


Abstract
Evaluating the impact of non-synonymous genetic variants is essential for uncovering disease associations and mechanisms of evolution. An in-depth understanding of sequence changes is also fundamental for synthetic protein design and stability assessments. However, the variant effect predictor performance gain observed in recent years has not kept up with the increased complexity of new methods. One likely reason for this might be that most approaches use similar sets of gene and protein features for modeling variant effects, often emphasizing sequence conservation. While high levels of conservation highlight residues essential for protein activity, much of the variation observable in vivo is arguably weaker in its impact, thus requiring evaluation at a higher level of resolution. Here, we describe functionNeutral/Toggle/Rheostatpredictor (funtrp), a novel computational method that categorizes protein positions based on the position-specific expected range of mutational impacts: Neutral (weak/no effects), Rheostat (function-tuning positions), or Toggle (on/off switches). We show that position types do not correlate strongly with familiar protein features such as conservation or protein disorder. We also find that position type distribution varies across different protein functions. Finally, we demonstrate that position types can improve performance of existing variant effect predictors and suggest a way forward for the development of new ones.
INTRODUCTION
Mapping molecular function or pathogenicity effects of genomic variation is crucial to our understanding of evolutionary, pharmacological, and disease mechanisms. Recent decades have seen significant advances in high-throughput experimentation, as well as growing sophistication in the analyses of the resulting data for research and medical purposes (1–3). However, our understanding of genomic variation is still lacking. For example, separate studies totalling over 7,500 individuals (4,5), have found that less than three percent of known disease-causing variants can actually be deemed actionable pathogenic variants. On the other hand, known disease-causing variants have been noted (6,7) in the (likely) healthy individuals of the 1000 Genome Project (8). Here, a key problem is the absence of an experimental gold standard for identifying disease-causing variants (4). Thus, identifying disease-association of the ∼10 000 protein sequence changing genetic variants of every individual (9) is like looking for the proverbial needle in a haystack.
Focusing on an arguably better-defined task of finding variants that alter protein function may help; however, variant effects are not all black and white, having a range of outcomes (10). While some variants may only marginally alter ligand affinity, others can induce drastic changes (11). Moreover, while subtle molecular modifications are difficult to detect, they can cause phenotypic changes if they occur in concert with other mutation-driven changes (12,13). Experimental techniques like deep mutational scanning (DMS) (14) allow for simultaneous assessment of the effects of hundreds of thousands of variants. DMS combines high throughput sequencing with the ability to create large protein libraries, i.e. uniting high throughput selection and high throughput sequencing methods. Still, large-scale mutant library generation is limited by a number of factors, such as bias in sequencing preparation, difficulties in designing accurate and meaningful screening methods (i.e. deciding which changes are evaluated), as well as significant time and cost requirements (15,16). Thus, it remains infeasible to experimentally assess, for example, the effects of all non-synonymous Single Nucleotide Polymorphisms (nsSNPs) of a given individual, much less a population. However, large-scale mutational fitness landscapes resulting from DMS analyses are an exciting resource for the development of new accurate computational variant effect predictors (17).
Single amino acid substitutions caused by nsSNPs are often associated with specific traits (18–20), diseases (21,22), and pharmacological responses (23). Moreover, targeted mutagenesis of specific protein sites is an essential tool in the synthetic biology toolkit (24). Given the broad range of their possible applications, it is not surprising that many computational algorithms for the prediction of single amino acid substitution effects have been developed (>200; as of January 2018). The different approaches range in algorithm complexity (e.g. random forests (25) or meta-servers (26)), training/development datasets (e.g. cancer (27) or stability changes (28)), and gene/protein features used (e.g. conservation or protein structure (29–31)). However, there is much room for progress (32–35), and despite their increasing number and complexity, there has, arguably, not been a significant improvement in prediction accuracy over the last decade.
Our collaborators have previously established a classification of protein sequence position types (36) - Toggle and Rheostat -in accordance with the effects of mutations in each position. Mutations in Toggle positions were mostly severely disruptive of protein function, while mutations in Rheostat positions had a broad range of effects. We further demonstrated (37) that existing computational predictors fall short of accurately differentiating between neutral (no-effect) and non-neutral (effect) mutations in the two position types. For example, at a Toggle position, mutations that have been experimentally shown to have no effect on protein function, were often computationally identified as having an effect by most predictors. We thus concluded that knowledge of position type could potentially improve prediction accuracy.
In an earlier work, protein sequence positions were characterized as Toggles or Rheostats on the basis of the distribution of their experimentally validated variant effects (38). However, experiments evaluating variant effects are still very limited even in comparison to the number of experiments annotating, for example, protein function or localization (e.g. 558 590 proteins in UniProtKB/Swiss-Prot (39), release September 2018). Moreover, trivially, for the purposes of computational variant effect predictors, once the variant effect is experimentally determined, its prediction becomes irrelevant. In other words, having to experimentally establish the position type precludes using it as a feature in a variant effect predictor.
Here, we present a new machine learning approach, functionNeutral/Toggle/Rheostatpredictor (funtrp), which identifies protein sequence position types using a curated set of sequence-based features. funtrp categorizes sequence positions based on the expected range of mutational impacts possible at each position; i.e. at Neutral positions most variation will have no or weak effect, at Rheostat positions a range of effects is possible (i.e. functional tuning) and at Toggle positions mostly strong effects are expected. We found that protein regions important for molecular functionality are enriched in Rheostats and Toggles, with the latter dominating crucial residues (e.g. catalytic sites). While these findings are in line with the conservation landscape, we observed lower than expected correlation between conservation and position types, particularly for Rheostats. Curiously, we found that the distribution of position types varied across protein classes, slightly differentiating enzymes from non-enzymes and distinguishing enzyme functional classes. We also showed that the predicted position types correlated with the manually curated experimental effect annotations for proteins extracted from the Protein Mutant Database (PMD) (40,41); i.e. we were able to fairly accurately predict the effects of variants in previously unseen proteins simply by considering their funtrp-predicted position type. Combining funtrp annotations with outputs of existing variant effect predictors further improved prediction accuracy.
These findings suggest that knowledge of position types is critical for evaluating functional effects of variants. Thus, funtrp predictions could aid the development of improved variant effect prediction methods.
MATERIALS AND METHODS
The funtrp training/development process is detailed in Figure 1. Training datasets are summarized in Supplementary Table S1. The DOIs for all funtrp source data, Python code, and Docker image of the final pipeline are listed in the Availability section below.
Figure 1.

Schematic overview of the funtrp pipeline. In training, experimentally measured variant effect scores are extracted for all residues present in the selected deep mutational scanning (DMS) datasets. These scores are used in the k-means clustering-based variant effect-labeling (VEL) step to initially label a subset of all positions(residues) as either Toggle or Neutral. Annotated with a computed set of sequence-based features, the subset of labeled positions is then used to train a Random Forest (RF) (52) classifier (ntModel) to predict the not yet labeled positions from the DMS datasets as either Toggle, Neutral, or Rheostat. After filtering, these are combined with the original VEL-positions and used with the same set of sequence-based features to train the final funtrpModel.
Training datasets and feature extraction
We extracted quantitative deep mutational scanning (DMS) amino acid substitution effect data for five proteins (Table 1) (42–46). For a given protein-coding gene, DMS generates a large set of mutations and their impact estimates. The DMS results for the five proteins used here for model training, were a subset of 41 studies, which we were able to identify in the literature. Studies were excluded from consideration for a number of reasons, including (i) unavailability of raw data (16 proteins), (ii) raw data that is difficult/impossible to extract due to formatting (e.g. pdf files, four proteins), (iii) experimental metrics incompatible with our approach (e.g. binary effect classifications or effect evaluation for multiple instead of single mutants) (six proteins) and (iv) missing information about wildtype and knockout functionality (three proteins). We also excluded studies whose reported measurements were not directly in line with our focus on functional effects, i.e. reporting changes at different expression levels or in different media types (three proteins). Finally, as the position types in the first step of our study were annotated based on the distribution of their variant effects, we also excluded proteins with the majority of positions having too few (<6) variants (four proteins). In total, our five selected proteins comprised 822 amino acids and 11 130 substitutions with measured effect scores. We removed from consideration the two unknown amino acids (labeled X in sequence), leaving 820 residues. Note that the number of available experimental scores per residue varied between and within datasets.
Table 1.
