

Abstract
Aging is a mysterious process, not only controlled genetically but also subject to random damage that can accumulate over time. While DNA damage and subsequent mutation in somatic cells were first proposed as drivers of aging more than 60 years ago, whether and to what degree these processes shape the neuronal genome in the human brain could not be tested until recent technological breakthroughs related to single-cell whole-genome sequencing. Indeed, somatic single-nucleotide variants (SNVs) increase with age in the human brain, in a somewhat stochastic process that may nonetheless be controlled by underlying genetic programs. Evidence from the literature suggests that in addition to demonstrated increases in somatic SNVs during aging in normal brains, somatic mutation may also play a role in late-onset, sporadic neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease. In this review, we will discuss somatic mutation in the human brain, mechanisms by which somatic mutations occur and can be controlled, and how this process can impact human health.
Introduction
The genome is under relentless attack by environmental and endogenous mutagens. Astonishingly, it is estimated that up to 120 000 chemical lesions occur within the ~ 6.5 gigabases of the human genome per day (1). Despite the highly efficient DNA repair pathways present in human cells, this onslaught sometimes results in somatic DNA mutations (Fig. 1). The idea that mutation load impacts the fitness of organisms traces back to Haldane (2), and in Escherichia coli, reproductive fitness decreases relative to controls in proportion to the number of random mutations induced in the E. coli genome (3). Mutations are dangerous because the genome is foundational to all programs that cells of the body must execute, and so mutations have the potential to rewrite the genetic information encoded in DNA, bestowing potentially beneficial, but more likely neutral or deleterious changes to that code. This presents a unique hazard in the human brain, where the primary functional cell type, the neuron, is postmitotic and cannot be replaced during life by a pool of stem cells.
Figure 1.
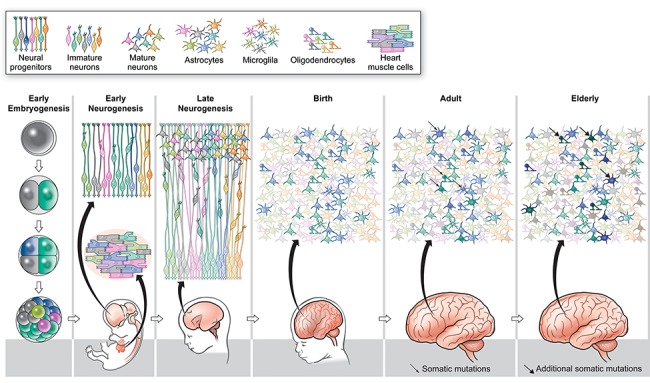
Somatic mutation generates mosaicism in the human brain. During early embryogenesis, cells of the embryo acquire somatic mutations generating identifiable clones of cells (green, blue, pink and lime cells). These early clones distribute across the body, but somatic mutations that arise in progenitors during early neurogenesis (yellow cell giving rise to an orange cell) are restricted to the brain. In late neurogenesis, marked progenitors divide asymmetrically to generate newborn neurons, which bear clonal somatic mutation reflecting their developmental origins. As a result, at birth the brain has a polyclonal architecture. In the adult and elderly brain, cells continue to accumulate somatic mutations (monochromatic green neurons become variegated shades of green, blues transition to shades of blue, etc.), such that each postmitotic cell has a unique genome.
Aging can be defined as ‘the progressive accumulation of changes with time that are associated with or responsible for the ever-increasing susceptibility to disease and death which accompanies advancing age’ (4). Late-onset, sporadic neurodegenerative diseases, such as Alzheimer’s disease (ad) and Parkinson’s disease (PD), share advanced age as their most common risk factor, yet the mechanisms by which age and disease risk interact are unknown, and the ultimate cause of neuronal loss in AD and PD remains elusive. Molecular mechanisms of aging are manifold (5) and can be broadly grouped in two categories: genetically encoded programs, for example genes that specify different life spans between organisms, the homologs of which may play a role in human longevity (6) and entropy, or random wear and tear of the systems of the body (7).
In this review, we will discuss the process of somatic mutation as it relates to aging in the brain and neurodegenerative disease. As a process, somatic mutation displays features of both random entropy and programmed structure, suggesting it may be involved in aging in many ways.
Classes of Somatic Mutation Identified in the Brain
The brain is profoundly mosaic, because several classes of mutation exist across the 100 billion neurons within each human brain (Table 1). Our understanding of mosaic mutations in individual cells has advanced rapidly in recent years, owing to advances in single-cell and targeted sequencing technologies (Table 2) and matching bioinformatic innovations (32–35). Each human neuron appears to be marked by somatic single-nucleotide variants (SNVs) (11), being born with several hundred (12,22). Most somatic SNVs present in newborn neurons would be expected to be ‘clonal’, since if they occurred during development in a dividing progenitor cell, all descendants of that mutated founder would share that same mark, forming an identifiable clone in the body. Recently, we showed that somatic SNVs accumulate during life independent of cell division in neurons, such that by old age neurons in the prefrontal cortex (PFC) and dentate gyrus of the hippocampus (DG) bear thousands of more mutations than neurons in the newborn brain (12). Postmitotic mutations would be expected to be ‘non-clonal’, unique to the non-dividing neuron in which they arose. Interestingly, young adult mouse neurons have low somatic SNV counts, ranging from 42 to 162 per cell (21), suggesting species differences in lifetime accumulation of mutations. These data suggest that specific mutational processes shape the somatic genome of the human brain.
Table 1.
