

The ability of human neutrophils to clear newly attached Staphylococcus aureus bacteria from a serum-coated glass surface was examined in vitro using time-lapse confocal scanning laser microscopy. Quantitative image analysis was used to measure the temporal change in bacterial biomass, neutrophil motility, and fraction of the surface area policed by neutrophils. In control experiments in which the surface was inoculated with bacteria but no neutrophils were added, prolific bacterial growth was observed.
KEYWORDS: biofilm, neutrophil, device-related infection, immune evasion, biomaterial, joint infections
ABSTRACT
The ability of human neutrophils to clear newly attached Staphylococcus aureus bacteria from a serum-coated glass surface was examined in vitro using time-lapse confocal scanning laser microscopy. Quantitative image analysis was used to measure the temporal change in bacterial biomass, neutrophil motility, and fraction of the surface area policed by neutrophils. In control experiments in which the surface was inoculated with bacteria but no neutrophils were added, prolific bacterial growth was observed. Neutrophils were able to control bacterial growth but only consistently when the neutrophil/bacterium number ratio exceeded approximately 1. When preattached bacteria were given a head start and allowed to grow for 3 h prior to neutrophil addition, neutrophils were unable to maintain control of the nascent biofilm. In these head-start experiments, aggregates of bacterial biofilm with areas of 50 μm2 or larger formed, and the growth of such aggregates continued even when multiple neutrophils attacked a cluster. These results suggest a model for the initiation of a biofilm infection in which a delay in neutrophil recruitment to an abiotic surface allows surface-attached bacteria time to grow and form aggregates that become protected from neutrophil clearance. Results from a computational model of the neutrophil-biofilm surface contest supported this conceptual model and highlighted the stochastic nature of the interaction. Additionally, we observed that both neutrophil motility and clearance of bacteria were impaired when oxygen tension was reduced to 0% or 2% O2.
INTRODUCTION
Implanted medical devices are vulnerable to localized infections (1, 2). These infections result when bacteria or yeast attach to the biomaterial surface and form a biofilm. In the biofilm state, microorganisms are protected from killing by disinfectants and antibiotics and they evade host defenses (3, 4). Consequently, device-related infections are recalcitrant to antimicrobial chemotherapy and are extremely difficult to resolve. Medical devices that are prone to foreign body infection include prosthetic joints, hernia meshes, orthopedic plates and screws, breast implants, artificial heart valves, catheters, and many others (1). As one specific example, consider knee and hip prostheses. In 2013, 27,000 revisions were performed for prosthetic joint infections, which are traumatic and expensive procedures (5). Because of the large number of patients who receive implanted materials in modern medicine, the collective burden to society, in terms of both economic cost and human suffering, is significant.
The innate immune system rapidly and effectively eliminates microorganisms from regions of the body where they are not normally resident; however, this process breaks down in the presence of a foreign body (6, 7). The reasons for this failure are not well understood, but this observation alone motivates exploration of approaches for restoring innate immune function in the presence of an implanted material. One mechanism of neutrophil compromise that was previously investigated is chronic activation of neutrophils that associate with a biomaterial (8–12). As the first responders to infection, neutrophils are of particular relevance and interest (13, 14).
We consider here six phenomena that may be important for successful clearance of a biomaterial adherent bacterial cell by a neutrophil. Poor clearance could result from defects at any one of these steps or from a combination of deficiencies at multiple steps. We emphasize that our focus is on the very early events after implantation, prior to the formation of mature biofilm structures that are known to be tolerant to antimicrobials and innate immunity (1–4). Here are six hypotheses for how a relatively small number of contaminating bacteria on an implanted medical device could evade clearance by neutrophils. (i) Recruitment of neutrophils to the implant surface is delayed, allowing attached bacteria a window of time in which to grow and form aggregates that are protected. (ii) Neutrophils are recruited to the biomaterial surface but do not move on or surveil the surface. (iii) Neutrophils are recruited to the implant surface and patrol it but fail to recognize attached bacteria even when encountered. (iv) Neutrophils are recruited to the surface and exhibit surveillance and discovery of attached bacteria but are unable to phagocytose adherent bacterial cells, even lone cells. (v) Neutrophils are recruited to the surface and exhibit motility, discovery, and phagocytosis of bacteria but fail to kill phagocytosed cells. (vi) Hypoxia near the biomaterial interface limits neutrophil motility and bactericidal efficacy.
Discriminating between these hypotheses likely requires direct microscopic observation of neutrophil-bacterium interactions at the single-cell level. To do this in vivo would require technically challenging, high-resolution intravital imaging. Such animal models for imaging individual neutrophil function on an abiotic surface have not yet been developed. We therefore turned to an in vitro system to perform direct microscopic video observation of human neutrophils interacting with surface-attached bacteria. This model gives access to hypotheses 2 to 6 described above but cannot measure the recruitment time of neutrophils to the implant surface (hypothesis 1). However, the in vitro model can be used to investigate the consequences of delayed recruitment by simply adding neutrophils at various time points after the bacterial attachment step.
In the research presented in this article, we have chosen to use Staphylococcus aureus as a model organism, as it is commonly implicated in prosthetic joint infections and has been the subject of extensive work on interaction with the innate immune system (14–17). Moreover, neutrophils are a key cell in eradicating S. aureus, as observed by the increase in susceptibility to S. aureus infections in individuals with neutrophil defects (18, 19). Much of the prior work investigating interaction of S. aureus biofilms with cells of the mammalian innate immune system has focused on mature biofilms (20–32). These studies collectively paint a picture of localized immune dysregulation, stasis, and sustained inflammation. They often implicate bacterial virulence factors and complex reciprocal signaling interactions between a bacterium and phagocyte. As we were particularly interested in the potential to prevent biofilm infections, we chose instead to focus on understanding the very early events, those that might occur in the first several hours following implantation of a contaminated biomaterial.
The goal of the work presented in this article was to characterize phenomena important in the interaction between human neutrophils and sparsely distributed bacteria adherent to an abiotic surface, simulating the situation on a medical device immediately following implantation. We used an in vitro system that allowed for independent experimental control of the densities of bacteria and neutrophils and also of the timing of neutrophil delivery. This microscopy-based system enabled direct observation of the dynamics of the two types of cells and extensive quantification of their behaviors by image analysis and suggested rate-limiting steps in the clearance of attached bacteria by these front-line leukocytes.
RESULTS
Neutrophils migrate on an abiotic surface and discover, phagocytose, and destroy single attached bacteria or very small bacterial aggregates.
