

Abstract

Biofilm formation is a key mechanism of antimicrobial resistance. We have recently reported two classes of orally bioavailable C-glycosidic inhibitors of the Pseudomonas aeruginosa lectin LecB with antibiofilm activity. They proved efficient in target binding, were metabolically stable, nontoxic, selective, and potent in inhibiting formation of bacterial biofilm. Here, we designed and synthesized six new carboxamides and 24 new sulfonamides for a detailed structure–activity relationship for two clinically representative LecB variants. Sulfonamides generally showed higher inhibition compared to carboxamides, which was rationalized based on crystal structure analyses. Substitutions at the thiophenesulfonamide increased binding through extensive contacts with a lipophilic protein patch. These metabolically stable compounds showed a further increase in potency toward the target and in biofilm inhibition assays. In general, we established the structure–activity relationship for these promising antibiofilm agents and showed that modification of the sulfonamide residue bears future optimization potential.
Introduction
Pseudomonas aeruginosa is an opportunistic Gram-negative bacterium with high clinical importance and classified as a critical priority 1 pathogen by the WHO in 2017.1−4 Especially for cystic fibrosis (CF) patients, chronic infections result in recurrent pneumonia, sepsis, and lung damage.5 Challenges in treating P. aeruginosa infections result from its intrinsic antimicrobial resistance and acquired resistances that often lead to multidrug-resistant MDR or XDR strains.6 In addition, the bacterium’s antimicrobial tolerance is further enhanced by the self-formation of biofilms, a protective enclosure against host immune defense and antibiotic treatment.7,8 Because bacteria residing in a biofilm are up to 1000-fold more resistant toward antibiotics,7 targeting biofilm formation has been an emerging therapeutic approach in recent years to overcome the resistance problem (reviewed in refs (9−11)).
The two virulence factors LecA12 and LecB13 (initially called PA-IL and PA-IIL14) are regulated by quorum sensing15 and have decisive roles in biofilm formation. It is currently anticipated that both tetravalent carbohydrate-binding proteins cross-link glycoconjugates on host cells or tissue with bacterial lipopolysaccharide and exopolysaccharides to stabilize the matrix and integrity of the biofilm.9,16 Thus, blocking this process with exogenous compounds could prevent the formation or even destroy established biofilms.
Both carbohydrate-binding proteins, so-called lectins, were first isolated from the clinical isolate P. aeruginosa PAO1 by Gilboa-Garber et al.14,17,18 One hurdle for therapeutic intervention is the fact that P. aeruginosa has a high genomic diversity among different isolates.19−21 The protein sequence of LecA is rather conserved among P. aeruginosa strains but for LecB isolates are grouped into either PAO1- or PA14-like LecB protein sequence families.22,23 Despite these sequence variations in the two LecB variants, they surprisingly have a conserved binding specificity for similar glycoconjugates, which paves the way for the simultaneous targeting of a broad range of clinical isolates with one single compound. In its quarternary structure, LecB forms noncovalent homotetramers where two Ca2+-ions are present in each monomer,23,24 mediating the recognition of its carbohydrate ligands, l-fucose and d-mannose (e.g., methyl α-d-mannoside (1), Figure 1). Because LecB is localized extracellularly,13 the Gram-negative bacterial cell envelope which usually imposes a stringent hurdle for many antibiotics with intracellular targets, is not problematic for targeting LecB.
Figure 1.

Design approach for C-glycosidic LecB inhibitors and extension of the structural space for extended SAR studies of compounds 4–37. Derivatives of methyl α-d-mannoside 1–3 and their inhibitory potency for the binding with LecBPAO1.36C-Glycosides simultaneously derived of d-mannosides and l-fucosides are hybrid-type LecB ligands 4 and 5 and 6 and 7.41
Besides its role in biofilm formation and bacterial adhesion, LecB was also shown to carbohydrate-dependently block human ciliary beating,25 interfere with tissue repair processes26 and, recently, activate B-cells.27 Furthermore, a direct involvement of LecA and LecB in infection and host colonization by P. aeruginosa using a murine infection model revealed the suitability of both lectins as therapeutic targets.28,29 Inhalation of an aerosol of fucose and galactose, the ligands of LecB and LecA, resulted in a reduction of bacterial load in human infected airways30,31 and in mice.32
Because fucosides display higher affinities for LecB than mannosides, inhibitor development generally centered around fucose-based inhibitors presented on a multivalent scaffold to further increase affinity/avidity.33,34 Following this strategy, multivalent glycopeptide dendrimers have been developed which efficiently inhibit the formation and disperse established biofilms of P. aeruginosa.35 Another example of multivalent fucosides on a calixarene scaffold showed very potent nanomolar affinities for LecB but surprisingly required millimolar concentrations (5 mM) for biofilm inhibition and, in contrast to the desired properties, the compound induced bacterial aggregation.28 Therefore, multivalent presentation of carbohydrates could mimic bacterial exopolysaccharides and, thus, stabilize the biofilm rather than inducing its desired disintegration. In addition, these multivalent presentations of native carbohydrates may be immunogenic and interfere with the patient’s immune system, e.g., by binding to the various innate immunity pattern recognition receptors.
To overcome these disadvantages intrinsic to multivalently displayed lectin ligands, we have embarked on the development of monovalent glycomimetic small molecules as competitive inhibitors of LecB.36−41 Our small molecules possess drug-like properties that resulted in oral bioavailability of the two tested C-glycosides with systemic distribution, which is impossible for the high molecular weight multivalent compounds.
We have started with the weak LecB ligand methyl α-d-mannoside (1) and transformed it into C-6 modified amide and sulfonamide derivatives that led to an increase in potency up to a factor of 20 (e.g., 2 and 3, Figure 1).36,38 These compounds showed good receptor binding kinetics and proved efficient in the prevention of bacterial adhesion. Because fucose and mannose are recognized by LecB, we merged the necessary functional groups that were shown to elicit attractive interactions with the protein into one molecule and obtained the first of our C-glycosides lacking the O-glycosidic linkage.37 After the exploration of additional interactions of heptose-39 or- fluoroglycomimetics40 with LecB, we then combined the initial C-glycosides with the amido- and sulfonamido substituents to obtain the glycomimetics 4–7.41 Especially the sulfonamides 6 and 7 displayed favorable profiles in target binding potency and selectivity, ADME/Tox parameters and oral bioavailability in a murine pharmacokinetics model. Importantly, 6 and 7 possessed excellent antibiofilm activity in a P. aeruginosa biofilm formation assay as monitored by confocal fluorescence light microscopy.
Here, we have significantly increased the number of derivatives of the C-glycoside ligands of LecB to 30 new derivatives yielding a detailed structure–activity relationship for this class of drug-like antibiofilm compounds. We have determined key interactions of the new more potent derivatives from crystallographic analyses, deduced the structural basis of the increased binding potency for the PA14 variant of LecB, and addressed a new binding pocket on the protein surface. The C-glycosidic dimethylthiophene sulfonamide 22 was identified as front runner with good potency in a biofilm assay and excellent ADME/Tox properties.
Results and Discussion
Design
C-Glycosides of the amide series, e.g., 4 and 5, and the sulfonamide series, e.g., 6 and 7, have previously been reported by us as potent glycomimetic inhibitors of LecB (Figure 1).41 To expand the structure–activity relationship of these potent compound classes, we aimed to further diversify the amide and sulfonamide substituents and explore their interaction with LecB.
In the amide series, the cinnamide and dimethoxycinnamide derivative of a mannoside and its C-glycoside analogue are better binders than benzamides or aliphatic amides.36,38,41 We therefore aimed at analyzing a potential rigidification of the cinnamide by ring closure between the ortho-position of the phenyl group and the α-carbon by introducing heteroatom linkers in benzothiophene-, benzofuran-, and indole-2-carboxamides (9–11). These substitutions affect rigidity, hydrogen-bond donating or accepting properties, and total polar surface area of the resulting molecules while maintaining the original cinnamide pharmacophore. Furthermore, we substituted the double bond in the cinnamoyl group with 5-membered heterocycles, i.e., a thiazole and a thiophene (12, 13).
To address the very potent sulfonamide series of C-glycosides and expand the highly orally bioavailable thiophenyl derivative 7, we included a number of different 5-membered heterocycles (14–28), e.g., furan, oxazole, pyrazoles, and the regioisomer of the original thiophene. In addition, various substituents were attached to those heterocycles, and the focus was set to methyl groups that showed good potency increase in the previous mannose-series (IC50s for LecBPA14: phenylsulfonamide 16 μM → trimethylphenylsulfonamide 1 μM, see Figure 2, compounds 47 and 3).36
Figure 2.

Competitive binding assay of inhibitors with LecBPAO1 and LecBPA14 based on fluorescence polarization. Means and standard deviations were determined from a minimum of three independent experiments. n.s.: not soluble at 1 mM in TBS/Ca containing 1% DMSO. IC50 values for 1–7, 38, and 41–46 with LecBPAO1 and LecBPA14 were previously published.23,36−38,41
Inspection of the crystal structure41 of the complex of 7 with LecB indicated a possible second shallow cleft between the two loops formed by residues Val69–Asp75 and Glu95–Ala105 within reach of the thiophene (Supporting Information, Figure S1). Therefore, we designed short spacers of directly attached to the thiophene moiety in position 5 with the aim to tether additional substituents targeting this cleft (29–37).
Synthesis of C-Glycoside LecB Inhibitors
For the synthesis of the β-C-glycosides 8–28, we obtained the precursor 39 from l-fucose (38) by Henry addition of nitromethane with in situ ring closure to the β-anomer followed by a reduction to yield amine 40 (Scheme 1).37,41,42 Then, diversification to give amide or sulfonamide substituted LecB antagonists was generated by coupling with different electrophiles (Scheme 1). The final coupling step yielded amides 8–13 and sulfonamides 14–28 in moderate to good yields (17–76%, over 2 steps).
Scheme 1. Synthesis of the Amides 8–13 and Sulfonamides 14–38.

Reagents and conditions: (a) MeNO2, DBU, molecular sieves 3 Å, 1,4-dioxane, 50 °C, 3 d; (b)Pt/C, H2, HCl, MeOH, rt, 2 d; (c) acyl/sulfonyl chloride or carboxylic acid/EDC·HCl, Et3N, DMF, 0 °C; (d) CuI, Pd(PPh3)2Cl2, RCCH, Et3N, DMF, 50 °C, 16–42 h; (e) 1 atm H2, Lindlar’s catalyst, quinoline, rt, 46 h. Yields for 8–28 are given over two steps from the nitro derivative 39.
To probe the potential additional binding pocket on LecB (see Supporting Information, Figure S1), the bromothiophene 28 was further transformed in palladium-catalyzed Sonogashira cross coupling reactions to the substituted alkyne derivatives 29–36 in acceptable yields of 24–74% for these protecting-group free syntheses. Z-Styryl 37 was obtained from the Sonogashira product 29 following hydrogenation with a Lindlar catalyst in 59% yield.
Inhibition of LecB Carbohydrate Binding Function
All synthesized structures were then analyzed for their capacity to inhibit both representative lectin variants of the two clinically relevant bacterial strain clades, LecBPAO1 and LecBPA14, using established competitive binding assays23,36 (Figure 2).
Previously, the impact of modifications at the cinnamide substituent in 2 was found negligible and a dimethoxy substitution (43, Figure 2) only marginally increased potency.38 Because rigidification and extension of the cinnamide to a naphthalene carboxamide was also tolerated by the protein for the mannose-series,38 we tested new derivatives of C-glycosidic cinnamide 4: benzothiophene (9), benzofuran (10), and indole (11). Despite the introduction of isosteric changes in polarity and hydrogen bonding properties, 9–11 showed similar or slightly decreased activities in this series with benzothiophene 9 as the best inhibitor of both LecB variants (IC50 4.28 and 2.34 μM, for LecBPAO1 and LecBPA14, respectively). A reduction in affinity which was especially pronounced for the PAO1-type lectin was also observed for carboxamide-linked thiazole 12. The corresponding thiophene derivative 13 was insoluble under the assay conditions.
In analogy to the mannose-series, C-glycosidic sulfonamide derivatives 6 and 7 showed superior affinities over the amide-group (4, 5). While a relatively extended SAR was described for the cinnamides in the mannose series,38 the previously synthesized mannose-derived sulfonamides36 occupy a narrow structural diversity: substitution of the phenyl moiety in 46 with methyl groups (→3) leads to increased affinities and isosteric replacement of the phenyl substituent by thiophene (46 → 45) improved potency toward LecB.
To extend the SAR of this potent LecB inhibitor class, we tested a set of C-glycosidic 5-membered heteroatom-substituted sulfonamides with additional substituents varying in constitution and heteroatom position (14–28). Despite the rather high structural diversity of the sulfonamide substituents, all tested compounds potently inhibited both LecB variants in the low micro- to nanomolar range (IC50[LecBPAO1] 1.32–7.01 μM; IC50[LecBPA14] 0.20–1.04 μM). While not affected by constitutional change of the heteroatom position (7 → 21, or 19 → 20), the affinity dropped slightly if the ring sulfur was substituted by oxygen in furan derivatives (7 → 14 or 22 → 15) or nitrogen in pyrazoles (18–20).
Enlargement of the aromatic core by addition of alkyl groups or halogen substituents in 7 generally had a beneficial impact on binding especially in the thiophene series with low micro- to nanomolar activities of 22–25 for both LecB variants. Dimethyl 22 and monomethyl 25 (IC50[LecBPAO1] 1.32–1.87 μM; IC50[LecBPA14] 0.20–0.33 μM) were the best ligands among these thiophenes, both presenting a methyl group in ortho-position to the sulfonamide linker. A similar phenomenon was observed for the pyrazoles, where the best compound 18 (IC50[LecBPAO1] 2.42 μM; IC50[LecBPA14] 0.68 μM) also had such an ortho-methyl substituent compared to the less active derivatives 19 and 20 (IC50[LecBPAO1] 5.88–7.01 μM; IC50[LecBPA14] 0.73–0.96 μM), whose methyl groups were in different positions.