Classes of somatic mutations demonstrated in the mammalian brain.
| Variant Class | Technique | Species | Rate per cell | Features | References |
|---|---|---|---|---|---|
| SNV | Single-cell MDA | Human | ~800-2000 in adults | Transcriptional damage and meCpG deamination | (11) |
| SCNT | Mouse | 62-142 | Transcriptional damage and APOBEC | (21) | |
| Single-cell MDA | Human | Age, region, and disease-specific | Developmental, age-related, and disease mutation signatures | (12) | |
| Clonal expansion | Human | 200-400 per mid-gestration NPC | Distinct early embryogeneis and late neurogenesis signatures | (22) | |
| CNV | MDA | Human | 0.13-0.41 | Enriched for deletions | (17) |
| DOP-PCR | Human | 0.69 | Enriched for deletions | (13) | |
| DOP-PCR | Human | 0.09 | Enrichement in repetitive elements, enriched for deletions | (15) | |
| MDA-RC-Seq | Human | 0.2-0.3 | Only SLAVs, which are CNVs near LINE1 loci, considered. | (14) | |
| Transposon Insertions | MDA-L1IP | Human | 0.07 | (10) | |
| MALBAC-RC-Seq | Human | 13.7 | Enrichment in transcribed regions | (19) | |
| MDA-Seq | Human | 0.18 | (9) | ||
| SCNT | Mouse | 0-4 | (21) | ||
| MALBAC-RC-Seq | Human | 0.14-0.25 | Re-analysis of Upton 2015 dataset | (32) | |
| MDA-RC-Seq | Human | 0.2-0.3 | (14) | ||
| Aneuploidy | MDA | Human | 0.027 | (17) | |
| DOP-PCR | Human | <0.035 | (13) | ||
| DOP-PCR | Human/ Mouse | 0.022/ 0.01 | (16) |
Abbreviations: MDA, multiple displacement amplification; MALBAC, multiple annealing and looping based amplification cycles; SCNT, somatic cell nuclear transfer; L1-IP, LINE1 insertion profiling; RC-Seq, retrotransposon capture sequencing.
Table 2.
Methods to analyze mosaicism in the brain.
| Technique | Pros | Cons | References | ||
|---|---|---|---|---|---|
| Single cell amplification | MDA | phi29 polymerase uses strand displacement to achieve highly processive amplification of genomic DNA in an isothermal reaction | Low error rate for SNVs; coverage of most of the genome in long (10–50kb) amplicons | High copy-number noise at megabase scale | (8–14) |
| DOP-PCR | Fragmentation of DNA followed by ligation of universal adapters and PCR | Even copy-number profile at small scale | High SNV error rate; small amplicon size | (13,15–17) | |
| Hybrid PCR/ isothermal | Quasilinear preamplification using random primers followed by PCR amplification | Even copy-number profile at small scale | High SNV error rate; Short (0.5-1.5kb) amplicons. | (18–20) | |
| Clonal expansion/somatic cell nuclear transfer (SCNT) | Cellular DNA replication is used to amplify the genome of a single cell of interest. Proliferative cells can be grown clonally in culture. For terminal differentiated cells, the nucleus can be reprogrammed using SCNT. | Cellular replication machinery operates at much higher fidelity than chemical methods. | Clonal growth in culture is limited to proliferative cells. SCNT has very low efficiency and is not amenable to human cells for technical and ethical reasons. | (21,22) | |
| Trio sequencing | Standard whole-genome sequencing with family information to identify germline de novo and somatic mutations in probands | Avoids whole-genome amplification | Lack of single-cell resolution; low-fraction mosaics hard to distinguish from errors | (23–25) | |
| Enrichment | Transposon insertion mapping | PCR-based techniques that target degenerate sequences in transposable elements to identify novel transposon insertions | Highly efficient method for capturing known and unknown insertion sites | Extensive validation is needed to confirm insertion sites, due to presence of false-positive signals | (10,14) |
| RC-Seq | DNA is fragmented and transposon-containing fragments are captured using hybridization to transposon-specific probes | Captures full-length transposon-containing loci | Extensive validation is needed to confirm insertion sites, due to presence of false-positive signals | (19) | |
| Panel sequencing | Capture a set of specified loci of interest using array-based | Can sequence relevant loci at ultra-high depth, providing accurate estimates of even low-level mosaicism | Information at loci not represented on the panel is lost | (11,26–31) |
Single-cell genome amplification, the use of deep sequencing of bulk DNA and techniques used to profile specific regions of the genome are described. Abbreviations: MDA, multiple displacement amplification; DOP-PCR, degenerate oligonucleotide primer polymerase chain reaction; L1IP, LINE1 insertion profiling; RC-Seq, retrotransposon capture sequencing. Information regarding each technique obtained from references in the table, as well as (92) and (32).
In addition to point mutations, several single-cell sequencing studies have detected large-scale somatic mosaic mutations in the brain, including somatic copy number variants (CNVs) and whole-chromosome gains and losses (13,14,16,17,36,37) and somatic mobile element insertions (9,10,14,19,21,32) (Table 2). While less common than somatic SNVs (most neurons lack these variants), their large size suggests they could be important for overall brain physiology.