We imaged the interaction of newly attached green fluorescent protein (GFP)-expressing S. aureus with human neutrophils on a surrogate biomaterial surface in vitro using time-lapse confocal scanning laser microscopy. Neutrophils were stained red, enabling continuous quantitative monitoring of neutrophil motility and interactions with bacteria. S. aureus was attached to the glass surface as single cells or aggregates of a few cells (Fig. 1A, C, and E). Between 5 × 104 and 2 × 105 CFU were initially seeded onto the surface, resulting in attached bacterial cell densities after rinsing to remove nonadherent bacteria ranging from approximately 5 × 102 to 8 × 104 objects cm−2 (1.55 × 104 ± 1.83 × 104 objects cm−2). Following attachment, 20% human serum in Hanks’ balanced salt solution (HBSS) was added to opsonize bacteria and condition the surface. After 30 min, HBSS was added to dilute the serum to 10%. In control experiments in which the surface was inoculated with bacteria but no neutrophils were added, the attached bacteria grew exponentially (Fig. 1A and B; see also Video S1 in the supplemental material). The measured maximum growth rate was 0.78 ± 0.15 h−1 (n = 17). Most of the bacteria remained sessile during the 4-h observation period. The green fluorescence from the plasmid-encoded GFP reliably correlated with bacterial growth, as we did not observe any outgrowth of non-GFP cells, even after 8 h in this system (see Videos S2 and S3). Additionally, after 8 h of growth, cells were scraped from the surface and plated on tryptic soy agar. All colonies remained fluorescent under UV light the following day, indicating plasmid retention.
FIG 1.

Neutrophil discovery and elimination of S. aureus attached to a serum-coated glass surface. Representative images from time 0 h (top row) and time 4 h (bottom row) from experiments with no neutrophils (A and B), low density of neutrophils (C and D), high density of neutrophils (E and F), and high density of neutrophils with preformed bacterial aggregate wherein bacteria were allowed to grow for 3 h prior to neutrophil addition (G and H) (see also Videos S1 and S4 to S6 in the supplemental material).
When human neutrophils were added to the system, they moved in random walk fashion over the surface (Fig. 1C to F and Videos S4 and S5). As neutrophils encountered bacterial cells, they appeared to quickly phagocytose the bacteria. The physical association of bacteria with the neutrophil was evident in that the previously stationary bacteria began to move with the leukocyte, which usually continued to migrate. When the neutrophil surface density was relatively low, not all the bacterial cells seeded on the surface were discovered by a neutrophil in the 4-h observation window, and undiscovered bacteria continued to grow and formed small aggregates. When more neutrophils were present, they discovered most or all the bacteria on the surface. After phagocytosis of a bacterial aggregate, the GFP signal decayed, suggesting loss of viability of the bacterial cell (33).
To confirm observations in Fig. 1 and Videos S4 and S5, image analysis was used to quantify the total area of green fluorescence, as a measure of viable bacterial biomass, in each field of view over time (Fig. 2). An increase in green fluorescence implied the net growth of bacteria, whereas a decrease in green fluorescence with time implied that neutrophil clearance of S. aureus was more rapid than microbial growth (Fig. 2A). To capture the overall outcome from a particular field of view, we calculated the log10 difference in GFP area between the time that neutrophils were added and the conclusion of the observation period 4 h later. To further probe the dynamics between the S. aureus-neutrophil interactions, we varied both the concentration of neutrophils added to the wells and the amount of bacteria initially attached to the surface (Fig. 2B). We observed increases in bacterial biomass of up to a log10 difference of 1.26 (in a control experiment with no neutrophils added) and reductions up to −2.35 (an experiment with neutrophils in which the GFP area was reduced to its limit of detection). The magnitude of these decreases in bacterial biomass is comparable to the results of neutrophil challenges to planktonic bacteria at similar neutrophil/bacterium ratios (34, 35). The dependence of bacterial biomass on the neutrophil/bacterium ratio plotted in Fig. 2B was statistically significant (P < 0.0001). Intuitively, better control of the nascent biofilm was observed when neutrophil densities were higher and bacterial densities were lower, as there were sufficient neutrophils to discover and phagocytose each bacterial aggregate. In this set of experiments, the multiplicity of infection (number of bacterial objects at the beginning of an experiment divided by the average number of neutrophils in the same field of view over the course of the experiment) ranged from 0.10 to 7, with a mean value of 1.58 ± 1.51.
FIG 2.

S. aureus bacterial dynamics during interaction with human neutrophils. (A) Representative experiments corresponding to bacterial growth in the absence of neutrophils, effective control of bacteria with a relatively high density of neutrophils, and an intermediate outcome at a lower density of neutrophils. Bottom line indicates the limit of detection (LOD), 1 pixel. (B) Net change in S. aureus bacterial area after a 4-h interaction with human neutrophils. Higher initial neutrophil numbers resulted in a net decrease in attached bacteria; n = 55 fields of view (17 bacteria only, 38 bacteria plus neutrophils) from 10 independent experiments.
The LysoBrite stain used to fluorescently label neutrophils did not adversely affect their ability to clear S. aureus. We challenged S. aureus with neutrophils that were either unstained or stained with LysoBrite and found that there were no significant differences in viable bacteria recovered from the surface (P = 0.25), total GFP area remaining on the surface (P = 0.89), or log differences in GFP area from a given field of view (P = 0.61) (see Fig. S1A to C).
Neutrophil movements over the abiotic surface are similar in the presence and absence of bacteria.
Neutrophils moved over the serum-coated surface similarly in the absence and presence of bacteria. The mean track length of a neutrophil during the 4-h observation period was 1,485 ± 514 μm in the presence of bacteria and 1,249 ± 307 μm in the absence of bacteria (P = 0.1368) (see Fig. S2). A neutrophil random motility coefficient was calculated to be 2.67 ± 0.75 × 10−8 cm2 s−1 by plotting the mean square displacement versus time during a 40-min window of linearity in the first hour of an experiment (see Table S1). Neutrophils gradually slowed during the 4-h experiment, and some stopped moving altogether (see Fig. S3). We hypothesized this was due to the depletion of a constituent of the medium or the accumulation of an inhibitive factor. To test for these effects, we conducted a set of experiments in which the medium was replenished halfway through an experiment (at t = 2 h) and the speed in the second 2-h interval was compared (Fig. S3). Linear regression was used to compare the speeds of neutrophils in control and replacement wells both before and after medium replacement. Prior to replacement, both the slopes and intercepts of the two groups were similar (P = 0.1587 and P = 0.7998, respectively). Following replacement of the medium, the slopes of the two curves were significantly different (P < 0.0001). Although there was a slight increase in the speed of polymorphonuclear leukocytes (PMNs) in wells where the medium was replaced, PMN speed was not restored to levels observed during the earliest time points of the experiment (n = 8 fields of view, 2 independent experiments), suggesting that depletion of a nutrient in the medium is not the sole driver of the observed decrease in PMN speed. The decrease in speed over time is likely in part due to the short life span of neutrophils ex vivo (36).
A measure of the directionality of neutrophil movement was calculated by dividing neutrophil displacement (distance between the starting location and its location 4 h later) by the total track length of the leukocyte. This measure is bounded from a minimum of zero (the neutrophil starts and ends at the same spot) to a maximum of one (the neutrophil moves in a straight line with no change in direction). The directionality coefficient was 0.26 ± 0.10 and was not significantly different between experiments with and without bacteria (P = 0.4453) (see Fig. S4). To further test for a possible role of chemotaxis in the discovery of attached bacteria, the fraction of bacteria discovered by and subsequently associated with a neutrophil was plotted as a function of the fraction of the field of view that had been patrolled by neutrophils (see Fig. S5). If the discovery of bacteria was purely random, one would anticipate that this relationship would be a straight line: when neutrophils had policed 20% of the surface they would encounter and associate with 20% of the bacteria on the surface. In experiments in which neutrophils covered 20% of the surface, they discovered and became associated with approximately 60% of the bacteria, suggesting a chemotactic component of the discovery of surface-attached bacteria. In some cases, we observed PMNs chemotaxing toward larger S. aureus aggregates (see Video S6). This suggests that chemotaxis may be occurring only when aggregates are sufficiently large or when neutrophils are already quite close to the aggregate, thus not significantly changing the overall directionality of the neutrophil.