Extension of the initial and unsubstituted thiophene 7 to address the second cleft on LecB yielded the set of 10 alkyne and alkene derivatives 29–37. Again, all compounds showed potent inhibition of both lectin variants in the submicromolar range for the PA14-type and a 1–3 μM affinity range for the PAO1-type. Phenylacetylene derivative 29 stands out as most potent inhibitor of both LecB variants (IC50[LecBPAO1] 1.52 μM; IC50[LecBPA14] 0.14 μM). Further modifications of 29 were tolerated by both proteins although with a moderate reduction in affinity of up to 2-fold: replacement of the phenyl with other groups (34, 35) or elongated spacing (36) was possible. The position of a methyl substituent at the phenyl group in ortho-, meta-, or para-position (30–32) did not have an influence on activity, and transformation of the acetylene into Z-alkene 37 was also not altering potency dramatically.
The thermodynamics of binding of the potent 2,5-dimethyl thiophene derivative 22 to both LecB variants was then further studied by isothermal titration calorimetry (Figure 3, Supporting Information, Table S1). The ligand showed a 1 ligand to 1 LecB monomer binding stoichiometry and affinities for LecB in the low micro- to nanomolar range, Kd = 1.2 μM for PAO1 and Kd = 0.32 μM for PA14, and thus confirmed the obtained IC50 data. As observed for 7,41 also this thiophene-containing ligand 22 showed an enhanced enthalpy driven binding (ΔH −47.3 to −40.4 kJ/mol) compared to the carbocyclic derivatives 4 and 6, which was partially compensated by disfavored entropic contributions (−TΔS 13.5–3.3 kJ/mol).
Figure 3.

Isothermal titration microcalorimetry of LecBPAO1 and LecBPA14 with dimethylthiophene 22. Means and standard deviations were determined from a minimum of three independent titrations. One representative titration graph is depicted for LecBPAO1 only.
Structure of LecB in Complex with Dimethylthiophene 22
To analyze the impact of the methyl substituents in this inhibitor class, we performed crystallization of dimethylthiophene 22 in complex with LecBPA14 using hanging drop cocrystallization. The complex of the lectin crystallized in the P61 space group with four protomers per asymmetric unit. The resulting structure was solved to 1.45 Å resolution, and all carbohydrate-binding sites were occupied with compound 22 (Figure 4, Supporting Information, Table S2).
Figure 4.

Crystal structure of LecBPA14 with C-glycoside ligand 22 (1.45 Å resolution, PDB 5MAZ), (A) Observed binding pose of 22 with crystal contacts. (B) Observed binding pose of 22 without crystal contacts. (C) Superposition of both observed binding poses. (D) Relevant binding pose of 22 with 2Fobs – Fcalc electron density displayed at 1σ. Ligands and amino acids of the carbohydrate recognition domain (CRD) are depicted as sticks colored by elements (C, gray; N, blue; O, red; S, yellow); protein surface in transparent blue and two Ca2+-ions in the binding sites are shown as green spheres.
In previous structures of LecB with sulfonamide ligands, we have observed two different rotamers for the sulfonamides resulting in two types of interaction: a specific interaction41 with the protein as described for 6 or 7 with the cleft enclosed between the two loops Val69–Asp75 and Asp96–Asp104 and a likely unspecific interaction36 induced by crystal contacts for the mannose-derivative 3. Interestingly, in the structure of LecB with dimethylthiophene derivative 22, both binding poses can be observed (Figure 4A,B,C). In this respect, one binding mode of 22 resembles the structure of LecB with thiophene 7 in a way that the thiophene residue is oriented between the same loops of the protein without disturbing crystal contacts (Figure 4B, D). This supports the previous argumentation for the relevance of this binding mode compared to the second observed pose of 22, which is similar to the reported structure36 of the mannose analogue 3 where crystal packing interactions likely favored the altered orientation of the sulfonamide substituent (Figure 4A).
Furthermore, the crystal structure of 7 in complex with LecBPA14 shows a tight coordination of the thiophene moiety to the CH2 group of Ser97 (S-CH2 distance 4.3 Å, or the calculated S-CH2 distance 3.3 Å, sum of H and S van der Waals radii is 3.05 Å). This Ser97 is part of the loop Asp96–Asp104 adjacent to the carbohydrate-binding site. A highly similar interaction is seen in the complex of 22 and LecBPA14 (S-CH2 distance 3.9 Å, or the calculated S-CH2 distance 3.2 Å, sum of H and S van der Waals radii is 3.05 Å). The methyl group of 22 in ortho-position to the sulfonamide linker enters deeply into this still rather shallow pocket and establishes numerous lipophilic contacts with Gly24, Val69, and the CH2 of Asp96. This extensive interaction pattern serves as an explanation for the beneficial effect on the binding affinity of the ortho-methyl group.
Coordination of the carbohydrate-derived ligands by LecBPAO1 and LecBPA14 is largely similar.23 Subtle differences are introduced by the sequence variations between those two type strains and result in the observed difference in activity of the LecB variants. Sulfonamides are generally recognized very well, and the PA14 variant of LecB has a very high affinity for the sulfonamides, 3–10-fold higher than the PAO1 variant. Interestingly, in the cinnamide series of the carboxamides, these differences are vanished and sometimes selectivity is even inverted to the benefit of LecBPAO1 (e.g., 20 vs 37 μM for 43 and 2.34 vs 2.8 μM for 5, respectively). An explanation for this observation results from the presence of Ser97 in LecBPA14 that introduces further steric bulk compared to Gly97 in its PAO1 relative.
The LecB structures reveal this Gly to Ser variation for LecBPA14 at position 97 pointing into the ligand binding site (Figure 5). In case of LecBPAO1, this serine is absent and a glycine moiety occupies its position. The position of the entire loop incorporating either serine or glycine is not affected by this substitution. Now, the complex of LecBPAO1 with the cinnamide of the mannose series (2, Figure 5B) shows a tight lipophilic interaction of the cinnamide moiety with the CH2 group of glycine.38 Furthermore, the cinnamide residue is additionally conformationally fixed through a hydrogen bond of the cinnamide NH with the side chain of Asp96.
Figure 5.

(A) Crystal structure of the complex of LecBPA14 with dimethylthiophene 22 (PDB 5MAZ) reveals the hydrophobic interaction of the ortho-methyl group attached to the thiophene residue with a hydrophobic patch on the protein surface. Furthermore, the thiophene interacts with the side chain CH2 of Ser97. (B) Crystal structure of the complex of LecBPAO1 with manno-cinnamide 2 (PDB 5A3O) reveals the hydrophobic interaction of the cinnamoyl group with a hydrophobic patch formed by the loop carrying Gly97 that lacks the serine side chain present in LecBPA14. (C) superposition of the two structures indicating the steric clash between carboxamide substituents, conformationally fixed through a hydrogen bond of their NH group with the carboxylate of Asp96, and the bulk of the side chain of Ser97 in LecBPA14. This serves as an explanation for the increased selectivity of the amides for LecBPAO1, which contrasts the selectivity of the sulfonamides for LecBPA14. Electron density 2Fobs – Fcalc is displayed at 1σ. Ligands are depicted as sticks colored by elements (C, gray; N, blue; O, red; S, yellow); protein surface in blue and two Ca2+-ions in the binding sites are shown as green spheres.
Because the carboxamides are conformationally fixed in their orientation through this hydrogen bond of the amide-NH with Asp96, they would experience a steric repulsion from this Ser97 in the PA14 variant of LecB and therefore possess the observed less pronounced affinity increase compared to the sulfonamides with LecBPA14 (see overlay in Figure 5C). The latter ones are positioned differently due to the different steric orientation of the linking sulfonamide (planar and trans for the carboxamides, Figure 5B, compared to three-dimensional and cis for the sulfonamides, Figure 5A). Therefore, the selectivity differences of LecBPA14 and LecBPAO1 for the cinnamide series of the carboxamides depend on the amino acid present at position 97.
Inhibition of Bacterial Biofilm Formation
The two most promising compounds selected from the competitive binding assay, dimethyl thiophene 22 and Sonogashira product 29, were then tested in a biofilm assay. We used genetically modified P. aeruginosa PA14 constitutively and intracellulary expressing the fluorescent protein mCherry from the pMP7605 plasmid,43 followed by quantification of fluorescence using confocal light scanning microscopy (CLSM) as previously described.41 The prerequisite for this in situ imaging assay that fluorescence intensity directly correlates with cell density was established earlier.41 The desired absence of bactericidal or bacteriostatic effects was also confirmed for these two selected compounds by measuring total fluorescence intensities of the bacterial cultures after growth in the presence of 100 μM compounds for 23 h. Bacteria reached comparable densities with compounds as the DMSO control (Figure 6).
Figure 6.

Bacterial growth of mCherry-expressing P. aeruginosa quantified by fluorescence intensity (FI) and normalized on the DMSO control in the presence of 100 μM lectin inhibitors 22 or 29.
For biofilm inhibition experiments, bacterial cultures were grown in the presence of 100 μM compounds for 48 h when biofilm mass was quantified by CLSM (Figure 7). The two sulfonamide C-glycosides 22 and 29 showed very potent inhibition of P. aeruginosa biofilm formation by >80% and >90%, respectively. This statistically significant reduction of biofilm formation compared to the DMSO control is contrasting the nonsignificant effect of the natural carbohydrate ligands methyl α-d-mannoside (1) and methyl α-l-fucoside (42), previously reported (data for the latter two compounds from Sommer et al.41). This inactivity of 1 and 42 is in contrast to their good biophysical protein binding activity and may result from continuous depletion in the complex biofilm experiment where bacterial factors could degrade these molecules and/or employ these natural glycosides as nutrients, whereas the C-glycosides remain active.
Figure 7.
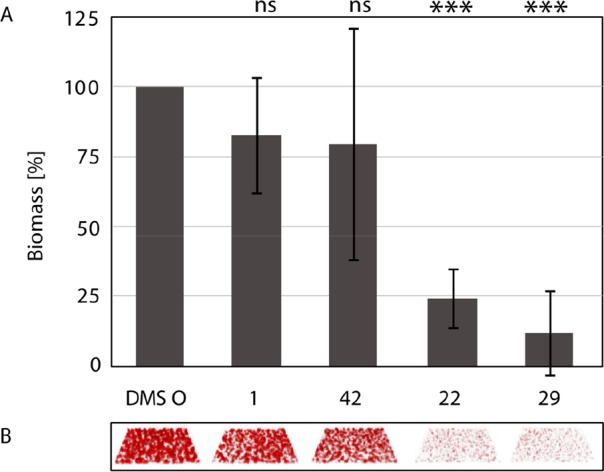
Inhibition of biofilm formation by P. aeruginosa after 48 h growth in the presence of compounds 22 or 29. Depicted data for methyl α-l-fucoside (42), 1, and the DMSO control have been published.41 (A) Quantification of biofilm biomass. Averages and standard deviations of biofilm formation from three independent assays. Statistical significance was calculated using the Student’s t test. (B) Raw data of confocal fluorescence microscopy 3D images show one representative z-stack per condition.
In Vitro Metabolic Stability and Toxicity
In vitro metabolic stability of this compound class was assessed for a number of sulfonamide-based LecB inhibitors in the presence of mouse or human liver microsomes and murine plasma (Table 1, Figure 8A, Supporting Information, Table S3). Compounds were selected based on structural diversity of the sulfonamide substituent and included oxazole 17, dimethylthiophene 22, methylthiophene 23, dichlorothiophene 27, and the Sonogashira product 29. A low intrinsic clearance (CLint) by mouse and human liver microsomes for all tested compounds with values at 10 μL/min/mg protein or below revealed a very high stability (Table 1). In murine plasma, those five compounds were also fully stable over a period of 120 min without any detectable degradation (Figure 8A). These data reveal a high degree of in vitro metabolic stability for this compound class and support our recent results on sulfonamide-linked C-glycosides bearing trimethylphenyl 6 and thiophene 7 that also showed a good metabolic stability against liver microsomes and murine plasma.41
Table 1. Metabolic Stability of Selected Sulfonamides against Mouse and Human Liver Microsomesa.
| CLint |
||
|---|---|---|
| compd | mouse | human |
| 17 | <10 | <10 |
| 22 | <10 | <10 |
| 23 | 10 | <10 |
| 27 | <10 | <10 |
| 29 | <10 | <10 |
Intrinsic clearance (CLint) in μL/min/mg.
Figure 8.

(A) Stability of LecB ligands in mouse plasma. The error bars show the standard deviation of minimum three assays. (B) Toxicity of LecB ligands to human liver Hep G2 cells. Measured OD560nm/670nm was normalized to the controls. Untreated cells served as negative control (normalized OD560nm/670nm = 1), and Triton X-100 treated cells served as positive control (normalized OD560nm/670nm = 0). The error bars show the standard deviation of minimum three assays.
The five thiophenes were further studied, and toxicity of compounds was assessed in vitro using the immortalized human hepatocyte cell line Hep G2 (Figure 8B). Only the alkyne derivative 29 displayed a weak toxicity at the highest concentration tested resulting in 64% hepatocyte viability. No toxicity was detected for all other compounds tested up to a concentration of 100 μM, which is consistent with previous in vitro data on 6 and 7 and allows to generally classify this compound class as nontoxic against Hep G2 cells. Additionally, the sulfonamides 6 and 7 are nontoxic in a murine pharmacokinetics model as reported.
