

Abstract
Background
The northern fowl mite (NFM), Ornithonyssus sylviarum, is an obligatory hematophagous ectoparasite of birds and one of the most important pests in the poultry industry on several continents. Although NFM poses a serious problem, it remains a neglected pest of poultry in China and other Asian countries. Therefore, a molecular analysis was conducted to provide baseline information on the occurrence, genetic diversity and emergence of NFM in poultry farms from China.
Methods
This study focused on morphological description and identification of adults based on electron microscopy, molecular sequencing of the mitochondrial cox1 gene and phylogenetic analysis. We have also used the DNA sequences of the cox1 gene to study the genetic diversity, population structure and demographic history. The neutrality tests were used to analyze signatures of historical demographic events.
Results
The mites collected were identified as the northern fowl mite Ornithonyssus sylviarum based on external morphological characterization using electron microscopy. Molecular analysis using a 756-bp long partial fragment of the cox1 gene revealed 99–100% sequence identity with NFM and phylogenetic inferences showed a bootstrap value of 99% indicating a well-supported monophyletic relationship. Molecular diversity indices showed high levels of haplotype diversity dominated by private haplotypes, but low nucleotide divergence between haplotypes. The Tajima’s D test and Fu’s Fs test showed negative value, indicating deviations from neutrality and both suggested recent population expansion of mite populations supported by a star-like topology of the isolates in the network analysis. Our genetic data are consistent with a single introduction of NFM infestations and the spread of NFM infestation in Hainan poultry farms and a private haplotype dominance, which suggest that infestations are recycled within the farms and transmission routes are limited between farms.
Conclusions
To our knowledge, this is the first time a molecular report of NFM in chicken from China including other Asian countries using DNA barcoding. The findings have potential implications with respect to understanding the transmission patterns, emergence and populations trends of parasitic infestations of poultry farms that will help for setting the parameters for integrated pest management (IPM) tactics against mite infestations.
Keywords: Ornithonyssus sylviarum, cox1 gene, Identification, Genetic diversity, Chicken
Background
The northern fowl mite (NFM), Ornithonyssus sylviarum, (Canestrini & Fanzago, 1877) (Mesostigmata: Macronyssidae), was first discovered in Pisa, Italy, in 1877 [1], but was first described in substantial detail and under controlled conditions by Sikes & Chamberlain [2]. It is one of the primary blood-feeding ectoparasites in poultry. Many North American records found that 72 species of wild birds from 26 bird families can serve as hosts for NFM [3]. Although NFM and another two species of mites, i.e. the tropical fowl mite (TFM) Ornithonyssus bursa and the poultry red mite (PRM) Dermanyssus gallinae, are largely considered as avian-specific ectoparasites, when natural host is absent, hungry mites will occasionally feed on a range of mammals, i.e. dogs [4], cats [5], horses [6], and humans particularly in people living or working in close proximity to poultry [7–10]. The poultry red mite D. gallinae (Mesostigmata: Dermanyssidae) was first identified by De Geer in 1778 and the tropical fowl mite, O. bursa (Berlese, 1888) (Mesostigmata: Macronyssidae) was first recognized as a potentially serious poultry pest by Wood [11]. Tropical fowl mite (TFM) and poultry red mite (PRM) both are also common blood-sucking avian ectoparasites found in the poultry industry [12, 13]. Superficially, NFM resembles PRM in gross appearance; however, there are morphological keys present to differentiate these species [14]. Unlike other mites of poultry, D. gallinae feeds mainly at night and for short periods of time. The rest of the time, mites reside in multi-stage colonies in cracks, crevices and hide away from the birds for the remainder of the time [15]. In contrast, NFM and TFM are permanent ectoparasites and complete their entire life-cycle on avian hosts [16, 17]. The nymphs and adults of TFM take blood meals, as opposed to only the protonymph and adult stages in NFM and PRM [2, 18]. NFM can cause severe damage and loss for chicken and turkey breeders, as well as egg degradation, and is considered a key pest for layers in North America, Brazil, China and Australia [19]. Although widespread, NFM is not a major problem in commercial poultry uniformly across the world [19]. PRM has a global distribution and is now at epidemic levels in many parts of the world [15]. The tropical fowl mite is widely distributed throughout subtropical and tropical countries [13]. However, there are some areas like Brazil where all three mite species have been collected from birds [20]. In China, Wang et al. [21] found that NFM prevalence was 47% in parent hens, whilst for the commercial layers the prevalence was 64% for PRM [21]. This suggests that these mites succeed differently depending on the production systems due to differences in their behavior and biology. At high infestation levels, NFM can cause up to 6% blood loss per day per hen, in addition to an activation of the of host’s immune responses. Signs of NFM infestation include restlessness, feather pecking, cannibalism, and anaemia that in severe infestations can cause death [22].
Although exact species identification is an important first step in any biological study, species that are closely related can be difficult to distinguish by the use of morphological characters alone because of similar gross morphology, unclear or inconsistent diagnostic morphological characters and high levels of morphological plasticity [23–25]. More than 40,000 species of mites have been described, and up to 1 million may grace our planet. These eight-legged arthropods (arachnids) represent one of the most successful animal radiations on land [26]. This calls for a need for specialists to be able to differentiate these from thousands of other mite species. Morphological identification may also be very labour-intensive if several specimens need to be identified. Some key characters may also only be present in certain life stages or only in females or males, limiting their usefulness across a variety of sample and sample types. Therefore, the use of sex- and life-stage independent approaches, such as using DNA-based methods could be key for successful identification at the species level. This approach can also provide additional information such as distribution patterns, origin, epidemiological and systematic investigations data for a particular species. Among different DNA regions used in molecular systematics, mitochondrial DNA is a suitable tool as a molecular marker for both intraspecific variation and species identification due to higher evolution rate of genes compared to those in nuclear DNA and their strictly maternal inheritance has been particularly useful at the intraspecific level [27, 28]. As the mitochondrial genome has a high copy number in most cells, molecular diagnostic tools targeting mitochondrial sequences may have the additional advantage of higher detection sensitivity than those targeting nuclear genes [27]. Among different mitochondrial DNA regions, the mitochondrial protein-coding gene cytochrome c oxidase subunit 1 (cox1) has been used as successful genetic marker in barcoding of mites. Many studies have demonstrated that cox1 can provide a powerful tool for phylogenetics at the deepest levels within the Acari [29–31], and for description of more shallow genetic relatedness, such as the movement of mites between farms and to describe industrial routes in larger geographical areas [32]. cox1 has been known as the ‘barcoding gene’ and it has proven successful for its use for species identification and discovery across a variety of animal organisms [33]. The usefulness of the marker is justified by the availability of abundant cox1 data generated for a variety of organisms in public databases. More importantly, the analysis of intraspecific mtDNA variation can also reveal information about the interconnectivity of populations and past demographic events such as population expansions [34].
