

Supplemental digital content is available in the text.
Key Words: pancreatic adenocarcinoma, drug response, characterization, HER2 overexpressed cell lines
Objective
The incidence of pancreatic adenocarcinoma (PA) approximates its prevalence, as the malignancy is almost consistently fatal within a year. Although the currently available adjuvant therapy seems to provide survival benefit, it is only moderate, and the standard regimen has not yet been established. Therefore, more biological resources to investigate the PA are needed.
Methods
Here, we established and characterized 10 human pancreatic cancer cell lines derived from primary tumor mass. Whole exome sequencing technique was used to identify driver mutations and aberrant pathways in each cell line.
Results
Five anticancer drugs were treated to find half maximal effective concentration (EC50), and the response was analyzed in reference to mutational status. Frame shift mutations in ARID1A gene and HER2 amplification were mutually related to better response to the anticancer drugs. In contrast, frame shift mutation in MSH6 gene was associated with resistance to anticancer drugs.
Conclusions
In summary, we established 10 pancreatic cancer cell lines and integrated various molecular aberrations and features of pancreatic cancer cells. Our biological resources are expected to contribute to facilitating research on PA.
Pancreatic cancer is relatively infrequent worldwide, comprising only 2.5% of all forms of cancer.1 Despite its low occurrence, its poor prognosis ranks pancreatic cancer as the fourth or fifth highest in malignancy mortalities.2,3 Surgery is still the mainstay of treatment for pancreatic cancer, but only 15% to 20% of pancreatic cancers are resectable at the time of diagnosis, and the prognosis after R0 resection is poor. Furthermore, although currently available adjuvant therapy seems to provide survival benefit, it is only moderate, and the standard regimen has not yet been established.4–6 Therefore, strategies to confirm therapeutic efficacy or identify additional options would be beneficial to both clinicians and patients. To address this need, we describe the establishment of a living biobank consisting of pancreatic cancer cell lines, which facilitates the integration of genomic data with drug screening of patients' tumor samples on an iterative platform. In previous work, we established and characterized 4 human pancreatic carcinoma cell lines.7 Here, we present 10 human pancreatic cancer cell lines (designated SNU-2466, SNU-2469, SNU-2485, SNU-2491, SNU-2543, SNU-2564, SNU-2570, SNU-2571, SNU-2608, and SNU-2617) and integrated various molecular aberrations and features of pancreatic cancer cells.
MATERIAL AND METHODS
Establishment of Cell Lines and Cell Culture Condition
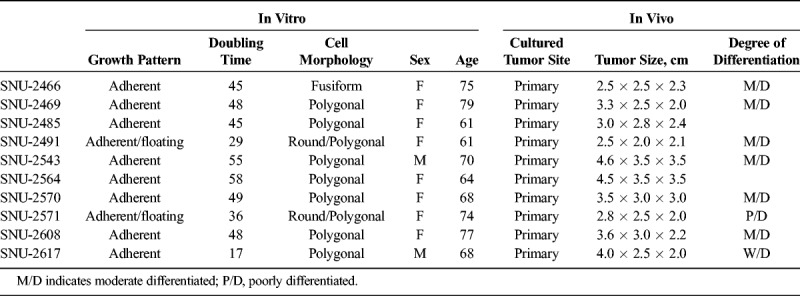
Cell lines were established from pathologically proven pancreatic carcinomas. Detailed procedure was described previously.8 The locations and stages of original tumors of newly established pancreatic cancer cell lines are listed in Table 1.
TABLE 1.
Clinicopathologic Characteristics of 10 Pancreatic Cancer Cell Lines
DNA Finger Printing
The genomic DNA from each cell line was amplified using the AmpFlSTR identifiler PCR amplification kit (Applied Biosystems, Foster City, Calif). Detailed procedure was described previously.8
Cell Growth Properties
Cell growth rate was determined by procedures that were described previously.8 Mycoplasma contamination was tested by the 16S-rRNA-gene–based polymerase chain reaction (PCR) amplification method using the e-Myco Mycoplasma PCR detection kit (Intron Biotechnology, Gyeonggi, South Korea).
Nucleic Acid Isolation and Complementary DNA Synthesis
Genomic DNA was extracted from the cell lines using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and RNA was extracted using the TRIzol (Life Technologies, Carlsbad, Calif) and RNeasy Plus Mini Kit (Qiagen) according to the manufacturer's protocol. For cDNA synthesis, detailed procedure was described previously.8
Quantitative Real-Time PCR
Detailed procedure was described previously.8 The primers that were used in this study are listed in the Supplementary Table 1, http://links.lww.com/MPA/A749.
Colony-Forming Assay
Cells were seeded using 300, 500, and 1000 cells onto 60-mm diameter tissue culture dishes containing 5 mL of culture medium and incubated for 7 days. The dishes were stained with cell stain solution (Cell Biolabs, Inc, San Diego, Calif), and the number of colonies containing more than 10 cells was counted. Gastric cancer cell line, SNU-1, was used as a control as it has proven clonogenic ability.
Protein Isolation and Western Blotting
Entire procedure was described previously.8 Antibodies used in Western blotting are listed in Supplementary Table 2, http://links.lww.com/MPA/A749.