Binding to plasma proteins reduces the amount of free drug, which is the necessary state for binding to the drug’s target. On the contrary, plasma protein binding can positively result in prolonged plasma half-lives (Figure 9). Therefore, balancing the plasma protein binding is necessary, and we tested this property for a set of selected LecB-targeting sulfonamides, the C-glycosides 6, 7, 17, 22, 23, and 29, and the O-glycosides 3 and 45 (which are the O-linked analogues of the C-glycosides 6 and 7). All tested compounds showed approx 70–80% plasma protein binding with the exception of the alkyne 29, which was highly bound by plasma proteins. The two sulfonamides trimethylphenyl 6 and thiophenyl 7 show a plasma protein binding of 73% and 81%, respectively. Both compounds had been analyzed for their pharmakokinetics in mice, and the compound with higher plasma protein binding, 7, also showed an increased plasma half-life of 34 min vs 17 min for 6.41
Figure 9.
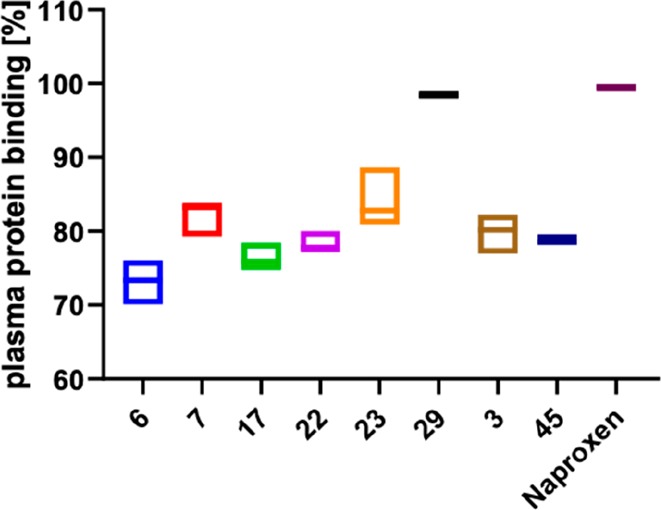
Plasma protein binding of LecB inhibitors C-glycosides 6–29 and the O-glycosides 3 and 45 and naproxen as a control. Box plots show mean and confidence intervals.
Conclusions
In summary, two clinically relevant variants of LecB were tested with >30 newly synthesized glycomimetic inhibitors to extend the known structure–activity relationship of C-glycosides. Potent inhibition for LecB of both strain types was observed, and sulfonamides generally performed better in inhibition experiments than the carboxamides. Interestingly, the affinity difference of the carboxamides for the two lectins variants was rather small and usually only approximately 2-fold better for LecBPA14. In contrast, the sulfonamides displayed a higher affinity increase for LecBPA14 over LecBPAO1, and the difference was up to 10-fold for the Sonogashira products 29–36.
The crystal structure of the dimethylthiophene 22 with LecBPA14 explains its increased potency due to a lipophilic interaction of the methyl group in ortho-position to the sulfonamide with hydrophobic protein residues. Furthermore, by comparison of this LecBPA14/22 structure with the previously reported structure of cinnamide 2 with LecBPAO1, the selectivity differences of LecBPA14 and LecBPAO1 for the cinnamide series of the carboxamides could be assigned to the amino acid present at position 97: serine in LecBPA14 induces a steric clash with the cinnamide residue that is absent in case of Gly97 for LecBPAO1.
The two promising compounds, dimethylthiophene 22 and phenylacetylene bearing thiophene 29, were further tested in a biofilm assay. Both compounds showed a strong inhibition of biofilm formation by P. aeruginosa. Further compound profiling for ADME and toxicity parameters showed that both compounds and others of this class had highest metabolic stability in murine plasma and with mouse and human liver microsomes. However, the dimethyl thiophene 22 was finally prioritized as the front-runner due to a very high plasma protein binding and mainly the moderate mammalian cytotoxicity observed at 100 μM for the otherwise very potent extended structure 29.
In general, C-glycosidic sulfonamides showed highest potency toward both LecB strain-types and their specific structure–activity relationship was rationalized by crystal structure analyses. Nevertheless, the surface exposed nature of the rather shallow binding site where the sulfonamide substituent resides on LecB allows a certain degree of modification at the sulfonamide substituents. This will be a beneficial trait for future optimization of compounds or attachment of other cargo molecules, e.g., imaging probes44 or antibiotics for targeted delivery.
Experimental Section
Chemical Synthesis
Thin layer chromatography (TLC) was performed on Silica Gel 60 coated aluminum sheets containing fluorescence indicator (Merck KGaA, Darmstadt, Germany) and developed under UV light (254 nm) and aqueous KMnO4 solution or a molybdate solution (a 0.02 M solution of ammonium cerium sulfate dihydrate and ammonium molybdate tetrahydrate in aqueous 10% H2SO4). Prepacked Silica Gel 60 columns from Interchim and a Teledyne Isco Combiflash Rf200 system were used for preparative medium pressure liquid chromatography (MPLC). Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III 500 UltraShield spectrometer or on a Bruker Avance III 400 UltraShield spectrometer at 500 MHz/400 MHz (1H), 126 MHz/101 MHz (13C), or 376 MHz (19F). Chemical shifts are given in parts per million (ppm) and were calibrated on residual solvent peaks as internal standard.45 Multiplicities were specified as s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). The signals were assigned with the help of 1H, 1H-COSY, and DEPT-135-edited 1H, 13C-HSQC experiments. Assignment numbering of the C-glycoside atoms and groups corresponds to the numbering in fucose. High resolution mass spectra (HRMS) were obtained on a Bruker maxis 4G hr-QqToF spectrometer, and the data were analyzed using DataAnalysis (Bruker Daltonics, Bremen, Germany). Commercial chemicals and solvents were used without further purification. Deuterated solvents were purchased from Eurisotop (Saarbrücken, Germany). C-Glycoside 39 was synthesized following the procedure described by Phiasivongsa et al.,42 and reduction toward amine 40 was described previously.37 The purity of the final compounds was further analyzed by HPLC-UV, and all UV active compounds had a purity of at least 95%. Chromatographic separation was performed on a Dionex Ultimate 3000 HPLC (Thermo Scientific, Germany) with UV detection at 254 nm using a RP-18 column (100/2 Nucleoshell RP18plus, 2.7 μm, from Machery Nagel, Germany) as stationary phase. LCMS grade distilled MeCN and double distilled H2O were used as mobile phases. In a gradient run, an initial concentration of 5% MeCN in H2O was increased to 95% during 7 min at a flow rate 600 μL/min. The injection volume was 10 μL of 1 mM compound in H2O/DMSO = 100:1.
General Procedure for Amide and Sulfonamide Couplings
β-l-fucopyranosyl methylamine (40) (1 equiv) and triethylamine (1.5 equiv) were dissolved in dry DMF (30 mL per gram substrate) and cooled to 0 °C. The corresponding chloride (1.2 equiv) dissolved in DMF (0.08 M) was added dropwise under nitrogen. In the case of carboxylic acids, EDC·HCl (1.2 equiv) was added. The reaction was allowed to warm to rt and was stirred for further 1–24 h. Saturated aqueous NH4Cl was added and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Unless otherwise indicated, the residue was purified by chromatography on silica (CH2Cl2 to CH2Cl2/EtOH = 10:1 or CH2Cl2/MeOH = 10:1).
N-β-l-Fucopyranosylmethyl benzamide (8)
Compound 8 was obtained following the general procedure from 40 and benzoyl chloride as a colorless solid (70.9 mg, 0.251 mmol, 55%). 1H NMR (500 MHz, MMeOH-d4) δ 7.82–7.81 (m, 2H, CHphenyl), 7.56–7.51 (m, 1H, ArCH), 7.78–7.44 (m, 2H, ArCH), 3.71 (m, 2H, CH2NH), 3.67–3.64 (m, 1H, H-4), 3.64–3.60 (m, 1H, H-5), 3.50–3.48 (m, 2H, H-1, H-3), 3.35–3.32 (m, 1H, H-2), 1.25 (d, J = 3.61 Hz, CH3). 13C NMR (126 MHz, MeOH-d4) δ 170.9 (C = O), 135.9 (ArC), 132.7 (ArCH), 129.6 (ArCH), 128.5 (ArCH), 80.0 (C-2), 76.3 (C-3), 75.8 (C-5), 73.7 (C-4), 69.9 (C-1), 42.6 (CH2), 17.2 (C-6) ppm. HRMS calcd C14H20NO5+: 282.1336, found 282.1345.
N-β-l-Fucopyranosylmethyl Benzo[b]thiophene-2- carboxamide (9)
Compound 9 was obtained following the general procedure from 40 and benzo[b]thiophene-2-carbonyl chloride as a yellowish solid (12 mg, 0.036 mmol, 21%). 1H NMR (500 MHz, MeOH-d4) δ 7.99 (s, 1H, ArCH), 7.93–7.88 (m, 2H, ArCH), 7.46–7.39 (m, 2H, ArCH), 3.77–3.72 (m, 1H, CH2NH), 3.70–3.61 (m, 3H, CH2NH, H-4, H-5), 3.54–3.46 (m, 2H, H-1, H-3), 3.38–3.33 (m, 1H, H-2), 1.28 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 165.4 (C=O), 142.6 (ArC), 140.9 (ArC), 140.0 (ArC), 127.6 (ArCH), 126.8 (ArCH), 126.4 (ArCH), 126.1 (ArCH), 123.3 (ArCH), 80.0 (C-2), 76.4 (C-3), 76.0 (C-5), 73.8 (C-4), 70.0 (C-1), 42.8 (CH2), 17.3 (CH3). HRMS calcd C16H20NO5S+: 338.1057, found 338.1045.
N-β-l-Fucopyranosylmethyl Benzo[b]furan-2-carboxamide (10)
Compound 10 was obtained following the general procedure from 40 and benzo[b]furan-2-carbonyl chloride as a colorless solid (32 mg, 0.10 mmol, 58%). 1H NMR (500 MHz, MeOH-d4) δ 7.74–7.71 (m, 1H, ArCH), 7.61–7.59 (m, 1H, ArCH), 7.50 (d, J = 0.80 Hz, olefin-H), 7.48–7.44 (m, 1H, ArCH), 7.34–7.30 (m, 1H, ArCH), 3.83–3.79 (m, 1H, CH2NH), 3.67–3.61 (m, 3H, CH2NH, H-4, H-5), 3.50–3.48 (m, 2H, H-1, H-3), 3.37–3.33 (m, 1H, H-2), 1.28 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 161.7 (C=O), 156.6 (ArC), 150.0 (ArC), 129.0 (ArC), 128.4 (ArCH), 125.0 (ArCH), 123.9 (ArCH), 113.0 (ArCH), 111.6 (olefin-CH), 80.0 (C-2), 76.4 (C-3), 76.0 (C-5), 73.8 (C-4), 70.3 (C-1), 42.2 (CH2) 17.3 (CH3). HRMS calcd C16H20NO6+: 322.1285, found 322.1300.
N-β-l-Fucopyranosylmethyl 1H-Indole-2-carboxamide (11)
Compound 11 was obtained following the general procedure from 40, 1H-indole-2-carboxylic acid and EDC·HCl as a colorless solid (28 mg, 0.09 mmol, 31%). 1H NMR (500 MHz, DMSO-d6) δ 11.57 (d, J = 2.1 Hz, 1H, NHindole), 8.41 (t, J = 5.7 Hz, 1H, NHCO), 7.60 (d, J = 8.0 Hz, 1H, ArCH), 7.43 (d, J = 8.3 Hz, 1H, ArCH), 7.29–7.12 (m, 2H, 2 × ArCH), 7.03 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H, ArCH), 4.96 (dd, J = 4.5, 1.5 Hz, 1H, OH), 4.68 (d, J = 5.2 Hz, 1H, OH), 4.39 (d, J = 4.9 Hz, 1H, OH-4), 3.70 (ddd, J = 13.9, 4.6, 2.4 Hz, 1H, CH2NH), 3.53–3.44 (m, 2H, H-5, H-4), 3.36–3.25 (m, 3H, CH2NH, H-2, H-3), 3.24–3.18 (m, 1H, H-1), 1.12 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (126 MHz, DMSO-d6) δ 161.5 (C = O), 136.4 (ArC), 131.6 (ArC), 127.1 (ArC), 123.3 (ArCH), 121.5 (ArCH), 119.7 (ArCH), 112.3 (ArCH), 102.9 (ArCH), 78.7 (C-1), 74.5 (C-2 or C-3), 73.8 (C-5), 71.6 (C-4), 68.7 (C-2 or C-3), 41.2 (CH2NH), 17.2 (C-6). HRMS calcd C16H21N2O5+: 321.1445, found 321.1429.
N-β-l-Fucopyranosylmethyl 2-Phenyl-1,3-thiazol-4-carboxamide (12)
Compound 12 was obtained following the general procedure from 40 and 2-phenyl-1,3-thiazol-4-carbonyl chloride as a yellowish solid (30 mg, 0.08 mmol, 49%). 1H NMR (500 MHz, MeOH-d4) δ 8.20 (s, 1H, CHthiazol), 8.05–8.02 (m, 2H, CHphenyl), 7.52–7.49 (m, 3H, CHphenyl), 3.90–3.85 (m, 1H, CH2NH), 3.68–3.63 (m, 2H, H-4, H-5), 3.62–3.56 (m, 1H, CH2NH), 3.52–3.49 (m, 2H, H-1, H-3), 3.38–3.32 (m, 1H, H-2), 1.29 (d, J = 6.62 Hz, CH3). 13C NMR (126 MHz, MeOH-d4) δ 170.0 (C=O), 163.8 (Cthiazol), 151.7 (Cthiazol), 134.3 (Cphenyl), 132.1 (CHphenyl), 130.4 (2 × C, CHphenyl), 127.9 (2 × C, CHphenyl), 124.9 (CHthiazol), 80.1 (C-2), 76.4 (C-3), 76.0 (C-5), 73.7 (C-4), 70.6 (C-1), 42.4 (CH2), 17.4 (CH3). HRMS calcd C17H21N2O5S+: 365.1166. found 365.1185.