Deep mutational scanning datasets used in funtrpModel training and testing
| Gene | Domain | Organism | Measured Activity | Set | Variants | Positions | SNP-p* | VEL** | funtrp*** |
|---|---|---|---|---|---|---|---|---|---|
| BRCA1 | RING | H. sapiens | Ubiquitin ligase activity | train | 3080 | 303 | 142 | 42 | 128 |
| PAB1 | RRM | S. cerevisiae | mRNA binding specificity | train | 1188 | 75 | 75 | 34 | 53 |
| UBE4B | U-box | H. sapiens | Ubiquitin ligase activity | train | 926 | 102 | 80 | 31 | 50 |
| TEM-1 | - | E. coli | Ampicillin resistance | train | 5469 | 286 | 282 | 95 | 163 |
| SPG1 | GB1 | Streptococcus sp. | Binding affinity to IgG | train | 467 | 56 | 14 | 5 | 9 |
| PTEN | - | H. sapiens | Protein stability | test | 3880 | 357 | 144 | 40 | 256 |
| TPMT | - | H. sapiens | Protein stability | test | 3756 | 241 | 169 | 40 | 189 |
| HSP90 | ATPase | S. cerevisiae | Yeast growth | test | 4231 | 210 | 207 | 159 | 201 |
(*) Positions with ≥6 SNP-possible variants (**) Variant effect-labeled (VEL) Neutral and Toggle positions used in ntModel training
(***) VEL- and ntModel labeled Neutral, Toggle, and Rheostat positions used in funtrpModel training
To test our model, we collected three additional DMS datasets, covering the PTEN, TPMT and HSP90 proteins (47,48) which were NOT used in training. Note, that for HSP90 the knockout variant effect measures were not directly available; we thus approximated knockout scores as the mean of the unnormalized effect scores (0.15) reported for variants in eight critical positions, i.e. those where any variant resulted in severe functional impact (R32, E33, N37, D40, G94, G118, G121, G123).
For each protein, the effects (scores) of substitutions (including the knockout variant scores) were then converted into absolute distances to wildtype, without differentiating beneficial and deleterious mutations (Equation 1).
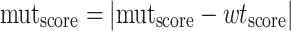 |
(1) |
We further computed ten sequence-based features (Table 2 and Supplementary Table S2) for each protein. Features were chosen based on biological relevance to reflect a broad range of properties associated with protein function. These features include basic amino acid properties, as well as structural properties generated using a virtualized (Docker) version of PredictProtein (with default parameters) (49). Note that for all homology searches, PredictProtein uses an 80% redundancy reduced database combining UniProt and the Protein Data Bank (PDB) (50) (see Availability section).
Table 2.
Set of sequence-based features used by prediction models
| id | Feature | Source | ReliefF** | Rank |
|---|---|---|---|---|
| 1 | Solvent Accessibility | PROF (*) | 0.18 | 3 |
| 2 | Secondary Structure | PROF (*) | 0.12 | 6 |
| 3 | Residue Flexibility | PROFbval (*) | 0.15 | 4 |
| 4 | Protein Disorder | MD (*) | 0.22 | 2 |
| 5 | Amino Acid | - | 5e-5 | 8 |
| 6 | Residue Size | - | 0 | 10 |
| 7 | Residue Charge | - | 1e-7 | 9 |
| 8 | SNP possible | - | 7e-4 | 7 |
| 9 | Conservation | ConSurf (*) | 0.34 | 1 |
| 10 | MSA Ratio | - | 0.14 | 5 |
(*) tools in the PredictProtein pipeline (Yachdav et al., 2014).
(**) Features ranked by importance using ReliefF (Kononenko, RobnikSikonja, & Pompe, 1996). Secondary structure scores were reported per position for helix, sheet, and loops (pH, pE, and pL). Feature descriptions and default parameters are detailed in Supplementary Table S2.
Toggle and Neutral variant effect-based labeling
For this stage in funtrp development we only considered SNP-possible substitutions, i.e. those amino acid substitutions requiring no more than one nucleotide change with respect to the wildtype. Note that we did NOT go back to the gene sequence to find the affected codon, but rather designated as SNP-possible any single nucleotide wildtype to mutant amino acid codon changes. As SNPs are more common than multi-nucleotide changes, using only the SNP-possible variants more closely mirrors natural selection patterns. Additionally, this approach excludes non-SNP-possible variants, i.e. the likely more severe results of multiple mutagenesis rounds. Note that only half of the variants in our set (5423 of 11 130) were SNP-possible. We set aside the 171 (21%) positions with three to five variants (FewVariants set) and removed from all further consideration the 56 (7%) positions with only one or two variants. We further used the remaining 593 positions (72% of 820) with at least six SNP-possible variants in our dataset (4769 variants across the five proteins; Table 1).
To each protein's set of experimental variant scores, the protein specific wildtype and knockout scores were added. K-means clustering (51) (with K = 3) was used to partition each individual protein variant set into three clusters. Variants assigned to the same cluster as the knockout score were labeled severe. Those assigned to the cluster containing the wildtype score were deemed neutral. All other variants were labeled intermediate.
We subdivided the protein sequence positions into Neutral and Toggle types on the basis of their variant labels. We previously defined Toggles (37) as positions intolerant of any change, while Neutrals are defined here as positions that can tolerate almost all substitutions without functional changes. Each sequence position x was classified (Equation 2) as Toggle or Neutral. If all but at most one variant at x were neutral, we labeled x Neutral (N; 153 positions). If all but at most two variants were severe, we labeled x a Toggle (T; 66 positions). If none of these two conditions held true, x was deemed unknown (374 positions; Unknown set).
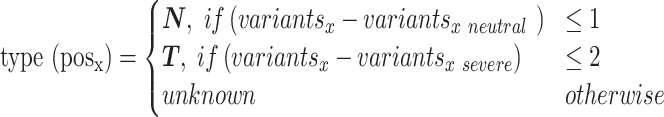 |
(2) |
We excluded all unknown positions from our set as well as six Toggle and six Neutral positions with a noticeably higher score variance and/or different score medians as compared to other positions of the same type. We thus retained a conservative training set of variant effect-labeled (VEL) Toggle and Neutral positions with comparable variance and medians of experimental variant scores (207 instances: 60 Toggles, 147 Neutrals).
ntModel and Neutral/Toggle scoring
Using the VEL set we trained a Random Forest (RF) (52) classifier (ntModel) to predict Toggle vs. Neutral position types on the basis of the ten features described above (Table 2). To account for the bias towards the Neutral type in the training set, we used over-sampling and trained our model on a balanced input set comprising 414 instances (200% of the unbalanced input). We evaluated the model performance using Leave-One-Out-Cross-Validation (LOO-CV). The model prediction scores were in the [0, 1] range, such that the sum of all type scores equaled 1. The LOO-CV predictions were used to determine prediction score type thresholds. We limited the number of false positive Toggle or Neutral predictions to ≤3% (Figure 2). The resulting thresholds were set at score ≤0.1 for Neutral and score ≥0.8 for Toggle predictions.
Figure 2.
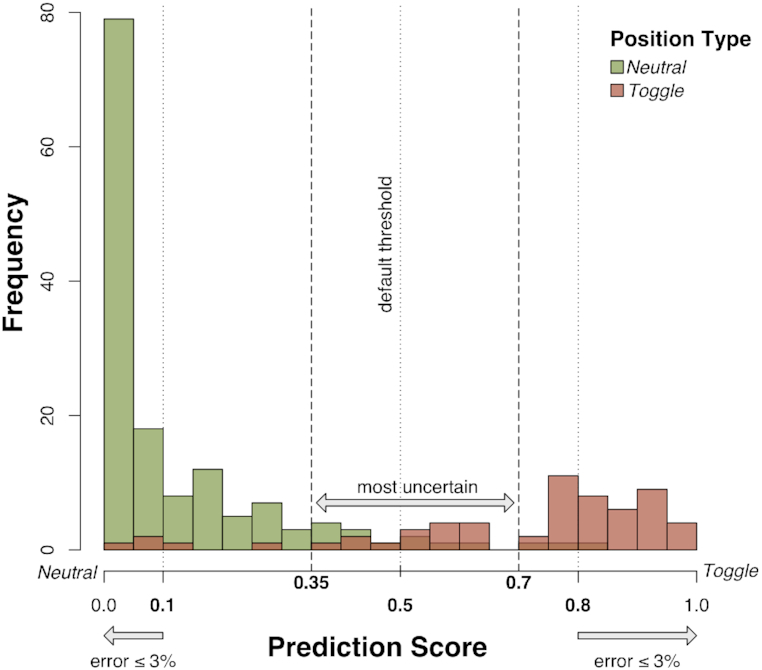
Determination of ntModel thresholds. LOO-CV predictions of the ntModel were used to determine individual position type prediction score thresholds. Positions with a Neutral label assigned in the VEL step are shown in green, those labeled as Toggle -in red. Thresholds were set at score ≤0.1 = Neutral and score ≥0.8 = Toggle, limiting the number of false positive Toggle or Neutral predictions to ≤3%. Positions with prediction scores in the range [0.35, 0.7] (containing 50% of all incorrect predictions of the ntModel) were defined as Rheostats.