Delaying neutrophil recruitment to the surface allows bacterial aggregates to form that are recalcitrant to clearance.
We noticed that bacteria that were not found by neutrophils early grew into discrete aggregates and that when these aggregates were eventually discovered by neutrophils, they appeared to be more difficult for neutrophils to destroy. To explore this effect further, “head-start” experiments were conducted, in which bacteria were allowed to grow for 1 to 3 h prior to neutrophil addition (Fig. 1G and H and Video S6). The average aggregate size within a field of view at the start of imaging was 6.54 ± 1.11 μm2, 11.36 ± 2.97 μm2, and 17.65 ± 4.66 μm2 after 1-, 2-, and 3-h head starts, respectively (see Fig. S6A). However, larger than average aggregates formed, and the maximum aggregate size in a given field of view was 12.08 ± 3.77 μm2, 31.05 ± 12.81 μm2, and 71.88 ± 23.59 μm2 after 1-, 2-, and 3-h head starts, respectively (Fig. S6B).
When neutrophils encountered bacterial aggregates that were similar in size or larger than the neutrophil itself, they often failed to achieve phagocytosis (Video S6). This outcome was evident in that the bacterial aggregate was not dislodged from the surface and did not move with neutrophil postencounter, as usually happened with single bacterial cells. In some cases, it was visibly apparent that the leukocyte failed to completely engulf the bacterial aggregate and the aggregate continued to grow. Occasionally, multiple neutrophils attacked the same aggregate, and in some instances, neutrophil engagement with the bacteria was strong enough to break the bacterial aggregate into smaller pieces. These observations suggest a range of outcomes, but in general, the control of bacteria by neutrophils was less effective when bacterial aggregates were preformed (Fig. 3A). The net change in bacterial area after neutrophil incubation for experiments in which neutrophil addition was delayed was a log10 difference of 0.46 ± 0.35 (a net increase in bacteria), whereas it was −0.65 ± 0.82 (a net decrease in bacteria) for experiments with no bacterial head start (P < 0.0001) (Fig. 3B). There was no significant difference between the neutrophil-to-bacterium ratio when comparing the head start and no head start data sets (data not shown, P = 0.6487). Thus, it appears that neutrophils must reach adherent S. aureus on a surface quickly, as even relatively small neutrophil-sized aggregates are recalcitrant to clearance by neutrophils.
FIG 3.

Larger S. aureus aggregates are recalcitrant to clearance by human neutrophils. (A) Net change in S. aureus bacterial area after a 4-h interaction with human neutrophils comparing outcomes from addition of neutrophils immediately following bacterial attachment (no head start) and after allowing bacteria to grow for 1, 2, or 3 h and form small aggregates prior to neutrophil addition (pooled head start); n = 38 fields of view without head starts from 9 independent experiments, n = 24 fields of view with head starts from 4 independent experiments (n = 8 fields of view per head start time). (B) Clearance of bacteria without a head start compared to pooled head starts of 1, 2, or 3 h (all time points included). Error bars represent standard deviations of the samples. ****, P < 0.0001 by an unpaired t test.
Hypoxia reduces neutrophil efficacy against attached S. aureus.
Inert biomaterial surfaces are not perfused by blood; therefore, oxygen availability may be reduced at the surface of an implant. Additionally, hypoxia is common in established biofilm infections and plausibly arises in the vicinity of a foreign body through the concerted consumption of oxygen by both leukocytes and bacteria in a diffusion-limited environment (37–41). We therefore measured the efficacy of human neutrophils in controlling attached S. aureus under conditions of reduced (2% O2) or no oxygen. Reduced oxygen tension did not affect the bacterial growth rate (P = 0.11) (Fig. 4A). The specific growth rate of S. aureus under anaerobic conditions has been reported to be 44% to 45% of the value under aerobic conditions (42, 43). Our reduced-oxygen conditions were not as a whole fully anoxic. The average neutrophil track length was decreased under reduced oxygen tension (P < 0.0001) (Fig. 4B). While there was some variability, on average, it appeared that a higher proportion of PMNs were stationary at the end of the experiment in the low oxygen (0% and 2%) environment than in the oxic (20% oxygen) environment, suggesting that the neutrophils are unable to effectively patrol the surface without sufficient oxygen (Fig. 4C) (P = 0.001). Low oxygen tension diminished the antibiofilm efficacy of neutrophils (Fig. 4D and E). The difference between the oxic and grouped low oxygen conditions was statistically significant (P < 0.0001). These data suggest that low oxygen conditions impair neutrophil motility, reducing the chance of discovering adherent S. aureus on a surface. Additionally, low oxygen tension may interfere with the oxidative burst produced by neutrophils, preventing them from effectively clearing S. aureus that is discovered and phagocytosed (44).
FIG 4.

Oxygen is required for effective motility and clearance of adherent S. aureus. (A) Maximum growth rate of S. aureus was not different between 20% oxygen and pooled 0% or 2% oxygen conditions; n = 17 fields of view from 8 independent experiments and n = 12 fields of view from 6 independent experiments for normal oxygen and low oxygen, respectively. (B and C) Neutrophils traverse less distance and a higher fraction of neutrophils are stationary in the last ∼5 min of an experiment in an environment with reduced oxygen tension; n = 32 from 5 independent experiments and n = 37 from 5 independent experiments for normal and low oxygen, respectively. (D and E) Net change in S. aureus bacterial area after 4 h under normal conditions (n = 32 from 8 independent experiments) or low-oxygen conditions (n = 33 from 3 independent experiments at 2% O2 and 3 at 0% O2). Error bars represent standard deviations of the samples. ***, P < 0.001; ****, P < 0.0001 by unpaired t tests.
Computational simulation of the neutrophil-bacterium surface interaction demonstrates variable outcomes governed by stochastic discovery of bacteria by neutrophils.
We constructed a mathematical model of the interaction of bacteria and neutrophils on a two-dimensional surface. The model integrates bacterial growth, bacterial elaboration and Fickian diffusion of a chemoattractant molecule, neutrophil movement by a combination of random motility and chemotaxis, and killing of bacteria upon discovery by a neutrophil. An illustrative example of a single simulation is provided in Fig. 5A, with the full video of this simulation available in the supplemental material (see Video S7). In such simulations, bacteria are stationary, while neutrophils move and may eventually discover the bacterial colonies. In the example in Fig. 5A, half of the bacterial colonies initially seeded in the simulation domain (12 total) were discovered and killed by a neutrophil, and the other half went undiscovered to the end of the 4-h simulation period. Every simulation is different, because the initial placements of both bacteria and neutrophils are random and also because the movement of each neutrophil is stochastic. This is illustrated with additional simulations of the case presented in Fig. 5A, in which 12 bacterial colonies and 8 neutrophils were initially placed in the simulation field (see Videos S8 and S9). We ran each case 156 times to illustrate the range of outcomes even when parameter values are fixed. Figure 5B shows 6 of the 156 outcomes for the case with 12 bacterial colonies initially and 8 neutrophils. In these 156 runs, the maximum number of bacterial colonies that survived was nine and the minimum was zero, with a mean value of 3.6. Because the undiscovered colonies continue to grow during the 4-h interval, they collectively contain an average of 40 viable bacteria at the end of the simulation, an increase from the 12 initially present. The average number of bacteria that survive the interaction with surface-associated neutrophils is predicted to be a strong linear function of the number of neutrophils present (Fig. 5C), recapitulating at least qualitatively the result shown in Fig. 2B.