N-β-l-Fucopyranosylmethyl 5-Phenylthiophene-2-carboxamide (13)
Compound 13 was obtained following the general procedure from 40 and 5-phenyl-2-thiophene carbonyl chloride as a colorless solid (26 mg, 0.07 mmol, 42%). 1H NMR (500 MHz, DMSO-d6) δ 8.53 (t, J = 5.36 Hz, 1H, NH), 7.83 (d, J = 3.78 Hz, 1H, CHthiophen), 7.70 (d, J = 7.25 Hz, 2H, CHphenyl), 7.53 (d, J = 4.10 Hz, 1H, CHthiophen), 7.44 (t, J = 7.57 Hz, 2H, CHphenyl), 7.36 (t, J = 7.25 Hz, 1H, CHphenyl), 4.89 (d, J = 4.73 Hz, 1H, OH), 4.68 (d, J = 5.36 Hz, 1H, OH), 4.37 (d, J = 4.73 Hz, 1H, OH), 3.71–3.65 (m, 1H, CH2NH), 3.51–3.43 (m, 2H, H-4, H-5), 3.34–3.15 (m, 4H, CH2NH, H-1, H-2, H-3), 1.12 (d, J = 6.62 Hz, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 161.4 (C=O), 147.4 (Cthiophen), 138.9 (Cthiophen), 133.2 (Cphenyl), 129.4 (CHthiophen), 129.3 (2 × C, CHphenyl), 128.6 (CHphenyl), 125.7 (2 × C, CHphenyl), 124.4 (CHhiophen), 78.5 (C-2), 74.5 (C-3), 73.8 (C-5), 71.6 (C-4), 68.7 (C-1), 41.5 (CH2), 17.2 (CH3). HRMS calcd C18H22NO5S+: 364.1213, found 364.1207.
N-β-l-Fucopyranosylmethyl 2-Furansulfonamide (14)
Compound 14 was obtained following the general procedure from 40 and furan-2-sulfonyl chloride as a colorless oil (45 mg, 0.273 mmol, 52%). 1H NMR (500 MHz, MeOH-d4) δ 7.74–7.73 (m, 1H, ArCH), 7.04 (dd, J = 3.47 Hz, J = 0.95 Hz, 1H, ArCH), 6.60–6.57 (m, 1H, ArCH), 3.62–3.60 (m, 1H, H-4), 3.54–3.49 (m, 1H, CH2NH, H-1, H-5), 3.43–3.38 (m, 3H, H-3), 3.35 (s, 1H), 3.18–3.13 (m, 1H, H-2), 3.12–3.07 (m, 1H, CH2NH), 1.21 (d, J = 6.62 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 150.8 (ArC), 147.7 (ArCH), 116.7 (ArCH), 112.3 (ArCH), 79.8 (C-2), 76.5 (C-3), 75.7 (C-5), 73.7 (C-4), 69.8 (C-1), 45.6 (CH2), 17.2 (C-6). HRMS calcd C11H18NO7S+ 308.0798, found 308.0800.
N-β-l-Fucopyranosylmethyl 2,5-Dimethyl-3-furansulfonamide (15)
Compound 15 was obtained following the general procedure from 40 and 2,5-dimethyl-3-furansulfonyl chloride as a colorless solid (10 mg, 0.03 mmol, 17%). 1H NMR (500 MHz, MeOH-d4) δ 6.19 (s, 1H, ArCH), 3.63–3.60 (m, 1H, H-4), 3.55–3.49 (m, 1H, H-5), 3.44–3.35 (m, 2H, H-1,H-3), 3.29–3.25 (m, 1H, CH2NH), 3.20–3.14 (m, 1H, H-2), 3.05–2.99 (m, 1H, CH2NH), 2.46 (s, 3H, furan-CH3), 2.25 (s, 3H, furan-CH3), 1.20 (d, 3H, J = 6.31, CH3). 13C NMR (126 MHz, MeOH-d4) δ 155.2 (ArC), 152.3 (ArC), 122.8 (ArC), 106.5 (ArCH), 79.8 (C-2), 76.5 (C-3), 75.7 (C-5), 73.8 (C-4), 69.9 (C-1), 45.5 (CH2NH), 17.2 (C-6), 13.2 (CH3), 13.0 (CH3). HRMS calcd C13H22NO7S+: 336.1111, found 336.1101.
N-β-l-Fucopyranosylmethyl 5-Methyl-2-trifluoromethyl-3-furansulfonamide (16)
Compound 16 was obtained following the general procedure from 40 and 5-methyl-2-trifluoromethyl-3-furansulfonyl chloride as a colorless oil (45 mg, 0.273 mmol, 52%). 1H NMR (500 MHz, MeOH-d4) δ 7.19–7.17 (m, 1H, ArCH), 3.62–3.60 (m, 1H, H-4), 3.52–3.47 (m, 1H, H-5), 3.41–3.35 (m, 3H, CH2NH, H-1, H-3), 3.20–3.13 (m, 1H, H-2), 3.10–3.05 (m, 1H, CH2NH), 2.59 (s, 3H, furan-CH3), 1.19 (d, J = 6.45 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 159.5 (ArC), 140.08 (q, 2JCF= 43.6 Hz, ArC), 124.6 (ArC), 120.0 (q, 1JCF = 266.4 Hz, CF3), 113.4 (q, 3JCF = 2.9 Hz, ArCH), 79.5 (C-2), 76.3 (C-3), 75.5 (C-5), 73.5 (C-4), 69.6 (C-1), 45.3 (CH2), 17.0 (C-6), 13.0 (CH3). 19F NMR (376 MHz, MeOH-d4) δ −66.5 (CF3). HRMS calcd C13H19F3NO7S+: 390.0829, found 390.0844.
N-β-l-Fucopyranosylmethyl 5-Methyl-4-isoxazolesulfonamide (17)
Compound 17 was obtained following the general procedure from 40 and 5-methyl-4-isoxazolesulfonyl chloride as a colorless oil (17.0 mg, 0.05 mmol, 19%). 1H NMR (500 MHz, MeOH-d4) δ 8.56 (d, J = 0.8 Hz, 1H, ArCH), 3.63–3.59 (m, 1H, H-4), 3.50 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.42–3.35 (m, 3H, H-1, H-3, CH2NH), 3.19–3.14 (m, 1H, H-2), 3.11 (dd, J = 12.9, 7.1 Hz, 1H, CH2NH), 2.66 (d, J = 0.7 Hz, 3H, CH3), 1.18 (d, J = 6.5 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 172.5 (ArC), 150.2 (ArCH), 119.5 (ArC), 79.5 (C-2), 76.3 (C-3), 75.5 (C-5), 73.5 (C-4), 69.6 (C-1), 45.3 (CH2NH), 17.0 (C-6), 11.8 (CH3). HRMS calcd C11H19N2O7S+: 323.0907, found 323.0902.
N-β-l-Fucopyranosylmethyl 1,3-Dimethyl-1-H-pyrazole-4-sulfonamide (18)
Compound 18 was obtained following the general procedure from 40 and 1,3-dimethyl-1-H-pyrazole-4-sulfonyl chloride as a colorless oil (24 mg, 0.07 mmol, 53%). 1H NMR (500 MHz, MeOH-d4) δ 7.97 (s, 1H, ArCH), 3.84 (s, 3H, NCH3), 3.61 (dd, J = 3.0, 1.1 Hz, 1H, H-4), 3.51 (qd, J = 6.4, 1.1 Hz, 1H, H-5), 3.45–3.36 (m, 2H, H-2, H-3), 3.31–3.27 (m, 1H, CH2NH), 3.15 (ddd, J = 9.2, 7.0, 2.6 Hz, 1H), 3.03 (dd, J = 13.0, 7.0 Hz, 1H), 2.36 (s, 3H), 1.20 (d, J = 6.5 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 148.6 (ArC), 135.7 (ArCH), 120.8 (ArC), 79.4 (C-2), 76.4 (C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-1), 45.3 (CH2NH), 39.1 (NCH3), 17.1 (C-6), 12.3 (CH3). HRMS calcd C12H22N3O6S+: 336.1224, found 336.1225.
N-β-l-Fucopyranosylmethyl 1-Methyl-1-H-pyrazole-3-sulfonamide (19)
Compound 19 was obtained following the general procedure from 40 and 1-methyl-1-H-pyrazole-3-sulfonyl chloride as a colorless oil (57 mg, 0.177 mmol, 63%). 1H NMR (500 MHz, MeOH-d4) δ 7.72 (d, J = 2.21 Hz, 1H, ArCH), 6.66 (d, J = 2.21 Hz, 1H, ArCH), 3.96 (s, 3H, NCH3), 3.63–3.61 (m, 1H, H-4), 3.56–3.50 (m, 1H, H-5), 3.47–3.39 (m, 3H, CH2NH, H-1, H-3), 3.23–3.18 (m, 1H, H-2), 3.16–3.10 (m, 1H, CH2NH), 1.21 (d, J = 6.62 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 152.1 (ArC), 134.0 (ArCH), 107.7 (ArCH), 79.9 (C-2), 76.5 (C-3), 75.7 (C-5), 73.8 (C-4), 69.8 (C-1), 45.8 (CH2), 39.9 (NCH3), 17.2 (C-6). HRMS calcd C11H20N3O6S+: 322.1067, found 322.1070.
N-β-l-Fucopyranosylmethyl 1-Methyl-1-H-pyrazole-4-sulfonamide (20)
Compound 20 was obtained following the general procedure from 40 and 1-methyl-1-H-pyrazole-4-sulfonyl chloride as a colorless solid (26 g, 0.08 mmol, 47%). 1H NMR (500 MHz, MeOH-d4) δ 8.08 (s, 1H, ArCH), 7.76 (s, 1H, ArCH), 3.93 (s, 3H, NCH3), 3.63–3.60 (m, 1H, H-4), 3.55–3.50 (m, 1H, H-5), 3.45–3.38 (m, 2H, H-1, H-3), 3.34–3.32 (m, 1H, CH2NH), 3.22–3.17 (m, 1H, H-2), 3.06–3.01 (m, 1H, CH2NH), 1.19 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 139.5 (ArCH), 133.8 (ArCH), 124.0 (ArC), 79.7 (C-2), 76.5 (C-3), 75.7 (C-5), 73.7 (C-4), 69.8 (C-1), 45.7 (CH2), 39.6 (NCH3), 17.2 (CH3). HRMS calcd C11H20N3O6S+: 322.1067, found 322.1064.
N-β-l-Fucopyranosylmethyl 3-Thiophenesulfonamide (21)
Compound 21 was obtained following the general procedure from 40 and 3-thiophenesulfonyl chloride as a colorless oil (41.6 mg, 0.13 mmol, 76%). 1H NMR (500 MHz, MeOH-d4) δ 8.07 (dd, J = 3.0, 1.3 Hz, 1H, ArCH), 7.59 (dd, J = 5.2, 3.0 Hz, 1H, ArCH), 7.38 (dd, J = 5.2, 1.3 Hz, 1H, ArCH), 3.60 (dd, J = 2.9, 1.1 Hz, 1H, H-4), 3.48 (qd, J = 6.4, 1.1 Hz, 1H, H-5), 3.44–3.33 (m, 3H, H-1, H-3, CH2NH), 3.17–3.11 (m, 1H, H-2), 3.04 (dd, J = 13.0, 7.2 Hz, 1H, CH2NH), 1.20 (d, J = 6.4 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 142.0 (ArC), 131.2 (ArCH), 129.2 (ArCH), 126.6 (ArCH), 79.5 (C-2), 76.3 (C-3), 75.5 (C-5), 73.6 (C-4), 69.7 (C-1), 45.6 (CH2NH), 17.1 (C-6). HRMS calcd for C11H16NO6S2+: 324.0570, found 324.0567.
N-β-l-Fucopyranosylmethyl 2,5-Dimethyl-3-thiophenesulfonamide (22)
Compound 22 was obtained following the general procedure from 40 and 2,5-dimethyl-2-thiophenesulfonyl chloride as a colorless oil (39.8 mg, 0.11 mmol, 67%). 1H NMR (500 MHz, MeOH-d4) δ 6.92 (q, J = 1.2 Hz, 1H, ArCH), 3.60 (dd, J = 3.1, 1.1 Hz, 1H, H-4), 3.46 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.44–3.29 (m, 3H, H-1, H-3, CH2NH), 3.14–3.07 (m, 1H, H-2), 3.02 (dd, J = 13.0, 7.3 Hz, 1H, CH2NH), 2.60 (s, 3H, thiophene-CH3), 2.39 (s, 3H, thiophene-CH3), 1.20 (d, J = 6.5 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 143.5 (ArC), 137.7 (ArC), 135.9 (ArC), 126.6 (ArCH), 79.4 (C-2), 76.3 (C-3), 75.6 (C-5), 73.6 (C-4), 69.8 (C-1), 45.3 (CH2NH), 17.1 (C-6), 14.8 (CH3), 14.2 (CH3). HRMS calcd for C13H22NO6S2+: 352.0883, found 352.0881.