Drug Sensitivity Test
At 70% to 80% of confluency, cells were detached from a T75 flask with 2× trypsin. After centrifugation, the cells were resuspended with a culture medium. According to various growth rates, 2 to 8 × 105 cells/mL were seeded on 96-well tissue culture plates in 80 μL of complete culture medium and incubated in humidified incubators at 37°C for 24 hours in an atmosphere of 5% CO2 and 95% air. Anticancer agents were serially diluted in DPBS and were then added to each well with a volume of 20 μL. After 72 hours of incubation, 10 μL of Ez-Cytox (Dea-Il biotech, Gyeonggi-do, South Korea) was added to each well. After 2 hours of incubation at 37°C, the optical density was measured at 450 nm using Multiskan FC Microplate Photometer (Thermo Fisher Scientific, Waltham, Mass).
Microsatellite Instability and Mutation Analyses of DNA Mismatch Repair Gene
For microsatellite instability analysis, BAT-25 and BAT-26 were evaluated using a capillary-based sequencing analysis. Polymerase chain reaction was performed as described previously, with the exception that the forward primers were labeled with a fluorescent dye, and the labeled samples were run on an ABI 3730 genetic analyzer (Applied Biosystems). GeneMapper software v4.0 (Applied Biosystems) was used to calculate the size of each fluorescent PCR product. For gel-based microsatellite instability analysis, the desired fragments were amplified in the presence of [a-P32] deoxycytidine triphosphate. The PCR products were denatured and separated on 6 M urea/7% polyacrylamide gels run at 60 W.
Statistical Analysis
Statistical analysis was performed using R program Version 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria) with various packages including maftools, PerformanceAnalytics, survminer, survival, iplot, gplot, and lattice. The relationship between gene mutation or expression and clinical data of pancreatic adenocarcinoma (PA) was analyzed by the χ2 test and Pearson correlation method. Fisher exact test was used to analyze Gene Ontology analysis of various genes. All tests were performed using bilateral 95% confidence intervals. A value of P < 0.05 was considered statistically significant.
RESULTS
General Characteristics of the Cell Lines
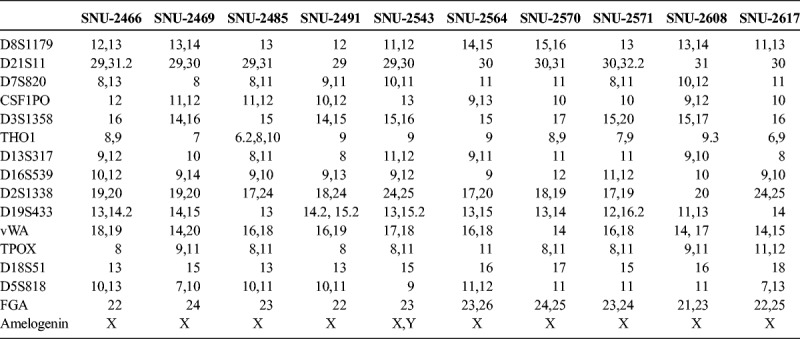
On in vitro cultivation, 8 cell lines (SNU-2466, SNU-2469, SNU-2485, SNU-2543, SNU-2564, SNU-2570, SNU-2608, and SNU-2617) grew as monolayer of substrate-adherent cells, and 2 cell lines (SNU-2491 and SNU-2571) formed floating and adherent aggregates. Most tumor cells displayed a polygonal shape and had round-to-oval nuclei with prominent single-to-double nucleoli (Fig. 1). Each cell line was passaged at least 3 times before characteristic analysis. Population doubling times ranged from 47 to 135 hours. Clinicopathologic information is listed in Table 1. Patients' history of preoperative/postoperative adjuvant therapy and overall survival are listed in Table 2. All cell lines were confirmed to be free of bacterial and mycoplasma contamination (Supplementary Fig. 1, http://links.lww.com/MPA/A749). Fifteen tetranucleotide repeat loci and Amelogen sex-determining markers were heterogeneously distributed in each cell line and were not cross-contaminated (Table 3).
FIGURE 1.
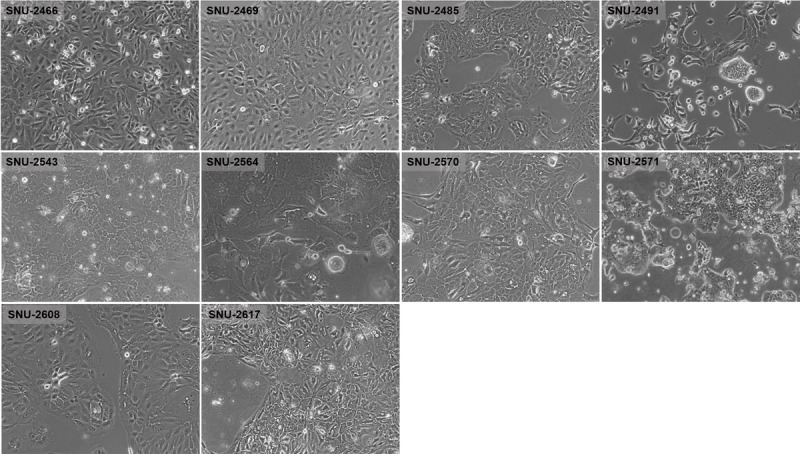
Phase-contrast microscopy of PA cell lines. On in vitro cultivation, 8 cell lines (SNU-2466, SNU-2469, SNU-2485, SNU-2543, SNU-2564, SNU-2570, SNU-2608, and SNU-2617) grew as monolayer of substrate-adherent cells, and 2 cell lines (SNU-2491 and SNU-2571) formed floating and adherent aggregates. Most tumor cells displayed a polygonal shape and had round-to-oval nuclei with prominent single-to-double nucleoli.