China produces more than 40% of all the eggs in the world, making it the worldʼs largest egg producer [35]. These levels of production are associated with the spread of intensive farming systems. The introduction of an ‘enriched cage’ system for rearing chickens could help parasitic mites to better survive and hide as these systems can provide excellent living areas for mite populations [36]. Economic costs have been estimated at €231 million annually due to PRM infestations in European countries alone, with more than 300 million hens in all production types suffering from infestations [37]. It is likely that losses now far exceed that level as an expanding global prevalence of mite infestation is reported [15]. The economic harm caused by NFM is well documented. A study by Mullens et al. [38] in the USA demonstrated that overall reductions in profit were calculated at US$1960–2800 for a 28,000 hen house due to reduced body weight gain, reduced egg weight (up to 2.2%), reduced feed conversion efficiency (by 5.7%) and reduced egg production (up to 4.0%). In addition to these serious welfare issues, mite infestation may significantly threaten the sustainability of food production and undermine the world’s capacity to meet their protein requirements. It is surprising that such an economically important pest for the poultry industry has been largely ignored in Asian countries including China, even from a very basic research standpoint. Similarly, in Europe, PRM research struggles with limited funding, even if there is an urgent need for effective and safe control strategies, sustainable therapy and treatment options, as well as developing solutions for integrated pest management for the poultry red mite control [15]. In order to be able to develop sustainable methods of pest control, we need to be able to understand more about the parasite in question, so that effective solutions, which may control the pests can be implemented. The first stage on this journey is to be able to identify the problem, hence the objectives of this study were specifically aimed towards developing and use of molecular and morphological tools for identification and characterization the NFM ectoparasites recovered from poultry farms in China. This study also sought to describe the intraspecific genetic variability of the cox1 gene and assess the population structure of NFM from different localities. A further aim of this study was to use molecular genetic approaches to gain insight into the patterns of emergence and spread of O. sylviarum infestation in Hainan poultry farms, providing proof of concept for a novel approach to the study of parasite epidemiology, and eventually supports the formulation of an effective control strategy.
Methods
Sampling methods and sampling effort
The sampling was carried out between May and November 2018, across seven different locations within Hainan Island (Fig. 1): Qionghai; Wenchang; Boao (a town of Qionghai); Danzhou; Wanning; Qiongzhong; and Dingan (for simplicity, the only three positive NFM infested farms reported in the study will be referred to as QA (Qionghai), WB (Wenchang) and BC (Boao). Mites were collected directly from the body surface and vent area of the chickens. Mite traps were used for further sampling of additional mites, with an average of 10 traps placed out in each poultry house. Briefly, the traps consisted of white tubes with inner and outer diameters of 15 mm and 17 mm, respectively, containing rolled corrugated 60 × 70 mm cardboard with thickness of 1 mm [39]. Mite traps were placed as close to chicken level and below feed troughs, inside fittings and fastening clips of cage supports, and under egg conveyor belts for four days (Fig. 2a), after which they were placed into self-sealing plastic bags and sent to the laboratory for mite recovery and then preserved in 70% ethanol at 4 °C until further use. Morphological identification to the species level was carried out using a dissection microscope; when necessary, specimens were slide-mounted, cleared in Hoyer’s medium, identified under a phase-contrast microscope and a scanning electron microscope (SEM) equipped with a digital camera. The identification was based on morphological features [14, 40] in accordance with the morphological keys of Moss [41]. Voucher specimens and corresponding DNA samples were preserved in the HU Vector Biology Lab. For the SEM study, mites were dehydrated in a graded ethanol series (70, 90 and 100%, for 10 min each). Specimens were subjected to critical point drying with CO2, sputter-coated with gold using an Anhui Beq SBC-12 coating apparatus, and examined using a field emission scanning electron microscope (S3000N, Hitachi, Berkshire, UK).
Fig. 1.

Map of Hainan Island, China showing locations of field sites for mite collections. Red colour indicates positive sites for northern fowl mite Ornithonyssus sylviarum
Fig. 2.
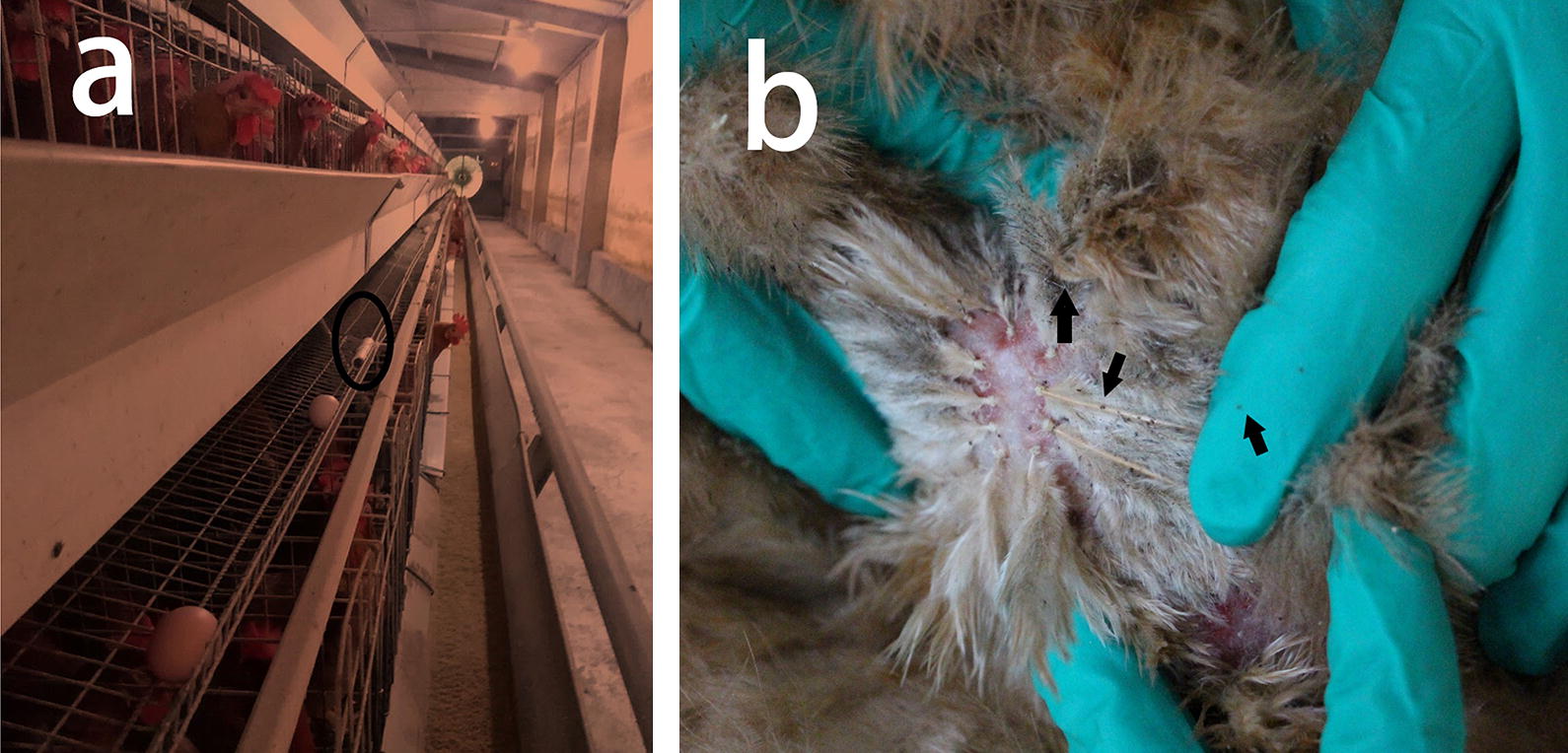
a Sampling of mites with trap (within black circle) in an area of mite aggregation, providing fast and easy way for monitoring mite infestations. b Northern fowl mites (Ornithonyssus sylviarum) on the vent region of an infested roster chicken. Mites are indicated by arrows
DNA extraction, primer design and PCR amplification
The number of samples for DNA extraction in each farm was set to a minimum 12 mites from each population; QA (n = 12); BC (n = 12); WB (n = 14). Total genomic DNA was extracted from individual mites using a QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany) and protocols therein with some modifications. DNA extraction was carried out following previously described protocols [42, 43], which allow preparation of specimen slides after DNA extraction. Individual mites were placed in a 1.5 ml microcentrifuge tube and the incubation period was extended to 6 hours, for maximum recovery of DNA. DNA was finally eluted in 20 μl of elution buffer and the concentration determined with a NanoPhotometer®N50 spectrophotometer (Wilmington, DE, USA). The exoskeleton of each mite was transferred from the bottom of the tube to the slide with a pipette and mounted in Hoyerʼs medium for morphological analyses. The DNA samples were stored at − 20 °C until used for further analysis. The cox1 