

Abstract
Capitalizing on the massive increase in sample concentrations which are produced by extremely low elution volumes, nanoliquid chromatography–electrospray ionization-tandem mass spectrometry (nano-LC–ESI-MS/MS) is currently one of the most sensitive analytical technologies for the comprehensive characterization of complex protein samples. However, despite tremendous technological improvements made in the production and the packing of monodisperse spherical particles for nanoflow high-pressure liquid chromatography (HPLC), current state-of-the-art systems still suffer from limits in operation at the maximum potential of the technology. With the recent introduction of the μPAC system, which provides perfectly ordered micropillar array based chromatographic support materials, completely new chromatographic concepts for optimization toward the needs of ultrasensitive proteomics become available. Here we report on a series of benchmarking experiments comparing the performance of a commercially available 50 cm micropillar array column to a widely used nanoflow HPLC column for the proteomics analysis of 10 ng of tryptic HeLa cell digest. Comparative analysis of LC–MS/MS-data corroborated that micropillar array cartridges provide outstanding chromatographic performance, excellent retention time stability, and increased sensitivity in the analysis of low-input proteomics samples and thus repeatedly yielded almost twice as many unique peptide and unique protein group identifications when compared to conventional nanoflow HPLC columns.
The field of proteomics aims at the qualitative and quantitative description of all proteins contained in complex biological samples. Currently, the most sensitive proteomics platforms are almost exclusively based on the combination of two key analytical methods: nanoflow high-performance liquid chromatography (nano-HPLC) and tandem mass-spectrometry (MS/MS), hyphenated by electrospray ionization (ESI). However, despite the tremendous improvements in ultrasensitive nano-HPLC–ESI-MS/MS-based proteomics workflows and instrumentation, which capitalize on the massive increase in sample concentrations produced by extremely low elution volumes, the comprehensive characterization of, e.g., single mammalian cells still challenges the sensitivity of currently available technologies.
Next to the development of dedicated low-input sample preparation methods,1−4 which aim at reducing sample losses prior to analysis, changes to the chromatographic support material have recently been identified key to the sensitive profiling of low-input proteomics samples by nano-HPLC–ESI-MS/MS.5,6
While nonporous particles have been demonstrated to hold the great potential of high chromatographic separation power and to further reduce on-column losses,5 their expedient integration into standard proteomics nanoflow HPLC systems has long suffered from their intrinsically low loading capacity.6 However, especially in the context of low-input proteomics, which aims at analyzing protein amounts in the nanogram to picogram-range, the capacity of these novel reversed-phase (RP) HPLC support materials is not a limiting factor. More importantly, the potential gain in sensitivity due to improved peak width and peak capacities provided by these new types of chromatographic columns makes them extremely attractive for ultrasensitive proteomics applications.
With the recent commercialization of perfectly ordered micropillar array-based nano-HPLC cartridges (μPAC, PharmaFluidics), we wished to explore potential benefits of this technology to the ultrasensitive analysis of low-input proteomics samples. Here we report on a series of benchmarking experiments comparing the performance of a 50 cm micropillar array nano-HPLC cartridge to a state-of-the-art, particle-based nano-HPLC column in the analysis of 10 ng of tryptic HeLa cell protein digest.