The Process of Somatic Mutation Displays Random and Programmed Features
Genosenium is the aging of the genome
Somatic SNVs accumulate in postmitotic neurons during life, a process termed genosenium, or genome aging (12). While DNA damage (38) and somatic mutation (39) were both hypothesized as drivers of aging over 60 years ago, this hypothesis could not be conclusively tested until the advent of single-cell, whole-genome sequencing (scWGS) and its application to human neurons of diverse ages. At birth, neurons of the PFC and DG average ~ 700 somatic SNVs per genome. A parallel study using clonal expansion of human neural progenitor cells (NPCs) estimated 200–400 somatic SNVs per genome at midgestation, strikingly dovetailing with results obtained by scWGS on postmortem brain tissue (22). During life, somatic SNVs slowly but inexorably accumulate in PFC and DG neurons in the normal brain, such that after age 80 PFC neurons bear on average ~ 2500 point mutations per genome, while DG neurons contain even more, ~ 4000 per cell. Obvious hot spots of somatic mutation—such as those that might be expected at specific gene promoters known to recurrently experience double-strand breaks (40)—have not been reported for somatic SNVs in the brain (11,12,21,22), nor for other classes of variant, suggesting a relatively even distribution of somatic mutations in the genome, albeit with a general predilection for more open areas of chromatin (11). Therefore, at the locus level, genosenium appears to be an entropic process whereby random damage throughout the genome generates somatic mutations over time, with any gene or intergenic region potentially falling victim to a permanent somatic mutation.
While selectively vulnerable or protected sites of the neuronal genome have yet been observed, analysis of molecular patterns of somatic mutations nominates specific damage and repair pathways as important mediators of somatic mosaicism. While PFC and DG neurons are born with overall similar numbers of somatic variants, somatic SNVs accumulate at different rates in PFC and DG during life, ~ 23 mutations per genome per year in PFC, but almost twice as fast, ~ 40 per year, in DG neurons (12). The PFC neurons analyzed were enriched for pyramidal subtypes, while DG experiments targeted granule neurons. Gene expression (41) and functional (42) differences between these cell types would suggest DNA damage and repair pathways that could mediate this difference. In two diseases characterized by progeria (accelerated aging) and early-onset neurodegeneration (loss of neurons in the brain), Cockayne syndrome type B (CS) Xeroderma pigmentosum (XP), somatic SNV rates were elevated (12), suggesting that in both normal individuals and in those afflicted by disease, somatic mutation rates and aging are tightly linked. CS and XP are both caused by germline loss-of-function mutations in specific nucleotide excision repair genes; the CS cases profiled by scWGS have confirmed mutations in CSB (ERCC6), while XP cases had mutations in either XPA or XPD (ERCC2). Since mutations were elevated in CS and XP neurons, we can conclude that CSB, XPA and XPD are part of a genetically encoded program that controls somatic mutation rates in neurons. Therefore, underlying biological differences across normal brain areas and between normal and diseased brains designate candidate genetic programs controlling somatic mutation rates.
Does genosenium occur in other areas of the body? Outside of the brain, single-cell and bulk DNA studies in the gastrointestinal tract (43,44), liver (43) and blood (45–47) have demonstrated an increase in somatic mutations during life in those organs. Single-neuron studies suggest that much of the accumulation of somatic mutations detected in these tissues, all of which actively proliferate or have the potential to, does not exclusively stem from errors during mitosis and may accumulate regardless of cell division.
Signature Analysis Allows for the Deconvolution of Mutational and Repair Processes
The biochemical reactions that cause mutations result in specific patterns of mutation in single cells, called mutational signatures (48). For example, exposure to mutagens present in tobacco smoke causes a specific cytosine to adenine (C>A) mutation signature in lung tumor genomes (49), while UV-light-induced di-pyrimidine tract C>T mutations mark the somatic genome in sun-exposed skin (50). Furthermore, DNA-damage repair (DDR) processes that prevent and repair somatic mutations also result in discernible mutation signatures. Transcription-coupled repair (TCR) is a DDR pathway that repairs DNA damage on the template strand actively transcribed loci, and because of the action of TCR, template strands accumulate mutations more slowly than non-template strands do in the germline (51) and somatic genome in cancer (52). Therefore, the analysis of mutational signatures is a way to uncover the causes of somatic mutations in the brain.
Somatic SNVs in the human brain are caused by at least several discreet processes. Using a mathematical technique called non-negative matrix factorization (NMF), the specific mutational signatures to contribute to the overall burden of mosaic variants in a dataset can be deduced (53). Applying NMF to the set of somatic SNVs discovered in 159 neurons from healthy and progeroid brains revealed three specific mutation signatures (12) (Fig. 2). Signature A was composed of primarily C>T and T>C mutations. Signature A mutation burden correlated with age, being infrequent in young neurons but more common in aged cells, and Signature A resembled a mutation signature in tumor genomes called Signature 5 (54), a clock-like mutation signature that correlates with tumor age-of-onset. A Signature-5-like signature was also observed in the normal human colon, small intestine and liver (43). Thus, a mutation clock is active not only in mitotic cells that give rise to tumors but also in postmitotic neurons, suggesting that Signature A/Signature 5 may identify clock-like mutations that ultimately may be found to operate independently of cell division.
Figure 2.