N-β-l-Fucopyranosylmethyl 5-Methyl-2-thiophenesulfonamide (23)
Compound 23 was obtained following the general procedure from 40 and 5-methyl-2-thiophenesulfonyl chloride as a colorless oil (40.3 mg, 0.12 mmol, 71%). 1H NMR (500 MHz, MeOH-d4) δ 7.40 (d, J = 3.7 Hz, 1H, ArCH), 6.88–6.79 (m, 1H, ArCH), 3.61 (dd, J = 3.0, 1.1 Hz, 1H, H-4), 3.51 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.46–3.33 (m, 3H, H-1, H-3, CH2NH), 3.21–3.15 (m, 1H, H-2), 3.06 (dd, J = 12.9, 7.1 Hz, 1H, CH2NH), 2.53 (d, J = 1.0 Hz, 3H, thiophene-CH3), 1.20 (d, J = 6.4 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 148.6 (ArC), 139.6 (ArC), 133.3 (ArCH), 126.9 (ArCH), 79.5 (C-2), 76.3 (C-3), 75.5 (C-5), 73.6 (C-4), 69.7 (C-1), 45.7 (CH2NH), 17.0 (C-6), 15.3 (CH3). HRMS calcd for C12H20NO6S2+: 338.0727, found 338.0724.
N-β-l-Fucopyranosylmethyl 4-Methyl-2-thiophenesulfonamide (24)
Compound 24 was obtained following the general procedure from 40 and 4-methyl-2-thiophenesulfonyl chloride as a colorless oil (26 mg, 0.078 mmol, 27%). 1H NMR (500 MHz, MeOH-d4) δ 7.44–7.42 (m, 1H, ArCH), 7.35–7.33 (m, 1H, ArCH), 3.63–3.60 (m, 1H, H-4), 3.54–3.48 (m, 1H, H-5), 3.44–3.35 (m, 3H, CH2NH, H-1, H-3), 3.21–3.16 (m, 1H, H-2), 3.10–3.04 (m, 1H, CH2NH), 2.28 (s, 3H, thiophene-CH3), 1.21 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 142.5 (ArC), 139.9 (ArC), 134.9 (ArCH), 128.6 (ArCH), 79.9 (C-2), 76.7 (C-3), 75.9 (C-5), 73.9 (C-4), 70.1 (C-1), 46.1 (CH2), 17.4 (C-6), 15.8 (CH3) ppm. HRMS calcd C12H20NO6S2+: 338.0727; found: 338.0720.
N-β-l-Fucopyranosylmethyl 3-Methyl-2-thiophenesulfonamide (25)
Compound 25 was obtained following the general procedure from 40 and 3-methyl-2-thiophenesulfonyl chloride as a colorless oil (24 mg, 0.07 mmol, 42%). 1H NMR (500 MHz, MeOH-d4) δ 7.58 (d, J = 5.04 Hz, 1H, ArCH), 7.00 (d, J = 5.04 Hz, 1H, ArCH), 3.61–3.58 (m, 1H, H-4), 3.50–3.44 (m, 1H, H-5), 3.43–3.32 (m, 3H, CH2NH, H-1, H-3), 3.13–3.03 (m, 2H, CH2NH, H-2), 2.47 (s, 3H, thiophene-CH3), 1.20 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 143.2 (ArC), 136.0 (ArC), 133.0 (ArCH), 130.7 (ArCH), 79.5 (C-2), 76.5 (C-3), 75.7 (C-5), 73.7 (C-4), 69.8 (C-1), 45.6 (CH2), 17.2 (CH3), 14.9 (thiophene-CH3). HRMS calcd C12H20NO6S2+: 338.0727, found 338.0732.
N-β-l-Fucopyranosylmethyl 5-Ethyl-2-thiophenesulfonamide (26)
Compound 26 was obtained following the general procedure from 40 and 5-ethyl-2-thiophenesulfonyl chloride as a colorless oil (32 mg, 0.091 mmol, 32%). 1H NMR (500 MHz, MeOH-d4) δ 7.43 (d, J = 3.78 Hz, 1H, ArCH), 6.87 (d, J = 3.78 Hz, 1H, ArCH), 3.62–3.60 (m, 1H, H-4), 3.54–3.48 (m, 1H, H-5), 3.45–3.34 (m, 3H, CH2NH, H-1, H-3), 3.20–3.15 (m, 1H, H-2), 3.08–3.03 (m, 1H, CH2NH), 2.91 (q, J = 7.57 Hz, 2H, thiophene-CH2CH3), 1.32 (t, J = 7.57 Hz, 3H, thiophene-CH2CH3), 1.19 (d, J = 6.31 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 156.2 (ArC), 139.4 (ArC), 133.3 (ArCH), 125.3 (ArCH), 79.7 (C-2), 76.5 (C-3), 75.7 (C-5), 73.7 (C-4), 69.9 (C-1), 45.9 (CH2), 24.6 (thiophene-CH2CH3), 17.2 (C-6), 16.3 (thiophene-CH2CH3). HRMS calcd C13H22NO6S2+: 352.0883, found 352.0899.
N-β-l-Fucopyranosylmethyl 4,5-Dichloro-2-thiophenesulfonamide (27)
Compound 27 was obtained following the general procedure from 40 and 4,5-dichloro-2-thiophenesulfonyl chloride as a colorless oil (42.0 mg, 0.11 mmol, 63%). 1H NMR (500 MHz, MeOH-d4) δ 7.51 (s, 1H, ArCH), 3. 63–3.60 (m, 1H, H-4), 3.51 (qd, J = 6.5, 1.1 Hz, 1H. H-5), 3.45–3.36 (m, 3H, H-1, H-3, CH2NH), 3.22–3.16 (m, 1H, H-2), 3.12 (dd, J = 12.9, 7.2 Hz, 1H, CH2NH), 1.19 (d, J = 6.5 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 140.5 (ArC), 131.5 (ArCH), 131.1 (ArC), 125.6 (ArC), 79.4 (C-2), 76.3 (C-3), 75.5 (C-5), 73.5 (C-4), 69.7 (C-1), 45.8 (CH2NH), 17.1 (C-6). HRMS calcd for C11H16Cl2NO6S2+: 391.9791, found 391.9787.
N-β-l-Fucopyranosylmethyl 5-Bromo-2-thiophenesulfonamide (28)
Compound 28 was obtained following the general procedure from 40 and 5-bromo-2-thiophenesulfonyl chloride as a colorless oil (43.6 mg, 0.11 mmol, 64%). 1H NMR (500 MHz, MeOH-d4) δ 7.40 (d, J = 3.9 Hz, 1H, ArCH), 7.18 (d, J = 4.0 Hz, 1H, ArCH), 3.64–3.58 (m, 1H, H-4), 3.51 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.43–3.36 (m, 3H, H-1, H-3, CH2NH), 3.22–3.15 (m, 1H, H-2), 3.08 (dd, J = 12.9, 7.2 Hz, 1H, CH2NH), 1.20 (d, J = 6.5 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 144.2 (ArC), 133.2 (ArCH), 131.9 (ArCH), 119.8 (ArC), 79.5 (C-2), 76.3 (C-3), 75.5 (C-5), 73.6 (C-4), 69.7 (C-1), 45.7 (CH2NH), 17.1 (C-6). HRMS calcd for C11H17BrNO6S2+: 401.9675, found 401.9671.
General Procedure for the Sonogashira Couplings
The solution of the bromothiophene 28, CuI, Pd(PPh3)2Cl2, the corresponding acetylene, and Et3N in DMF (0.03 M) was degassed (100 mbar) at 0 °C. The reaction mixture was stirred under argon atmosphere at 50 °C for 16–42 h, monitored by LCMS, and poured into H2O (4 mL). The organic phase was separated, and the aqueous phase was extracted with EtOAc (7 × 3 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed in vacuo.
N-β-l-Fucopyranosylmethyl 5-Phenylethynyl-2-thiophenesulfonamide (29)
A solution of thiophene 28 (30 mg, 0.07 mmol), CuI (0.7 mg, 0.004 mmol), Pd(PPh3)2Cl2 (1.3 mg, 0.002 mmol), phenylacetylene (0.01 mL, 0.09 mmol), and Et3N (20 μL, 0.15 mmol) in DMF (2.5 mL) was degassed (100 mbar) at 0 °C. The reaction mixture was stirred under argon atmosphere at 50 °C for 3 h and monitored by LCMS. Phenylacetylene (0.01 mL, 0.09 mmol) was added, and the reaction mixture was stirred at 50 °C for 14 h and poured into H2O (4 mL). The organic phase was separated, and the aqueous phase was extracted with EtOAc (7 × 3 mL). The combined organic layers were dried over Na2SO4, and the solvent was removed in vacuo. The residue was purified by HPLC (VP250/10 Nucleodur C-18 Gravity SB, 5 μm from Macherey Nagel, 8 mL/min; eluent A, H2O; eluent B, MeCN, 0–40 min 30%–50% B). After lyophilization, 29 was obtained as a colorless solid (18.4 mg, 0.043 mmol, 59%). 1H NMR (500 MHz, MeOH-d4) δ 7.57–7.50 (m, 3H, CHthiophene, CHphenyl), 7.41 (dd, J = 5.2, 2.0 Hz, 3H, CHphenyl), 7.29 (d, J = 3.8 Hz, 1H, CHthiophene), 3.64–3.59 (m, 1H, H-4), 3.52 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.45–3.38 (m, 3H, H-2, H-3, CH2NH), 3.20 (ddd, J = 9.3, 7.2, 2.2 Hz, 1H, H-1), 3.11 (dd, J = 12.9, 7.2 Hz, 1H, CH2NH), 1.21 (d, J = 6.4 Hz, 3H, CH3). 13C NMR (126 MHz, MeOH-d4) δ 143.5 (ArC), 133.0 (ArC), 132.7 (ArC), 132.6 (ArC), 130.4 (ArC), 130.1 (ArC), 129.7 (ArC), 123.2 (ArC), 96.7 (Cethynyl), 81.7 (Cethynyl), 79.6 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 45.8 (CH2NH), 17.1 (C-6). HRMS calcd C19H22NO6S2+: 424.0883, found 424.0884.
N-β-l-Fucopyranosylmethyl 5-(o-Methyl-phenylethynyl)-2-thiophenesulfonamide (30)
Compound 30 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (1.3 mg, 0.002 mmol), CuI (0.7 mg, 0.004 mmol), Et3N (20 μL, 0.14 mmol), and (o-methylphenyl)acetylene (17 μL, 0.136 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O + 0.1% HCOOH; eluent B, MeCN + 0.1% HCOOH, 0–4 min 5%–30% B, 4–25 min 30% B, 25–39 min 30%–40% B). After lyophilization, 30 was obtained as a colorless solid (24 mg, 0.05 mmol, 74%). 1H NMR (500 MHz, MeOH-d4) δ 7.53 (d, J = 3.9 Hz, 1H, CHaromatic), 7.49–7.45 (m, 1H, CHaromatic), 7.33–7.26 (m, 3H, CHaromatic), 7.23–7.18 (m, 1H, CHaromatic), 3.63–3.59 (m, 1H, H-4), 3.52 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.47–3.38 (m, 3H, H-2, H-3, CH2NH), 3.24–3.17 (m, 1H, H-1), 3.11 (dd, J = 12.9, 7.3 Hz, 1H, CH2NH), 2.47 (s, 3H, CH3), 1.21 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 143.4 (Caromatic), 141.4 (Caromatic), 132.9 (CHaromatic), 132.7 (CHaromatic), 132.7 (CHaromatic), 130.7 (CHaromatic), 130.5 (Caromatic), 130.3 (CHaromatic), 127.0, (CHaromatic) 122.8 (Caromatic), 95.7 (Cethynyl), 85.6 (Cethynyl), 79.5 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 45.8 (CH2NH), 20.7 (CH3), 17.1 (C-6). HR-MS calcd C20H24NO6S2+: 438.1040, found 438.1028.
N-β-l-Fucopyranosylmethyl 5-(m-Methyl-phenylethynyl)-2-thiophenesulfonamide (31)
Compound 31 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (2.6 mg, 0.004 mmol), CuI (1.4 mg, 0.007 mmol), Et3N (20 μL, 0.14 mmol), and (m-methylphenyl)acetylene (23 μL, 0.176 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O; eluent B, MeCN, 0–4 min 5%–30% B, 4–45 min 30% B). After lyophilization, 31 was obtained as a colorless solid (18 mg, 0.04 mmol, 55%). 1H NMR (500 MHz, MeOH-d4) δ 7.52 (d, J = 3.9 Hz, 1H, CHaromatic), 7.37–7.34 (m, 1H, CHaromatic), 7.34–7.30 (m, 1H, CHaromatic), 7.30–7.28 (m, 1H, CHaromatic), 7.27 (d, J = 3.9 Hz, 1H, CHaromatic), 7.25–7.22 (m, 1H, CHaromatic), 3.63–3.59 (m, 1H, H-4), 3.52 (qd, J = 6.4, 1.1 Hz, 1H, H-5), 3.46–3.37 (m, 3H, H-2, H-3, CH2NH), 3.23–3.17 (m, 1H, H-1), 3.11 (dd, J = 12.9, 7.2 Hz, 1H, CH2NH), 2.35 (d, J = 0.7 Hz, 3H, CH3), 1.21 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 143.4 (Caromatic), 139.7 (Caromatic), 133.0 (CHaromatic), 132.9 (CHaromatic), 132.7 (CHaromatic), 131.2 (CHaromatic), 130.3 (Caromatic), 129.7 (CHaromatic), 129.6 (CHaromatic), 123.0 (Caromatic), 97.0 (Cethynyl), 81.3 (Cethynyl), 79.6 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 45.8 (CH2NH), 21.2 (CH3), 17.1 (C-6). HR-MS calcd C20H24NO6S2+: 438.1040, found 438.1041.