TABLE 2.
Patients' History of Preoperative/Postoperative Adjuvant Therapy
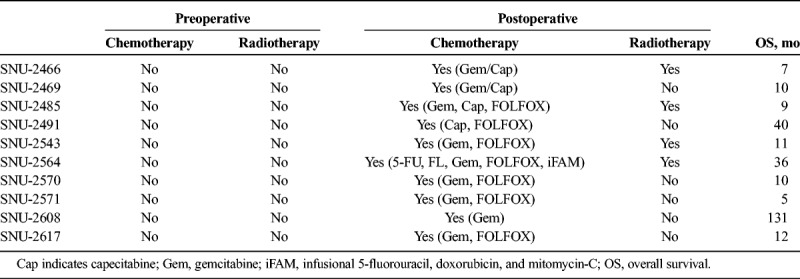
TABLE 3.
Short Tandem Repeat Profile of 10 Pancreatic Cancer Cell Lines
Whole Exome Sequencing Analysis
To establish the mutational context of the established pancreatic cancer cell lines, whole exome sequencing (WES) was performed. To further analyze WES data, 434 genes that have been involved in PA were selected (Supplementary Table 3, http://links.lww.com/MPA/A749), and mutations that occurred in the sorted genes were screened. The general information, such as variant classification and single nucleotide variants class, are summarized in Figure 2A. SNU-2491 had the largest number of variants, whereas SNU-2571 had the smallest number of variants. The median number of variants per sample was 176.5. Mutations were further analyzed for gene set enrichment analysis to find representative pathways that were aberrated in the established PA cell lines. Genes consisting of MAPK family signaling cascade and interleukin-20 family signaling were mostly mutated (Fig. 2B). The prevalence of aberrations in key driver genes is categorized into 5 groups as indicated in Figure 2C. The mutational statuses and proposed functions of such genes are summarized in Supplementary Table 4, http://links.lww.com/MPA/A749. Many such driver genes in cancer are co-occurring, or show exclusiveness in their mutation patterns, and can be detected using somatic interactions function in Maftools, which performs pair-wise Fisher exact test to detect such significant pair of genes. For instance, mutations in PIK3R1 and CDH1 genes are co-occurring, whereas mutations in HIF-1A and BIRC5 genes are exclusive (Fig. 2D). Mutational signatures characterized by a specific pattern of nucleotide substitutions were extracted by decomposing a matrix of nucleotide substitutions and were then compared with the public database presented by Alexandrov et al.9 Newly established pancreatic cancer cell lines showed a pattern of signature 5 (Fig. 2E). Drug-gene interactions and gene druggability information can be extracted from drug-gene interaction database using drug interactions function in Maftools. The result showed that kinase and DNA repair pathways were potential druggable gene categories (Fig. 2F).
FIGURE 2.
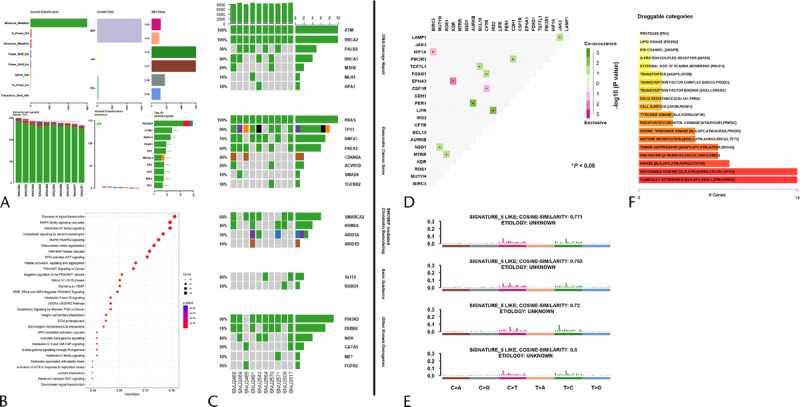
Mutational context of the established pancreatic cancer cell lines. A, Summarization of variants. B, Gene set enrichment analysis to find representative pathways that were aberrated in the established PA cell lines. C, The prevalence of aberrations in key driver genes with 5 categories. D, Co-occurring or exclusiveness in the mutation patterns of pancreatic cancer cell lines. E, Mutational signatures characterized by a specific pattern of nucleotide substitutions. F, Targetable genes were identified in reference with the number of genes that are involved in drug target pathways.