For these experiments, we installed the respective pre- and analytical columns (i.e., μPAC RP18, 50 cm, PharmaFluidics; PepMap C18, 3 μm, 75 μm × 50 cm, Thermo) in identical LC–ESI-MS/MS setups, all comprising an Ultimate 3000 RSLCnano LC system (Dionex–Thermo) operated at 50 °C, coupled to the exact same Q Exactive HF-X mass-spectrometer (Thermo) using a 360 μm capillary fitting (C360UFS2, Vici) connected to 12 cm fused silica electrospray emitters (part number FS3602010N20C12, 360 μm o.d., 20 μm i.d., nominal tip i.d. = 10 μm, uncoated). The column outlet of the PepMap C18 system was connected to the emitter via a 10 cm silica capillary (20 μm i.d. × 360 μm o.d., Polymicro Technologies), μPAC cartridges were connected using their preinstalled connection tubings. One new electrospray emitter was used for all analytical runs on each column setup. The samples (10 ng/μL HeLa digest, Pierce; in 0.1% formic acid) were injected using a 1 μL sample-loop at full loop injection, were trapped on a precolumn, and then separated by developing two-step linear gradients of increasing length, at a fixed flow-rate of 250 nL/min: from 2% to 20% acetonitrile in 0.1% formic acid in 45, 90, and 135 min, followed by 20–32% acetonitrile in 0.1% formic acid within 15, 30, and 45 min (i.e., 60, 120, or 180 min total gradient time), respectively. All three gradient programs were completed by a final gradient step from 32 to 78% acetonitrile in 0.1% formic acid, within 5 min.
The mass-spectrometer was operated in positive mode and set to the following acquisition parameters: MS1 resolution = 60 000, MS1 AGC-target = 1 × 106, MS1 maximum inject time = 60 ms, MS1 scan range = 350–1500 m/z, MS2 resolution = 15 000, 45 000, or 60 000, MS2 AGC-target = 2 × 105, maximum inject time = 105 ms, TopN = 10, isolation window = 0.7 m/z, MS2 scan range = “dynamic first mass”, normalized collision energy = 28, minimum AGC target = 1 × 104, intensity threshold 9.5 × 104, precursor charge states = 2–6, peptide match = preferred, exclude isotopes = ON, dynamic exclusion = 45 s, “if idle...” = do not pick others. All experiments were performed in technical triplicates.
Subsequently, all LC–MS/MS raw-data were processed and identified using Proteome Discoverer (version 2.3.0.523, Thermo Scientific). For this, MS/MS spectra were extracted from the raw-files and searched against the Swissprot protein database, restricting taxonomy to Homo sapiens and including common contaminant protein sequences (20 341 sequences; 1 1361 548 residues) using MSAmanda (Engine version v2.0.0.12368).7 The search engine parameters were set as follows: peptide mass tolerance = ±7 ppm, fragment mass tolerance = 15 ppm, cleavage specificity = trypsin, missed cleavage sites = 2, fixed modifications = carbamidomethylation of cysteine, variable modifications = oxidation of methionine. Results of the MS/MS search engine were filtered to 1% FDR on protein and peptide level using the Elutator algorithm,8 implemented as a node to Proteome Discoverer 2.3. Identified peptide features were extracted from the raw-files and quantified, using the in-house-developed Proteome Discover-node apQuant.9
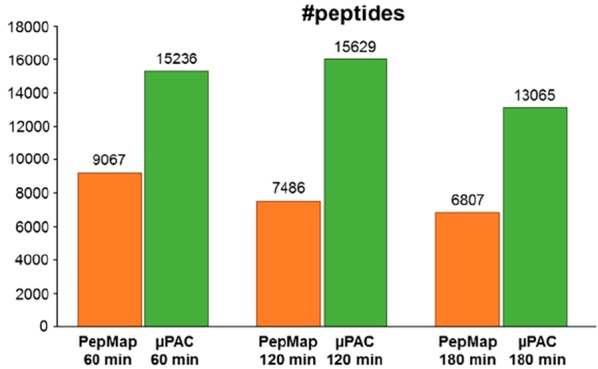
Comparative analysis of the MS/MS data (Figure 1A,B) highlighted that μPAC cartridges repeatedly yielded almost twice as many unique peptide identifications (e.g., 15 629, at 120 min gradient length) and unique protein groups (e.g., 2 743, at 120 min gradient length), when compared to the PepMap C18 system (e.g., 7 364 unique peptides and 1 500 unique protein groups, at a gradient length of 120 min). Of note, enabling the second search-option available in MS Amanda7 yielded approximately 30% more peptide spectrum matches and approximately 20% more peptide identifications, irrespective of the chromatographic system used (data not shown). An overlay of the identifications showed excellent agreement with respect to a core-set of peptides (Figure 1C) and protein groups (Figure 1D), which were identified with both nano-HPLC setups. Remarkably, however, although >90% of all peptide and protein group identified with the PepMap C18 system were also found in the μPAC setup, the latter allowed for the additional identification of 80% more peptides and 90% more protein groups (Figure 1C,D). This important increase in peptide and protein group identifications specifically provided by the μPAC system, were primarily due to almost twice as many MS/MS spectra triggered in the respective experiments (e.g., 26 358 and 14 027, at 120 min gradient length, for μPAC and PepMap, respectively), corroborating increased sensitivity. Of note, the maximum TopN number of 10 precursor ion-masses to be scheduled for MS/MS analysis was hardly reached in the μPAC experiments (e.g., in 10 duty-cycles, at 120 min gradient length) and never exceeded more than 6 precursor ion-masses to be scheduled for MS/MS in the PepMap experiments at 120 min gradient length.