Defining Rheostats
Here we defined Neutral and Toggle positions for the development of our methods as positions containing mostly neutral and mostly severe variants, respectively. Rheostats, however, are positions of functional tuneability, encompassing positions that could contain a wide range of neutral, intermediate, and severe effects. Thus, they can not be defined explicitly by the number and type of variants they house. We also can not simply define them ‘by exclusion’ as non-(Toggle or Neutral) positions, as our VEL-driven Toggle/Neutral position classification was based on the analysis of incomplete experimental data. Effect mislabeling of even a single variant and/or missing variants, could easily transform Rheostats defined ‘by exclusion’ into Toggles or Neutrals.
Our ntModel was able to precisely distinguish Toggles and Neutrals. The few incorrect predictions that it did make were a likely indication of initial mislabeling of Rheostat positions as one of the other two types. We thus hypothesized that the ntModel predictions in the [0.35, 0.7] range (containing ∼50% Neutral/Toggle mispredictions) were enriched in Rheostat positions.
funtrpModel and remaining residue labeling
The FewVariants (171 positions) and the Unknown (374 positions) sets comprised 545 (66% of 820) yet-unlabeled positions. We ran the ntModel and used score thresholds as defined above to assign the N, R and T predictions to each position. Note that we excluded ntModel predictions in ranges 0.1 to 0.35 and 0.7 to 0.8, leaving only the most reliably predicted positions.
Within each protein, Toggle and Neutral position variant score distributions were compared between ntModel and (variant effect-labeled) VEL-based assignments (Figure 3 and Supplementary Figure S1). We retained only those ntModel-Neutral positions whose variant experimental score medians were less than or equal to the highest median score of the VEL-Neutrals. Similarly, the ntModel-Toggles were retained only if their experimental score medians were more than or equal to the lowest median score of VEL-Toggles. We retained only those Rheostats whose medians were in-between the highest VEL-Neutral and lowest VEL-Toggle median scores. The resulting positions (72 Neutrals, 20 Toggles, 104 Rheostats) were added to the VEL set to form the funtrpTraining set (403 positions: 219 Neutrals, 80 Toggles, 104 Rheostats).
Figure 3.

Distributions of experimental effect scores for E. coli TEM-1 protein positions. Positions are colored by assigned position type (green = Neutral, blue = Rheostat, red = Toggle) and ordered by the median of the associated variant score distribution. VEL-classified positions are shown in solid color. Those predicted by the ntModel are translucent - darker when the number of variants per position is ≥6 and lighter otherwise. The dashed horizontal lines represent data set-specific knockout (red) and wildtype (green) scores. Positions removed during manual and automatic filtering steps are not shown. Details for the remaining four proteins are available in Supplementary Figure S1.
The funtrpTraining set was used to build funtrpModel, a RF classifier trained as described above for ntModel, i.e. using the same ten features, over-sampling-based class balancing (806 instances; 200% of the unbalanced input set), and LOO-CV evaluation.
The per position prediction score for the funtrpModel was in the [0,1] range for each position type (N, R or T), such that the sum total of all type scores equaled 1. By default, each position was assigned to the highest-scoring type.
Measuring model performance
Performance for both ntModel and funtrpModel was reported as accuracy, precision, recall, and F-measure (F1 score, Equation 3). For each position type (N, R or T) at every score cutoff, true positives (TP) were the correctly predicted position types. For each type (e.g. N), false positives (FP) were the positions incorrectly predicted to be of that type (e.g. R or T positions, predicted as N), while false negatives (FN) were that type positions incorrectly predicted as something else (e.g. N positions, predicted as R or T).
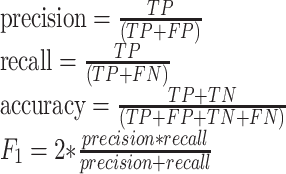 |
(3) |
To establish a random baseline for funtrpModel performance we generated random predictions at each position in our funtrpTraining set. Specifically, three scores were randomly sampled from a uniform distribution in the [0,1] range and each was divided by the sum of the three, resulting in Neutral, Rheostat and Toggle predictions that add up to 1, analogous to our model. The highest score determined the predicted position type for the random predictor.
funtrp pipeline implementation
We used a Java based implementation of the Random Forest Classifier (WEKA, version 3.8) (52,53) to build funtrpModel and R (version 3.3.3) (54) for K-Means clustering, performance and significance evaluations, and for visualizations. Protein features were computed using a Docker image of the PredictProtein (49) pipeline. The funtrp prediction pipeline, which runs all necessary funtrpModel feature extractions and the model itself, requires Python version 3.6 or later and is available as stand-alone version and as a webservice. Docker image DOIs as well as those of the current software release, source code and datasets can be found in the Availability section.
Predicting position types in protein sets
Neutral, Rheostat, and Toggle position types were predicted for various sets of proteins (Supplementary Table S3). All 20 410 manually curated (Swiss-Prot) human proteins were extracted from the UniProt Knowledgebase (UniProtKB release September 2018) (39). For 5% of these proteins (909; 32 enzymes and 877 non-enzymes) the required set of funtrp input features in Table 2 could not be computed due to PredictProtein pipeline problems or compute limitations. The remaining 19 501 sequences were processed with funtrp using clubber (55) to distribute computation among multiple High-Performance Cluster (HPC) environments. The subsets of this data were as follows:
The EXPV set: 1250 Swiss-Prot enzymes with experimentally validated, unique, unambiguous EC (Enzyme Commission) numbers (56).
All human enzymes with catalytic site annotations from the M-CSA database (57), which also have binding site annotations in UniProt (94 proteins; 419 catalytic and 214 binding sites).
A set of sahle spheres (crucial for metal binding; defined as all residues within a 15Å radius sphere from the geometric center of the metal ligand (58)) extracted from 231 transition metal binding protein structures in the PDB (Bromberg et al., unpublished data). PDB structures were mapped to UniProt; funtrp predictions were available for 230 of these.
Disordered (6309) versus ordered (13 192) Swiss-Prot proteins (labeled disordered if at least 50% of residues were predicted disordered by the MetaDisorder predictor; MD score threshold of ≥0.5 (59)).
Proteins containing variants with experimental effect annotations in the PMD database (40,41) were also collected. We extracted 16 038 variants in 1224 proteins, along with their SNAP (30), SIFT (29) and PolyPhen-2 (31) predictions of effect from (60). We also extracted precomputed effect predictions of Envision (17), a recently developed method trained on DMS datasets. Note, that Envision predictions where transformed into a binary effect classification by labeling predictions with scores ≥0.9 as no-effect and those <0.9 as effect. funtrp predictions were available for 1220 of these proteins (the remaining four sequences failed feature extraction). Within this set variants are experimentally labeled as either benign (neutral), or having an intermediate (mild, moderate) or strong effect (severe) on protein function. We further extracted from the VarCards database (61) the binary effect (deleterious) versus no-effect (benign) predictions of 23 computational predictors. VarCards predictions could be determined for 8,800 PMD variants in 1042 proteins. For variants with fewer than 23 predictions available, we assigned random prediction scores (uniformly distributed) and generated random binary predictions (>0.5 = effect, ≤0.5 = no-effect). We defined the per variant Ensemble Prediction Ratio as the number of effect predictions divided by 23 (the number of predictors). Thus, a ratio above 0.5 (≥12/23) results in an ensemble prediction of effect, while a ratio below 0.5 (≤11/23) results in a no-effect prediction. Finally, we defined predictions based on the Ensemble Prediction Ratio as either correct (the ensemble prediction agreed with the annotated PMD effect) or as incorrect (ensemble prediction in disagreement with PMD effect annotation). Note, that Envision effect predictions were not available for 34 of 8800 variants (overlap of VarCards and PMD).
Statistical evaluations
We calculated the standard error of the mean across proteins in different subsets for all three position types. For each subset, we randomly resampled 50% of the proteins (without replacement) 100 times. For each protein we extracted the fraction of each position type (N, R or T) and computed the type means and standard errors across protein subsets for the individual position (Equation 4; σ = standard deviation of means across subsets; N = total sampling iterations = 100).
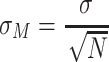 |
(4) |
Distributions of feature scores for the three position types were analyzed for similarity using the one-way analysis of variance (ANOVA) test.