N-β-l-Fucopyranosylmethyl 5-(p-Methyl-phenylethynyl)-2-thiophenesulfonamide (32)
Compound 32 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (1.3 mg, 0.002 mmol), CuI (0.7 mg, 0.004 mmol), Et3N (20 μL, 0.14 mmol), and (p-methylphenyl)acetylene (20 μL, 0.14 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O; eluent B, MeCN, 0–4 min 5%–30% B, 4–45 min 30% B). After lyophilization, 32 was obtained as a colorless solid (21 mg, 0.05 mmol, 64%). 1H NMR (500 MHz, MeOH-d4) δ 7.51 (d, J = 3.9 Hz, 1H, CHaromatic), 7.43–7.39 (m, 2H, CHaromatic), 7.25 (d, J = 3.9 Hz, 1H, CHaromatic), 7.25–7.20 (m, 2H, CHaromatic), 3.63–3.59 (m, 1H, H-4), 3.51 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.45–3.37 (m, 3H, H-2, H-3, CH2NH), 3.23–3.16 (m, 1H, H-1), 3.11 (dd, J = 12.9, 7.3 Hz, 1H, CH2NH), 2.37 (s, 3H, CH3), 1.21 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 143.2 (Caromatic), 141.0 (Caromatic), 132.7 (CHaromatic), 132.7 (CHaromatic), 132.5 (CHaromatic), 130.5 (Caromatic), 130.4 (CHaromatic), 120.2 (Caromatic), 97.0 (Cethynyl), 81.1 (Cethynyl), 79.6 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 45.8 (CH2NH), 21.6 (CH3), 17.1 (C-6). HR-MS calcd C20H24NO6S2+: 438.1040, found 438.1040.
N-β-l-Fucopyranosylmethyl 5-(p-Methoxy-phenylethynyl)-2-thiophenesulfonamide (33)
Compound 33 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (2.6 mg, 0.004 mmol), CuI (1.4 mg, 0.007 mmol), Et3N (20 μL, 0.14 mmol), and (p-methoxyphenyl)acetylene (12 mg, 0.09 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O + 0.1% HCOOH; eluent B, MeCN + 0.1% HCOOH, 0–4 min 5%–30% B, 4–45 min 30% B). After lyophilization, 33 was obtained as a colorless solid (15 mg, 0.03 mmol, 44%). 1H NMR (500 MHz, MeOH-d4) δ 7.50 (d, J = 3.8 Hz, 1H, CHaromatic), 7.48–7.44 (m, 2H, CHaromatic), 7.23 (d, J = 3.9 Hz, 1H, CHaromatic), 6.98–6.93 (m, 2H, CHaromatic), 3.83 (s, 3H, OCH3), 3.63–3.59 (m, 1H, H-4), 3.52 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.45–3.36 (m, 3H, H-2, H-3, CH2NH), 3.22–3.16 (m, 1H, H-1), 3.11 (dd, J = 12.9, 7.3 Hz, 1H, CH2NH), 1.21 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 162.0 (Caromatic), 142.8 (Caromatic), 134.2 (CHaromatic), 132.7 (CHaromatic), 132.4 (CHaromatic), 130.8 (Caromatic), 115.4 (CHaromatic), 115.1 (Caromatic), 97.1 (Cethynyl), 80.4 (Cethynyl), 79.6 (C-1), 76.4 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 55.9 (OCH3), 45.8 (CH2NH), 17.1 (C-6). HR-MS calcd C20H24NO7S2+: 454.0989, found 454.0975.
N-β-l-Fucopyranosylmethyl 5-Cyclopropylethynyl-2-thiophenesulfonamide (34)
Compound 34 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (1.3 mg, 0.002 mmol), CuI (0.7 mg, 0.004 mmol), Et3N (20 μL, 0.14 mmol), and cyclopropylacetylene (16 μL, 0.19 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O + 0.1% HCOOH; eluent B, MeCN + 0.1% HCOOH, 0–40 min 5%–50% B). After lyophilization, 34 was obtained as a colorless solid (10 mg, 0.03 mmol, 35%). 1H NMR (500 MHz, MeOH-d4) δ 7.43 (d, J = 3.9 Hz, 1H, CHaromatic), 7.07 (d, J = 3.9 Hz, 1H, CHaromatic), 3.63–3.59 (m, 1H, H-4), 3.50 (qd, J = 6.4, 1.1 Hz, 1H, H-5), 3.42–3.35 (m, 3H, H-2, H-3, CH2NH), 3.21–3.14 (m, 1H, H-1), 3.07 (dd, J = 12.9, 7.3 Hz, 1H, CH2NH), 1.53 (tt, J = 8.3, 5.0 Hz, 1H, CH), 1.20 (d, J = 6.5 Hz, 3H, H-6), 0.99–0.92 (m, 2H, CH2), 0.81–0.76 (m, 2H, CH2). 13C NMR (126 MHz, MeOH-d4) δ 141.9 (Caromatic), 132.5 (CHaromatic), 132.1 (CHaromatic), 131.5 (Caromatic), 102.1 (Cethynyl), 79.6 (C-1), 76.4 (C-2 or C-3), 75.6 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 68.2 (Cethynyl), 45.8 (CH2NH), 17.1 (C-6), 9.3 (CH2), 0.9 (CH). HR-MS calcd C16H22NO6S2+: 388.0883, found 388.0873.
N-β-l-Fucopyranosylmethyl 5-(3-Butyn-2-ol,2-methyl)-2-thiophenesulfonamide (35)
Compound 35 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (2.6 mg, 0.004 mmol), CuI (1.4 mg, 0.007 mmol), Et3N (20 μL, 0.14 mmol), and (1-hydroxy-1-methylethyl)acetylene (17 μL, 0.18 mmol). The residue was purified by MPLC (CH2Cl2 + 0.1% MeOH to CH2Cl2 + 0.1% MeOH/MeOH = 9:1). 35 was obtained as a colorless solid (15 mg, 0.04 mmol, 50%). 1H NMR (500 MHz, MeOH-d4) δ 7.47 (d, J = 3.9 Hz, 1H, CHaromatic), 7.17 (d, J = 3.9 Hz, 1H, CHaromatic), 3.62–3.59 (m, 1H, H-4), 3.50 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.43–3.36 (m, 3H, H-2, H-3, CH2NH), 3.20–3.15 (m, 1H, H-1), 3.08 (dd, J = 12.9, 7.3 Hz, 1H, CH2NH), 1.55 (s, 6H, COH(CH3)2), 1.20 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 143.2 (Caromatic), 132.8 (CHaromatic), 132.5 (CHaromatic), 130.1 (Caromatic), 102.2 (Cethynyl), 79.6 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 74.3 (Cethynyl), 73.6 (C-4), 69.7 (C-2 or C-3), 65.9 (COH(CH3)2), 45.8 (CH2NH), 31.3 (COH(CH3)2), 17.1 (C-6). HR-MS calcd [M + H+–H2O] C16H22NO6S2+: 388.0883, found 388.0887.
N-β-l-Fucopyranosylmethyl 5-(Benzene,3-butyn-1-yl)-2-thiophenesulfonamide (36)
Compound 36 was obtained following the general procedure for Sonogashira coupling from 28 (30 mg, 0.075 mmol), Pd(PPh3)2Cl2 (2.6 mg, 0.004 mmol), CuI (1.4 mg, 0.007 mmol), Et3N (20 μL, 0.14 mmol), and (3-butynyl)benzene (13 μL, 0.09 mmol). The residue was purified by RP-MPLC (RediSep column: C18 15.5 g of gold, 30 mL/min; eluent A, H2O + 0.1% HCOOH; eluent B, MeCN + 0.1% HCOOH, 0–5 min 5%–20% B, 5–45 min 20%–45% B). After lyophilization, 36 was obtained as a colorless solid (8 mg, 0.02 mmol, 24%). 1H NMR (500 MHz, MeOH-d4) δ 7.43 (d, J = 3.9 Hz, 1H, CHaromatic), 7.33–7.25 (m, 4H, CHaromatic), 7.23–7.18 (m, 1H, CHaromatic), 7.05 (d, J = 3.9 Hz, 1H), 3.63–3.58 (m, 1H, H-4), 3.49 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.41–3.36 (m, 3H, H-2, H-3, CH2NH), 3.21–3.13 (m, 1H, H-1), 3.07 (dd, J = 13.0, 7.3 Hz, 1H, CH2NH), 2.90 (t, J = 7.2 Hz, 2H, CH2), 2.75 (dd, J = 7.5, 6.9 Hz, 2H, CH2), 1.19 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 142.2 (Caromatic), 141.6 (Caromatic), 132.5 (CHaromatic), 132.0 (CHaromatic), 131.2 (Caromatic), 129.6 (CHaromatic), 129.4 (CHaromatic), 127.4 (CHaromatic), 98.2 (Cethynyl), 79.5 (C-1), 76.3 (C-2 or C-3), 75.6 (C-5), 74.1 (Cethynyl), 73.6 (C-4), 69.7 (C-2 or C-3), 45.8 (CH2NH), 35.6 (CH2), 22.6 (CH2), 17.1 (C-6). HR-MS calcd C21H26NO6S2+: 452.1196, found 452.1200.
N-β-l-Fucopyranosylmethyl 5-[(1Z)-2-Phenylethenyl]-2-thiophenesulfonamide (37)
The acetylene 29 (17 mg; 0.04 mmol) was dissolved in dry MeOH (0.4 mL). Lindlar catalyst (4 mg) and quinoline (6 μL; 0.05 mmol) was added, and the reaction mixture was stirred at rt for 46 h under H2 atmosphere (1 bar). The suspension was filtrated over Celite, and the volatiles were removed in vacuo. The residue was purified by HPLC (VP250/10 Nucleodur C-18 Gravity SB, 5 μm from Macherey Nagel, 8 mL/min; eluent A, H2O + 0.05% HCOOH; eluent B, MeCN + 0.05% HCOOH, 0–40 min 20%–60% B). After lyophilization, 37 was obtained as a colorless solid (10 mg, 0.02 mmol, 59%). 1H NMR (500 MHz, MeOH-d4) δ 7.42–7.36 (m, 3H, CHaromatic, CHDB), 7.36–7.29 (m, 3H, CHaromatic), 6.98 (dd, J = 3.9, 0.6 Hz, 1H, CHDB), 6.83–6.72 (m, 2H, CHaromatic), 3.62–3.58 (m, 1H, H-4), 3.47 (qd, J = 6.5, 1.1 Hz, 1H, H-5), 3.39–3.36 (m, 2H, H-2, H-3), 3.33–3.27 (m, 1H, CH2NH), 3.15–3.10 (m, 1H, H-1), 2.99 (dd, J = 12.9, 7.3 Hz, 1H), CH2NH, 1.18 (d, J = 6.5 Hz, 3H, H-6). 13C NMR (126 MHz, MeOH-d4) δ 147.0 (Caromatic), 141.5 (Caromatic), 137.9 (Caromatic), 133.4 (CHaromatic), 132.2 (CHDB), 130.0 (CHaromatic), 129.6 (CHaromatic), 129.4 (CHaromatic), 129.3 (CHaromatic), 123.6 (CHDB), 79.5 (C-1), 76.4 (C-2 or C-3), 75.5 (C-5), 73.6 (C-4), 69.7 (C-2 or C-3), 45.7 (CH2NH), 17.1 (C-6). HR-MS calcd C19H24NO6S2+: 426.1040; found: 426.1038.
Competitive Binding Assay
According to previously described procedures,23,36 20 μL of a stock solution of LecBPAO1 (225 nM) or LecBPA14(150 nM) and fluorescent reporter ligand N-(fluorescein-5-yl)-N′-(α-l-fucopyranosyl ethylen)thiocarbamide (15 nM) in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2) were mixed with 10 μL of serial dilutions (1 mM to 12.8 nM) of testing compounds in TBS/Ca in triplicates in black 384-well microtiter plates (Greiner Bio-One, Germany, catalogue no. 781900). After incubation at rt in a humidity chamber for 22–24 h, fluorescence emission parallel and perpendicular to the excitation plane was measured on a PheraStar FS (BMG Labtech, Germany) plate reader (excitation filters, 485 nm; emission filters, 535 nm). The data were analyzed using BMG Labtech MARS software. After reduction of the measured intensities by buffer values, the fluorescence polarization was calculated and fitted according to the four parameter variable slope model. Bottom and top plateaus were defined by the standard compounds l-fucose (39) and methyl α-d-mannoside (1), respectively, and the data was reanalyzed with these values fixed. A minimum of three independent measurements of triplicates each was performed for every ligand.
Isothermal Titration Calorimetry (ITC)
ITC was performed on a Microcal ITC200 (General Electric) at 25 °C. A solution of ligands in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.3 supplemented with 1 mM CaCl2) was titrated into a LecB solution in the same buffer. A molar extinction coefficient of 6990 M–1 cm–1 (obtained from ProtParam46) was used to calculate the concentration of the monomer of LecBPAO1 or LecBPA14 from the absorbance determined by UV spectroscopy. Concentrations of ligands and protein are given in Supporting Information, Table S1. The collected data were analyzed according to the one site binding model using the Microcal Origin software. A minimum of three independent titrations was performed for each protein/ligand combination.