Figure 1.

Comparative analysis of the LC–MS/MS data generated from 10 ng of HeLa digest, using the μPAC RP18 or the PepMap C18 setup. (A) Average number of unique peptide and (B) unique protein groups identified, at different gradient lengths, across three technical repeats, each. Overlay of repeatedly identified (C) peptide sequences and (D) protein groups identified in at least two technical repeats at a gradient length of 120 min. Comparative “box-and-whiskers” plots of precursor-ion specific chromatographic peak areas of (E) peptides identified in both nano-HPLC setups and (F) peptides which were exclusively identified in either of the two nano-HPLC setups in at least two technical repeats at a gradient length of 120 min. All data processing, chromatographic peak detection, and peak area calculations were performed using Proteome Discoverer 2.3, MS Amanda, and apQuant.
Further investigating the substantial μPAC-specific gain in peptide and protein identifications, we extracted peptide precursor-ion specific chromatographs and calculated the respective chromatographic peak areas, using apQuant. Comparison of the data highlighted that peptide precursor-ion intensities were on average 2-fold higher when using the μPAC cartridges (Figure 1E). Clearly warranting further investigations, we speculate that the “core-shell”-type architecture of the μPAC micropillar arrays (as opposed to the porous bead structure of the PepMap material) effectively reduces on-column losses of low-input samples and results in a global increase in peptide precursor-ion signal. This hypothesis is further corroborated by the observation that, in contrast to a steady decline in peptide identifications with increasing gradient length observed for the PepMap column (Figure 1A), sample/signal-dilution by peak-broadening appeared to be partially compensated for by reduced on-column losses in μPAC cartridges even at 120 min gradient length (Figure 1A). At 180 min gradient length, however, this compensatory effect of the μPAC system appeared to become exhausted by peak-broadening and resulted in a decline in peptide/protein identifications also for the μPAC-cartridge. Similarly, also μPAC-only identifications (i.e., 8 977 peptides and 1 343 proteins; Figure 1C) predominantly derived from low-abundance peptide precursor-ions (Figure 1F). The sparse PepMap-only identified peptides, however, were detected within a precursor-ion intensity range which was similar to common peptide identifications.
Additionally, we comparatively analyzed the chemical characters of peptides uniquely identified by either of the two nano-LC–MS/MS systems. The data revealed subtle, yet mechanistically insightful, differences on the separation characteristics of the two chromatographic systems compared in our study (please see Supplementary Figure 1). Most importantly, this analysis provides clear indications that the use of μPAC cartridges do indeed allow for a more sensitive detection, specifically of longer tryptic peptides, when compared to the pepMap columns (Supplementary Figure 1C). By contrast, uniquely identified peptide sequences, do not show appreciable differences with respect to pI-value or GRAVY-index (please see Supplementary Figure 1B,C). Similarly, GO-term enrichment analysis of uniquely identified proteins on either system did not provide substantial insight into the chromatographic merit of either system (data not shown).