RESULTS AND DISCUSSION
funtrp accurately recognizes position types
Both RF classifier models were evaluated using LOO-CV (Equation 3; Supplementary Table S4 A,B) using the label with the highest score as the prediction. The ntModel achieved an overall accuracy of 92.3% while the funtrpModel accuracy was 85.1% (Table 3 and Figure 4; also better than random accuracy of 55.5%). Interestingly, the funtrpModel differentiated Toggles and Neutrals better (97.8% accuracy) than the ntModel. However, its performance decreased when considering the additional Rheostats. Note that the higher prediction scores of the funtrpModel correlated with higher precision (reliability) of the predictions, albeit lower recall.
Table 3.
Performance of funtrp models for training and independent test sets
| ntModel | funtrpModel | random | PTEN | TPMT | HSP90 | ||
|---|---|---|---|---|---|---|---|
| Precision | Neutral | 0.94 | 0.90 | 0.53 | 0.84 | 0.91 | 0.90 |
| Rheostat | - | 0.73 | 0.28 | 0.70 | 0.64 | 0.34 | |
| Toggle | 0.88 | 0.88 | 0.20 | 0.87 | 0.88 | 0.73 | |
| Recall | Neutral | 0.95 | 0.91 | 0.32 | 0.87 | 0.90 | 0.73 |
| Rheostat | - | 0.77 | 0.36 | 0.78 | 0.71 | 0.62 | |
| Toggle | 0.85 | 0.80 | 0.35 | 0.73 | 0.83 | 0.67 | |
| Overall Accuracy | 92.3% | 85.1% | 55.5% | 79.3% | 84.1% | 70% | |
Model performance was evaluated separately for training sets using Leave-One-Out-Cross-Validation (LOO-CV) and three independent test sets not seen before. Performance measures were calculated using Equation (3).
Figure 4.
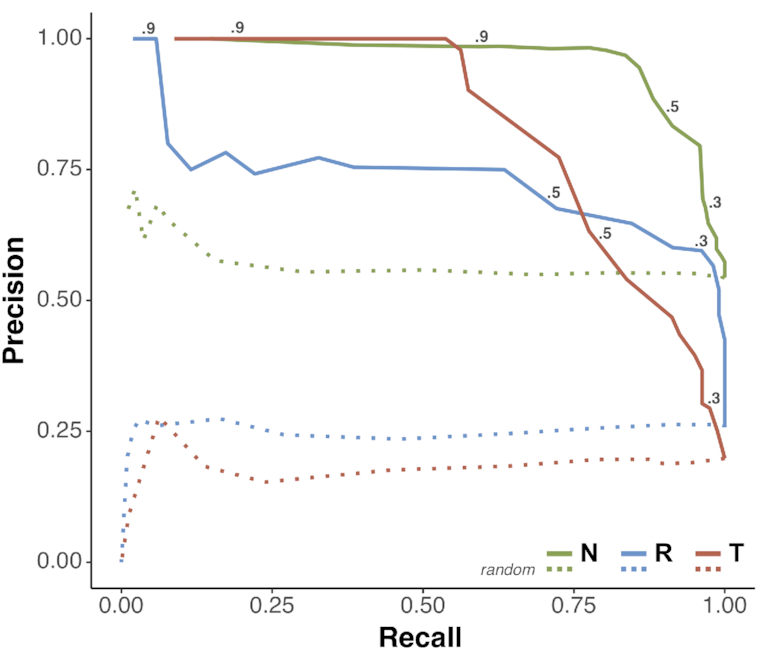
funtrp type prediction performance. Precision-Recall curves for LOO-CV predictions of Neutral (green), Rheostat (blue) and Toggle (red) positions for the funtrpModel (solid lines) and random predictions (dashed lines). The funtrpModel performance for all three position types is indicated at different cutoffs. Performance was calculated per position type vs. the other two types combined.
A funtrpModel performance discrepancy among position types was also expected. After all, while Toggles and Neutrals are explicitly defined, Rheostats are not. As such, they encompass a much larger range/variability in residue properties. In our training set, a position containing three intermediate variants would be as much a Rheostat if it additionally contained three neutral variants or three severe ones. Additionally, truly neutral mutations are often subjective (as opposed to mild effect variants) and always more difficult to identify, experimentally or computationally, than severe ones. Thus, the differentiation between Rheostat and Neutral positions is arguably more complex even when using experimental data. Indeed, the majority (80%; 16 of 20) of the incorrectly predicted Rheostats were labeled Neutral and more than half of these predictions were unreliable (scores in the 0.4–0.49 range). Similarly, of the incorrectly predicted Neutral positions, 80% (19 of 24) were labeled as Rheostats.
Position type labeling is robust in additional proteins
Our position labels are not experimentally derivable and we, thus, do not have a gold standard set of Neutrals, Rheostats, and Toggles to truly evaluate funtrp performance. How could we then know whether our method performs well for proteins that it has not seen? To address this concern, we assumed that our initial position labeling is correct, i.e. SNP-possible filtering plus VEL and ntModel (Materials and Methods) results can be used as the ground truth for new DMS datasets never used in training (PTEN, TPMT and HSP90). We found that the distribution of identified position types across these sets (646 positions: 51% Neutrals, 27% Toggles, 22% Rheostats) was very similar to that of funtrpTraining (54% Neutrals, 20% Toggles, 26% Rheostats). The minor discrepancy in the type ratios between sets was likely due to the absence of filtering for the test set. Note that filtering the training dataset (on the basis of variant experimental effect variance; Materials and Methods) affected Toggles the most (74% excluded) while Rheostats were affected least (31% excluded) (Supplementary Table S1).
We further used funtrp to predict the test set position types. We found that funtrp predictions could re-create the (VEL- and ntModel) position labels with an average accuracy of 75.9% over all three datasets. The PTEN and TPMT DMS datasets both reporting structural stability, could be predicted with accuracies of 79.3% and 84.1%, respectively, comparable to the overall funtrp performance of 85.1% (Table 3). This performance on new data suggests, that the DMS datasets selected for training were sufficiently diverse for our model training. Larger training sets are often preferred as they include a larger number of feature-observation combinations, resulting in more complete models. However, adding more data can also introduce new biases and increase noise, and potential errors. Thus, as expected, the results of the re-prediction of position types for both PTEN and TPMT suggest that adding either protein to training would not drastically change our model performance.
Unfortunately, for HSP90 funtrp was only 70% accurate (Table 3). We note that as opposed to directly measuring the effect of mutations on protein function and/or structure, the HSP90 dataset reflects how mutants in Hsp90, a chaperone protein, impact the growth rate of budding yeast. This reduced performance is thus very likely due to two critical experimental design decisions. First, chaperones assist in correct (un)folding of other proteins. The measured and reported effect of yeast growth is therefore not directly coupled to the mutagenized protein function, but rather to its ability to facilitate function of other proteins. Second, the reported fitness measure of overall growth rate is more removed from direct variant effect than what was evaluated for other proteins in our set, i.e. ligand binding affinity or protein stability. These observations can likely explain why the re-labeling of HSP90 could lead to the observed model variability and highlight the need for standardized selection of experimental training data.
Position types are not ticks on the same effect scale
The concept and characteristics of Rheostat and Toggle positions were first introduced in earlier work by our collaborators (36). The key finding of that work highlighted Rheostats as positions that can be altered to produce variation in protein functionality. We further elaborated on the associated problem of computationally predicting variant effects across different position types (37). Computational methods were unable to differentiate effect severity, as opposed to effect vs. no-effect: in Toggles variants are nearly guaranteed to be of severe effect, while in Rheostats the effect can be varied. Thus, they mispredicted most neutral variants in Toggle positions and many of the mild effect variants in Rheostat positions. Here we additionally introduced the Neutral position type, aiming to highlight positions where most variants are neutral. Note that using a single scale of protein position types was not possible because (i) if the scale enumerated the number of variants of any effect size, Rheostats and Toggles would be grouped together opposite of Neutrals and (ii) if the scale represented the number of severe effect variants, both Neutrals and Rheostats would be at one end and Toggles at another. A totality-of-variant-effect scale, as opposed to counting variants, could run from 0 (Neutral) to 1 (Toggle) but raises the question of how to evaluate the scores in-between. At 0.5, for example, half of the variants may have a severe effect and half be neutral OR all variants can have an intermediate effect. The first situation is indeed halfway between Neutral (most variants are neutral) and Toggle (most have a severe effect) - but is, arguably, biologically rare. The second, on the other hand, indicates a true Rheostat, but does not even belong on the same scale as intermediate effects are not part of Toggle or Neutral understanding. That is, a position that scores 0.4 is just as much a Rheostat as the one that scores 0.6, as opposed to being more Neutral in the first case and more Toggle in the other. Instead of using a single scale, our three-state classification scheme allows for a measure of prediction reliability of each position type, i.e. a Rheostat with a prediction score of 0.6 (75% precision; Figure 4), is more likely truly a Rheostat than one with a score of 0.4 (47% precision).