Anticancer Drug Response With Mutational Contexts
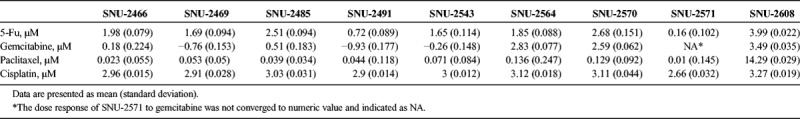
Five anticancer drugs (5-Fu, cisplatin, paclitaxel, gemcitabine, and cetuximab) were used to estimate the drug sensitivities of the established pancreatic cancer cell lines. SNU-2617 was excluded because of its tardy growth rate. The SNU-2571 and SNU-2491 cell lines were relatively sensitive to 5-Fu. Of the remaining 8 cell lines, SNU-2608 was the most resistant to 5-Fu, where its half maximal inhibitory concentration (IC50) was 3.993 μM. The remaining cell lines had IC50 of 5-Fu ranging from 1.653 to 2.678 μM (Fig. 3A). IC50 for cisplatin was relatively consistent in the established pancreatic cancer cell lines where its range was from 2.664 to 3.265 μM. Similar to 5-Fu, the most sensitive and resistant cell lines for cisplatin were SNU-2571 and SNU-2608, respectively (Fig. 3B). IC50 for paclitaxel was also relatively constant, except for SNU-2608. Similar to 5-Fu and cisplatin, the most sensitive and resistant cell lines for paclitaxel were SNU-2571 and SNU-2608, respectively (Fig. 3C). Cells were clustered in 2 groups for sensitivities of gemcitabine. SNU-2570, SNU-2564, and SNU-2608 were comparatively resistant, whereas SNU-2466, SNU-2469, SNU-2485, SNU-2491, and SNU-2543 were relatively sensitive (Fig. 3D). The sigmoidal dose response of SNU-2571 to gemcitabine was not obtained because its EC50 was not converged to numeric value. The results are summarized in Table 4. IC50 value for cetuximab was not able to be computed because all cell lines except for SNU-2491 did not respond to the maximum concentration of cetuximab. Therefore, IC50 unit was converted to area under curve to compare different molar concentration of 5 anticancer drugs. Mutational status of 20 genes that have been reported to be involved in PA were screened with regard to the calculated response of 5 anticancer drugs. Interestingly, SNU-2491 and SNU-2571 cell lines had frameshift mutation in ARID1A gene and were sensitive to all anticancer drugs tested in this study. In contrast, SNU-2608 cell line had frameshift mutation in MSH6 gene and was the most resistant cell line for all anticancer drugs tested in this study (Fig. 3E).
FIGURE 3.
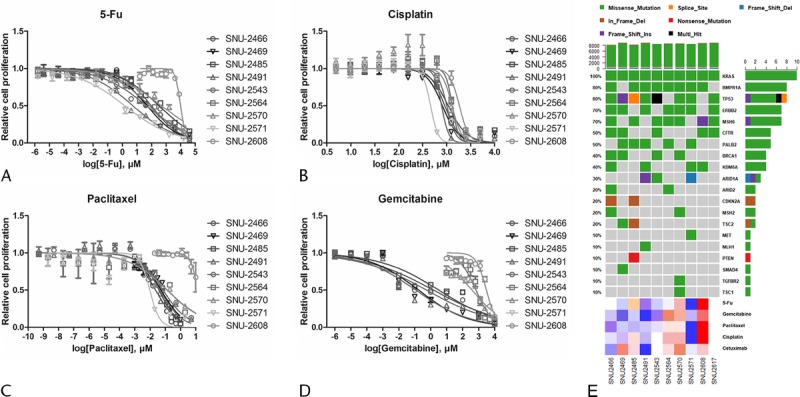
Anticancer drug response with mutational contexts. Four anticancer drugs (5-Fu [A], cisplatin [B], paclitaxel [C], and gemcitabine [D]) were used to estimate drug response of the established pancreatic cancer cell lines. SNU-2564, SNU-2570, and SNU-2608 cell lines were the most resistant group for the anticancer agents. SNU-2608 was the most resistant cell line for all anticancer drugs tested in this study. E, Mutational status of 20 genes that have been reported to be involved in PA was screened with regard to the calculated response of 5 anticancer drugs.
TABLE 4.
