [Google Scholar]
- Rosenblat M., Aviram M. Oxysterol-induced activation of macrophage NADPH-oxidase enhances cell-mediated oxidation of LDL in the atherosclerotic apolipoprotein E deficient mouse: inhibitory role for vitamin E. Atherosclerosis. 2002;160:69–80. doi: 10.1016/s0021-9150(01)00563-9. [DOI] [PubMed] [Google Scholar]
- Sarachana T., Hu V.W. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol. Autism. 2013;4:14. doi: 10.1186/2040-2392-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A., Macosko E.Z., Wysoker A., Goldman M., Krienen F.M., De Rivera H., Bien E., Baum M., Bortolin L., Wang S.Y. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–1030.e16. doi: 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M. Ataxia with vitamin E deficiency. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., Amemiya A., editors. GeneReviews((R)) University of Washington; 1993. [PubMed] [Google Scholar]
- Servitja J.M., Masgrau R., Pardo R., Sarri E., Picatoste F. Effects of oxidative stress on phospholipid signaling in rat cultured astrocytes and brain slices. J. Neurochem. 2000;75:788–794. doi: 10.1046/j.1471-4159.2000.0750788.x. [DOI] [PubMed] [Google Scholar]
- Sotelo C., Changeux J.P. Transsynaptic degeneration 'en cascade' in the cerebellar cortex of staggerer mutant mice. Brain Res. 1974;67:519–526. doi: 10.1016/0006-8993(74)90499-5. [DOI] [PubMed] [Google Scholar]
- Thompson R.F. The neurobiology of learning and memory. Science. 1986;233:941–947. doi: 10.1126/science.3738519. [DOI] [PubMed] [Google Scholar]
- Traber M.G., Atkinson J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007;43:4–15. doi: 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulatowski L., Manor D. Vitamin E trafficking in neurologic health and disease. Annu. Rev. Nutr. 2013;33:87–103. doi: 10.1146/annurev-nutr-071812-161252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulatowski L., Parker R., Warrier G., Sultana R., Butterfield D.A., Manor D. Vitamin E is essential for Purkinje neuron integrity. Neuroscience. 2014;260:120–129. doi: 10.1016/j.neuroscience.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D., Furlan A., Islam S., Abdo H., Lonnerberg P., Lou D., Hjerling-Leffler J., Haeggstrom J., Kharchenko O., Kharchenko P.V. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- Vaarmann A., Gandhi S., Abramov A.Y. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J. Biol. Chem. 2010;285:25018–25023. doi: 10.1074/jbc.M110.111450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kumar N., Solt L.A., Richardson T.I., Helvering L.M., Crumbley C., Garcia-Ordonez R.D., Stayrook K.R., Zhang X., Novick S. Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands. J. Biol. Chem. 2010;285:5013–5025. doi: 10.1074/jbc.M109.080614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.Z., Pan H.L. High voltage-activated Ca(2+) channel currents in isolectin B(4)-positive and -negative small dorsal root ganglion neurons of rats. Neurosci. Lett. 2004;368:96–101. doi: 10.1016/j.neulet.2004.06.067. [DOI] [PubMed] [Google Scholar]
- Xie L., Dolai S., Kang Y., Liang T., Xie H., Qin T., Yang L., Chen L., Gaisano H.Y. Syntaxin-3 binds and regulates both R- and L-type calcium channels in insulin-secreting INS-1 832/13 cells. PLoS One. 2016;11:e0147862. doi: 10.1371/journal.pone.0147862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Sastry B.R. Induction of hippocampal long-term potentiation by alpha-tocopherol. Brain Res. 1993;604:173–179. doi: 10.1016/0006-8993(93)90365-t. [DOI] [PubMed] [Google Scholar]
- Xie Z., Sastry B.R. Impairment of long-term potentiation in rats fed with vitamin E-deficient diet. Brain Res. 1995;681:193–196. doi: 10.1016/0006-8993(95)00271-q. [DOI] [PubMed] [Google Scholar]
- Yokota T., Igarashi K., Uchihara T., Jishage K., Tomita H., Inaba A., Li Y., Arita M., Suzuki H., Mizusawa H., Arai H. Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. U S A. 2001;98:15185–15190. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T., Shiojiri T., Gotoda T., Arai H. Retinitis pigmentosa and ataxia caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N. Engl. J. Med. 1996;335:1770–1771. doi: 10.1056/NEJM199612053352315. [DOI] [PubMed] [Google Scholar]
- Yokota T., Uchihara T., Kumagai J., Shiojiri T., Pang J.J., Arita M., Arai H., Hayashi M., Kiyosawa M., Okeda R., Mizusawa H. Postmortem study of ataxia with retinitis pigmentosa by mutation of the alpha-tocopherol transfer protein gene. J. Neurol. Neurosurg. Psychiatry. 2000;68:521–525. doi: 10.1136/jnnp.68.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudin Y., Rohacs T. Inhibitory G(i/O)-coupled receptors in somatosensory neurons: potential therapeutic targets for novel analgesics. Mol. Pain. 2018;14 doi: 10.1177/1744806918763646. 1744806918763646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang G.Z., Keeler B., Grant J., Bianchi L., Fu E.S., Zhang Y.P., Erasso D.M., Cui J.G., Wiltshire T., Li Q. Carbonic anhydrase-8 regulates inflammatory pain by inhibiting the ITPR1-cytosolic free calcium pathway. PLoS One. 2015;10:e0118273. doi: 10.1371/journal.pone.0118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg J.M. Vitamin E: a role in signal transduction. Annu. Rev. Nutr. 2015;35:135–173. doi: 10.1146/annurev-nutr-071714-034347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.