FIG 5.
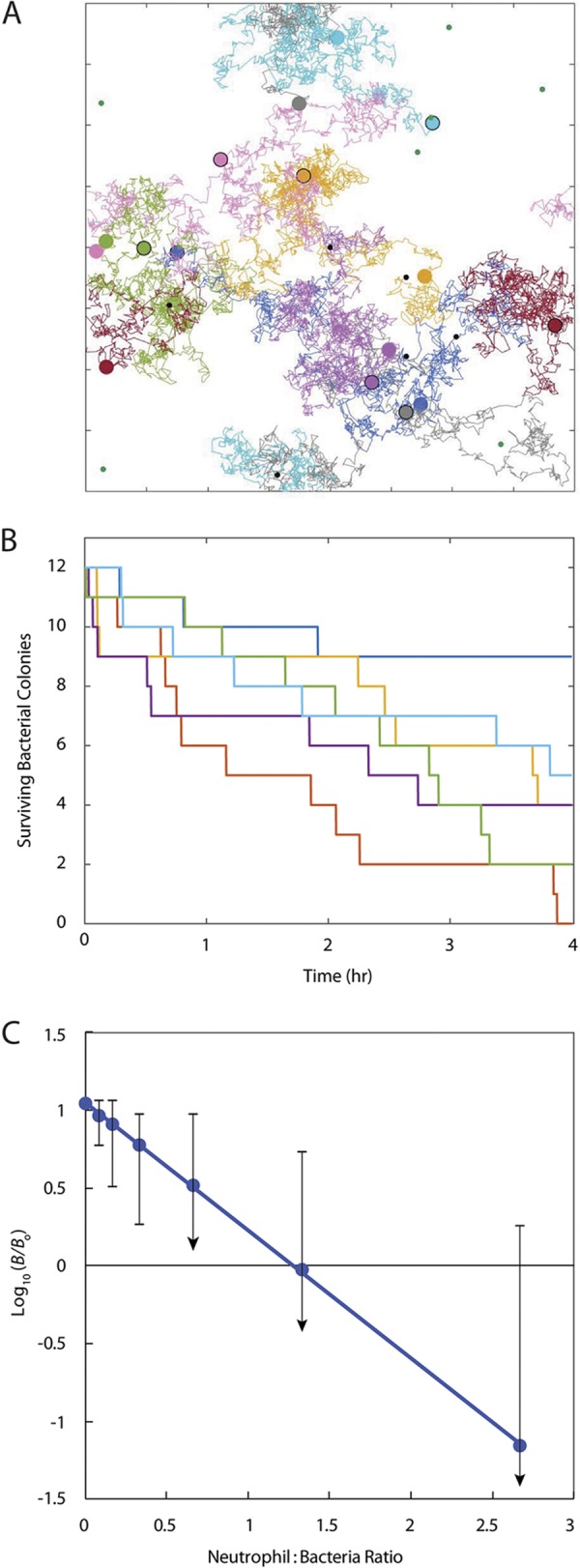
Mathematical model illustrates stochastic outcomes of neutrophil-S. aureus interactions. (A) Sample computational result showing neutrophil (larger variously colored circles) initial (with black outline) and final (no outline) locations and tracks (colors correspond to the individual neutrophil) during the course of a 4-h simulation. Bacterial colonies (small circles) are shown in green where they have escaped detection by neutrophils and remain viable or in black where they have been discovered and killed by a neutrophil. Tick marks are 100 μm apart. See full video of the simulation in the supplemental material (Video S7). (B) Decay of the number of surviving bacterial colonies for 6 of 156 repeated simulations. All simulations used the same initial number of bacterial colonies (12) and same number of neutrophils (8), but the initial distributions of these two cell types were randomly determined for each individual repetition. Shown are simulations resulting in the maximum (dark blue) and minimum (red) number of residual bacterial colonies and four others to illustrate the range of outcomes. (C) Change in total viable bacterial numbers as a function of the neutrophil/bacterium ratio (mean from 156 repetitions of each case). Simulations all used the same initial number of bacterial colonies (12) with neutrophil numbers of 0, 1, 2, 4, 8, 16, and 32 (blue circles). Error bars denote the ranges from maximum to minimum outcomes with a downward arrow signifying complete elimination of viable bacteria as the minimum. The dotted line is a least-squares linear regression.
DISCUSSION
How do a few microorganisms contaminating an implanted biomaterial evade host defenses long enough to establish a mature and protected biofilm? To gain insight into specific phenomena that may compromise neutrophil clearance of attached bacteria, we imaged and quantified the interaction of human neutrophils with a nascent S. aureus biofilm on a surrogate biomaterial surface in vitro. A critical conclusion from these observations is that neutrophils are able to discover, phagocytose, and destroy surface-adherent bacteria when the microorganism is present as single cells or very small aggregates and when a sufficient number of neutrophils are present. This is important because it suggests that neutrophil-mediated innate immunity has the capacity to eliminate a nascent S. aureus biofilm. The approximate areal density needed for protective surveillance of the surface in our experiments was on the order of 104 to 105 neutrophils per cm2. This surface density of neutrophils must be present at the outset of the contest in order for the leukocytes to be able to discover and destroy bacteria.
When neutrophil discovery of bacteria was delayed, either because the surface density of neutrophils was too low or because neutrophils were added after giving the bacteria a head start, undiscovered bacteria grew and formed aggregates. These relatively young bacterial biofilm aggregates were much less susceptible to clearance by neutrophils than were lone bacterial cells. This result suggests that for neutrophils to prevent biofilm formation, the time for their recruitment to the abiotic surface must be shorter than the time for the bacteria to form neutrophil-sized aggregates. Thus, there are two critical time scales that are important to quantify in vivo: the characteristic time for neutrophil recruitment to a biomaterial surface and the time necessary for bacteria to form aggregates that are approximately 10 μm in diameter.
The time scale for aggregate formation by S. aureus, at least in the in vitro system used here, was 3 to 4 h. This corresponds to approximately 4 doubling times at the in vitro growth rate measured in this work of 0.78 h−1. In vivo, this time scale may be different, likely longer, depending on the in vivo doubling time of the bacteria. There are very few measurements of staphylococcal growth rate in vivo, especially in a niche where an implanted device could dwell. Data from Fig. 1B in the article by Dastgheyb et al. (45) show a growth rate of S. aureus in human synovial fluid of 0.46 ± 0.08 h−1; 4 doublings at this specific growth rate would take 6 h. In triplicate measurements of S. aureus growth in our early biofilm system, we found the specific growth rate to be 0.63 ± 0.42 h−1 in pure bovine serum and 0.56 ± 0.09 h−1 in pure bovine synovial fluid. The time scale for neutrophil recruitment to an implanted abiotic surface is also uncertain. Animal studies of neutrophil dynamics at sites of infection indicate that the time scale for neutrophil recruitment may range from an hour to a day (46–48).