forward primer OS6F (5′-GTG CAG GGA CTG GAT GAA CT-3′) was designed in the present study using NFM cox1 sequences available on GenBank (FN432541.1; KF218580.1; FN432548.1). Sequences were downloaded and aligned by ClustalW algorithm using the EMBL site [44] and visually checked for conserved regions. Primers targeting the conserved regions of the genes were then designed using Primer-BLAST. Primer properties such as melting temperature, self-complementarity, heterodimer formation, and hairpin structures of the designed primers were further analyzed using Primer-BLAST [45]. The reverse primer CO1R (5′-TAC AGC TCC TAT AGA TAA AAC-3′) was previously reported [46, 47]. The PCR reactions were performed in a 50 μl volume containing 25 μl of 2xEco Taq PCR® SuperMix (TransGen Biotech, Beijing, China), 1.0 μl of each primer (0.4 μM of each primer), 22 μl water and 1.0 μl of genomic DNA as template. A touchdown cycling protocol was used with the following cycling conditions: an initial denaturation step at 95 °C for 4 min; 10 cycles of 95 °C for 30 s, 62 °C for 30 s (reduced by 1 °C each cycle), and 71 °C for 1 min; followed by 26 cycles of 95 °C for 30 s, 52 °C for 30 s, and 71 °C for 1 min; and a final extension step at 71 °C for 8 min. Amplicons were visualized by UV light in a 1.0% agarose gel following electrophoresis and staining with ethidium bromide. The PCR protocol generated a ≈800-bp fragment. The amplified DNA with expected sizes were excised from gels and purified using a SanPrep Column PCR Product Purification Kit (Sangon, Shanghai, China) according to the manufacturer’s instructions. Purified PCR products were subsequently cloned into the pMD 18-T Vector (TaKaRa, Kusatsu, Japan) and transformed into Escherichia coli strain DH5-Alpha competent cells, according to the manufacturer’s instructions. Plasmids from positive colonies were isolated using SanPrep Column Plasmid Mini-Preps Kit (Sangon, Shanghai, China) according to the manufacturer’s instructions and subjected to Sanger sequencing on an ABI PRISM 3730 XL DNA Analyzer, using a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Both strands were sequenced for all PCR products to ensure good quality of the sequences used in the subsequent analysis. This sequence protocol generated a 756-bp fragment from Ornithonyssus sylviarum to be used in the analysis (without primer region).
Sequencing, phylogenetic and network analysis
The obtained chromatograms were imported into Contig express module in Vector NTI (ThermoFisher Scientific, Waltham MA, USA) and any flanking sequence containing PCR primers were manually removed before sequence alignment and phylogenetic analysis. Forward and reverse chromatograms were assembled in pairs for each sample. First, all sequences generated were aligned by a ClustalW algorithm [48] using the software in MEGA7 [49], and then AlignX module using Vector NTI 9.0 software (Thermo Fisher Scientific). This enabled haplotype assessment, and a total of 32 haplotypes were identified by the guide tree and arbitrarily named (CNFM_1 to CNFM_32). Secondly, additional reference sequences retrieved from GenBank were included in the alignment. All aligned sequences were subjected to phylogenetic analyses using Neighbour-Joining (NJ) algorithms with the Kimura 2-parameter nucleotide substitution model and Maximum Likelihood (ML) algorithms with the Kimura 2-parameter model in the MEGA version 7.0 [49]. For the NJ and ML analyses, robustness of nodes was assessed by including 500 bootstrap replicates. The tree was rooted using a sequence from Chlaenius pusillus (GenBank: DQ059793). However, traditional phylogenetic methods have poor statistical power because intraspecific data sets have small genetic distances between individuals [50]. Therefore, relationships between haplotypes were estimated based on the Median-Joining method using PopART [51]. This haplotype network analysis is useful for intraspecific data in revealing multiple connections between haplotypes and indicating possible missing mutational connections. In addition, MEGA version 7.0 was used to calculate numbers of variable sites, singleton sites, parsimony-informative sites and nucleotide composition. Sequences were translated into predicted amino acid sequences using the invertebrate mitochondrial genetic code with publicly available databases using ExPASy tool [52]. Sequences obtained during this study and used in phylogenetic analyses have been deposited in GenBank database under the accession numbers MK993338–MK993375.
Genetic divergence, diversity, structure, tests of neutrality and estimates of population expansion
To confirm our study in the context of divergence within the species, analyses on divergence of cox1 sequence (DNA barcoding marker) were performed using MEGA version 7.0 [49] based on the same model used to infer the phylogenetic relationships. Population diversity indices such as numbers of segregating sites (S), haplotypes number (nh), haplotype diversity (Hd), the mean number of pairwise difference (k) and nucleotide diversity (π) for each location were calculated using DnaSP 4.0 [53]. Haplotype diversity (also known as gene diversity) represents the probability that two randomly sampled alleles are different, while nucleotide diversity is defined as the average number of nucleotide differences per site in pairwise comparisons among DNA sequences [54]. Arlequin 3.5.1 was used to test for population structure of genetic variation within and between populations [55]. The significance of population pairwise comparisons of the fixation indices (Fst) was tested by 1000 permutations of the data matrix. The neutrality indices of Tajimaʼs D and Fuʼs Fs in each population were also calculated using Arlequin 3.5.1 version, with P-values generated using 1000 simulations under a model of selective neutrality [55]. Tajima’s D uses the frequency of segregating nucleotide sites, while Fu’s Fs uses the distribution of alleles or haplotypes [56, 57]. Both tests are based on the principle that a sudden population expansion that is associated with a non-neutral process will show a shift in the allele frequency spectrum compared to a neutral Wright-Fisher model consistent with population expansion under neutral evolution.