Next, we analyzed chromatographic performance parameters of the two systems. For this, we automatically extracted peptide-specific retention times at the peak apex, determined full-width at half-maximum (fwhm), and calculated peak asymmetry parameters (i.e., skewness), using apQuant. The comparison of peptide-specific peak widths (i.e., fwhm) revealed broader peaks on the PepMap columns (i.e., mean = 12.9 and 10.2 s, for a 120 min gradient on PepMap and μPAC, respectively; Figure 2A) and suggested a 25% increase in peak capacity for the μPAC system (i.e., estimated peak capacity of 560 and 705, at 120 min gradient length, for PepMap and μPAC, respectively). More importantly, however, while 90% of all peptides identified with μPAC cartridges had a fwhm of <12 s, the same proportion of peptides were detected with a fwhm of <16 s on the PepMap C18 system (Figure 2B). Additionally, while both nano-LC setups performed similarly at the 60 min gradient length (Figure 2C), the μPAC system provided superior peak-width at extended gradient times (i.e., 180 min; Figure 2D). Chromatographic peak symmetry, as determined by calculating peptide-specific peak skewness, was found similar in both setups (Figure 2E).
Figure 2.

Chromatographic performance parameters of the μPAC RP18 and the PepMap C18 nano-HPLC systems. (A) Relative comparison of the density distribution of peptide-specific peak widths (fwhm). (B) Cumulative plot of peptide specific fwhm at 120 min, (C) 60 min, and (D) 180 min. For all plots, fwhm bin width = 1 s. (E) Comparison of chromatographic peak asymmetry at 120 min gradient length, as calculated by “skewness” (bin width = 0.2). All chromatographic peak detection and feature calculations were performed using apQuant for Proteome Discoverer 2.3.
Most interestingly, however, close investigation of the LC–MS/MS data generated in the course of this study revealed an unprecedented degree of retention-time stability of μPAC cartridges (Figure 3). For this, we calculated the deviation from the mean retention time of the identified peptide features across three technical replicates. While 95% of all features identified on the PepMap system were found to elute within a retention time window of approximately 44 s, at 120 min gradient length, the same proportion of features identified on the μPAC setup eluted within in a time window of approximately 4 s at 120 min gradient length and did not exceed 10 s at 180 min gradient length. This outstanding retention-time stability and precision of μPAC cartridges clearly warrants future applications of this ultrasensitive nano-HPLC setup in time-dependent LC–MS and LC–MS/MS data-acquisition regimes (e.g., scheduled tSIM or PRM workflows). Of note, when compared to packed-bead columns, micropillar arrays still suffer from an inherently reduced maximum loading capacity (i.e., vendor recommendations suggest not to exceed a sample load of 1 μg of tryptic HeLa digest for a 50 cm μPAC cartridge) which might in some cases preclude their application in, e.g., clinical high-input proteomics workflows.
Figure 3.

Comparison of retention time stability and precision.
Taken together, our results highlight impressive improvements in performance in the analysis of low-input proteomics samples by the application of μPAC RP18 cartridges over current state-of-the-art nano-HPLC systems. Not only did we yield almost twice as many unique peptide and unique protein group identifications, when compared to conventional nanoflow HPLC columns, but we also observed unprecedented retention-time stability on the micropillar array-based nano-HPLC μPAC cartridges.
Owing to the flexibility in design and the great potential to even further optimize chromatographic support materials and formats, this first commercial implementation of the μPAC concept clearly provides very attractive new strategies for increased sensitivity in nano-HPLC–ESI-MS/MS based proteomics platforms toward the ultrasensitive analysis of single mammalian cells.