Individual sequence-based features are not sufficient to describe position types
Using the ReliefF (62) feature selection algorithm we ranked the importance of funtrp features for labeling sequence positions in Swiss-Prot (Table 2). As expected, evolutionary conservation was ranked most important. However, the assigned weight was only slightly higher than other important features: protein disorder, solvent accessibility, and residue flexibility. These results suggest that none of these features alone can explain the predicted position types.
Conservation is widely used as an approximation for residue importance (63,64); i.e. the more conserved a residue is, the higher the likelihood that its substitution by another amino acid will result in function/structure disruption. We compared position-specific conservation scores defined by ConSurf (65) across all positions of experimentally verified enzymes (EXPV); by default, ConSurf uses up to 150 homologous sequences to build its multiple sequence alignments (MSA). As expected, these scores were significantly different between the three position types (Figure 5; medians in bold; ANOVA P-value < 2e−16). ConSurf scores are normalized by default, so that the average score over all residues of one protein is zero, and the standard deviation is one; here, lower scores indicate more conserved residues. Toggle positions were predominantly conserved while Neutral positions were mostly non-conserved. Rheostats, however, were in-between the other position types and often showed similarly high conservation as the Toggles. Moreover, on average, significantly fewer sequences were aligned at Neutral positions (107) compared to Rheostats (125) and Toggles (128).
Figure 5.
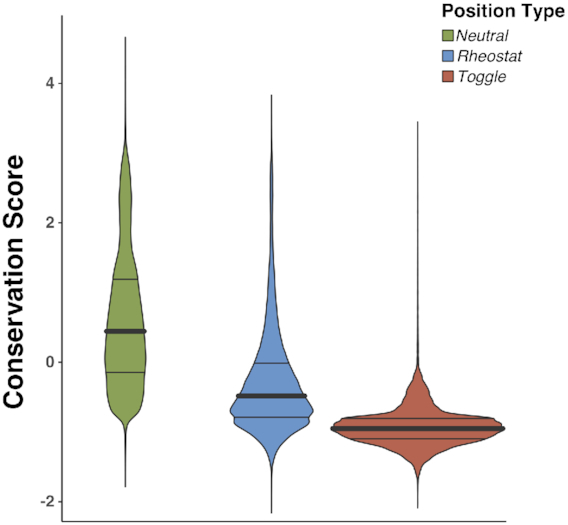
Sequence conservation does not fully reflect position types. Violin plots of sequence conservation (ConSurf) compared across position types; distribution medians are highlighted in bold. ConSurf scores are by default normalized, such as 0 depicts the average score over the entire protein and the standard deviation is 1.
To further establish how well a predictor of position types could perform using conservation alone, we computed the number of positions in Swiss-Prot proteins that could be correctly identified as a funtrp Rheostat, Toggle or Neutral at a fixed cutoff. The lowest cutoff (lower score = more conserved) for Neutrals was selected by taking the mean of the distribution medians of Neutral and Rheostat conservation scores. Similarly, the highest cutoff for Toggles was at the mean of Rheostat and Toggle conservation score medians. Rheostats were assigned all other conservation scores. The overall accuracy for this thresholding was 61% (Neutrals = 0.80/0.70, Toggles = 0.45/0.80, Rheostats = 0.44/0.39 precision/recall, respectively; Supplementary Table S5). Note, that we observed the same trends for the funtrpTraining dataset (Supplementary Figure S2).
Thus, evolutionary conservation - despite being the highest-ranking feature - was not solely representative of position types. Furthermore, none of the remaining features was likely to perform better than conservation indicated by their consistently lower ReliefF rankings (Table 2). Moreover, arguably, for a given position in a given protein establishing the conservation thresholds for each of the three types would be infeasible as the number of available homologs used for an alignment, as well as evolutionary distances between them, vary by protein family.
Position type profiles differ across protein classes
Swiss-Prot (Figure 6A) enzymes had proportionately more Toggle and fewer Neutral positions than non-enzymes. However, there was no difference in the number of Rheostats between enzymes and non-enzymes. As Rheostats allow for functional and evolutionary flexibility while adapting to different environments, the latter result is expected. The increase of Toggles in enzymatic proteins, i.e. positions critical for defining protein activities: active sites, ligand specificity, etc., is very likely due to enzymes having evolved to implement a set of very specific functionalities. These mutation sensitive key positions are thus enriched in comparison with non-enzymatic proteins. Note that this increase in Toggles at the expense of the reduction in Neutral sites is unlikely due to resolution limits of the funtrp predictor, as this would likely produce fewer (more Toggle-similar) Rheostats.
Figure 6.

Distribution of position types per protein class. Distributions are based on the entire Swiss-Prot (A) and EXPV sets (B). Colors are according to position type (green = Neutral, blue = Rheostat, red = Toggle). Percentages in (A) are rounded to the nearest integer and thus do not add up to 100%. Fractions in (B) are averaged on a per-protein basis and differ significantly among enzyme classes.
We further compared the mean per-protein fraction of position types between the six main enzyme classes of the experimentally annotated EXPV set: Oxidoreductases (EC 1), Transferases (EC 2), Hydrolases (EC 3), Lyases (EC 4), Isomerases (EC 5) and Ligases (EC 6). Although the general trend of more Neutrals than Rheostats than Toggles was maintained across all enzyme classes, the classes differed significantly in the actual fractions per position type (Figure 6B). For all EC classes, Toggles made up less than a quarter of all positions per protein, suggesting that enzymes are fairly robust to mutation. We observed similar trends for the full Swiss-Prot dataset (Supplementary Figure S3), with slight differences in position type distributions likely explained by the latter dataset sequence redundancy. Note that the EXPV proteins are experimentally annotated and, thus, tend to be less redundant (98% of the sequences are <90% sequence similar).
Distribution of position types varies by residue function
We compared the distribution of position types for catalytic sites, binding sites, and other residues in Swiss-Prot enzymes (Figure 7A). Note, that here we included only the 47 proteins containing both binding and catalytic site annotations, which were non-overlapping, i.e. annotated in different positions of the protein.
Figure 7.

Distribution of position types across protein sites. (A) Enzymes, (B) Metal binding proteins. Colors are according to position type (green = Neutral, blue = Rheostat, red = Toggle). Percentages are rounded to the nearest integer and thus do not add up to 100%.
As expected, the majority of catalytic sites were Toggles and only 1% were Neutral. Note, that Rheostat and Neutral prediction scores were rather low on average (0.54 and 0.6 respectively), suggesting that these are unreliable. Note, however, there is sufficient evidence in the literature highlighting variation that is compatible with persistent, albeit changed, functionality of enzymes (66,67). Thus, it is entirely possible that some of the catalytic sites are in fact Rheostats by our definition. Binding sites were less frequently Toggles than catalytic sites, but much more frequently so than the other residues, which were predominantly Neutral. Curiously, unlike fractions of Neutrals and Toggles, the fraction of Rheostats across catalytic sites, binding sites, and the other residues set was similar, likely indicating the presence of allosterically relevant residues present outside critical binding/active sites.
Notably the catalytic site primary actors - the charged amino acids (D, E, R, K, H; Supplementary Figure S4) (68) were unexpectedly low among the Toggles and Rheostats of the other residues; i.e. they were very important in functional sites, but not as relevant elsewhere in the protein. This finding is particularly interesting in the light of the generic assumptions made about irreplaceability of charged residues. Outside the functional sites, the more commonly structure-relevant large hydrophobic amino acids (C, W, Y, M, F) were most often Toggles, while the smaller (A, I, L, V) were enriched in Rheostats (Supplementary Figure S4).
Distribution of position types varies by metal-ligand binding proximity
We evaluated the composition of position types of residues located in the proximity of metal-containing ligands (sahle 3D-structure spheres, Methods) for Swiss-Prot proteins. As for functional sites above, we defined three sets of residues: those annotated in Swiss-Prot as metal binding, sahle sphere residues within 15Å of the ligand center, and other residues (Figure 7B). We excluded from consideration any residues annotated as metal binding and not located within a sahle sphere.
Metal binding residues showed a similar distribution of position types as catalytic sites (80% Toggle, 5% Neutral). Notably, sahle spheres were more enriched in Rheostats (38%) than were the binding sites described above (26%). However, the latter were more frequently Toggles (59%) than the former (44%). This result suggests that binding sites are critical features of function, while sahle spheres encompass residues relevant to functional flexibility. Moreover, outside of sahle spheres Toggles were the least abundant and more than half of the residues were Neutral, suggesting that most of the other residues are significantly less involved in protein function.