Data analysis
All data were entered in Microsoft Excel 2016. The significance level was set to P < 0.05 for all statistical analyses. Molecular diversity indices data were expressed as the mean ± standard deviation (SD).
Results
Mite infestations
A total of seven farms were sampled representing a total of 281,000 birds, of which two farms were for parent birds (representing a total of 170,000 birds), five farms were for commercial layer and broiler birds (which covered a total of 111,000 birds). Three farms were infested with NFM representing a total of 120,000 birds, of which one farm was for parent layer birds and the other two were commercial layer farms (Fig. 2b).
Morphological analysis
Although NFM is very similar to TFM in life history and appearance, it can be distinguished by the dorsal plate and number of setae on the sternal shield (Figs. 3, 4). A prominent characteristic for differentiating NFM is that adult females have two pairs of setae on the sternal plate while TFM have three pairs of setae on the sternal plate (Figs. 3d, 4a). The posterior end tapers acutely and the posteriorly narrowly rounded genitoventral shield in NFM (vs tapering more evenly in TFM; Fig. 3a–c). Superficially, NFM also resembles another common and economically most important haematophagous ectoparasite of the poultry industry worldwide, the poultry red mite (Dermanyssus gallinae). The main feature differentiating NFM from PRM is the shape of the anal plate and the presence of elongate chelicerae in females with movable digit present in NFM but absent in PRM (Fig. 4d). The anal plate of adult females is clearly different, with NFM having a teardrop-shaped anal plate at the terminus of the abdomen and anal opening at the anterior end (vs square or keystone-shaped structure of the anal shield in PRM; Fig. 4b–c). Additionally, in PRM the dorsal surface has a shield with prominent lateral margins tapering towards the rear, but these do not reach the distal end of the body and are truncated at the end (Fig. 3e). Measurements (average lengths and widths) of unfed adults of NFM, TFM and PRM were taken to compare sizes. Specimens of D. gallinae are generally the largest of the three species. The average lengths and widths of adult unfed mites PRM, NFM and TFM were 900 × 510 μm; 570 × 345 μm and 720 × 465 μm, respectively. Following these data, the mite was morphologically identified as Ornithonyssus sylviarum.
Fig. 3.

Comparative SEM of dorsal and ventral view of NFM, TFM and PRM. a Dorsal view of NFM with a posterior end tapering acutely (arrow). b The posterior end tapers more evenly in TFM (arrow). c Ventral view of NFM with the genitoventral (epigynal) shield attenuate or narrowly rounded posteriorly and a distinct sternal shield (st) with two pairs of setae (arrows). d Ventral view of the NFM with two pairs of setae on the sternal plate (arrows) and a third pair of setae on the unsclerotized integument (arrowhead). e Dorsal view of PRM, showing the idiosoma broadly rounded posteriorly (arrow) and prominent shoulder (arrowhead); adults PRM are generally larger in size than NFM and TFM. Scale-bars: a, c, e, 300 μm; b, d, 200 μm. Abbreviations: gn, gnathostoma; st, sternal plate; ao, anal opening; as, anal shield
Fig. 4.

Comparative SEM of NFM, TFM and PRM. a Two pairs of setae on the sternal plate of NFM (arrows) whereas TFM have three pairs of setae on the sternal plate (not shown). b Female PRM with a keystone-shaped anal plate. c The anal plate is distinct, with female NFM being tear-drop shaped with three anal setae (arrows). d Female NFM has elongated chelicerae with well-developed and distinct, fixed and movable digit, whereas adult female PRM have a smooth “whip-like” celicerae (not shown). Scale-bars: a, c, d, 50 μm; b, 500 μm. Abbreviations: ch, chelicera; fd, fixed digit; md, movable digit; Pa, pedipalp; ao, anal opening; as, anal shield; st, sternal plate; (LI–IV), leg; gs, genitoventral shield
Molecular analysis
DNA concentration on the mite extracts ranged from 13.2 to 38.6 ng/μl. PCR amplification was successful for 99% of the samples. A nucleotide BLAST (megablast/blastn) of the generated sequences in NCBI GenBank database confirmed the identity of the species as all top hits were of O. sylviarum cox1 sequences. Four sequences (GenBank: MK993346, MK993351, MK993365, MK993369) showed 100% similarity to the Swedish strain (GenBank: KF218580) whereas the others showed more than 99% identity. The coverage score was however not high, as no available NFM sequences in the database covered the complete regions described in our dataset. There was< 86% sequence identity to Ornithonyssus bacoti (GenBank: FM179677.2) and< 80% identity to D. gallinae (GenBank: LC029547.1). A combined length of 756 bp of the cox1 gene for a total of 38 isolates from three different farms were used to determine intraspecies sequence variability. The overall nucleotide compositions were 42.1% T, 28.7% A, 16.6 % C and 12.5% G. This high A-T content (28.7% A, 42.1% T) is consistent with the general feature of the cox1 mitochondrial DNA region in arthropods, and is similar to cox1 data from other studies on insects and other mite species [32, 47]. The sequences revealed 55 polymorphic sites, with 59 transitions and 6 tranversions. These polymorphic sites contained 44 (5.82%) singleton variable sites with two variants and 11 (1.45%) parsimony informative sites with two variants at the following positions: 16, 23, 42, 134, 164, 391, 403, 478, 481, 624 and 693. No insertions or deletions were observed in the examined sequences and none of the sequences exhibited any unusual mutations (i.e. false stop-codons, frameshift-causing insertions/deletions). Using the invertebrate translation table, sequences were confirmed to be in frame, and the substitution occurred at the third codon position as expected.
Phylogenetic and network analyses
An alignment using the shorter GenBank NFM entries included in the phylogenetic calculations, and flanking sites were not included in the analysis. Neighbor-Joining and Maximum Likelihood trees had virtually equivalent topologies. The results of NJ analysis with bootstrap values are shown in Figs. 5 and 6. To further depict the phylogenetic and geographical relationships among the identified sequences, haplotype networks were constructed using the Median-Joining method in PopART software (Fig. 7). The resultant network exhibited a star-like pattern surrounding haplotype H9, which was the most common haplotype in all three populations (QA, BC and WB).
Fig. 5.