IC50 Values of 10 Established Pancreatic Cancer Cell Lines for 4 Anticancer Drugs
Anticancer Drug Response With Expression of Molecular Markers
The mRNA expressions of various genes were verified by reverse transcription PCR (Figs. 4A, B). The functional roles of the examined genes are categorized into the following 4 groups: angiogenesis, cell cycle regulation, epithelial to mesenchymal transition (EMT), and stemness. MIA PaCa-2 and PANC-1 cell lines were obtained from the Korean Cell Line Bank to compare gene expression in widely used pancreatic cancer cell lines. The mRNA levels of vascular endothelial growth factor A (VEGF-A), VEGF-C, and hypoxia-inducible factor 1α (HIF-1α) were screened to examine the angiogenesis-related ability of each cell line. Vascular endothelial growth factor A was weakly or not expressed in all pancreatic cancer cell lines. The expression patterns of VEGF-C and HIF-1α were analogous in all cell lines with the exception of the SNU-2608 and SNU-2617 cell lines. The SNU-2564, SNU-2570, and SNU-2571 cell lines did not express all angiogenesis-related genes tested. The mRNA levels of cyclin A, cyclin B1, and cyclin D were screened to examine the cell cycle–related genes. Cyclin A was strongly expressed in MIA PaCa-2 and PANC-1, whereas newly established pancreatic cell lines had weak expression. Cyclin B1 was not expressed in the SNU-2543 and SNU-2564 cell lines. All cell line expressed Cyclin D with the exception of SNU-2608. Among the EMT markers, N-cadherin was expressed in all cell lines with the exception of SNU-2570. The mRNA levels of E-cadherin and N-cadherin did not have inverse correlation. Various stemness-related genes were also examined. Markedly, SOX9 was expressed in all established pancreatic cancer cell lines, whereas SOX4 was not expressed in all established pancreatic cancer cell lines. The expression of EMT-related proteins was examined by Western blotting (Fig. 4C). Vimentin was exclusively expressed in SNU-2466. The protein expressions of epidermal growth factor receptor (EGFR), HER2, ERK1, ERK2, pERK1/2, PTEN, and SMAD4 were also screened. Among the 10 pancreatic cancer cell lines, HER2 was exceptionally expressed in SNU-2491. The patterns of mRNA expression were analyzed in comparison with drug response, and the mRNA expressions of VEGF-C and HIF-1A were inversely related to overall drug response (Fig. 4D).
FIGURE 4.
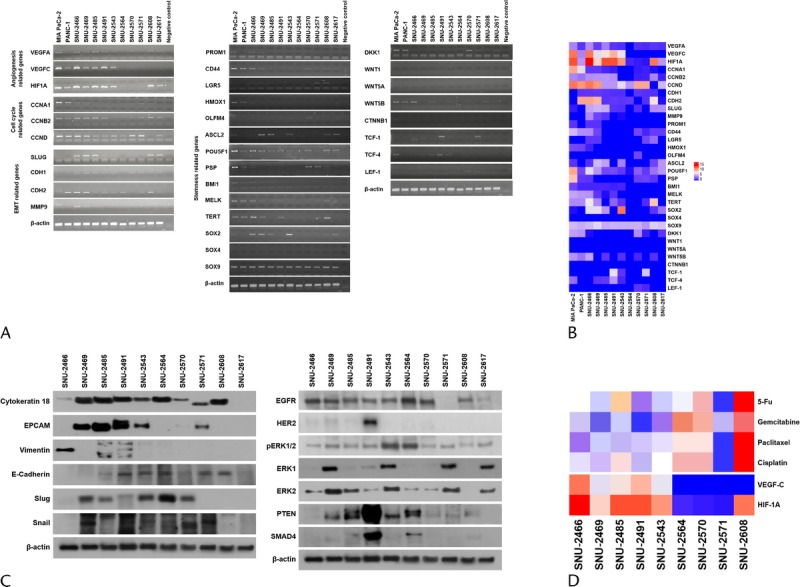
Gene expression patterns of PA cell lines. A, The mRNA levels of various fundamental genes that account for angiogenesis, cell cycle regulation, EMT, and stemness were examined. B, The mRNA level is converted to numeric value to be expressed in heatmap. C, The expression of EMT-related proteins and key proteins to develop pancreatic cancer were examined by Western blotting. D, mRNA expressions of VEGF-C and HIF-1A were inversely related to overall drug response.
erbB2 Amplification Influences Colony-Forming Ability and Response to Cetuximab in Pancreatic Cancer Cells
Since the exclusive erbB2 amplification (Figs. 4C, 5B), positive response to cetuximab (Figs. 3E, 5C) and negative response to trastuzumab (Fig. 5D) of SNU-2491 cell line were intriguing, we further screened whether there is phenotypical difference between SNU-2491 and other cell lines. Each cell line was tested for anchorage-dependent colony-forming assay. The SNU-1 cell line was obtained from Korean Cell Line Bank as the positive control for the colony formation. Among the 10 pancreatic cancer cell lines, only SNU-2491 formed an average of 17 colonies 28 days after seeding (Fig. 5A). Anchorage-dependent colony-forming assay was performed again with maximum concentrations of trastuzumab (100 μg/mL) and cetuximab (1000 μg/mL) to consolidate the positive response of SNU-2491 to cetuximab instead of trastuzumab. SNU-2491-containing PBS yielded an average of 41 colonies 45 days after seeding. When treated with trastuzumab, there were an average of 35 colonies. When treated with cetuximab, there were an average of 13 colonies. Dual treatment of cetuximab and trastuzumab yielded only 6 colonies (Fig. 5E).
FIGURE 5.