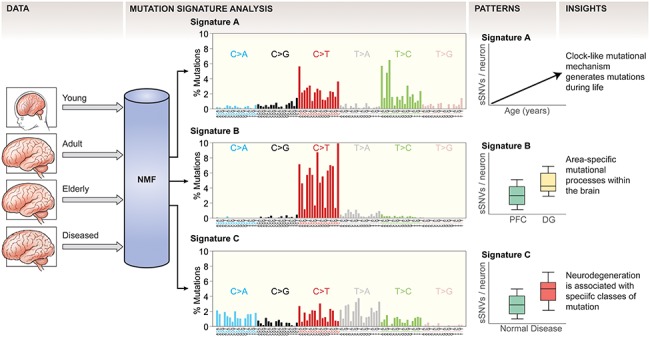
Mutation signature analysis identifies mutation causing pathways in the brain. Somatic mutation data from different brain samples is inspected by mutation signature analysis using NMF, which decomposes the complex spectrum of identified mutations in specific signatures. Each mutation signature is composed of 96 features, consisting of each of the six possible mutation types, subdivided into the 16 possible combinations of 5′ and 3′ base relative to the mutation. By analyzing the patterns by which these signatures distribute across different samples, biological insights can be derived regarding what processes cause somatic mutations. For example, the observation that Signature A mutations increase during aging suggests it is a mutational clock active in neurons. PFC, prefrontal cortex; DG, dentate gyrus of the hippocampus; sSNV, somatic single-nucleotide variant.
A second prominent signature found in neuronal SNVs, called Signature B, was comprised almost exclusively C>T mutations and was somewhat enriched in DG relative to PFC neurons. C>T transitions are a common artifact mode in whole-genome amplification (WGA), which is a necessary step prior to scWGS, so it must be interpreted with caution (11,33,55,56). However, C>T mutations are generally abundant in post-zygotic mutations discovered by non-WGA-based analyses of mutations in various tissues, for example the blood (57,58), skin (59), male reproductive tract (60), digestive tract (43,61) and liver (43), suggesting that C>T variants likely play a prominent role in shaping the somatic genome. Signature B mutation burden was constant during life in the PFC, but in the DG specifically Signature B mutations increased during aging. Thus, the mechanism that generates Signature B mutations is active across the brain prenatally and then becomes restricted to the DG after birth. Interestingly, single NPCs analyzed by clonal expansion (without chemical amplification) revealed a mutation signature that broadly resembled Signature B, in that it was enriched for C>T mutations, albeit in slightly different trinucleotide contexts (22), in agreement with Signature B mutations being at least partially developmental. Signature B was enriched in PFC neurons in CS patients but not in XP, suggesting that the CSB protein may be important for regulating the accumulation of mutations during development, while XPA and XPD are dispensable for that process.
Signature C mutations comprised many substitution types and contexts, but notably contained C>A mutations, which were largely absent in Signatures A and B. C>A mutations are a classic mark of DNA damage by reactive oxygen species (ROS), and oxidative stress is a hallmark of aging across the body (5). While Signature C weakly but significantly correlated with age in normal cells, Signature C mutations were highly elevated in the early-onset neurodegenerative disease neurons isolated from CS- and XP-patient brains, suggesting that the repair of ROS-mediated mutations may be compromised in these diseases. Further experimentation as to the exact cause of Signature C mutations is needed to validate that hypothesis, especially given the broad contribution of several mutation classes to Signature C. A second signature discovered using clonal expansion of human NPCs by Bae et al. was enriched for C>A mutations, specifically in those mutations that were found in single cells but did not validate in bulk tissues. These mutations were hypothesized to be late developmental mutations, potentially indicating that Signature C mutations begin to accumulate before birth.
Clonal Mutations, Non-Clonal Mutations and Late-Onset Neurodegeneration
AD and PD are debilitating neurodegenerative disorders that have a tremendous impact on the lives of affected individuals and their families. ad is the most common neurodegenerative disorder in the United States, affecting ~ 50% of adults by age 85 (62). A small proportion (1–2%) of ad cases result from fully penetrant, dominantly acting variants in PSEN1, PSEN2 and APP, which increase the amount of amyloid β peptide, which in turn forms pathogenic oligomers (63–66). More commonly, dosage of the APOE ε4 allele confers significantly increased risk of the disease (67). PD is the second most common late-onset neurodegenerative disorder with 2 and 1.3% lifetime risk frequencies for American men and women over age 40, respectively (68). PD is also primarily sporadic, with genetic familial forms accounting a small fraction (<5%) of cases, reflecting mutations at 19 different loci, importantly including autosomal dominant mutations in the GTPase LRRK2 and α-synuclein (SNCA), the primary component of Lewy bodies, the pathological hallmark of PD (69).
Several neurological disorders have been shown to be caused by clonal mosaic mutations of germline risk genes (70,71), suggesting that in principle mosaic mutations in ad and PD risk genes could cause those disorders. One case of early-onset ad has been conclusively shown to be associated with a mosaic variant, a PSEN1 mutation marking 8% of lymphocytes and 14% of cells in the cerebral cortex of the affected individual (72), demonstrating that neurodegeneration could be caused by a mosaic mutation present in as little as 14% of the cells in the brain, possibly less. However, deep panel and exome sequencing has failed to identify as significant a differential burden of pathogenic somatic mutations in known ad or PD genes relative to controls (73–78), which may suggest that larger-scale, higher-powered studies are needed to measure the potentially minor impact clonal somatic mutation has on ad or PD (Fig. 3).
Figure 3.