Acknowledgments
We thank all members of our laboratories for helpful discussions. We acknowledge G. Krššáková for method development. This work has been supported by EPIC-XS, Project Number 823839, funded by the Horizon 2020 Program of the European Union and the Austrian Science Fund by the ERA-CAPS I 3686 International Project. J.S. is a Wittgenstein Prize Fellow and funded by the T. von Zastrow Foundation. J.M.P. is supported by grants from IMBA, the Austrian Academy of Sciences, the T. von Zastrow Foundation, an ERC Advanced Grant, and an Era of Hope Innovator award.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b02899.
Supplementary Figure 1, Comparison of chemical properties of peptides uniquely identified peptides by either of the two nanoLC–MS/MS systems: (A) peptide length as number of amino acids per peptide, (B) pI value of uniquely identified peptide sequences, as calculated using the “isoelectric point calculator” (http://isoelectric.org), and (C) GRAVY-index of the respective peptide sequences, as calculated using the GRAVY calculator (http://www.gravy-calculator.de) (PDF)
The authors declare the following competing financial interest(s): G.V.R. and J.O.D.B. are employees of PharmaFluidics, and P.J. is one of the co-founders of PharmaFluidics.
Notes
All mass spectrometry-based proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier [PXD014124]. For review purpose please query the data using the following Reviewer account details: Username: reviewer67592@ebi.ac.uk. Password: k8kv5Isu
Supplementary Material
References
- Zhu Y.; Piehowski P. D.; Zhao R.; Chen J.; Shen Y.; Moore R. J.; Shukla A. K.; Petyuk V. A.; Campbell-Thompson M.; Mathews C. E.; Smith R. D.; Qian W. J.; Kelly R. T. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9 (1), 882. 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Gaffrey M. J.; Zhu Y.; Chrisler W. B.; Fillmore T. L.; Yi L.; Nicora C. D.; Zhang T.; Wu H.; Jacobs J.; Tang K.; Kagan J.; Srivastava S.; Rodland K. D.; Qian W. J.; Smith R. D.; Liu T.; Wiley H. S.; Shi T. Carrier-Assisted Single-Tube Processing Approach for Targeted Proteomics Analysis of Low Numbers of Mammalian Cells. Anal. Chem. 2019, 91 (2), 1441–1451. 10.1021/acs.analchem.8b04258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.; Gaffrey M. J.; Fillmore T. L.; Nicora C. D.; Yi L.; Zhang P.; Shukla A. K.; Wiley H. S.; Rodland K. D.; Liu T.; Smith R. D.; Qian W. J. Facile carrier-assisted targeted mass spectrometric approach for proteomic analysis of low numbers of mammalian cells. Commun. Biol. 2018, 1, 103. 10.1038/s42003-018-0107-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19 (1), 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima Y.; Ohara O. Development of a NanoLC-MS/MS System Using a Nonporous Reverse Phase Column for Ultrasensitive Proteome Analysis. Anal. Chem. 2018, 90 (21), 12334–12338. 10.1021/acs.analchem.8b03382. [DOI] [PubMed] [Google Scholar]
- De Malsche W.; Eghbali H.; Clicq D.; Vangelooven J.; Gardeniers H.; Desmet G. Pressure-driven reverse-phase liquid chromatography separations in ordered nonporous pillar array columns. Anal. Chem. 2007, 79 (15), 5915–26. 10.1021/ac070352p. [DOI] [PubMed] [Google Scholar]
- Dorfer V.; Pichler P.; Stranzl T.; Stadlmann J.; Taus T.; Winkler S.; Mechtler K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13 (8), 3679–84. 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfer V.; Maltsev S.; Winkler S.; Mechtler K. CharmeRT: Boosting Peptide Identifications by Chimeric Spectra Identification and Retention Time Prediction. J. Proteome Res. 2018, 17 (8), 2581–2589. 10.1021/acs.jproteome.7b00836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doblmann J.; Dusberger F.; Imre R.; Hudecz O.; Stanek F.; Mechtler K.; Dürnberger G. apQuant: Accurate Label-Free Quantification by Quality Filtering. J. Proteome Res. 2018, 18 (1), 535–541. 10.1021/acs.jproteome.8b00113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.