Preferred residues for metal binding are C, H, D and E (69), which is confirmed by our data (Supplementary Figure S5). Toggles were the dominant position type for all of these amino acids except glutamic acid (E), which were mostly Neutrals or Rheostats. One possible explanation for the observation that the variants affecting glutamic acids only slightly impact protein function, is the length of its side chain, which introduces greater flexibility and allows for a larger range of possible substitutions than a more rigid structure.
Position type profiles enable identification of disordered proteins
Based on MetaDisorder predictions (Methods) we labeled 6309 Swiss-Prot proteins as disordered and 13 192 as ordered and compared the ratios of position types between these sets. The two classes of proteins were clearly separable by distribution of position types (Supplementary Figure S6).
Ordered proteins contained more than twice as many Toggles as disordered proteins (19% vs. 8%), while disordered proteins were preferentially Neutral (68% versus 46%). Of the 668 proteins, where Neutrals made up over 80% of all residues, 94% (650) were disordered. This result is, to a certain extent, expected due to frequent modulation of function, i.e. Rheostatic activity, achieved via structural changes; e.g. changes in residue solvent accessibility or secondary structure may, and often do, modulate functionality (70). However, this finding may also indicate that disordered proteins are poorly predicted by funtrp, as our method relies on structural features. Another hypothesis based on this observation may be that our definition of position types is not directly applicable to disordered proteins, where changes in functionality may be harder to objectively measure and evaluate.
Experiments focus on high impact variants
We evaluated the relationship of position types with experimental annotations of variant effects extracted from the literature (PMD effect annotations as reported in (60)). Note that the number of PMD variants was the same across position types, i.e. 33% affecting Neutrals, 33% Rheostats, and 34% Toggles. Note that, as mentioned above, position type ratios per protein are not at all similar, i.e. Swiss-Prot proteins had, on average, ∼53% Neutral positions, ∼33% Rheostats and ∼14% Toggles. This emphasis on Toggles in variant distribution suggests a strong preference in experimental studies towards evaluating the most likely severe variants and/or the most likely functionally or structurally important regions, in contrast to the unguided DMS approach.
Based on PMD effect annotations, variants could be categorized into three main experimentally-defined impact groups: neutral, mild/moderate, and severe (Supplementary Figure S7). In line with the above reasoning, there were more severe variants (43%) than mild/moderate variants (36%), and significantly more of either than of the neutral variants (20%). We further evaluated the PMD variant-affected position types. As expected, most of the Toggle positions (90%) had variants of at least some impact (severe or mild/moderate; 58% severe only). The fraction of Rheostat positions having non-neutral variants was slightly lower (80% any effect; 50% severe only). However, even as much as a third (35%) of the Neutral positions had severe variants (67% any impact). This high level of variant impacts across all evaluated protein positions underlines the exaggerated specific selection in experimental studies for expected-to-be-observed impact.
These observations of the bias in the reported variant impacts highlight the need for variant effect predictors to take into account or, at least, be mindful of, their effect-focused training/testing/development data.
Position types can improve variant effect prediction
Changing the perspective to examine variant localization per position we observed that roughly half (52%) of 3254 experimentally defined neutral variants were in funtrp predicted Neutral positions, while 18% affected Toggles (Supplementary Figure S7). Of the severe (6872) variants 41% affected Toggles and were least abundant in Neutrals (25%). The variants in the mild/moderate group were nearly evenly distributed (33%, 32%, 35% Neutrals, Rheostats, Toggles, respectively) across all three position types. Note that finding some neutral variants in Toggle positions and some severe variants in Neutral positions is not unexpected, as our position type definitions allow for some variety of effects. However, because funtrp has not been trained to recognize variant effects, the dominant trend of finding variants of expected impact in the right position types, i.e. neutral variants in Neutrals and severe variants in Toggles, highlights our method's ability to recognize functionally relevant protein positions.
The VarCards Ensemble Prediction Ratio/Score (Methods), which reflects the agreement of commonly used variant effect prediction tools, correlated with the severity of PMD impacts (Figure 8) across position types; i.e. there were more experimentally defined non-neutral (mild/moderate and severe) variants at higher ensemble prediction scores than at lower ones, while the opposite was true for experimental neutral variants. The no-effect recall of the ensemble predictor was highest at Neutral positions (0.66 in Neutral, 0.44 in Rheostats, and 0.23 in Toggles; Figure 8 and Supplementary Table S6). In Toggles, the ensemble predictor more frequently incorrectly identified the experimentally non-neutral variants as being no-effect than the experimentally neutral variants (0.30 no-effect precision). This suggests that no-effect predictions at Toggle positions are less reliable than at Neutral positions (F1no-effect = 0.48 and = 0.26, in Neutrals and Toggles respectively; Equation 3). As expected, the prediction of neutral variants at Rheostat positions is less reliable than in Neutrals, but more reliable than in Toggle positions (F1no-effect = 0.39 in Rheostats).
Figure 8.

VarCards ensemble predictions correlate with PMD variant experimental effects in corresponding funtrp position types. Each column reflects the fraction (y-axis) of the correct (blue) and incorrect (red) ensemble predictions (x-axis) per PMD variant effect group (light = neutral, medium = mild/moderate, dark = severe). The ensemble prediction ratio signifies the fraction of tools predicting variant effect (no-effect prediction = 0/23, all methods predict effect = 23/23). Thus, more correct no-effect predictions are made in Neutral positions, while effect predictions are better at Toggle positions.
Due to the limited number of experimental neutrals in PMD, they made up only a small fraction of all effect predictions per position type (effect precision 0.93 in Toggles, 0.89 in Rheostats, and 0.80 in Neutrals). However, effect recall was still significantly higher in Toggles than elsewhere (0.95 in Toggles, 0.85 in Rheostats, and 0.56 in Neutrals), suggesting that effect predictions at Toggle positions are more reliable than at Rheostat or Neutral positions (F1effect = 0.66, = 0.87, = 0.94 in Neutrals, Rheostats, and Toggles respectively)
To demonstrate the potential impact of position type knowledge on individual variant effect predictors we evaluated the per position type performance for effect and no-effect predictions of SNAP, SIFT, PolyPhen-2 and Envision. As with VarCards scores, effect predictions were consistently better at Toggle compared to Neutral positions (Figure 9, left panel) while no-effect predictions were better at Neutral compared to Toggle positions (Figure 9, right panel). Notably, this was the case for all three traditional variant effect predictions methods as well as for Envision, i.e. the more recent DMS data trained approach. These findings unambiguously show that incorporating position types leads to much more reliable variant effect prediction.
Figure 9.
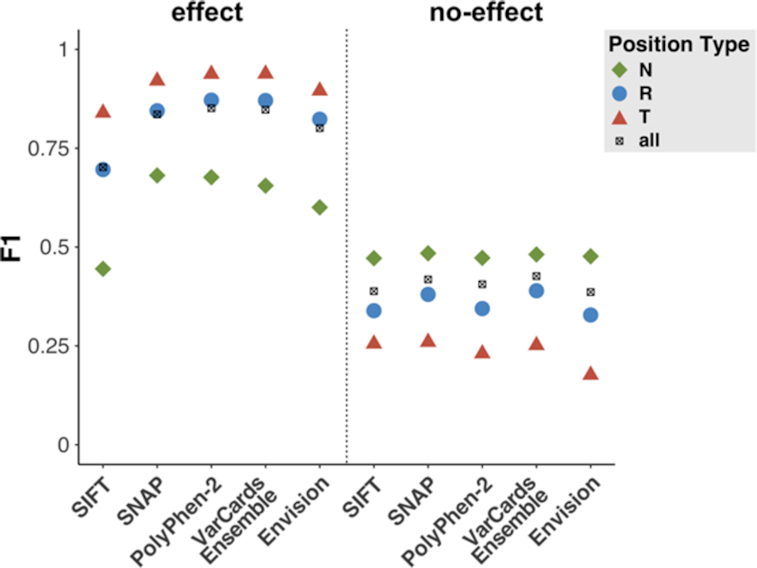
Performance of variant effect predictors significantly improves when considering affected position type. Performance (F1 score) of five variant effect predictors for effect (left panel) and no-effect (right panel) predictions of PMD variants, evaluated overall (black crosses = all) and per position type (green = Neutral, blue = Rheostat, red = Toggle).