Crystallization and Structure Determination
LecBPA14 (10 mg mL–1 in water) was incubated in a 9:1 ratio with a 10 mM solution of 22 in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.3 supplemented with 1 mM CaCl2) 1 h prior to crystallization. Hanging drop vapor diffusion method using 1 μL of protein plus ligand + 1 μL of reservoir solution at 19 °C in a 24-well plate yielded crystals after 1 or 2 days. For the LecBPA14–22 complex, data were collected from a broken rod crystal obtained with 22% PEG 8K, 50 mM (NH4)2SO4, and 0.1 M Tris-HCl pH 8.5. The crystal was transferred in a solution where 20% glycerol was added for cryoprotection prior mounting in a cryoloop and flash-freezing in liquid nitrogen. All diffraction data were collected at 100 K at the European Synchrotron Radiation Facility (Grenoble, France) on beamline ID23-2 using a Pilatus detector. The data were processed using XDS.47 All further computing was performed using the CCP4 suite.48 Five percent of the observations were set aside for cross-validation analysis, and hydrogen atoms were added in their riding positions and used for geometry and structure-factor calculations. The structures were solved by molecular replacement using PHASER.49 For the complex LecBPA14–22, the coordinates of the 5A6Q tetramer were used as a search model to search for one tetramer in the asymmetric unit. The structures were refined with restrained maximum likelihood refinement using REFMAC 5.850 iterated with manual rebuilding in Coot.51 Ligand libraries were created using Sketcher and JLigand. The ligands were introduced after inspection of the 2Fo – DFc weighted maps. Water molecules, introduced automatically using Coot, were inspected manually. The stereochemical quality of the models was assessed with the PDB Validation Server, and coordinates were deposited in the Protein Data Bank under accession code 5MAZ. Data quality and refinement statistics are summarized in Supporting Information, Table S2.
Biofilm Assay
P. aeruginosa PA14 wt (DSM19882) carrying mCherry expression plasmid pMP760543 was constructed previously.41 Bacterial precultures were inoculated from single colonies in 5 mL of LB and grown at 37 °C and 180 rpm overnight to stationary phase. For the biofilm assay, the bacterial precultures were diluted to an OD600 nm of 0.02 in fresh M63 minimal medium. Then, 200 μL of bacterial culture were transferred to each well of a 24-well imaging plate (catalogue no. 3231, Zell-Kontakt GmbH, Germany). Compounds were resolved in DMSO (100 mM) and diluted to 200 μM in M63 minimal medium. Then, 200 μL of compound solution were transferred per well to the 24-well plate to reach a final concentration of 100 μM. Plates were incubated at 30 °C for 48 h. Biofilms of mCherry-labeled bacteria were analyzed using a confocal laser scanning microscope (Leica TCS Sp8 CLSM). Focal planes were acquired starting from the bottom of the plate (position 0) with an interplane distance (z-step size) of 2 μm (using a 25× numerical-aperture water objective). mCherry was excited with a 561 nm laser. Images were batch-processed using ImageJ52 and Comstat 2.1.53
To obtain total fluorescence intensities, bacteria were grown in black 96-well plates (F-bottom, Greiner Bio-One no. 655077) in the presence of 100 μM compounds. Fluorescence intensities were recorded with the BMG FluoStar Omega plate reader using a 6 × 6 matrix per well, 10 flashes per point, and excitation at 584 nm and emission at 620 nm.
Microsomal Stability Assay
Tested compounds (1 μM in 0.1 M phosphate buffer at pH 7.4) were preincubated with microsomes (0.5 mg/mL, human liver microsomes: UltraPool HLM 150 Mixed Gender, Corning, no. 452117; mouse liver microsomes, male C57BL/6J, Corning, no. 457247) in 96-well plates (polypropylene, 1200 μL, Treff Lab, 96.8996.9.01) for 10 min and cofactor NADPH (the NADPH regenerating system consists of 30 mM glucose-6-phosphate disodium salt hydrate; 10 mM NADP, 30 mM MgCl2·6H2O, and 5 mg/mL glucose-6-phosphate dehydrogenase (Roche Diagnostics) in 0.1 M potassium phosphate buffer pH 7.4) was added to start the enzymatic reaction. After incubation for 1, 3, 6, 9, 15, 25, 35, and 45 min at 37 °C on a TECAN automated liquid handling system (Tecan Group Ltd., Switzerland), aliquots were removed and quenched with 1:3 (v/v) MeCN containing internal standards (2-(8-aminotetralin-5-yl)-1,1,1,3,3,3-hexafluoro-propan-2-ol, CAS number 625837-83-4, 100 ng/mL in MeCN). Four enzyme markers (midazolam for CYP3A4, buproprion for CYP2B6, dextromethorphan for CYP2D6, diclofenac for CYP2C9) were used for assessing the metabolic activity of the microsomes incubations as quality control. Samples were cooled (4 °C) and centrifuged (3000 rpm) before analysis of the supernatant by LC-MS/MS. Data was analyzed by fitting a linear fit of the Ln peak area ratios (test compound peak area/internal standard peak area) against incubation time. The slope of the fit was used to calculate the intrinsic clearance: Clint (μL/min/mg protein) = −slope (min–1) × 1000/[protein concentration (mg/mL)].
Plasma Stability Assay
All test compounds were dissolved in DMSO and added to mouse plasma (pH 7.4, 37 °C) to yield a final concentration of 25 μM. In addition, procaine, propoxycaine, and procainamide (dissolved in DMSO) were added to mouse plasma (pH 7.4, 37 °C) to yield a final concentration of 250 μM. Procaine and propoxycaine served as positive controls as they are known to be unstable in mouse plasma. Procainamide served as negative control, as it is known to be stable in mouse plasma. The samples were incubated for 0, 15, 30, 60, 90, and 120 min at 37 °C. At each time point, 7.5 μL of the respective sample was extracted with 22.5 μL of methanol containing an internal standard for 5 min at 2000 rpm on a shaking incubator (Eppendorf). Then, samples were centrifuged for 2 min at 13000 rpm, and the supernatants were transferred to HPLC glass vials. For procaine, propoxycaine, and procainamide samples, 100 ng/mL naproxen was used as internal standard for HPLC-MS. For the other compounds, 100 ng/mL glipizide was used as internal standard for HPLC-MS. All samples were analyzed via HPLC-MS using an Agilent 1290 HPLC system equipped with a diode array UV detector and coupled to an AB Sciex QTrap 6500 mass spectrometer. HPLC conditions were as follows: column, Agilent Zorbax Eclipse Plus C18, 50 mm × 2.1 mm, 1.8 μm; temperature 30 °C; injection volume 1 μL; flow rate 700 μL/min; solvent A, H2O + 0.1% HCOOH; solvent B, CH3CN + 0.1% HCOOH; gradient 99% A at 0 min, 99% to 0% A from 0.1 to 5.50 min, 0% A until 6.00 min, then 99% A postrun for 2 min; UV detection 190–400 nm. Mass spectrometric conditions were as follows: Scan type, Q1MS; scan rate, 1000 Da/s; scan start, 100 Da; scan stop, 1000 Da. Procaine, procainamide, and propoxycaine samples were detected in positive scan mode. The other compounds were detected in negative scan mode. Peak areas of each compound and of the internal standard were analyzed using MultiQuant 3.0 software (AB Sciex). Peaks were quantified using indicated m/z search windows (Supporting Information, Table S3). Peak areas of the respective compound were normalized to the internal standard peak area and to the respective peak areas at time point 0 min, (C/D)/(A/B); with A, peak area of the respective compound at time point 0 min; B, peak area of the respective internal standard at time point 0 min; C, peak area of the respective compound at the respective time point; D, peak area of the respective internal standard at the respective time point. Every experiment was at least repeated three times independently.
Plasma Protein Binding
Plasma protein binding was assessed using the rapid equilibrium device (RED) system from ThermoFisher. Compounds were dissolved in DMSO. Naproxen served as control as it shows high plasma protein binding. Compounds were diluted in murine plasma (from CD-1 mice, pooled) to a final concentration of 100 μM. Dialysis buffer and plasma samples were added to the respective chambers according the manufacturer’s protocol. The RED plate was sealed with a tape and incubated at 37 °C for 2 h on an Eppendorf MixMate vortex Mixer at 800 rpm. Then, samples were withdrawn from the respective chambers. To 25 μL of each dialysis sample, 25 μL of plasma, and to 25 μL of plasma sample, 25 μL of dialysis buffer was added. Then 150 μL of ice-cold extraction solvent (CH3CN/H2O (90:10) containing 12.5 ng/mL caffeine as internal standard) was added. Samples were incubated for 30 min on ice. Then, samples were centrifuged at 4 °C at 1000 rpm for 10 min. Supernatants were transferred to Greiner V-bottom 96-well plates and sealed with a tape. Samples were analyzed using an Agilent 1290 Infinity II HPLC system coupled to an AB Sciex QTrap 6500plus mass spectrometer. LC conditions were as follows: column, Agilent Zorbax Eclipse Plus C18, 50 mm × 2.1 mm, 1.8 μm; temperature, 30 °C; injection volume, 5 μL; flow rate, 700 μL/min; solvent A, H2O + 0.1% HCOOH; solvent B, 95% CH3CN/5% H2O + 0.1% HCOOH; gradient, 99% A at 0 min, 99% A until 0.10 min, 99% to 0% A from 0.1 to 3.50 min, 0% A until 3.70 min, 0% to 99% A from 3.70 to 3.80 min, 99% A until 4.00 min.
Mass transitions for naproxen and compounds are depicted in Supporting Information, Table S4. The percentage of bound compound was calculated as follows:
MTT Assay (Hepatocytes Toxicity)
The epithelial cell line Hep G2 (ATCC HB-8065TM) was cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% heat-inactivated fetal calf serum (FCS) at 37 °C and 5% CO2. Hep G2 cells were seeded into a 96-well plate (Nunc, Roskilde, Denmark) and grown to 75% confluency. Every compound was dissolved in DMSO and diluted in PBS (final DMSO concentration in the cell assay: 0.1%). Hep G2 cells were incubated with the respective compound in concentrations ranging from 0.01 to 100 μM for 24 h at 37 °C and 5% CO2. Hep G2 cells treated with vehicle only (DMSO diluted in PBS, final DMSO concentration in the cell assay: 0.1%) served as a negative control. Furthermore, pure medium (DMEM + 10% FCS) and completely damaged cells served as positive controls. To damage cells, Hep G2 cells were treated with 0.5% Triton X-100 1 h prior to addition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich). After 24 h, Hep G2 cells were washed twice with DMEM + 10% FCS. MTT diluted in PBS (stock solution 5 mg/mL) was added to the wells at a final concentration of 1 mg/mL. The cells were incubated for 3 h at 37 °C and 5% CO2. Medium was removed, and 0.04 M HCl in 2-propanol was added. The cells were incubated at room temperature for 15 min. Then the supernatant was transferred to a 96-well plate. The absorbance of the samples was measured at 560 nm and at 670 nm as a reference wavelength on a Tecan Sunrise ELISA reader using Magellan software. Data was normalized using the following formula: (A–B)/(C–B) with “A” as the respective data point, “B” as the value of the Triton X-100-treated control, and “C” as the vehicle control. The experiment was repeated at least three times. The error bars indicate the standard deviation.
Acknowledgments
We are grateful to Dr. Stefan Böttcher for initial MS studies and Dr. Michael Hoffmann for HRMS measurements (all HIPS Saarbrücken); we acknowledge technical assistance from Janine Schreiber (HZI, Braunschweig). Crystal data collection was performed at the European Synchrotron Radiation Facility, Grenoble, France, and we are grateful for access and technical support to beamline ID23-2. A.I. and A.V. acknowledge support from the ANR projects Glyco@Alps (ANR-15-IDEX-02) and Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). We further thank the Helmholtz Association (grant no. VH-NG-934, to A.T.), EU COST action BM1003 (to R. S.), the Deutsche Forschungsgemeinschaft (to A.T., grant no Ti756/2-1), and the European Research Council for an ERC starting grant (to A.T., Sweetbullets) for financial support.
Glossary
Abbreviations Used
- WHO
World Health Organization
- CF
cystic fibrosis
- XDR
extensively drug resistant
- P. aeruginosa
Pseudomonas aeruginosa
- EDC·HCl
N-(3-(fimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- PDB
Protein Data Bank
- CLSM
confocal light scanning microscopy
- FI
fluorescence intensity
- CLint
intrinsic clearance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01120.
1H, 13C, and 19F NMR spectra of new compounds; ITC measurements with 22; data collection and refinement statistics for LecBPA14 structure in complex with 22; m/z search window for plasma stability assay; mass transitions of compounds; rationale for the extension of the thiophene moiety in 7 to target an additional patch or a subpocket on the surface of LecB; purity of key compounds 17, 22, 23, 27, and 29 by HPLC-UV (PDF)
Molecular formula strings for all new compounds (8–37) (CSV)
Author Contributions
R.S., S.H., S.N., and D.H. synthesized C-glycosides; R.S. performed LecB binding assays and ITC; R.S., A.V., and A.I. performed and analyzed protein crystallography experiments; S.W. performed and analyzed the biofilm assay; T.R. analyzed metabolic stability in liver microsomes; K.R. and T.A. performed and analyzed plasma stability, plasma protein binding, and toxicity assays; A.I., M.B., and R.W.H. analyzed data and provided conceptual advice; R.S., K.R., S.W., and A.T., conceived the study; R.S. and A.T. wrote the paper with input from all coauthors.
The authors declare no competing financial interest.