Neighbor-Joining phylogenetic tree based on Ornithonyssus sylviarum cox1 gene sequences. Sequence data generated in this study are presented in QA, BC, WB with GenBank accession numbers, species and country names. The optimal tree with the sum of branch length of 0.82084573 is shown. Bootstrap values (> 70%) are displayed as circles on the branches. Circle sizes at nodes are the proportion of 500 bootstrap resamplings that support the topology shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. This analysis involved 60 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + noncoding. All ambiguous positions were removed for each sequence pair (pairwise deletion option). Evolutionary analyses were conducted in MEGA. Clades are coloured according to the species: O. sylviarum (mite: red), Chlaenius pusillus (insect: black), Rhipicephalus microplus (tick: black) and Dermanyssus gallinae, Dermanyssus longipes, Dermanyssus hirsutus (mites: blue). Abbreviations: Ch, China; Ban, Bangladesh; USA, United States of America; Fr, France; Sw, Sweden; Is, Israel; Nr, Norway
Fig. 6.

The evolutionary history was inferred using the Neighbor-Joining method based on Chinese isolates generated in this study. Sequence data generated in this study are presented in QA, BC, WB with GenBank accession numbers, species and country names. Bootstrap values (> 70%) are displayed as circles on the branches. Circle sizes at nodes are the proportion of 500 bootstrap resamplings that support the topology shown. The optimal tree with the sum of branch length of 0.43874121 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. This analysis involved 40 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + noncoding. All ambiguous positions were removed for each sequence pair (pairwise deletion option). Evolutionary analyses were conducted in MEGA. Clades are coloured according to the species: Ornithonyssus sylviarum (mite: red), Chlaenius pusillus (insect: black) and Rhipicephalus microplus (tick: black). Abbreviations: Ch, China; Ban, Bangladesh; USA, United States of America
Fig. 7.

Median-joining haplotype network of mt cox1 gene of Ornithonyssus sylviarum. Circle size is relative to number of haplotype copies present in the dataset. The most common haplotype (H9) was observed in all populations. A branch represents a single nucleotide change, slashes on branches indicate inferred missing haplotypes (providing a relative estimate of genetic divergence between haplotypes)
Population diversity indices and structure
A total of 32 haplotypes were identified in the 38 samples. Among these identified haplotypes, 29 haplotypes were detected as singletons (unique to a single population) and three haplotypes (H9, H13 and H14) were most widely shared. Haplotype 9 was the most common in each population (8.33%, 8.33% and 7.14% in QA, BC and WB, respectively). The relative frequencies of the cox1 haplotypes in each Hainan population are shown in Table 1. The number of haplotypes was large in each population. On the other hand, nucleotide diversity was low for each population. The mean haplotype diversity and nucleotide sequence diversity in the three populations were 0.9900 and 0.00521, respectively. Overall, the haplotype and nucleotide diversity were similar between populations. High haplotype diversity (Hd) and low nucleotide diversity (π) are shown in Table 2. The pairwise Fst values for genetic differentiation varied from 0.02625 to 0.01299. The pairwise Fst values among QA, BC and WB populations were relatively low and with non-significant P-values, except for the comparison between QA and WB, indicating very little genetic differentiation. The pairwise Fst with P-values between the locations are shown in Table 3. There was no considerable variation in haplotype diversity (1.00000 to 0.9848) and in nucleotide diversity (0.00569 to 0.004869) in each population, which was reflected in non-significant differences in pairwise comparisons of the Fst values.
Table 1.
Relative haplotype frequencies for three NFM populations
| Haplotype number | QA | BC | WB |
|---|---|---|---|
| Hap1 | 0.0833 | 0 | 0 |
| Hap2 | 0.0833 | 0 | 0 |
| Hap3 | 0.0833 | 0 | 0 |
| Hap4 | 0.0833 | 0 | 0 |
| Hap5 | 0.0833 | 0 | 0 |
| Hap6 | 0.0833 | 0 | 0 |
| Hap7 | 0.0833 | 0 | 0 |
| Hap8 | 0.0833 | 0 | 0 |
| Hap9 | 0.0833 | 0.0833 | 0.0714 |
| Hap10 | 0.0833 | 0 | 0 |
| Hap11 | 0.0833 | 0 | 0 |
| Hap12 | 0.0833 | 0 | 0 |
| Hap13 | 0 | 0.0833 | 0.0714 |
| Hap14 | 0 | 0.0833 | 0.0714 |
| Hap15 | 0 | 0.0833 | 0 |
| Hap16 | 0 | 0.0833 | 0 |
| Hap17 | 0 | 0.0833 | 0 |
| Hap18 | 0 | 0.0833 | 0 |
| Hap19 | 0 | 0.0833 | 0 |
| Hap20 | 0 | 0.0833 | 0 |
| Hap21 | 0 | 0.0833 | 0 |
| Hap22 | 0 | 0.167 | 0 |
| Hap23 | 0 | 0 | 0.0714 |
| Hap24 | 0 | 0 | 0.0714 |
| Hap25 | 0 | 0 | 0.0714 |
| Hap26 | 0 | 0 | 0.143 |
| Hap27 | 0 | 0 | 0.0714 |
| Hap28 | 0 | 0 | 0.0714 |
| Hap29 | 0 | 0 | 0.0714 |
| Hap30 | 0 | 0 | 0.0714 |
| Hap31 | 0 | 0 | 0.0714 |
| Hap32 | 0 | 0 | 0.0714 |
| Nt | 12 | 11 | 13 |
Abbreviations: Hap, haplotype; QA, Qionghai; BC, Boao; WB, Wenchang; Nt, number of haplotypes per population
Table 2.
Summary of diversity indices based on mt cox1 sequences
| Population | n | S | nh | Hd ± SD | κ | π ± SD |
|---|---|---|---|---|---|---|
| QA | 12 | 25 | 12 | 1.0000 ± 0.0340 | 4.30303 | 0.005692 ± 0.003411 |
| BC | 12 | 18 | 11 | 0.9848 ± 0.0403 | 3.75758 | 0.004970 ± 0.003034 |
| WB | 14 | 22 | 13 | 0.9890 ± 0.0314 | 3.68132 | 0.004869 ± 0.002940 |
| Total | 38 | 65 | 36 | 0.9900 ± 0.009 | 3.93883 | 0.00521 ± 0.00233 |
Abbreviations: n, number of sequences examined; S, number of segregating sites; nh, number of haplotypes; Hd, haplotype diversity; SD, standard deviation; k, average nucleotide difference; π, nucleotide diversity per population; QA, Qionghai; BC, Boao; WB, Wenchang
Table 3.
Pairwise Fst values for cox1 sequences of Ornithonyssus sylviarum (lower diagonal) and associated P-values (upper diagonal)
| Population | QA | BC | WB |
|---|---|---|---|
| QA | – | P = 0.2060 | P = 0.0214 |
| BC | 0.01299 | – | P = 0.3818 |
| WB | 0.02625 | 0.00194 | – |
Abbreviations: QA, Qionghai; BC, Boao; WB, Wenchang
Demographic history
The results of Tajima’s D test and Fu’s Fs test are presented in Table 4, including the associated simulated P-values. Tajima’s D values were significantly negative for all populations, indicating an excess of rare nucleotide site variants compared to the expectation under a neutral model of evolution. The results of Fu’s Fs test, which is based on the distribution of haplotypes, also showed highly significant negative values for all populations, indicating an excess of rare haplotypes over what would be expected under neutrality. Following this test, the hypothesis of neutral evolution was rejected for all populations.