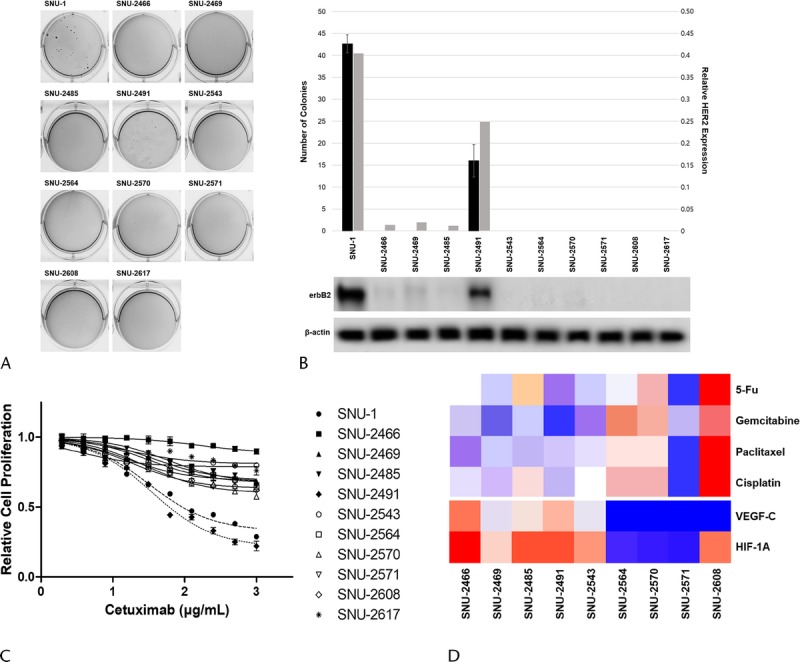
erbB2 amplification influences colony-forming ability and response to cetuximab in pancreatic cancer cells. A, Among 10 pancreatic cancer cell lines, only SNU-2491 formed average 17 colonies 28 days after seeding. B, SNU-2491 was the only cell line that had evidently upregulated HER2 protein level compared with other established cell lines. C, Each cell lines were tested for the response of cetuximab. SNU-2491 was the only cell line that showed significantly inhibited growth rate in accordance with cetuximab treatment. D, SNU-2491 did not respond to trastuzumab despite of amplified HER2. E, The efficiency of cetuximab compared with trastuzumab in SNU-2491 was re-encapsulated with anchorage-dependent colony-forming assay.
DISCUSSION
The incidence of PA approximates its prevalence, as the malignancy is almost consistently fatal, frequently within a year.10 This high mortality rate is mostly due to its aggressiveness, resistance to chemotherapy, and difficulty in early detection. The disease often progresses to an advanced stage at the time of diagnosis, making surgical resection impractical.11 Moreover, the molecular basis of the aggressiveness remains elusive because of its biological complexity, and more detailed investigation of PA is indispensable to improve strategies for prevention and treatment.12 A major predicament in researching PA biology arises from the peculiar morphologic characters of the tumor. In vivo, PA stimulates desmoplastic reactions that widen the spaces among tumor cells with reactive nontumor elements, such as inflammatory cells and fibroblasts, and consequently reduce the number of viable tumor cells available per single macroscopic field.13,14 With this biological nature, there are only 53 pancreatic cancer cell lines available in the research field. Besides, those available cell lines are somewhat outdated. For example, the SW-1990 line, which has been widely used in PA study, was established in 1978 from a spleen metastasis of a grade II PA and passaged numerous times. Accumulation of genetic aberrations of cancer cell lines that occurs with increasing passage numbers has limited their clinical correlation. Genetically altered cancer cell lines under in vitro conditions do not truly represent clinical scenarios. There is a wide range of variability in patient response toward the same drugs used on tumors that are identical in their genetic aberration. The derivation and short-term culture of primary cells from solid tumors have thus gained significant importance in personalized cancer therapy. Therefore, newly established pancreatic cancer cell lines may provide adequate models for studying a wider spectrum of biological and molecular characters of PA.15 Here, we present 10 new human pancreatic cancer cell lines (designated SNU-2466, SNU-2469, SNU-2485, SNU-2491, SNU-2543, SNU-2564, SNU-2570, SNU-2571, SNU-2608, and SNU-2617) derived from a primary tumor mass. All cell lines introduced in this study will be deposited to Korean Cell Line Bank (http://cellbank.snu.ac.kr) at initial passages to be distributed to researchers worldwide.
Various molecular aberrations have been documented in PA involving several oncogenes and oncosuppressor genes, such as KRAS, TP53, CDKN2A, SMAD4, and ARID1A.10,16 Classical progression of pancreatic carcinogenesis has been classified into early (telomere shortening and activating mutations in KRAS), intermediate (inactivating mutations or epigenetic silencing of CDKN2A2), and late (inactivating mutations of TP53 and SMAD4) events.17 Activating mutations of KRAS is frequently reported in PA. Most (>98%) KRAS mutations in PA are point mutations at codon G12, whereas codon G13 and Q61 comprise lower portions.18,19 All of our PA cell lines harbored point mutations at codon G12. Forty-nine percent of pancreatic cancers in the COSMIC (Catalogue Of Somatic Mutations In Cancer) database harbor TP53 mutations.20 Except for SNU-2608, our PA cell lines had TP53 mutation either in the splice site or in the exonic region. All missense mutations and one frameshift insertion in the TP53 gene were previously reported in COSMIC. CDKN2A is known to be a tumor suppressor gene,21 and germline mutation of this gene is associated with increased occurrence of PA.22 SNU-2466 had a p.Thr18_Ala20del in-frame deletion, and SNU-2485 harbored p.Pro70_Asn71del disruptive in-frame deletion on the CDKN2A gene, and both mutations were homozygous. Average survival time of 10 enrolled pancreatic cancer patients was 27 months. Follow-up record of SNU-2466 and SNU-2485 patients was lost after 7 months and 9 months, respectively, because of the markedly aggressive relapse. This suggested that the in-frame deletion of CDKN2A may affect the recurrence rate after the surgery.