To delve further into the possibility of delayed neutrophil recruitment to an implant surface, we extracted quantitative estimates of neutrophil recruitment times from published reports in murine models (see Fig. S7, Table S2, and related methods in the supplemental material). When a chemical stimulus was used (e.g., macrophage inflammatory protein 2 [MIP-2], zymosan, lipopolysaccharide [LPS], and peptidoglycan), the mean neutrophil recruitment time and standard deviation were 3.6 ± 2.6 h. When staphylococci were inoculated, the recruitment time was 12 ± 10.1 h. In two investigations in which an uninoculated implant (titanium or stainless steel) was examined, the neutrophil recruitment time was 21 h. While these values range widely, they appear to be consistent with the possibility of delayed recruitment. Clearly, additional research is needed to better define the characteristic times for both bacterial growth and neutrophil recruitment to a biomaterial surface in vivo.
This work raises the possibility that, in the earliest stage of biofilm formation, neutrophil efficacy is limited by the discovery of bacteria rather than by bacterial virulence. The stochastic nature of this interaction and the survival of occasional undiscovered bacteria are reinforced by computational modeling. Unlike the scenario of bacteria and neutrophils interacting in a homogenous well-mixed suspension (15), the interaction on a sparsely contaminated implant surface is inherently heterogeneous and probabilistically determined. If neutrophil discovery limits bacterial clearance from the surface of a biomaterial in vivo, then it might be possible to prevent device-related infections by boosting the recruitment and activity of neutrophils in a window of time shortly following implantation. Our in vitro results suggest that for such a strategy to succeed, both a sufficient number of neutrophils need to be delivered to the surface and their recruitment must be relatively rapid.
Of the six hypotheses presented above for how bacteria contaminating an abiotic surface evade neutrophil clearance, the two that are most consistent with our in vitro results are delayed neutrophil recruitment (hypothesis 1) and hypoxia (hypothesis 6). Hypotheses 2 to 5 are contradicted by our observation of efficient and consistent neutrophil surveillance, discovery, phagocytosis, and destruction of attached single bacteria or very small aggregates.
We hypothesized that delayed recruitment of neutrophils to a biomaterial surface provides a window of time for contaminating microorganisms to grow and establish a protected biofilm (hypothesis 1). A biomaterial is compromised in at least two important ways that could delay leukocyte recruitment. Biomaterial surfaces are not vascularized: they have no built-in system to rapidly deliver immune cells. In addition, there may be local disruption of vascularized tissue around the device as a result of the surgical implantation procedure. To ensure complete surveillance and discovery of all contaminating microorganisms, neutrophils must migrate from vascularized tissue to all the surfaces of the biomaterial. A second deficiency of a biomaterial with respect to neutrophil recruitment is that the material itself is mute in the sense that it cannot communicate with the host. Unlike vital tissue that can release chemokines in response to the presence of bacteria and thereby initiate neutrophil recruitment, abiotic implants lack any signaling capacity. The time required for neutrophils to arrive at a biomaterial surface likely depends on the anatomical site of the implant, the proximity of specific locations on the implant to perfused tissue, and the number of contaminating bacteria.
We also investigated the possibility that local hypoxia in the vicinity of the implant could diminish neutrophil efficacy (hypothesis 6). The same issues mentioned above—lack of vasculature on the biomaterial surface and trauma to neighboring tissue during implantation—could reduce provision of oxygen around a biomaterial. Whereas hypoxia is widely recognized to be likely in an established biofilm infection, data on oxygen availability in the vicinity of a newly implanted biomaterial are lacking.
Improved understanding of how innate immune cell function at biomaterial surfaces is compromised and what mechanisms limit leukocyte efficacy on the surface of a foreign body may someday lead to alternative strategies for preventing device-related infections (24, 49–59). The work presented here provides insight into the pathogenesis of foreign body infections and a previously unexplored mechanism of immune evasion. We have identified that a relatively brief time window, dependent on the in vivo bacterial growth rate and neutrophil recruitment dynamics, exists, wherein adherent bacteria are susceptible to effective clearance by neutrophils. This short time period could possibly explain how small aggregates of contaminating bacteria on an implant surface are able to develop into biofilm infections that are recalcitrant to immune clearance and antibiotic therapy.
Areas of opportunity for future work include (i) development and application of animal models using intravital imaging to observe neutrophil-bacterium interactions with single-cell resolution, (ii) measurement of the distribution of neutrophil recruitment times to a bacterially contaminated implant surface in vivo, (iii) measurement of lag times and specific growth rates of surface-attached microorganisms in vivo, and (iv) elucidation of mechanisms of immune evasion by small (neutrophil-sized) bacterial aggregates.
MATERIALS AND METHODS
Bacterial and neutrophil preparation.
S. aureus strain AH2547 (HG001+pCM29, courtesy of Alex Horswill), a known biofilm-forming strain with constitutive expression of green fluorescent protein (GFP), was grown overnight in tryptic soy broth supplemented with 10 μg/ml chloramphenicol (60). Overnight cultures were centrifuged for 5 min at 4,000 rpm, rinsed, resuspended in phosphate-buffered saline (PBS), and serially diluted. Cells were attached to a 4-chambered glass bottom petri dish (Cellvis, Mountain View, CA, USA) to facilitate live-cell imaging. To attach cells, between 25 and 100 μl of the 10−3 dilution was added to the surface, and PBS was added to a final volume of 500 μl. After 30 min of incubation at 37°C, unattached bacteria were gently rinsed from the surface with PBS. Each chamber of the petri dish was filled with 500 μl of 20% fresh human serum in Hanks’ balanced salt solution (HBSS) to simultaneously coat the surface with serum and opsonize bacteria, and the dish was incubated at 37°C for 30 min. Prior to imaging, the serum concentration in each chamber was then diluted to ∼10% with additional HBSS. Human neutrophils (polymorphonuclear leukocytes, or PMNs) were isolated from heparinized venous blood obtained from healthy donors according to a standard institutional review board (IRB)-approved protocol, as described previously (61). PMNs were kept on ice until they were stained with LysoBrite Red (AAT Bioquest, Sunnyvale, CA, USA) according to the manufacturer’s instructions.
Microscopy.
A Leica SP5 inverted confocal scanning microscope was utilized for all imaging. GFP-tagged bacteria and PMNs were excited with the 488-nm and 561-nm laser lines, respectively. A LiveCell environmental chamber system (Pathology Devices, San Diego, CA, USA) was utilized to maintain 5% CO2, 20% O2, 50% humidity, and 37°C for sample incubation during imaging. Image stacks with 1-μm z-slices were taken sequentially at 1- to 2-min intervals over a 4-h time course using a 20× dry lens objective. At least two fields of view from each chamber of the dish were imaged per experiment.
Image analysis.