Table 4.
Results of Tajima’s D and Fu’s Fs neutrality tests including associated P-values
| Population | Tajima’s D | P-value | Fu’s Fs | P-value |
|---|---|---|---|---|
| QA | − 2.13951 | P < 0.01 | -9.03579 | P < 0.0001 |
| BC | − 1.61094 | P = 0.0520 | -6.93339 | P < 0.0001 |
| WB | − 1.96944 | P < 0.05 | -9.69639 | P < 0.0001 |
Abbreviations: QA, Qionghai; BC, Boao; WB, Wenchang
Discussion
The northern fowl mite, O. sylviarum spends its entire life attached to its host, whereas poultry red mites are nocturnal active ectoparasites which typically (though not exclusively) during short visits suck the birds’ blood during periods of darkness and hide themselves in all kinds of gaps and cracks during the daytime [12, 19]. This behaviour calls for different treatment approaches for controlling these mite populations. Correct mite identification is therefore essential for successful integrated control management because dual mite infestation is possible and treatment strategies are likely to differ between these species. In this study, the integration of comparative morphological and molecular methods allowed precise species identification and description of haplotypes from Hainan. Data from the present and previous studies therefore suggest that NFM is geographically dispersed not only in temperate regions but also in tropical areas, and has a wide distribution in China [21]. To the best of the author’s knowledge, this report represents the first molecular evidence of NFM infestation in China. The farm prevalence for NFM infestation in chickens was 42.8%, which is higher than reported in a survey from 11 provinces in China (22.7%) [21]. A range of several factors, including sampling season, diagnostic techniques, poultry management system, biosecurity and housing differences, bird density, poultry rearing system, geography, anthropogenic and environmental factors, may be responsible for the differences in mite population levels observed in different areas of China [58, 59].
It is not surprising that the data in this study reported a high degree of variability within the cox1 gene. This molecular marker is known to be highly polymorphic within species, hence its widespread use in analyses of intraspecific variation [60]. According to Avise [34], intraspecific divergences in mitochondrial genes are rarely greater than 2% and most are less than 1%. The genetic distance value obtained from the analysis of the Kimura-2 parameter (K2P) method shows a range of genetic distance values between 0–1.1% (data not shown), suggesting that our result is in the context of divergences within species. This level of intraspecific variation is similar to that found by other researchers for NFM [61]. The pairwise uncorrected genetic distance (p-distance) approach was used by Jansson et al. [61] to estimate sequence variation within the range of 0–2.8%. Neighbor-joining and Maximum Likelihood trees generated in this study indicated a close relationship between the Chinese isolates, as expected when compared with the GenBank entries of NFM from Europe/USA etc. included in the analysis. All the Hainan samples clustered into one group which was supported by a high bootstrap value; this is evidence that the mites studied belong to a single species and that mites from Hainan have a close genetic relationship.
A major finding of this study was the high haplotype diversity and the low nucleotide diversity, indicating only small differences between haplotypes. This was also evident from the Median-Joining network, which showed mostly single nucleotide differences between haplotypes (Fig. 7). In addition, the combination of high haplotype and low nucleotide diversity, as observed in the present study, can be a signature of a rapid population expansion from a small effective population size [34]. The Median-Joining network construction showed a dominant central haplotype (H9) surrounded by several satellite haplotypes; this pattern is considered to represent a recent origin and rapid subsequent population expansion [62]. This finding fits with a major population bottleneck followed by population expansion and the idea that these mites were introduced with a few individuals and then expanded into a much larger population. These results are similar to those described in another feather mite species, Zachvatkinia isolata [63], and is consistent with bottleneck or founder event followed by population size expansion during bird-to-bird feather mite transmission. A number of statistical tests have been developed to test selective neutrality of mutations and they are used to detect such population growth [64]. In this study, both tests (Tajima’s D and Fu’s Fs test) were negative with highly significant P-values, particularly for the Fu’s Fs test for all populations. The overall negative values resulting from both tests indicate that there is an excess of rare mutations in the populations, which imply a recent population expansion. An analysis including additional neutral nuclear DNA markers could give a more complete perspective on the neutral population structure of these mite populations. Population expansion from a small effective population can be influenced by the following factors. First, the entire life-cycle is short, and large populations can develop rapidly on the birds just after 5 to 12 days [2, 65]. Consequently, weekly doubling of populations is possible under optimal conditions. In addition to movement of the mites by wild birds, the mites can be transmitted by contaminated egg trays, flats, chicken crates and conveyor equipment. Another possible explanation for population expansion might be the result of poor acaricide coverage by spraying and dusting procedures currently used on those farms. These results might explain why NFM maintains a relatively high genetic diversity and a clear signature of historical population expansion. The ability of mite populations to build up quickly means that low mite infestations may rapidly develop into severe infestations debilitating the chickens. An understanding of the demographic history is useful in reflecting population trends in order to integrate multiple control tactics against mite infestations.
In terms of population structure, the Fst pairwise values between two populations (BC and WB) were very low (0.00194) with a high P-value, three shared haplotypes and four shared branches in the network, indicating no divergence between populations (Table 3, Fig. 7). Similarly, the populations QA and BC showed relatively very little divergence (0.01299) with a non-significant P-value, one shared haplotype and one shared branch in the analysis. In contrast, the Fst pairwise values between two populations (QA and WB) were relatively high (0.02625) with a significant P-value and no shared branches in the analysis, but still shared a common haplotype with the other populations. The distance between the two farms is nearly 120 km (these are the most geographically distant populations in the analysis). For these two populations, the genetic distance value indicated that the geographical distance is one of the factors causing differences in genetic distance values, the farther the distance between populations, the higher the genetic distance value. It is not surprising that the overall Fst values are relatively low since they probably came from the same source and a bottleneck population (i.e. all share one common haplotype at the center of the haplotype network and not have any obvious geographically separated clusters in the network) and are still expanding. Furthermore, a star-like gene tree of this part of the network indicates very little geographical structure. Finally, although we identified high genetic diversity, there was little evidence for differentiation between populations, indicating a single panmictic population.