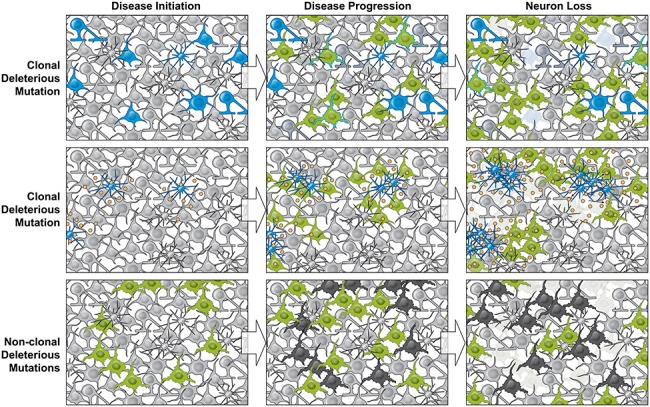
Three mechanisms by which somatic mutation can impact neurodegenerative diseases. Top row, a gene linked to disease, for example PSEN1 in AD, is mutated after fertilization resulting in a clonal somatic mutation in the brain, marking a subset of cells (blue). Such mutations at mosaic fractions as low as 14% (72) can be responsible for disease initiation. This small fraction of cells is sufficient to disease progression cell autonomously and cell non-autonomously (green), ultimately leading to neuron loss (faded cells). Middle row, clonal SNVs present in microglial cells (blue) could cause microglial proliferation and overproduction of inflammatory cytokines (yellow), causing disease (green) and neurodegeneration (faded cells), as has been shown in mouse (79). Bottom row, known mechanisms causing neurodegenerative disease, such as germline genetic risk, environmental exposure or advanced age, could initiate disease (green), resulting in genotoxic stress and the accumulation of deleterious somatic mutations in essential genes (black), resulting in neurodegeneration (faded cells).
Clonal somatic mutations could cause neurodegeneration by mechanisms other than somatic SNVs in known ad or PD genes. In the mouse, clonal somatic mutation of BRAF in microglial progenitors during development caused a neurodegenerative phenotype in the adult (79). The mutant allele generated a BRAF V600E protein, a known oncogene, in this case increasing microglial activation instead of causing tumorigenesis. BRAF V600E is likely not compatible with life if inherited in the germline, suggesting it may only play a role in neurodegenerative disease when mutated somatically (Fig. 3). Array-based and in situ-hybridization-based approaches suggest that APP CNV gains in ad patients (80,81) and SCNA CNV gains in PD cases (82) might be more common in ad and PD than controls, respectively, although these studies have yet to be confirmed by sequence-based approaches.
An interesting possibility, which remains untested, is that damage to neuronal genomes may occur at higher rates in late-onset degenerative disease such as ad and PD, resulting in a pathogenic burden of non-clonal somatic mutations in ad and PD neurons. ROS can damage DNA by several mechanisms, causing mutations (83), and both PD (69,84–86) and ad (87,88) are associated with increased oxidative stress. scWGS in two progeroid, early-onset neurodegenerative diseases, CS and XP, suggested that non-clonal somatic mutations linked to oxidative stress are associated with accelerated aging and neuron loss in the human brain (12). Thus, increased levels of oxidative stress on the genome may suggest a common thread connecting early-onset genetic neurodegeneration in diseases like CS and XP with late-onset, predominantly sporadic neurodegenerative diseases like PD and ad (Fig. 3). Whether permanent somatic mutations are elevated in PD and ad patient neurons is currently an open question, one that could in principle be addressed by examining scWGS of PD and ad patient brains.
The generation of non-clonal somatic mutations in neurons could conceivably interfere with neuronal function or even lead to cell death. Indeed, germline de novo mutation load in humans increases with paternal age, and with it so does the risk of sporadic neuropsychiatric diseases like autism spectrum disorder (ASD) and schizophrenia (89). ASD probands have been shown to have a higher de novo somatic mutation load than unaffected siblings (90), suggesting that increased mutations increase leads to an increase in the chance of disrupting important neuronal functions. The probability of generating deleterious mutations at both alleles of the same locus increases exponentially during the linear accumulation of mutations during the life of a normal neuron, with ~ 1 in 1000 neurons likely a biallelic knockout for at least one gene in elderly brains (12). Importantly, this analysis assumed a completely unmutated germline genome, when in reality the typical human germline genome contains 149–182 protein-truncating variants, 10 000–12 000 peptide-sequence-altering variants and 459 000–565 000 variant sites overlapping known regulatory regions (91), suggesting that germline and somatic mutations could together have an important impact on cell function.
Conclusions
The human brain is a mosaic, because somatic variants of several classes constantly accumulate in primitive cells during development, generating clonal mutations, and in postmitotic neurons cells in the adult, generating non-clonal mutations. Somatic mutation displays both programmed and random features and is linked to aging and age-related disease. Late-onset neurodegenerative diseases like AD and PD have several features suggesting somatic mutation involvement, including close relationships between disease risk and age, and signatures of oxidative stress, which should be investigated in the future.
Funding
K99 AG054749 (to M.A.L.); National Institute of Neurological Disorders and Stroke (R01 NS35129, R01 NS032457 to C.A.W.); National Institute of Mental Health (U01MH106883 to C.A.W.); Allen Discovery Center program through The Paul G. Allen Frontiers Group (to C.A.W.)
Acknowledgements
Figures were illustrated by Ken Probst of Xavier Studio with input from the authors. We thank Rachel Rodin for contributions to Figure 1 and 1 and Tables 2. C.A.W. is an Investigator of The Howard Hughes Medical Institute.