MetaMorph (Molecular Devices) image analysis software was used to measure the change in bacterial biomass by quantifying the thresholded area of bacterial green fluorescence over a period of 4 h. Maximum growth rates were calculated by linear regression of all data within exponential phase. Movies were prepared with Imaris version 8.0 (Bitplane). To quantify neutrophil motion, the “ImarisTrack” module that tracks three-dimensional (3D) objects over time, displays their path, and analyzes their motion was utilized. Neutrophils were automatically identified as “spots” on the basis of size and manually edited and confirmed when needed. Motion was tracked using the Brownian motion algorithm. To quantify the fraction of a field of view “patrolled” by a neutrophil over 4 h, neutrophil tracks determined by the ImarisTrack module were set to be cylinders 10.87 μm in diameter. The image was then imported into MetaMorph, and the percentage of the field of view covered by a neutrophil track was determined by thresholding. To determine the average track length for PMNs in a given field of view, the average speed of each neutrophil in the field of view was determined using the ImarisTrack module. The average speed for all neutrophils in a field of view was calculated for each frame, and the average track length was subsequently determined by integrating with respect to time using the trapezoidal method. This method decreased the bias of neutrophils that may have been in the field of view for only a small number of frames as opposed to simply using the track length generated by Imaris. The fraction of neutrophils moving in a given field of view was calculated by binning neutrophils that were moving greater than 0.01 μm/s. An average from the final three frames of an experiment (∼5 to 7 min) was used to compare the numbers of neutrophils moving at the end of an experiment for the high- versus low-oxygen environment assays.
Bacterium-neutrophil surface dynamics.
To assay how PMNs interact with bacteria on a surface, we varied both the density of attached S. aureus aggregates on the surface and the number of neutrophils added to the surface (n = 10 independent experiments, with up to 8 fields of view per experiment). Experimental conditions included wells with S. aureus only, PMNs only, and S. aureus and PMNs at various multiplicities of infection (MOIs).
Head-start experiments.
S. aureus cells were attached to the surfaces of separate chambers of the dish as described above at staggered time points of 3.5, 2.5, or 1.5 h prior to the addition of PMNs. After 0.5 h, unattached S. aureus cells were gently rinsed from the surface, and 10% tryptic soy broth (TSB) was added to the dish. After 2.5, 1.5, or 0.5 h of incubation, the medium was gently removed with a pipette and replaced with 20% human serum in HBSS for conditioning and opsonization. Half of an hour later, the serum concentration was diluted to 10%, PMNs were added, and the dishes were imaged for 4 h. Results from the head-start experiments (n = 4) were pooled and compared to data obtained during previous experiments wherein bacteria were not given a head start.
Oxygen limitation experiments.
Low-oxygen conditions were attained by adding N2 to the environmental chamber to maintain O2 levels of either 0% or 2% (n = 3 experiments each). Results for both the 0% and 2% conditions were pooled and compared to data obtained during previous standard experiments (20% O2).
Directionality and mean square displacement analysis.
Neutrophil directionality was calculated by dividing neutrophil track displacement by total track length using tracking data generated using the ImarisTrack module. Mean square displacement was plotted as a function of time, and an approximately 40-min-long period of linearity in the curve was used to calculate a random motility coefficient based on the slope of the curve.
Computational modeling of the neutrophil-bacterium surface interaction.
Neutrophil-bacterium interactions were mathematically modeled in a three-dimensional spatial domain lying above a flat plate. The domain was periodic in the horizontal directions x and y and open above the surface of a plate (z = 0) in the vertical direction (z ≥ 0). Spatial periodicity was set at 800 μm in both the x and y directions. The simulation time was 240 min in all computations. Model elements consisted of a set of nonmotile, growing bacterial colonies that emit chemoattractant (at a rate proportional to colony size) which freely diffuses in the region where z was ≥0 as well as a set of neutrophils which move on the plate (z = 0) with a combination of random and directed (via the gradient of the chemoattractant concentration) movement as in a Keller-Segel type model (62). When a neutrophil encountered a bacterial colony, the colony was eliminated and ceased to emit chemoattractant.
In each simulation, 12 bacterial colonies, each with an initial population of 1, were randomly seeded with uniform probability on the 800-μm2 plate. Coordinates (xj = [xj, yj, 0], j = 1 to 12) designated colony locations for any given simulation. Note that due to horizontal periodicity, there was effectively an infinite tiling of the plane at a z of 0 with “image” colonies at locations (xj, yj, 0) + (800k, 800l, 0), j = 1 to 12, where k and l are arbitrary integers. Prior to an encounter with a neutrophil, any given colony grew at specific rate r such that colony j had population cj(t) = ert.
Each colony, including the images, emitted chemoattractant at rate , until it had an encounter with a neutrophil at, for example, t = Tj, at which time it ceased to emit chemoattractant and cj(t) was 0 for t of ≥Tj. The chemoattractant concentration, denoted by u(x, t) = u(x, y, z, t), satisfies the following diffusion (with sources) equation:
where δ is the Dirac delta function, with initial and boundary conditions
In fact, this system can be solved analytically for u(x, t) (see supplemental material).
In addition, a number N (with N = 1, 2, 4, 8, 16, or 32) neutrophils are also (initially) randomly seeded with uniform probability on the plate at locations X(j) (t = 0) =, j = 1 to N. Neutrophil locations X(j) (t) are modeled by stochastic differential equations
where is a standard 2-dimensional Wiener process (63), and is the restriction of the analytical solution to the chemoattractant equations to the z of 0 plane. Solutions were approximated numerically using the Euler-Maruyama scheme (63). We assumed that there is a maximum speed Vmax at which a neutrophil can move in response to the chemoattractant, imposed using a Monod-type function in the drift coefficient with half-saturation Km. Note, again, that as a consequence of periodicity, there are effectively a doubly periodically infinite set of neutrophils. For computational purposes, we only needed to track one set: each time a neutrophil crossed the boundary of the model 800-μm2 plate, it effectively re-entered across the corresponding periodic boundary. Simulation parameters and sources were as follows: D1 = 0.036 mm2 min−1 (comparison to raffinose, molecular weight [MW] 504 g/mole [64]); D2 = 0.00016 mm2 min−1 (see Table S1 in the supplemental material); Km = 1,000 concentration mm−1 (assumed); Vmax = 0.012 mm min−1 (see Fig. S2); β = 0.01 concentration min−1 per bacteria (assumed); r = 0.01 min−1 (see “Neutrophils migrate on an abiotic surface and discover, phagocytose, and destroy single attached bacteria or very small bacterial aggregates” above).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences (award R01GM109452) and the National Institute of Allergy and Infectious Diseases (award 1R56AI135039-01A1). Imaging was made possible by microscope facilities at the Center for Biofilm Engineering, which were supported by funding from the National Science Foundation MRI Program and the M. J. Murdock Charitable Trust.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00745-19.