The molecular toolbox is largely unexplored regarding Ornithonyssus spp. mites [19]. At present, there have been no reports of cox1 gene sequencs for NFM from Asian countries, as all information is originated from European countries, USA or Canada. To our knowledge, the present study reports, for the first time, mitochondrial cox1 sequence data for O. sylviarum from China which could provide essential information on the genetic structure of NFM populations circulating in Hainan Island. A further aim of the present study was to use novel molecular genetic approaches to gain insight into the emergence and spread of NFM infestation in Hainan. At the farm level, infestation may emerge singly or on multiple occasions through animal movement [66]. The identification of a single common haplotype in all populations suggests a single emergence of NFM infestation on the farms from which the parasitic populations were derived. The statistical Median-Joining haplotype network provided a clear indication that a single introduction of a mite population and a subsequent spread of the parasites has occurred. A similar situation has been reported for the poultry red mite, D. gallinae; mostly single haplotypes were found on most farms, suggesting that infestations are recycled within the farms and the transmission routes are few between farms [32]. In contrast, multiple common haplotypes in one population is considered as evidence for multiple introductions of infestation. A recent study reported multiple introductions of Calicophoron daubneyi infection in United Kingdom cattle herds, as multiple common haplotypes were identified in 26 populations [66]. It is also noted that there is a chance that our sampling (12–14 mites per farm) may have left some haplotypes undetected, thus underestimating the haplotype diversity per farm. Increased number of samples in some farms in future studies could address this issue. Given that our dataset correctly mirrors the actual diversity per farm, it seems to reflect similar haplotype diversity, and a relatively homogeneous population structure in each farm, which was also found for PRM in Europe [32].
Conclusions
This study highlights the importance of an integrative approach in taxonomic studies, combining morphology and molecular features. To our knowledge, this study describes for the first time key comparative morphological features for differentiating three mite species sharing the same host species and environment. Our genetic data also provide the first insight into the emergence and the spread of O. sylviarum in China. These findings demonstrate a single introduction of a mite population at the farm level followed by extreme population expansion. Our dataset is limited, so further research is needed in order to understand possible transmission routes of this parasite, and to access the significance of its genetic variation and diversity in understanding the epidemiology and biology of this parasite.
Acknowledgments
We are grateful to Professor Rihui Yan, Dr Jochen Wilhelm, Dr Magne Kjerulf Hansen, Dr Mohammad Noor Amal Azmai, George Lohay, Min Chen, Kaidong Wang, Ling Fun,Tang Yu, Wang Quihui, Kawser Ayon, and all of our laboratory members for sharing ideas, discussions and statistical analysis. Special thanks to Dr Max R. Bangs (Florida State University) for helpful comments on the manuscript.
Abbreviations
- NFM
northern fowl mite
- TFM
tropical fowl mite
- PRM
poultry red mite
- QA
Qionghai
- WB
Wenchang
- BC
Boao
- HU
Hainan University
- K2P
Kimura-2 parameter
Authorsʼ contributions
QH, BB and JZ conceived and designed the study protocol and participated in writing of the manuscript. BB performed molecular analysis, statistical analysis, interpretation, and was a major contributor in writing the manuscript. JZ and ØØ participated in study conception and design, sequencing analysis, and edited the manuscript. TB, CL and LZ conducted the study, assisting sample collection and performed molecular analysis in the laboratory. The manuscript was reviewed by all authors. All authors read and approved the final manuscript.
Funding
The work was funded by the Key Research and Development Program of Hainan Province (ZDYF2019073), and Hainan University Research Fund (hdkytg201702).
Availability of data and materials
All data generated or analyzed during this study are included within the article.
Ethics approval and consent to participate
The care and use of chickens in this study was approved by Hainan University Institutional Animal Care and Use Committee. Informed consent is not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Biswajit Bhowmick, Email: biswajit.bhowmick@hotmail.com.
Jianguo Zhao, Email: 973878018@qq.com.
Øivind Øines, Email: oivind.oines@vetinst.no.
Tianlin Bi, Email: 2966154798@qq.com.
Chenghong Liao, Email: liaochh77@163.com.
Lei Zhang, Email: rayzhang007@foxmail.com.
Qian Han, Email: qianhan@hainanu.edu.cn.
References
- 1.Cameron D. The northern fowl mite (Liponyssus sylviarum C. & F., 1877) investigations at MacDonald College, Que, with a summary of previous work. Can J Res. 1938;16:230–254. doi: 10.1139/cjr38d-015. [DOI] [Google Scholar]
- 2.Sikes RK, Chamberlain RW. Laboratory observations on three species of bird mites. J Parasitol. 1954;40:691–697. doi: 10.2307/3273713. [DOI] [PubMed] [Google Scholar]
- 3.Knee W, Proctor H. Host records for Ornithonyssus sylviarum (Mesostigmata: Macronyssidae) from birds of North America (Canada, United States and Mexico) J Med Entomol. 2007;44:709–713. doi: 10.1093/jmedent/44.4.709. [DOI] [PubMed] [Google Scholar]
- 4.Cafiero Maria Assunta, Barlaam Alessandra, Camarda Antonio, Radeski Miroslav, Mul Monique, Sparagano Olivier, Giangaspero Annunziata. Dermanysuss gallinae attacks humans. Mind the gap! Avian Pathology. 2019;48(sup1):S22–S34. doi: 10.1080/03079457.2019.1633010. [DOI] [PubMed] [Google Scholar]
- 5.Di Palma A, Leone F, Albanese F, Beccati M. A case report of Dermanyssus gallinae infestation in three cats. Vet Dermatol. 2018;29:348. doi: 10.1111/vde.12547. [DOI] [PubMed] [Google Scholar]
- 6.Mignon B, Losson B. Dermatitis in a horse associated with the poultry mite (Dermanyssus gallinae) Vet Dermatol. 2008;19:38–43. doi: 10.1111/j.1365-3164.2007.00646.x. [DOI] [PubMed] [Google Scholar]
- 7.George DR, Finn RD, Graham KM, Mul M, Maurer V, Valiente Moro C, Sparagano OA. Should the poultry red mite Dermanyssus gallinae be of wider concern for veterinary medical science. Parasit Vectors. 2015;8:178. doi: 10.1186/s13071-015-0768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exp Dermatol. 2000;25:129–131. doi: 10.1046/j.1365-2230.2000.00594.x. [DOI] [PubMed] [Google Scholar]
- 9.Mullens BA, Kuney DR, Hinkle NC, Szijj CE. Producer attitudes and control practices for northern fowl mites in southern California. J Appl Poult Res. 2004;13:488–492. doi: 10.1093/japr/13.3.488. [DOI] [Google Scholar]
- 10.Pezzi M, Leis M, Chicca M, Roy L. Gamasoidosis caused by the special lineage L1 of Dermanyssus gallinae (Acarina: Dermanyssidae): a case of heavy infestation in a public place in Italy. Parasitol Int. 2017;66:666–670. doi: 10.1016/j.parint.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Wood HP. Tropical fowl mite in the United States: with notes on life history and control. Washington: U.S. Dept. of Agriculture; 1920. [Google Scholar]
- 12.Sparagano OA, George DR, Harrington DW, Giangaspero A. Significance and control of the poultry red mite, Dermanyssus gallinae. Annu Rev Entomol. 2014;59:447–466. doi: 10.1146/annurev-ento-011613-162101. [DOI] [PubMed] [Google Scholar]
- 13.Murthy GSS, Panda R. Prevalence of Dermanyssus and Ornithonyssus species of mites in poultry farms of Vikarabad area of Hyderabad. J Parasit Dis. 2016;40:1372–1375. doi: 10.1007/s12639-015-0693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Palma A, Giangaspero A, Cafiero MA, Germinara GS. A gallery of the key characters to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae) Parasit Vectors. 2012;5:104. doi: 10.1186/1756-3305-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomley FM, Sparagano O. Spotlight on avian pathology: red mite, a serious emergent problem in layer hens. Avian Pathol. 2018;47:533–535. doi: 10.1080/03079457.2018.1490493. [DOI] [PubMed] [Google Scholar]
- 16.Proctor H, Owens I. Mites and birds: diversity, parasitism and coevolution. Trends Ecol Evol. 2000;15:358–364. doi: 10.1016/S0169-5347(00)01924-8. [DOI] [PubMed] [Google Scholar]
- 17.Owen JP, Mullens BA. Influence of heat and vibration on the movement of the northern fowl mite (Acari: Macronyssidae) J Med Entomol. 2004;41:865–872. doi: 10.1603/0022-2585-41.5.865. [DOI] [PubMed] [Google Scholar]
- 18.Denmark HA, Cromroy HL. Tropical fowl mite, Ornithonyssus bursa (Berlese) (Arachnida: Acari: Macronyssidae). 2003. http://entnemdept.ufl.edu/creatures/livestock/tropical_fowl_mite.htm. Accessed 24 Feb 2019.