Among the cell lines, only SNU-2491 formed a colony during the anchorage-dependent colony-forming assay. Colony formation may imply stronger tumorigenic behavior, which can lead to further speculation of poorer prognosis.23 The patient developed liver metastasis only 43 days after surgery, which may reflect the aggressive behavior of SNU-2491. Chemotherapy (Gemzar and Xeloda) was performed, but the disease continuously progressed and folinic acid, fluorouracil, and oxaliplatin (FOLFOX) was additionally administered with little success. The follow-up record of the patient after 6 months was lost. Although the prognosis of this patient seems unfavorable, additional cases need to be investigated to draw a conclusion on the clinical implication of colony formation assay.
It was observed that HER2 was exclusively amplified in SNU-2491. HER2 amplification is known to occur in 2% of PA and has distinct features, such as a preponderance of lung and brain metastases.24 Although the connection between augmented HER2 expression and colony-forming efficacy has not been documented in pancreatic cancer cells, Alajati et al25 reported that an amplified HER2 increased the colony-forming capacity of breast cancer cells. Our data suggested that HER2 amplification in PA may cause more aggressive behavior.
The exclusive HER2 amplification and colony-forming ability rendered the SNU-2491 as the suitable model for the trastuzumab (a monoclonal antibody that targets the HER2 receptor) treatment. Against expectations, inhibition of HER2 with trastuzumab did not impede proliferation of SNU-2491 as in other PA cell lines (Fig. 5D). Remarkably, cetuximab, a monoclonal antibody that targets anti-EGFR, seemed exclusively effective on the SNU-2491 compared with other cell lines (Fig. 5B), although the protein level of EGFR was analogous among the PA cell lines (Fig. 4C). The positive response of cetuximab was confirmed again with anchorage-dependent colony-forming assay (Fig. 5E). The poor response of pancreatic cancer cells to trastuzumab had been reported in multiple studies.26–28 Besides, the use of cetuximab in treating PA has been previously reported.29 Our data reinforced that the use of cetuximab in HER2-overexperssed pancreatic cancer has better outcomes than the use of trastuzumab. Nevertheless, the clinical importance and the molecular inhibitory mechanism of cetuximab in HER2-amplified pancreatic cancer still need to be elucidated.
Five generally used anticancer drugs (5-Fu, cisplatin, paclitaxel, gemcitabine, and cetuximab) in pancreatic cancer30 were used for estimating the drug sensitivities of 10 newly established pancreatic cancer cell lines. The SNU-2564, SNU-2570, and SNU-2608 cell lines were the most resistant group for the anticancer agents. Mutations that were exclusively present in 3 drug-resistant cell lines were screened (Supplementary Table 5, http://links.lww.com/MPA/A749). Among them, the MYL1 gene had frameshift mutation at the very first codon in all 3 drug-resistant cell lines. Further investigations regarding exclusive mutations are suggested. SNU-2608 was the most resistant cell line for all anticancer drugs tested in this study. Interestingly, SNU-2608 had relatively low numbers of driver mutations compared with other established cell lines (Figs. 2C, 3E), and the survival time of SNU-2608 patient after surgery was 131 months, which was longest among the 10 enrolled patients in this study.
Impaired drug delivery pathways have been shown to be another reason for the drug resistance in pancreatic cancer.31 It is believed that solid tumor masses appear within pancreatic tumors that lack adequate vasculature with intensive desmoplastic reaction.25 The patterns of mRNA expression were analyzed in comparison with drug response, and the mRNA expressions of VEGF-C and HIF-1A were inversely related to overall drug response (Fig. 4D). It has been reported that the expression of HIF-1A and VEGF-C was directly related to lymphatic vessel density,32 and level of lymphatic vessel density influences direct targeting of tumor vasculature in the clinic with Food and Drug Administration-approved drugs, such as bevacizumab, sorafenib, and sunitinib that target kinase activity of VEGF family receptors.33 Our data may suggest that mRNA expressions of VEGF-C and HIF-1A affect other anticancer drugs, such as 5-Fu, cisplatin, paclitaxel, and gemcitabine. Nevertheless, drawing a correlation between the HIF-1A and VEGF-C mRNA expression and drug resistance on a 2-dimensional model was inadequate. The approach using the 3-dimensional culture model should be accessed to further investigate the stroma marker expression of the tumor and its effect on drug delivery.
Supplementary Material
Footnotes
This study was supported by the Korean Cell Line Research Foundation, the Priority Research Centers Program (2009-0093820), and the Basic Science Research Program (2012R1A1A3010709) through the National Research Foundation of Korea funded by the MSIP. This was also supported by the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation funded by the Ministry of Science and ICT (NRF-2017M3C9A5031591). S.-C.K. received a scholarship from the BK21-plus education program provided by the National Research Foundation of Korea. For the remaining authors, none were declared.