REFERENCES
- 1.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 2.Parsek MR, Singh PK. 2003. Bacterial biofilms: an emerging link to disease pathogenesis. Annu Rev Microbiol 57:677–701. doi: 10.1146/annurev.micro.57.030502.090720. [DOI] [PubMed] [Google Scholar]
- 3.Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138. doi: 10.1016/s0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 4.Stewart PS. 2015. Antimicrobial tolerance in biofilms. Microbiol Spectr 3:MB-0010-2014. doi: 10.1128/microbiolspec.MB-0010-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J. 2012. Economic burden of periprosthetic joint infection in the United States. J Arthroplasty 27:61–65. doi: 10.1016/j.arth.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerli W, Waldvogel FA, Vaudaux P, Nydegger UE. 1982. Pathogenesis of foreign body infection: description and characteristics of an animal model. J Infect Dis 146:487–497. doi: 10.1093/infdis/146.4.487. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerli W, Lew PD, Waldvogel FA. 1984. Pathogenesis of foreign body infection. Evidence for a local granulocyte defect. J Clin Invest 73:1191–1200. doi: 10.1172/JCI111305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan SS, Basford RE, Kormos RL, Hardesty RL, Simmons RL, Mora EM, Cardona M, Griffith BL. 1990. Biomaterial associated impairment of local neutrophil function. ASAIO Trans 36:M172–M175. [PubMed] [Google Scholar]
- 9.Sapatnekar S, Kao WJ, Anderson JM. 1997. Leukocyte-biomaterial interactions in the presence of Staphylococcus epidermidis: flow cytometric evaluation of leukocyte activation. J Biomed Mater Res 35:409–420. doi:. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan SS, Heine RP, Simmons RL. 1999. Defensins impair phagocytic killing by neutrophils in biomaterial-related infection. Infect Immun 67:1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atel JD, Krupka T, Anderson JM. 2007. iNOS-mediated generation of reactive oxygen and nitrogen species by biomaterial-adherent neutrophils. J Biomed Mater Res A 80:381–390. doi: 10.1002/jbm.a.30907. [DOI] [PubMed] [Google Scholar]
- 12.Tautenhahn J, Meyer F, Buerger T, Schmidt U, Lippert H, Koenig W, Koenig B. 2010. Interactions of neutrophils with silver-coated vascular polyester grafts. Langenbecks Arch Surg 395:143–149. doi: 10.1007/s00423-008-0439-7. [DOI] [PubMed] [Google Scholar]
- 13.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 14.Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. 2017. Epic immune battles of history: neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol 7:286. doi: 10.3389/fcimb.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Karlin A, Loike JD, Silverstein SC. 2002. A critical concentration of neutrophils is required for effective bacterial killing in suspension. Proc Natl Acad Sci U S A 99:8289–8294. doi: 10.1073/pnas.122244799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulido L, Ghanem E, Joshi A, Purtill J, Parvizi J. 2008. Periprosthetic joint infection: the incidence, timing, and predisposing factors. Clin Orthop Relat Res 466:1710–1715. doi: 10.1007/s11999-008-0209-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricciardi BF, Muthukrishnan G, Masters E, Ninomiya M, Lee CC, Schwarz EM. 2018. Staphylococcus aureus evasion of host immunity in the setting of prosthetic joint infection: biofilm and beyond. Curr Rev Musculoskelet Med 11:389–400. doi: 10.1007/s12178-018-9501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lekstrom-Himes JA, Gallin JI. 2000. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med 343:1703–1714. doi: 10.1056/NEJM200012073432307. [DOI] [PubMed] [Google Scholar]
- 19.Roos D, de Boer M. 2014. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol 175:139–149. doi: 10.1111/cei.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Günther F, Wabnitz GH, Stroh P, Prior B, Obst U, Samstag Y, Wagner C, Hänsch GM. 2009. Host defence against Staphylococcus aureus biofilms infection: phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol Immunol 46:1805–1813. doi: 10.1016/j.molimm.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Stroh P, Günther F, Meyle E, Prior B, Wagner C, Hänsch GM. 2011. Host defence against Staphylococcus aureus biofilms by polymorphonuclear neutrophils: oxygen radical production but not phagocytosis depends on opsonisation with immunoglobulin G. Immunobiology 216:351–357. doi: 10.1016/j.imbio.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186:6585–6596. doi: 10.4049/jimmunol.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guggenberger C, Wolz C, Morrissey JA, Heesemann J. 2012. Two distinct coagulase-dependent barriers protect Staphylococcus aureus from neutrophils in a three-dimensional in vitro infection model. PLoS Pathog 8:e1002434. doi: 10.1371/journal.ppat.1002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanke ML, Heim CE, Angle A, Sanderson SD, Kielian T. 2013. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J Immunol 190:2159–2168. doi: 10.4049/jimmunol.1202348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heim CE, Vidlak D, Kielian T. 2015. Interleukin‐10 production by myeloid‐derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol 98:1003–1013. doi: 10.1189/jlb.4VMA0315-125RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, Kielian T. 2015. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol 194:3861–3872. doi: 10.4049/jimmunol.1402689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherr TD, Hanke ML, Huang O, James DB, Horswill AR, Bayles KW, Fey PD, Torres VJ, Kielian T. 2015. Staphylococcus aureus biofilms induce macrophage dysfunction through leukocidin AB and alpha-toxin. mBio 6:e01021-15. doi: 10.1128/mBio.01021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, Torres VJ, Wozniak DJ. 2018. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A 115:7416–7421. doi: 10.1073/pnas.1721949115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He L, Le KY, Khan BA, Nguyen TH, Hunt RL, Bae JS, Kabat J, Zheng Y, Cheung GYC, Li M, Otto M. 2019. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat Microbiol 4:1114–1119. doi: 10.1038/s41564-019-0413-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heim CE, West SC, Ali H, Kielian T. 2018. Heterogeneity of Ly6G+ Ly6C+ myeloid-derived suppressor cell infiltrates during Staphylococcus aureus biofilm infection. Infect Immun 86:e00684-18. doi: 10.1128/IAI.00684-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le KY, Villaruz AE, Zheng Y, He L, Fisher EL, Nguyen TH, Ho TV, Yeh AJ, Joo HS, Cheung GYC, Otto M. 2019. Role of phenol-soluble modulins in Staphylococcus epidermidis biofilm formation and infection of indwelling medical devices. J Mol Biol 431:3015–3027. doi: 10.1016/j.jmb.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutierrez Jauregui R, Fleige H, Bubke A, Rohde M, Weiss S, Förster R. 2019. IL-1β promotes Staphylococcus aureus biofilms on implants in vivo. Front Immunol 10:1082. doi: 10.3389/fimmu.2019.01082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz J, Leidal KG, Femling JK, Weiss JP, Nauseef WM. 2009. Neutrophil bleaching of GFP-expressing staphylococci: probing the intraphagosomal fate of individual bacteria. J Immunol 183:2632–2641. doi: 10.4049/jimmunol.0804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voyich JM, Vuong C, DeWald M, Nygaard TK, Kocianova S, Griffith S, Jones J, Iverson C, Sturdevant DE, Braughton KR, Whitney AR, Otto M, DeLeo FR. 2009. The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J Infect Dis 199:1698–1706. doi: 10.1086/598967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sward EW, Fones EM, Spaan RR, Pallister KB, Haller BL, Guerra FE, Zurek OW, Nygaard TK, Voyich JM. 2018. Staphylococcus aureus SaeR/S-regulated factors decrease monocyte-derived tumor necrosis factor-α to reduce neutrophil bactericidal activity. J Infect Dis 217:943–952. doi: 10.1093/infdis/jix652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. 