To further highlight the relationship between funtrp position type predictions and annotated variant effects, we calculated the experimental neutral vs. non-neutral ratios per type across a range of funtrp prediction scores (Figure 10). In line with the above results, we found that reliably predicted Toggle positions were more likely to have more non-neutral variants (a lower ratio of neutral to non-neutral variants), while reliably predicted Neutrals had more neutral variants (a higher ratio).
Figure 10.
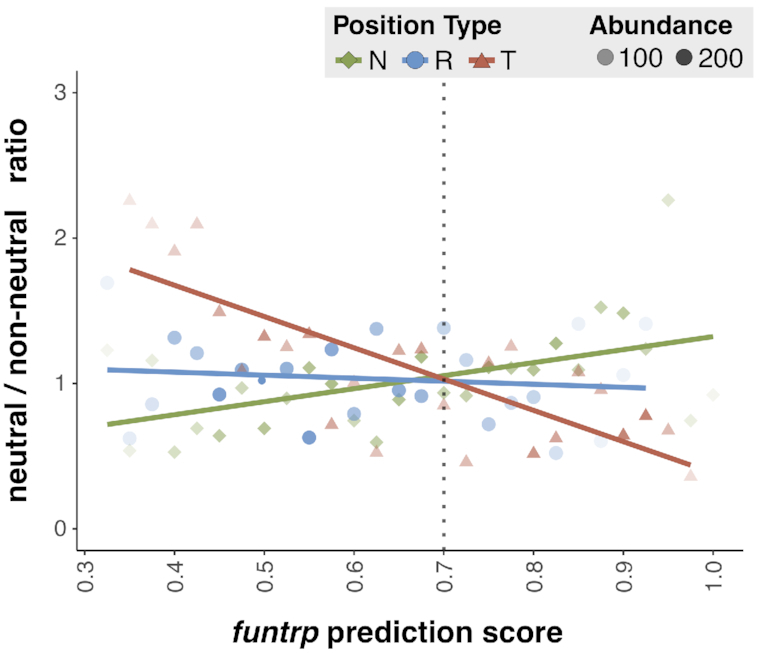
funtrp predictions correlate with PMD effect annotations. Ratio of experimentally neutral vs. non-neutral PMD variants (y-axis) per position type (green = Neutral, blue = Rheostat, red = Toggle) at respective funtrp prediction scores (x-axis). Higher number of positions at a certain score is represented by more opaque dots. Trendlines are shown in the position type color scheme.
Thus, we suggest that variant effect predictors could improve significantly if trained/developed separately for each funtrp position-type and/or accounting for the reliability of position type prediction. We expect that prediction could be most improved for Rheostats, where increased resolution is likely once the obvious Toggle and Neutral variants are no longer the main focus. We note that Rheostat positions are the most likely proverbial evolutionary fireplaces (60), i.e. locations where a multitude of tiny changes optimize a functionality best fit to the particular environment. Tracing the conversion of Rheostats into Neutrals or Toggles across homologs can likely highlight the evolutionary paths taken or currently in place for any given molecular functionality. Thus, our new definition of position types will likely contribute to the understanding not only of biophysics of protein folding and related epistatic mutation effects, but will also highlight prime candidate residues for directed evolutionary pathways, and help shine light on pathogenicity mechanisms.
DATA AVAILABILITY
The Java based implementation of Random Forest Classification is part of the WEKA library (53) and can be found at https://sourceforge.net/projects/weka.
R is a free software environment for statistical computing and graphics and can be downloaded at https://www.r-project.org.
The PredictProtein Docker image used for feature generation can be found at https://doi.org/10.5281/zenodo.3018245. The latest release is available via Docker Hub (bromberglab/predictprotein). The source code is accessible at https://bitbucket.org/bromberglab/predictprotein.
The Big80 sequence database used in the PredictProtein pipeline is available at http://rostlab.org/rost-db-data/big, prefix big_80.
The funtrp prediction pipeline Docker image used in this work can be found at https://doi.org/10.5281/zenodo.3020352. The latest release is available via Docker Hub (bromberglab/funtrp). The source code is accessible at https://bitbucket.org/bromberglab/funtrp.
funtrp training data can be found at https://doi.org/10.5281/zenodo.3066344.
funtrp is also available as webservice at https://services.bromberglab.org/funtrp.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Liskin Swint-Kruse (University of Kansas Medical Center) for all help and comments that made this work possible. We are also grateful to Dr Predrag Radivojac (Northeastern University), Dr Jay A. Tischfield, Dr Gary Heiman, Dr Chengsheng Zhu, Dr Yannick Mahlich, Yanran Wang and Zishuo Zeng (all Rutgers University) for all discussions. We further would like to thank the LZ (Munich) and Dr Sonakshi Bhattacharjee (Columbia University) for discussions and help with the manuscript. We would also like to express gratitude to all people who deposit their data into publicly available databases and to those who maintain them.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the National Institutes of Health [U01 GM115486 to M.M. and Y.B., R01 MH115958 01 to M.M]; and the NASA Astrobiology Institute CAN-8 [NNH17ZDA003C to Y.B.]. P.C.K. acknowledges anonymous private support. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bruse S., Moreau M., Bromberg Y., Jang J.H., Wang N., Ha H., Picchi M., Lin Y., Langley R.J., Qualls C. et al.. Whole exome sequencing identifies novel candidate genes that modify chronic obstructive pulmonary disease susceptibility. Hum. Genomics. 2016; 10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ellinghaus D., Zhang H., Zeissig S., Lipinski S., Till A., Jiang T., Stade B., Bromberg Y., Ellinghaus E., Keller A. et al.. Association between variants of PRDM1 and NDP52 and Crohn's disease, based on exome sequencing and functional studies. Gastroenterology. 2013; 145:339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turner T.N., Hormozdiari F., Duyzend M.H., McClymont S.A., Hook P.W., Iossifov I., Raja A., Baker C., Hoekzema K., Stessman H.A. et al.. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am. J. Hum. Genet. 2016; 98:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dorschner M.O., Amendola L.M., Turner E.H., Robertson P.D., Shirts B.H., Gallego C.J., Bennett R.L., Jones K.L., Tokita M.J., Bennett J.T. et al.. Actionable, pathogenic incidental findings in 1,000 participants' exomes. Am. J. Hum. Genet. 2013; 93:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amendola L.M., Dorschner M.O., Robertson P.D., Salama J.S., Hart R., Shirts B.H., Murray M.L., Tokita M.J., Gallego C.J., Kim D.S. et al.. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015; 25:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cassa C.A., Tong M.Y., Jordan D.M.. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum. Mutat. 2013; 34:1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N. et al.. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012; 91:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Birney E., Soranzo N.. Human genomics: The end of the start for population sequencing. Nature. 2015; 526:52–53. [DOI] [PubMed] [Google Scholar]
- 9. Bromberg Y. Building a genome analysis pipeline to predict disease risk and prevent disease. J. Mol. Biol. 2013; 425:3993–4005. [DOI] [PubMed] [Google Scholar]
- 10. Swint-Kruse L. Using evolution to guide protein engineering: the devil is in the details. Biophys. J. 2016; 111:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walker I.H., Hsieh P.C., Riggs P.D.. Mutations in maltose-binding protein that alter affinity and solubility properties. Appl. Microbiol. Biotechnol. 2010; 88:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zabalza R., Nurminen A., Kaguni L.S., Garesse R., Gallardo M.E., Bornstein B.. Co-occurrence of four nucleotide changes associated with an adult mitochondrial ataxia phenotype. BMC Res. Notes. 2014; 7:883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kowarsch A., Fuchs A., Frishman D., Pagel P.. Correlated mutations: a hallmark of phenotypic amino acid substitutions. PLoS Comput. Biol. 2010; 6:e1000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fowler D.M., Araya C.L., Fleishman S.J., Kellogg E.H., Stephany J.J., Baker D., Fields S.. High-resolution mapping of protein sequence-function relationships. Nat. Methods. 2010; 7:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fowler D.M., Fields S.. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014; 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Araya C.L., Fowler D.M.. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 2011; 29:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gray V.E., Hause R.J., Luebeck J., Shendure J., Fowler D.M.. Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Syst. 2018; 6:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duffy D.L., Montgomery G.W., Chen W., Zhao Z.Z., Le L., James M.R., Hayward N.K., Martin N.G., Sturm R.A.. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am. J. Hum. Genet. 2007; 80:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Box N.F., Wyeth J.R., OGorman L.E., Martin N.G., Sturm R.A.. Characterization of melanocyte stimulating hormone receptor variant alleles in twins with red hair. Hum. Mol. Genet. 1997; 6:1891–1897. [DOI] [PubMed] [Google Scholar]
- 20. Shastry B.S. SNPs: impact on gene function and phenotype. Methods Mol. Biol. 2009; 578:3–22. [DOI] [PubMed] [Google Scholar]
- 21. de Ligt J., Veltman J.A., Vissers L.E.. Point mutations as a source of de novo genetic disease. Curr. Opin. Genet. Dev. 2013; 23:257–263. [DOI] [PubMed] [Google Scholar]
- 22. Kumar R., Arioz C., Li Y., Bosaeus N., Rocha S., Wittung-Stafshede P.. Disease-causing point-mutations in metal-binding domains of Wilson disease protein decrease stability and increase structural dynamics. Biometals. 2017; 30:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halushka M.K., Walker L.P., Halushka P.V.. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin. Pharmacol. Ther. 2003; 73:122–130. [DOI] [PubMed] [Google Scholar]