Supplementary Material
References
- Nagao M.; Iinuma Y.; Igawa J.; Saito T.; Yamashita K.; Kondo T.; Matsushima A.; Takakura S.; Takaori-Kondo A.; Ichiyama S. Control of an outbreak of carbapenem-resistant Pseudomonas aeruginosa in a haemato-oncology unit. J. Hosp Infect 2011, 79, 49–53. 10.1016/j.jhin.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Rice L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- Tsutsui A.; Suzuki S.; Yamane K.; Matsui M.; Konda T.; Marui E.; Takahashi K.; Arakawa Y. Genotypes and infection sites in an outbreak of multidrug-resistant Pseudomonas aeruginosa. J. Hosp Infect 2011, 78, 317–322. 10.1016/j.jhin.2011.04.013. [DOI] [PubMed] [Google Scholar]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed; World Health Organization: Geneva, 2017; http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/ (accessed May 2017)
- Hauser A. R., Rello J., Eds. Severe Infections Caused by Pseudomonas aeruginosa; Kluwer Academic Publishers Group: Boston, MA, 2003. [Google Scholar]
- Poole K. Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2011, 2, 65. 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming H.-C.; Wingender J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- Davies D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discovery 2003, 2, 114–122. 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- Sommer R.; Joachim I.; Wagner S.; Titz A. New approaches to control infections: anti-biofilm strategies against gram-negative bacteria. Chimia 2013, 67, 286–290. 10.2533/chimia.2013.286. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Sommer R.; Hinsberger S.; Lu C.; Hartmann R. W.; Empting M.; Titz A. Novel strategies for the treatment of Pseudomonas aeruginosa infections. J. Med. Chem. 2016, 59, 5929–5969. 10.1021/acs.jmedchem.5b01698. [DOI] [PubMed] [Google Scholar]
- Calvert M. B.; Jumde V. R.; Titz A. Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections. Beilstein J. Org. Chem. 2018, 14, 2607–2617. 10.3762/bjoc.14.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle S. P.; Stacey R. E.; Dodd C.; Cámara M.; Williams P.; Winzer K. The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ. Microbiol. 2006, 8, 1095–1104. 10.1111/j.1462-2920.2006.001001.x. [DOI] [PubMed] [Google Scholar]
- Tielker D.; Hacker S.; Loris R.; Strathmann M.; Wingender J.; Wilhelm S.; Rosenau F.; Jaeger K.-E. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 2005, 151, 1313–1323. 10.1099/mic.0.27701-0. [DOI] [PubMed] [Google Scholar]
- Gilboa-Garber N. Pseudomonas aeruginosa lectins. Methods Enzymol. 1982, 83, 378–385. 10.1016/0076-6879(82)83034-6. [DOI] [PubMed] [Google Scholar]
- Winzer K.; Falconer C.; Garber N. C.; Diggle S. P.; Camara M.; Williams P. The Pseudomonas aeruginosa lectins PA-IL and PA-IIL are controlled by quorum sensing and by RpoS. J. Bacteriol. 2000, 182, 6401–6411. 10.1128/JB.182.22.6401-6411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong J. N. C.; Yildiz F. H. Biofilm matrix proteins. Microbiol. Spectrum 2015, 3, MB-0004-2014. 10.1128/microbiolspec.MB-0004-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa-Garber N.; Mizrahi L.; Garber N. Mannose-binding hemagglutinins in extracts of Pseudomonas aeruginosa. Can. J. Biochem. 1977, 55, 975–981. 10.1139/o77-145. [DOI] [PubMed] [Google Scholar]
- Gilboa-Garber N. Purification and properties of hemagglutinin from Pseudomonas aeruginosa and its reaction with human blood cells. Biochim. Biophys. Acta, Gen. Subj. 1972, 273, 165–173. 10.1016/0304-4165(72)90204-8. [DOI] [PubMed] [Google Scholar]
- Klockgether J.; Munder A.; Neugebauer J.; Davenport C. F.; Stanke F.; Larbig K. D.; Heeb S.; Schöck U.; Pohl T. M.; Wiehlmann L.; Tümmler B. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J. Bacteriol. 2010, 192, 1113–1121. 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether J.; Cramer N.; Wiehlmann L.; Davenport C. F.; Tümmler B. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2011, 2, 150. 10.3389/fmicb.2011.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dötsch A.; Schniederjans M.; Khaledi A.; Hornischer K.; Schulz S.; Bielecka A.; Eckweiler D.; Pohl S.; Häussler S. The Pseudomonas aeruginosa Transcriptional Landscape Is Shaped by Environmental Heterogeneity and Genetic Variation. mBio 2015, 6, e00749-15 10.1128/mBio.00749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukerb A. M.; Decor A.; Ribun S.; Tabaroni R.; Rousset A.; Commin L.; Buff S.; Doléans-Jordheim A.; Vidal S.; Varrot A.; Imberty A.; Cournoyer B. Genomic rearrangements and functional diversification of lecA and lecB lectin-coding regions impacting the efficacy of glycomimetics directed against Pseudomonas aeruginosa. Front. Microbiol. 2016, 7, 811. 10.3389/fmicb.2016.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer R.; Wagner S.; Varrot A.; Nycholat C. M.; Khaledi A.; Haussler S.; Paulson J. C.; Imberty A.; Titz A. The virulence factor LecB varies in clinical isolates: consequences for ligand binding and drug discovery. Chem. Sci. 2016, 7, 4990–5001. 10.1039/C6SC00696E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell E.; Houles C.; Sudakevitz D.; Wimmerova M.; Gautier C.; Pérez S.; Wu A. M.; Gilboa-Garber N.; Imberty A. Structural basis for oligosaccharide-mediated adhesion of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Nat. Struct. Biol. 2002, 9, 918–921. 10.1038/nsb865. [DOI] [PubMed] [Google Scholar]
- Adam E. C.; Mitchell B. S.; Schumacher D. U.; Grant G.; Schumacher U. Pseudomonas aeruginosa II lectin stops human ciliary beating: therapeutic implications of fucose. Am. J. Respir. Crit. Care Med. 1997, 155, 2102–2104. 10.1164/ajrccm.155.6.9196121. [DOI] [PubMed] [Google Scholar]
- Cott C.; Thuenauer R.; Landi A.; Kühn K.; Juillot S.; Imberty A.; Madl J.; Eierhoff T.; Römer W. Pseudomonas aeruginosa lectin LecB inhibits tissue repair processes by triggering β-catenin degradation. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863, 1106–1118. 10.1016/j.bbamcr.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm I.; Levit-Zerdoun E.; Jakob J.; Villringer S.; Frensch M.; Übelhart R.; Landi A.; Müller P.; Imberty A.; Thuenauer R.; Claudinon J.; Jumaa H.; Reth M.; Eibel H.; Hobeika E.; Römer W. Carbohydrate-dependent B cell activation by fucose-binding bacterial lectins. Sci. Signaling 2019, 12, eaao7194 10.1126/scisignal.aao7194. [DOI] [PubMed] [Google Scholar]
- Boukerb A. M.; Rousset A.; Galanos N.; Méar J.-B.; Thepaut M.; Grandjean T.; Gillon E.; Cecioni S.; Abderrahmen C.; Faure K.; Redelberger D.; Kipnis E.; Dessein R.; Havet S.; Darblade B.; Matthews S. E.; de Bentzmann S.; Guéry B.; Cournoyer B.; Imberty A.; Vidal S. Anti-adhesive properties of glycoclusters against Pseudomonas aeruginosa lung infection. J. Med. Chem. 2014, 57, 10275–10289. 10.1021/jm500038p. [DOI] [PubMed] [Google Scholar]
- Chemani C.; Imberty A.; de Bentzmann S.; Pierre M.; Wimmerová M.; Guery B. P.; Faure K. Role of LecA and LecB lectins in Pseudomonas aeruginosa-induced lung injury and effect of carbohydrate ligands. Infect. Immun. 2009, 77, 2065–2075. 10.1128/IAI.01204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bismarck P.; Schneppenheim R.; Schumacher U. Successful treatment of Pseudomonas aeruginosa respiratory tract infection with a sugar solution--a case report on a lectin based therapeutic principle. Klin. Paediatr. 2001, 213, 285–287. 10.1055/s-2001-17220. [DOI] [PubMed] [Google Scholar]
- Hauber H.-P.; Schulz M.; Pforte A.; Mack D.; Zabel P.; Schumacher U. Inhalation with fucose and galactose for treatment of Pseudomonas aeruginosa in cystic fibrosis patients. Int. J. Med. Sci. 2008, 5, 371–376. 10.7150/ijms.5.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucior I.; Abbott J.; Song Y.; Matthay M. A.; Engel J. N. Sugar administration is an effective adjunctive therapy in the treatment of Pseudomonas aeruginosa pneumonia. Am. J. Physiol Lung Cell Mol. Physiol 2013, 305, L352–L363. 10.1152/ajplung.00387.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecioni S.; Imberty A.; Vidal S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 2015, 115, 525–561. 10.1021/cr500303t. [DOI] [PubMed] [Google Scholar]
- Bernardi A.; Jiménez-Barbero J.; Casnati A.; De Castro C.; Darbre T.; Fieschi F.; Finne J.; Funken H.; Jaeger K.-E.; Lahmann M.; Lindhorst T. K.; Marradi M.; Messner P.; Molinaro A.; Murphy P. V.; Nativi C.; Oscarson S.; Penadés S.; Peri F.; Pieters R. J.; Renaudet O.; Reymond J.-L.; Richichi B.; Rojo J.; Sansone F.; Schäffer C.; Turnbull W. B.; Velasco-Torrijos T.; Vidal S.; Vincent S.; Wennekes T.; Zuilhof H.; Imberty A. Multivalent glycoconjugates as anti-pathogenic agents. Chem. Soc. Rev. 2013, 42, 4709–4727. 10.1039/C2CS35408J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E. M. V.; Crusz S. A.; Kolomiets E.; Buts L.; Kadam R. U.; Cacciarini M.; Bartels K.-M.; Diggle S. P.; Cámara M.; Williams P.; Loris R.; Nativi C.; Rosenau F.; Jaeger K.-E.; Darbre T.; Reymond J.-L. Inhibition and dispersion of Pseudomonas aeruginosa biofilms by glycopeptide dendrimers targeting the fucose-specific lectin LecB. Chem. Biol. 2008, 15, 1249–1257. 10.1016/j.chembiol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Hauck D.; Joachim I.; Frommeyer B.; Varrot A.; Philipp B.; Möller H. M.; Imberty A.; Exner T. E.; Titz A. Discovery of two classes of potent glycomimetic inhibitors of Pseudomonas aeruginosa LecB with distinct binding modes. ACS Chem. Biol. 2013, 8, 1775–1784. 10.1021/cb400371r. [DOI] [PubMed] [Google Scholar]
- Sommer R.; Exner T. E.; Titz A. A biophysical study with carbohydrate derivatives explains the molecular basis of monosaccharide selectivity of the Pseudomonas aeruginosa lectin LecB. PLoS One 2014, 9, e112822 10.1371/journal.pone.0112822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer R.; Hauck D.; Varrot A.; Wagner S.; Audfray A.; Prestel A.; Möller H. M.; Imberty A.; Titz A. Cinnamide derivatives of D-mannose as inhibitors of the bacterial virulence factor LecB from Pseudomonas aeruginosa. ChemistryOpen 2015, 4, 756–767. 10.1002/open.201500162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann A.; Sommer R.; Hauck D.; Stifel J.; Göttker-Schnetmann I.; Titz A. Synthesis of mannoheptose derivatives and their evaluation as inhibitors of the lectin LecB from the opportunistic pathogen Pseudomonas aeruginosa. Carbohydr. Res. 2015, 412, 34–42. 10.1016/j.carres.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Beshr G.; Sommer R.; Hauck D.; Siebert D. C. B.; Hofmann A.; Imberty A.; Titz A. Development of a competitive binding assay for the Burkholderia cenocepacia lectin BC2L-A and structure activity relationship of natural and synthetic inhibitors. MedChemComm 2016, 7, 519–530. 10.1039/C5MD00557D. [DOI] [Google Scholar]
- Sommer R.; Wagner S.; Rox K.; Varrot A.; Hauck D.; Wamhoff E.-C.; Schreiber J.; Ryckmans T.; Brunner T.; Rademacher C.; Hartmann R. W.; Brönstrup M.; Imberty A.; Titz A. Glycomimetic, orally bioavailable LecB inhibitors block biofilm formation of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2018, 140, 2537–2545. 10.1021/jacs.7b11133. [DOI] [PubMed] [Google Scholar]
- Phiasivongsa P.; Samoshin V.; Gross P. Henry condensations with 4,6-O-benzylidenylated and non-protected D-glucose and L-fucose via DBU-catalysis. Tetrahedron Lett. 2003, 44, 5495–5498. 10.1016/S0040-4039(03)01328-5. [DOI] [Google Scholar]
- Lagendijk E. L.; Validov S.; Lamers G. E. M.; de Weert S.; Bloemberg G. V. Genetic tools for tagging Gram-negative bacteria with mCherry for visualization in vitro and in natural habitats, biofilm and pathogenicity studies. FEMS Microbiol. Lett. 2010, 305, 81–90. 10.1111/j.1574-6968.2010.01916.x. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Hauck D.; Hoffmann M.; Sommer R.; Joachim I.; Müller R.; Imberty A.; Varrot A.; Titz A. Covalent lectin inhibition and application in bacterial biofilm imaging. Angew. Chem., Int. Ed. 2017, 56, 16559–16564. 10.1002/anie.201709368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR Chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Wilkins M. R.; Gasteiger E.; Bairoch A.; Sanchez J. C.; Williams K. L.; Appel R. D.; Hochstrasser D. F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1998, 112, 531–552. 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydorn A.; Nielsen A. T.; Hentzer M.; Sternberg C.; Givskov M.; Ersbøll B. K.; Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 2000, 146, 2395–2407. 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.