References
- 1. Bauer N.C., Corbett A.H. and Doetsch P.W. (2015) The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res, 43, 10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haldane J.B.S. (1937) The effect of variation on fitness. The American Naturalist, 71, 337–349. [Google Scholar]
- 3. Elena S.F. and Lenski R.E. (1997) Test of synergistic interactions among deleterious mutations in bacteria. Nature, 390, 395–398. [DOI] [PubMed] [Google Scholar]
- 4. Harman D. (1981) The aging process. Proc Natl Acad Sci USA, 78, 7124–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopez-Otin C., Blasco M.A., Partridge L., Serrano M. and Kroemer G. (2013) The hallmarks of aging. Cell, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kenyon C.J. (2010) The genetics of ageing. Nature, 464, 504–512. [DOI] [PubMed] [Google Scholar]
- 7. Hayflick L. (2007) Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet, 3, e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dean F.B., Nelson J.R., Giesler T.L. and Lasken R.S. (2001) Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res, 11, 1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evrony G.D., Lee E., Mehta B.K., Benjamini Y., Johnson R.M., Cai X., Yang L., Haseley P., Lehmann H.S., Park P.J. et al. (2015) Cell lineage analysis in human brain using endogenous retroelements. Neuron, 85, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evrony G.D., Cai X., Lee E., Hills L.B., Elhosary P.C., Lehmann H.S., Parker J.J., Atabay K.D., Gilmore E.C., Poduri A. et al. (2012) Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell, 151, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lodato M.A., Woodworth M.B., Lee S., Evrony G.D., Mehta B.K., Karger A., Lee S., Chittenden T.W., D'Gama A.M., Cai X. et al. (2015) Somatic mutation in single human neurons tracks developmental and transcriptional history. Science, 350, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C. et al. (2018) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science, 359, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cai X., Evrony G.D., Lehmann H.S., Elhosary P.C., Mehta B.K., Poduri A. and Walsh C.A. (2014) Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep, 8, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erwin J.A., Paquola A.C., Singer T., Gallina I., Novotny M., Quayle C., Bedrosian T.A., Alves F.I., Butcher C.R., Herdy J.R. et al. (2016) L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat Neurosci, 19, 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knouse K.A., Wu J. and Amon A. (2016) Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res, 26, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knouse K.A., Wu J., Whittaker C.A. and Amon A. (2014) Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci USA, 111, 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McConnell M.J., Lindberg M.R., Brennand K.J., Piper J.C., Voet T., Cowing-Zitron C., Shumilina S., Lasken R.S., Vermeesch J.R., Hall I.M. et al. (2013) Mosaic copy number variation in human neurons. Science, 342, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zong C., Lu S., Chapman A.R. and Xie X.S. (2012) Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science, 338, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Upton K.R., Gerhardt D.J., Jesuadian J.S., Richardson S.R., Sanchez-Luque F.J., Bodea G.O., Ewing A.D., Salvador-Palomeque C., Knaap M.S., Brennan P.M. et al. (2015) Ubiquitous L1 mosaicism in hippocampal neurons. Cell, 161, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chronister W.D., Burbulis I.E., Wierman M.B., Wolpert M.J., Haakenson M.F., Smith A.C.B., Kleinman J.E., Hyde T.M., Weinberger D.R., Bekiranov S. et al. (2019) Neurons with complex karyotypes are rare in aged human neocortex. Cell Rep, 26(825–835), e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hazen J.L., Faust G.G., Rodriguez A.R., Ferguson W.C., Shumilina S., Clark R.A., Boland M.J., Martin G., Chubukov P., Tsunemoto R.K. et al. (2016) The complete genome sequences, unique mutational spectra, and developmental potency of adult neurons revealed by cloning. Neuron, 89, 1223–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bae T., Tomasini L., Mariani J., Zhou B., Roychowdhury T., Franjic D., Pletikos M., Pattni R., Chen B.J., Venturini E. et al. (2018) Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science, 359, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lim E.T., Uddin M., De Rubeis S., Chan Y., Kamumbu A.S., Zhang X., D'Gama A.M., Kim S.N., Hill R.S., Goldberg A.P. et al. (2017) Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci, 20, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dou Y., Yang X., Li Z., Wang S., Zhang Z., Ye A.Y., Yan L., Yang C., Wu Q., Li J. et al. (2017) Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum Mutat, 38, 1002–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krupp D.R., Barnard R.A., Duffourd Y., Evans S.A., Mulqueen R.M., Bernier R., Riviere J.B., Fombonne E. and O'Roak B.J. (2017) Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet, 101, 369–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jamuar S.S., Lam A.T., Kircher M., D'Gama A.M., Wang J., Barry B.J., Zhang X., Hill R.S., Partlow J.N., Rozzo A. et al. (2014) Somatic mutations in cerebral cortical malformations. N Engl J Med, 371, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. D'Gama A.M., Geng Y., Couto J.A., Martin B., Boyle E.A., LaCoursiere C.M., Hossain A., Hatem N.E., Barry B.J., Kwiatkowski D.J. et al. (2015) Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol, 77, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. D'Gama A.M., Pochareddy S., Li M., Jamuar S.S., Reiff R.E., Lam A.N., Sestan N. and Walsh C.A. (2015) Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron, 88, 910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jansen L.A., Mirzaa G.M., Ishak G.E., O'Roak B.J., Hiatt J.B., Roden W.H., Gunter S.A., Christian S.L., Collins S., Adams C. et al. (2015) PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain, 138, 1613–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mirzaa G.M., Conti V., Timms A.E., Smyser C.D., Ahmed S., Carter M., Barnett S., Hufnagel R.B., Goldstein A., Narumi-Kishimoto Y. et al. (2015) Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol, 14, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D'Gama A.M., Woodworth M.B., Hossain A.A., Bizzotto S., Hatem N.E., LaCoursiere C.M., Najm I., Ying Z., Yang E., Barkovich A.J. et al. (2017) Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep, 21, 3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evrony G.D., Lee E., Park P.J. and Walsh C.A. (2016) Resolving rates of mutation in the brain using single-neuron genomics. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong X., Zhang L., Milholland B., Lee M., Maslov A.Y., Wang T. and Vijg J. (2017) Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bohrson C.L., Barton A.R., Lodato M.A., Rodin R.E., Luquette L.J., Viswanadham V.V., Gulhan D.C., Cortes-Ciriano I., Sherman M.A., Kwon M. et al. (2019) Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nat Genet, 51, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sherman M.A., Barton A.R., Lodato M.A., Vitzhum C., Coulter M.E., Walsh C.A. and Park P.J. (2017) PaSD-qc: quality control for single cell whole-genome sequencing data using power spectral density estimation. bioRxiv, in press. [DOI] [PMC free article] [PubMed]
- 36. Knouse K.A., Wu J. and Amon A. (2016) Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res, 26, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gole J., Gore A., Richards A., Chiu Y.J., Fung H.L., Bushman D., Chiang H.I., Chun J., Lo Y.H. and Zhang K. (2013) Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol, 31, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szilard L. (1959) On the nature of the aging process. Proc Natl Acad Sci USA, 45, 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Failla G. (1958) The aging process and cancerogenesis. Ann N Y Acad Sci, 71, 1124–1140. [DOI] [PubMed] [Google Scholar]
- 40. Madabhushi R., Gao F., Pfenning A.R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T.X., Yamakawa H., Pao P.C. et al. (2015) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell, 161, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lake B.B., Ai R., Kaeser G.E., Salathia N.S., Yung Y.C., Liu R., Wildberg A., Gao D., Fung H.L., Chen S. et al. (2016) Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science, 352, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greig L.C., Woodworth M.B., Galazo M.J., Padmanabhan H. and Macklis J.D. (2013) Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci, 14, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blokzijl F., Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P. et al. (2016) Tissue-specific mutation accumulation in human adult stem cells during life. Nature, 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martincorena I., Fowler J.C., Wabik A., Lawson A.R.J., Abascal F., Hall M.W.J., Cagan A., Murai K., Mahbubani K., Stratton M.R. et al. (2018) Somatic mutant clones colonize the human esophagus with age. Science, 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jacobs K.B., Yeager M., Zhou W., Wacholder S., Wang Z., Rodriguez-Santiago B., Hutchinson A., Deng X., Liu C., Horner M.J. et al. (2012) Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet, 44, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laurie C.C., Laurie C.A., Rice K., Doheny K.F., Zelnick L.R., McHugh C.P., Ling H., Hetrick K.N., Pugh E.W., Amos C. et al. (2012) Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet, 44, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang L., Dong X., Lee M., Maslov A.Y., Wang T. and Vijg J. (2019) Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc Natl Acad Sci USA, 116, 9014–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alexandrov L.B. and Stratton M.R. (2014) Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev, 24, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pleasance E.D., Stephens P.J., O'Meara S., McBride D.J., Meynert A., Jones D., Lin M.L., Beare D., Lau K.W., Greenman C. et al. (2010) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 463, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saini N., Roberts S.A., Klimczak L.J., Chan K., Grimm S.A., Dai S., Fargo D.C., Boyer J.C., Kaufmann W.K., Taylor J.A. et al. (2016) The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet, 12, e1006385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Polak P. and Arndt P.F. (2008) Transcription induces strand-specific mutations at the 5′ end of human genes. Genome Res, 18, 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pleasance E.D., Cheetham R.K., Stephens P.J., McBride D.J., Humphray S.J., Greenman C.D., Varela I., Lin M.L., Ordonez G.R., Bignell G.R. et al. (2010) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature, 463, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S. and Stratton M.R. (2015) Clock-like mutational processes in human somatic cells. Nat Genet, 47, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hou Y., Song L., Zhu P., Zhang B., Tao Y., Xu X., Li F., Wu K., Liang J., Shao D. et al. (2012) Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell, 148, 873–885. [DOI] [PubMed] [Google Scholar]
- 56. Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C. et al. (2019) Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell, 176(1282–1294), e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Genovese G., Kahler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. et al. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med, 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ju Y.S., Martincorena I., Gerstung M., Petljak M., Alexandrov L.B., Rahbari R., Wedge D.C., Davies H.R., Ramakrishna M., Fullam A. et al. (2017) Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature, 543, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M. et al. (2015) Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science, 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Francioli L.C., Polak P.P., Koren A., Menelaou A., Chun S., Renkens I., Duijn C.M., Swertz M., Wijmenga C., Ommen G. et al. (2015) Genome-wide patterns and properties of de novo mutations in humans. Nat Genet, 47, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Behjati S., Huch M., Boxtel R., Karthaus W., Wedge D.C., Tamuri A.U., Martincorena I., Petljak M., Alexandrov L.B., Gundem G. et al. (2014) Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature, 513, 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bishop N.A., Lu T. and Yankner B.A. (2010) Neural mechanisms of ageing and cognitive decline. Nature, 464, 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tanzi R.E., Gusella J.F., Watkins P.C., Bruns G.A., St George-Hyslop P., Van Keuren M.L., Patterson D., Pagan S., Kurnit D.M. and Neve R.L. (1987) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science, 235, 880–884. [DOI] [PubMed] [Google Scholar]