- 24. Sun X.J., Hu Z., Chen R., Jiang Q.Y., Song G.H., Zhang H., Xi Y.J.. Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci. Rep-UK. 2015; 5:10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D. et al.. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Capriotti E., Altman R.B., Bromberg Y.. Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics. 2013; 14(Suppl. 3):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Douville C., Carter H., Kim R., Niknafs N., Diekhans M., Stenson P.D., Cooper D.N., Ryan M., Karchin R.. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013; 29:647–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Capriotti E., Fariselli P., Casadio R.. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005; 33:W306–W310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ng P.C., Henikoff S.. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bromberg Y., Rost B.. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007; 35:3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X.. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015; 24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mahmood K., Jung C.H., Philip G., Georgeson P., Chung J., Pope B.J., Park D.J.. Variant effect prediction tools assessed using independent, functional assay-based datasets: implications for discovery and diagnostics. Hum. Genomics. 2017; 11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monzon A.M., Carraro M., Chiricosta L., Reggiani F., Han J., Ozturk K., Wang Y., Miller M., Bromberg Y., Capriotti E. et al.. Performance of computational methods for the evaluation of Pericentriolar Material 1 missense variants in CAGI-5. Hum. Mutat. 2019; 49:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller M., Wang Y., Bromberg Y.. What went wrong with variant effect predictor performance for the PCM1 challenge. Hum. Mutat. 2019; 40:1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meinhardt S., Manley M.W. Jr, Parente D.J., Swint-Kruse L.. Rheostats and toggle switches for modulating protein function. PLoS One. 2013; 8:e83502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller M., Bromberg Y., Swint-Kruse L.. Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci. Rep. 2017; 7:41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hodges A.M., Fenton A.W., Dougherty L.L., Overholt A.C., Swint-Kruse L.. RheoScale: A tool to aggregate and quantify experimentally determined substitution outcomes for multiple variants at individual protein positions. Hum. Mutat. 2018; 39:1814–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The UniProt, C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishikawa K., Ishino S., Takenaka H., Norioka N., Hirai T., Yao T., Seto Y.. Constructing a protein mutant database. Protein. Eng. 1994; 7:733. [DOI] [PubMed] [Google Scholar]
- 41. Kawabata T., Ota M., Nishikawa K.. The protein mutant database. Nucleic Acids Res. 1999; 27:355–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Starita L.M., Young D.L., Islam M., Kitzman J.O., Gullingsrud J., Hause R.J., Fowler D.M., Parvin J.D., Shendure J., Fields S.. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics. 2015; 200:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melamed D., Young D.L., Gamble C.E., Miller C.R., Fields S.. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013; 19:1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Starita L.M., Pruneda J.N., Lo R.S., Fowler D.M., Kim H.J., Hiatt J.B., Shendure J., Brzovic P.S., Fields S., Klevit R.E.. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Firnberg E., Labonte J.W., Gray J.J., Ostermeier M.. A comprehensive, high-resolution map of a gene's fitness landscape. Mol. Biol. Evol. 2014; 31:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu N.C., Olson C.A., Sun R.. High-throughput identification of protein mutant stability computed from a double mutant fitness landscape. Protein Sci. 2016; 25:530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pejaver V., Babbi G., Casadio R., Folkman L., Katsonis P., Kundu K., Lichtarge O., Martelli P.L., Miller M., Moult J. et al.. Assessment of methods for predicting the effects of PTEN and TPMT protein variants. Hum. Mutat. 2019; 40:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mishra P., Flynn J.M., Starr T.N., Bolon D.N.A.. Systematic mutant analyses elucidate general and client-specific aspects of Hsp90 function. Cell Rep. 2016; 15:588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Honigschmid P., Schafferhans A., Roos M., Bernhofer M. et al.. PredictProtein–an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014; 42:W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The protein data bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lloyd S.P. Least-squares quantization in PCM. IEEE Trans. Inform. Theory. 1982; 28:129–137. [Google Scholar]
- 52. Breiman L. Random forests. Mach Learn. 2001; 45:5–32. [Google Scholar]
- 53. Smith T.C., Frank E.. Introducing machine learning concepts with WEKA. Methods Mol. Biol. 2016; 1418:353–378. [DOI] [PubMed] [Google Scholar]
- 54. R Core Team R Foundation for Statistical Computing. 2015; Vienna. [Google Scholar]
- 55. Miller M., Zhu C., Bromberg Y.. clubber: removing the bioinformatics bottleneck in big data analyses. J Integr. Bioinform. 2017; 14:doi:10.1515/jib-2017-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mahlich Y., Steinegger M., Rost B., Bromberg Y.. HFSP: high speed homology-driven function annotation of proteins. Bioinformatics. 2018; 34:i304–i312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ribeiro A.J.M., Holliday G.L., Furnham N., Tyzack J.D., Ferris K., Thornton J.M.. Mechanism and Catalytic Site Atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2018; 46:D618–D623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Senn S., Nanda V., Falkowski P., Bromberg Y.. Function-based assessment of structural similarity measurements using metal co-factor orientation. Proteins. 2014; 82:648–656. [DOI] [PubMed] [Google Scholar]
- 59. Schlessinger A., Punta M., Yachdav G., Kajan L., Rost B.. Improved disorder prediction by combination of orthogonal approaches. PLoS One. 2009; 4:e4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bromberg Y., Kahn P.C., Rost B.. Neutral and weakly nonneutral sequence variants may define individuality. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14255–14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li J., Shi L., Zhang K., Zhang Y., Hu S., Zhao T., Teng H., Li X., Jiang Y., Ji L. et al.. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018; 46:D1039–D1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kononenko I., RobnikSikonja M., Pompe U.. ReliefF for estimation and discretization of attributes in classification, regression, and ILP problems. Fr. Art. Int. 1996; 35:31–40. [Google Scholar]
- 63. Shakhnovich E., Abkevich V., Ptitsyn O.. Conserved residues and the mechanism of protein folding. Nature. 1996; 379:96–98. [DOI] [PubMed] [Google Scholar]
- 64. Capra J.A., Singh M.. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007; 23:1875–1882. [DOI] [PubMed] [Google Scholar]
- 65. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N.. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016; 44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Silva F.S., Moss B.. Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: catalytic site mutations reduce virulence but not virus replication in cultured cells. J. Virol. 2003; 77:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Song J., Zhang Z., Hu W., Chen Y.. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J. Biol. Chem. 2005; 280:40122–40129. [DOI] [PubMed] [Google Scholar]
- 68. Bartlett G.J., Porter C.T., Borkakoti N., Thornton J.M.. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002; 324:105–121. [DOI] [PubMed] [Google Scholar]
- 69. Cao X., Hu X., Zhang X., Gao S., Ding C., Feng Y., Bao W.. Identification of metal ion binding sites based on amino acid sequences. PLoS One. 2017; 12:e0183756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Studer R.A., Dessailly B.H., Orengo C.A.. Residue mutations and their impact on protein structure and function: detecting beneficial and pathogenic changes. Biochem. J. 2013; 449:581–594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Java based implementation of Random Forest Classification is part of the WEKA library (53) and can be found at https://sourceforge.net/projects/weka.
R is a free software environment for statistical computing and graphics and can be downloaded at https://www.r-project.org.
The PredictProtein Docker image used for feature generation can be found at https://doi.org/10.5281/zenodo.3018245. The latest release is available via Docker Hub (bromberglab/predictprotein). The source code is accessible at https://bitbucket.org/bromberglab/predictprotein.
The Big80 sequence database used in the PredictProtein pipeline is available at http://rostlab.org/rost-db-data/big, prefix big_80.
The funtrp prediction pipeline Docker image used in this work can be found at https://doi.org/10.5281/zenodo.3020352. The latest release is available via Docker Hub (bromberglab/funtrp). The source code is accessible at https://bitbucket.org/bromberglab/funtrp.
funtrp training data can be found at https://doi.org/10.5281/zenodo.3066344.
funtrp is also available as webservice at https://services.bromberglab.org/funtrp.