The authors declare no conflict of interest.
Supplemental digital contents are available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.pancreasjournal.com).
REFERENCES
- 1.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. [DOI] [PubMed] [Google Scholar]
- 2.Olson SH, Kurtz RC. Epidemiology of pancreatic cancer and the role of family history. J Surg Oncol. 2013;107:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an update. Dig Dis. 2010;28:645–656. [DOI] [PubMed] [Google Scholar]
- 4.Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–277. [DOI] [PubMed] [Google Scholar]
- 5.Stocken DD, Büchler MW, Dervenis C, et al. Meta-analysis of randomised adjuvant therapy trials for pancreatic cancer. Br J Cancer. 2005;92:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neoptolemos JP, Dunn JA, Stocken DD, et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet. 2001;358:1576–1585. [DOI] [PubMed] [Google Scholar]
- 7.Ku JL, Yoon KA, Kim WH, et al. Establishment and characterization of four human pancreatic carcinoma cell lines. Genetic alterations in the TGFBR2 gene but not in the MADH4 gene. Cell Tissue Res. 2002;308:205–214. [DOI] [PubMed] [Google Scholar]
- 8.Kim SC, Hong CW, Jang SG, et al. Establishment and characterization of paired primary and peritoneal seeding human colorectal cancer cell lines: identification of genes that mediate metastatic potential. Transl Oncol. 2018;11:1232–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heestand GM, Kurzrock R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget. 2015;6:4553–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chifenti B, Morelli M, Zavaglia M, et al. Establishment and characterization of 4 new human pancreatic cancer cell lines: evidences of different tumor phenotypes. Pancreas. 2009;38:184–196. [DOI] [PubMed] [Google Scholar]
- 12.Dunne RF, Hezel AF. Genetics and biology of pancreatic ductal adenocarcinoma. Hematol Oncol Clin North Am. 2015;29:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pothula SP, Xu Z, Goldstein D, et al. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016;381:194–200. [DOI] [PubMed] [Google Scholar]
- 14.Iacobuzio-Donahue CA, Ryu B, Hruban RH, et al. Exploring the host desmoplastic response to pancreatic carcinoma: gene expression of stromal and neoplastic cells at the site of primary invasion. Am J Pathol. 2002;160:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitra A, Mishra L, Li S. Technologies for deriving primary tumor cells for use in personalized cancer therapy. Trends Biotechnol. 2013;31:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malesci A, Laghi L, Bianchi P, et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res. 2007;13:3831–3839. [DOI] [PubMed] [Google Scholar]
- 17.Nakata B, Wang YQ, Yashiro M, et al. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin Cancer Res. 2002;8:2536–2540. [PubMed] [Google Scholar]
- 18.Laghi L, Beghelli S, Spinelli A, et al. Irrelevance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS One. 2012;7:e46002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goggins M, Offerhaus GJ, Hilgers W, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998;152:1501–1507. [PMC free article] [PubMed] [Google Scholar]
- 20.Ghimenti C, Tannergård P, Wahlberg S, et al. Microsatellite instability and mismatch repair gene inactivation in sporadic pancreatic and colon tumours. Br J Cancer. 1999;80:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomaszewska R, Okoń K, Stachura J. Expression of the DNA mismatch repair proteins (hMLH1 and hMSH2) in infiltrating pancreatic cancer and its relation to some phenotypic features. Pol J Pathol. 2003;54:31–37. [PubMed] [Google Scholar]
- 22.Yamamoto H, Itoh F, Nakamura H, et al. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001;61:3139–3144. [PubMed] [Google Scholar]
- 23.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J. 2012;18:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alajati A, Guccini I, Pinton S, et al. Interaction of CDCP1 with HER2 enhances HER2-driven tumorigenesis and promotes trastuzumab resistance in breast cancer. Cell Rep. 2015;11:564–576. [DOI] [PubMed] [Google Scholar]
- 26.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 27.Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takai E, Yachida S. Genomic alterations in pancreatic cancer and their relevance to therapy. World J Gastrointest Oncol. 2015;7:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McWilliams RR, Wieben ED, Rabe KG, et al. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. Eur J Hum Genet. 2011;19:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franken NA, Rodermond HM, Stap J, et al. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. [DOI] [PubMed] [Google Scholar]
- 31.Chou A, Waddell N, Cowley MJ, et al. Clinical and molecular characterization of HER2 amplified-pancreatic cancer. Genome Med. 2013;5:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larbouret C, Gaborit N, Chardès T, et al. In pancreatic carcinoma, dual EGFR/HER2 targeting with cetuximab/trastuzumab is more effective than treatment with trastuzumab/erlotinib or lapatinib alone: implication of receptors' down-regulation and dimers' disruption. Neoplasia. 2012;14:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larbouret C, Robert B, Bascoul-Mollevi C, et al. Combined cetuximab and trastuzumab are superior to gemcitabine in the treatment of human pancreatic carcinoma xenografts. Ann Oncol. 2010;21:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.