- 64. St George-Hyslop P., Haines J., Rogaev E., Mortilla M., Vaula G., Pericak-Vance M., Foncin J.F., Montesi M., Bruni A., Sorbi S. et al. (1992) Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet, 2, 330–334. [DOI] [PubMed] [Google Scholar]
- 65. Schellenberg G.D., Bird T.D., Wijsman E.M., Orr H.T., Anderson L., Nemens E., White J.A., Bonnycastle L., Weber J.L., Alonso M.E. et al. (1992) Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science, 258, 668–671. [DOI] [PubMed] [Google Scholar]
- 66. Levy-Lahad E., Wijsman E.M., Nemens E., Anderson L., Goddard K.A., Weber J.L., Bird T.D. and Schellenberg G.D. (1995) A familial Alzheimer’s disease locus on chromosome 1. Science, 269, 970–973. [DOI] [PubMed] [Google Scholar]
- 67. Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L. and Pericak-Vance M.A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261, 921–923. [DOI] [PubMed] [Google Scholar]
- 68. Ascherio A. and Schwarzschild M.A. (2016) The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol, 15, 1257–1272. [DOI] [PubMed] [Google Scholar]
- 69. Reed X., Bandres-Ciga S., Blauwendraat C. and Cookson M.R. (2019) The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol Dis, 124, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rodin R.E. and Walsh C.A. (2018) Somatic mutation in pediatric neurological diseases. Pediatr Neurol, 87, 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. D'Gama A.M. and Walsh C.A. (2018) Somatic mosaicism and neurodevelopmental disease. Nat Neurosci, 21, 1504–1514. [DOI] [PubMed] [Google Scholar]
- 72. Beck J.A., Poulter M., Campbell T.A., Uphill J.B., Adamson G., Geddes J.F., Revesz T., Davis M.B., Wood N.W., Collinge J. et al. (2004) Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum Mol Genet, 13, 1219–1224. [DOI] [PubMed] [Google Scholar]
- 73. Proukakis C., Houlden H. and Schapira A.H. (2013) Somatic alpha-synuclein mutations in Parkinson’s disease: hypothesis and preliminary data. Mov Disord, 28, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Proukakis C., Shoaee M., Morris J., Brier T., Kara E., Sheerin U.M., Charlesworth G., Tolosa E., Houlden H., Wood N.W. et al. (2014) Analysis of Parkinson’s disease brain-derived DNA for alpha-synuclein coding somatic mutations. Mov Disord, 29, 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sala Frigerio C., Lau P., Troakes C., Deramecourt V., Gele P., Van Loo P., Voet T. and De Strooper B. (2015) On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimers Dement, 11, 1265–1276. [DOI] [PubMed] [Google Scholar]
- 76. Keogh M.J., Wei W., Aryaman J., Walker L., Ameele J., Coxhead J., Wilson I., Bashton M., Beck J., West J. et al. (2018) High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat Commun, 9, 4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nicolas G., Acuna-Hidalgo R., Keogh M.J., Quenez O., Steehouwer M., Lelieveld S., Rousseau S., Richard A.C., Oud M.S., Marguet F. et al. (2018) Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer’s disease. Alzheimers Dement, 14, 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Park J.S., Lee J., Jung E.S., Kim M.H., Kim I.B., Son H., Kim S., Kim S., Park Y.M., Mook-Jung I. et al. (2019) Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat Commun, 10, 3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mass E., Jacome-Galarza C.E., Blank T., Lazarov T., Durham B.H., Ozkaya N., Pastore A., Schwabenland M., Chung Y.R., Rosenblum M.K. et al. (2017) A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature, 549, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bushman D.M., Kaeser G.E., Siddoway B., Westra J.W., Rivera R.R., Rehen S.K., Yung Y.C. and Chun J. (2015) Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife, 4. [DOI] [PMC free article] [PubMed]
- 81. Lee M.H., Siddoway B., Kaeser G.E., Segota I., Rivera R., Romanow W.J., Liu C.S., Park C., Kennedy G., Long T. et al. (2018) Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature, 563, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mokretar K., Pease D., Taanman J.W., Soenmez A., Ejaz A., Lashley T., Ling H., Gentleman S., Houlden H., Holton J.L. et al. (2018) Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain, 141, 2419–2431. [DOI] [PubMed] [Google Scholar]
- 83. De Bont R. and Larebeke N. (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis, 19, 169–185. [DOI] [PubMed] [Google Scholar]
- 84. Mariani E., Polidori M.C., Cherubini A. and Mecocci P. (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci, 827, 65–75. [DOI] [PubMed] [Google Scholar]
- 85. Dias V., Junn E. and Mouradian M.M. (2013) The role of oxidative stress in Parkinson's disease. J Parkinsons Dis, 3, 461–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu Z., Zhou T., Ziegler A.C., Dimitrion P. and Zuo L. (2017) Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med Cell Longev, 2017, 2525967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Huang X., Moir R.D., Tanzi R.E., Bush A.I. and Rogers J.T. (2004) Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann N Y Acad Sci, 1012, 153–163. [DOI] [PubMed] [Google Scholar]
- 88. Galimberti D. and Scarpini E. (2011) Inflammation and oxidative damage in Alzheimer’s disease: friend or foe? Front Biosci (Schol Ed), 3, 252–266. [DOI] [PubMed] [Google Scholar]
- 89. Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Wong W.S. et al. (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature, 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Iossifov I., O'Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E. et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. 1000 Genomes Project, C, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bourcy C.F., De Vlaminck I., Kanbar J.N., Wang J., Gawad C. and Quake S.R. (2014) A quantitative comparison of single-cell whole genome amplification methods. PLoS One, 9, e105585. [DOI] [PMC free article] [PubMed] [Google Scholar]