2002. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci U S A 99:6901–6906. doi: 10.1073/pnas.092148299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Döring G. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109:317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lone AG, Atci E, Renslow R, Beyenal H, Noh S, Fransson B, Abu-Lail N, Park JJ, Gang DR, Call DR. 2015. Staphylococcus aureus induces hypoxia and cellular damage in porcine dermal explants. Infect Immun 83:2531–2541. doi: 10.1128/IAI.03075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James GA, Ge Zhao A, Usui M, Underwood RA, Nguyen H, Beyenal H, deLancey Pulcini E, Agostinho Hunt A, Bernstein HC, Fleckman P, Olerud J, Williamson KS, Franklin MJ, Stewart PS. 2016. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen 24:373–383. doi: 10.1111/wrr.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Maltesen RG, Larsen LH, Schønheyder HC, Le VQ, Nielsen JL, Nielsen PH, Thomsen TR, Nielsen KL. 2016. In vivo gene expression in a Staphylococcus aureus prosthetic joint infection characterized by RNA sequencing and metabolomics: a pilot study. BMC Microbiol 16:80. doi: 10.1186/s12866-016-0695-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Y, Klapper I, Stewart PS. 2018. Hypoxia arising from concerted oxygen consumption by neutrophils and microorganisms in biofilms. Pathog Dis 76:fty043. doi: 10.1093/femspd/fty043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belay N, Rasooly A. 2002. Staphylococcus aureus growth and enterotoxin A production in an anaerobic environment. J Food Prot 65:199–204. doi: 10.4315/0362-028X-65.1.199. [DOI] [PubMed] [Google Scholar]
- 43.Ferreira MT, Manso AS, Gaspar P, Pinho MG, Neves AR. 2013. Effect of oxygen on glucose metabolism: utilization of lactate in Staphylococcus aureus as revealed by in vivo NMR studies. PLoS One 8:e58277. doi: 10.1371/journal.pone.0058277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, Anwar S, Willcocks LC, Whyte MKB, Condliffe AM, Chilvers ER. 2011. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol 186:453–463. doi: 10.4049/jimmunol.1002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dastgheyb S, Parvizi J, Shapiro IM, Hickok NJ, Otto M. 2015. Effect of biofilms on recalcitrance of staphylococcal joint infection to antibiotic treatment. J Infect Dis 211:641–650. doi: 10.1093/infdis/jiu514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pribaz JR, Bernthal NM, Billi F, Cho JS, Ramos RI, Guo Y, Cheung AL, Francis KP, Miller LS. 2012. Mouse model of chronic post-arthroplasty infection: noninvasive in vivo bioluminescence imaging to monitor bacterial burden for long-term study. J Orthop Res 30:335–340. doi: 10.1002/jor.21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harding MG, Zhang K, Conly J, Kubes P. 2014. Neutrophil crawling in capillaries; a novel immune response to Staphylococcus aureus. PLoS Pathog 10:e1004379. doi: 10.1371/journal.ppat.1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stavrakis AI, Niska JA, Shahbazian JH, Loftin AH, Ramos RI, Billi F, Francis KP, Otto M, Bernthal NM, Uslan DZ, Miller LS. 2014. Combination prophylactic therapy with rifampin increases efficacy against an experimental Staphylococcus epidermidis subcutaneous implant-related infection. Antimicrob Agents Chemother 58:2377–2386. doi: 10.1128/AAC.01943-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rózalska B, Ljungh A, Paziak-Domańska B, Rudnicka W. 1996. Effects of granulocyte-macrophage colony stimulating factor (GM-CSF) on biomaterial-associated staphylococcal infection in mice. Microbiol Immunol 40:931–939. doi: 10.1111/j.1348-0421.1996.tb01162.x. [DOI] [PubMed] [Google Scholar]
- 50.Boelens J, van der Poll T, Dankert J, Zaat S. 2000. Interferon-γ protects against biomaterial-associated Staphylococcus epidermidis infection in mice. J Infect Dis 181:1167–1171. doi: 10.1086/315344. [DOI] [PubMed] [Google Scholar]
- 51.Schlöbe A, Schnitzler N, Schweizer K, Rohde D. 2003. Granulocyte colony-stimulating factor for the treatment of biomaterial-associated staphylococcal infections in vitro. Urol Res 30:394–398. doi: 10.1007/s00240-002-0289-7. [DOI] [PubMed] [Google Scholar]
- 52.Li B, Jiang B, Boyce B, Lindsey B. 2009. Multilayer polypeptide nanoscale coatings incorporating IL-12 for the prevention of biomedical device-associate infections. Biomaterials 30:2552–2558. doi: 10.1016/j.biomaterials.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agalar C, Eroglu E, Sari M, Sari A, Daphan C, Agalar F. 2005. The effect of G-CSF in an experimental MRSA graft infection in mice. J Invest Surg 18:227–231. doi: 10.1080/08941930500248581. [DOI] [PubMed] [Google Scholar]
- 54.Franz S, Rammelt S, Scharnweber D, Simon JC. 2011. Immune responses to implants - a review of the implications for the design of immunomodulatory biomaterials. Biomaterials 32:6692–6709. doi: 10.1016/j.biomaterials.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 55.Bryers JD, Giachelli CM, Ratner BD. 2012. Engineering biomaterials to integrate and heal: the biocompatibility paradigm shifts. Biotechnol Bioeng 109:1898–1911. doi: 10.1002/bit.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vishwakarma A, Bhise NS, Evangelista MB, Rouwkema J, Dokmeci MR, Ghaemmaghami AM, Vrana NE, Khademhosseini A. 2016. Engineering immunomodulatory biomaterials to tune the inflammatory response. Trends Biotechnol 34:470–482. doi: 10.1016/j.tibtech.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 57.Kelly SH, Shores LS, Votaw NL, Collier JH. 2017. Biomaterial strategies for generating therapeutic immune responses. Adv Drug Deliv Rev 114:3–18. doi: 10.1016/j.addr.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selders GS, Fetz AE, Radic MZ, Bowlin GL. 2017. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater 4:55–68. doi: 10.1093/rb/rbw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang C, Li J, Zhu C, Zhang Q, Yu J, Wang J, Wang Q, Tang J, Zhou H, Shen H. 2019. Advanced antibacterial activity of biocompatible tantalum nanofilm via enhanced local innate immunity. Acta Biomater 89:403–418. doi: 10.1016/j.actbio.2019.03.027. [DOI] [PubMed] [Google Scholar]
- 60.Pabst B, Pitts B, Lauchnor E, Stewart PS. 2016. Gel-entrapped Staphylococcus aureus bacteria as models of biofilm infection exhibit growth in dense aggregates, oxygen limitation, antibiotic tolerance, and heterogenous gene expression. Antimicrob Agents Chemother 60:6294–6301. doi: 10.1128/AAC.01336-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Saïd-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, Musser JM, DeLeo FR. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol 175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- 62.Keller EF, Segel LA. 1971. Model for chemotaxis. J Theor Biol 30:225–234. doi: 10.1016/0022-5193(71)90050-6. [DOI] [PubMed] [Google Scholar]
- 63.Kloeden P, Platen E. 1999. Numerical solution of stochastic differential equations. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 64.Stewart PS. 2003. Diffusion in biofilms. J Bacteriol 185:1485–1491. doi: 10.1128/jb.185.5.1485-1491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.