- 19.Murillo AC, Mullens BA. A review of the biology, ecology, and control of the northern fowl mite, Ornithonyssus sylviarum (Acari: Macronyssidae) Vet Parasitol. 2017;246:30–37. doi: 10.1016/j.vetpar.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Rezende LDC, Cunha LM, Teixeira CM, Oliveira PRD, Martins NRDS. Mites affecting hen egg production: some considerations for Brazilian farms. Cienc Rural. 2013;43:1230–1237. doi: 10.1590/S0103-84782013005000088. [DOI] [Google Scholar]
- 21.Wang FF, Wang M, Xu FR, Liang DM, Pan BL. Survey of prevalence and control of ectoparasites in caged poultry in China. Vet Rec. 2010;167:934–937. doi: 10.1136/vr.c6212. [DOI] [PubMed] [Google Scholar]
- 22.DeLoach JR, DeVaney JA. Northern fowl mite ingests large quantities of blood from white leghorn hens. J Med Entomol. 1981;18:374–377. doi: 10.1093/jmedent/18.5.374. [DOI] [PubMed] [Google Scholar]
- 23.Laumann M, Norton RA, Weigmann G, Scheu S, Maraun M, Heethoff M. Speciation in the parthenogenetic oribatid mite genus Tectocepheus (Acari, Oribatida) as indicated by molecular phylogeny. Pedobiologia. 2007;51:111–122. doi: 10.1016/j.pedobi.2007.02.001. [DOI] [Google Scholar]
- 24.Heethoff M, Domes K, Laumann M, Maraun M, Norton RA, Scheu S. High genetic divergences indicate ancient separation of parthenogenetic lineages of the oribatid mite Platynothrus peltifer (Acari, Oribatida) J Evol Biol. 2007;20:392–402. doi: 10.1111/j.1420-9101.2006.01183.x. [DOI] [PubMed] [Google Scholar]
- 25.Roy L, Dowling APG, Chauve CM, Lesna I, Sabelis MW, Buronfosse T. Molecular phylogenetic assessment of host range in five Dermanyssus species. Exp Appl Acarol. 2009;48:115–142. doi: 10.1007/s10493-008-9231-1. [DOI] [PubMed] [Google Scholar]
- 26.Sharma PP. Chelicerates. Curr Biol. 2018;28:774–778. doi: 10.1016/j.cub.2018.05.036. [DOI] [PubMed] [Google Scholar]
- 27.Cruickshank RH. Molecular markers for the phylogenetics of mites and ticks. Syst Appl Acarol. 2002;7:3–14. [Google Scholar]
- 28.Bucklin A, Steinke D, Blanco-Bercial L. DNA Barcoding of marine Metazoa. Annu Rev Mar Sci. 2011;3:471–508. doi: 10.1146/annurev-marine-120308-080950. [DOI] [PubMed] [Google Scholar]
- 29.Toda S, Osakabe M, Komazaki S. Interspecific diversity of mitochondrial COI sequences in Japanese Panonychus species (Acari: Tetranychidae) Exp Appl Acarol. 2000;24:821–829. doi: 10.1023/A:1006484018547. [DOI] [PubMed] [Google Scholar]
- 30.Ballard JWO, Rand DM. The population biology of mitochondrial DNA and its phylogenetic implications. Annu Rev Ecol Evol Syst. 2005;36:621–642. doi: 10.1146/annurev.ecolsys.36.091704.175513. [DOI] [Google Scholar]
- 31.Roy L, Dowling APG, Chauve CM, Buronfosse T. Delimiting species boundaries within Dermanyssus Dugès, 1834 (Acari: Dermanyssidae) using a total evidence approach. Mol Phylogenet Evol. 2009;50:446–470. doi: 10.1016/j.ympev.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Øines Ø, Brännström S. Molecular investigations of cytochrome c oxidase subunit I (COI) and the internal transcribed spacer (ITS) in the poultry red mite, Dermanyssus gallinae, in northern Europe and implications for its transmission between laying poultry farms. Med Vet Entomol. 2011;25:402–412. doi: 10.1111/j.1365-2915.2011.00958.x. [DOI] [PubMed] [Google Scholar]
- 33.International Barcode of life.https://ibol.org/. Accessed 27 Jan 2019.
- 34.Avise JC. Phylogeography: the history and formation of species. Cambridge, MA: Harvard University Press; 2000. p. 447. [Google Scholar]
- 35.Yang Z, Rose SP, Yang HM, Pirgozliev V, Wang ZY. Egg production in China. Worlds Poult Sci. 2018;74:1–10. doi: 10.1017/S0043933918000144. [DOI] [Google Scholar]
- 36.Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239–245. doi: 10.1016/S0304-4017(98)00167-8. [DOI] [PubMed] [Google Scholar]
- 37.Flochlay AS, Thomas E, Sparagano O. Poultry red mite (Dermanyssus gallinae) infestation, a broad impact parasitological disease that still remains a significant challenge for the egg-laying industry in Europe. Parasit Vectors. 2017;10:357. doi: 10.1186/s13071-017-2292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullens BA, Owen JP, Kuney DR, Szijj CE, Klingler KA. Temporal changes in distribution, prevalence and intensity of northern fowl mite (Ornithonyssus sylviarum) parasitism in commercial caged laying hens, with a comprehensive economic analysis of parasite impact. Vet Parasitol. 2009;160:116–133. doi: 10.1016/j.vetpar.2008.10.076. [DOI] [PubMed] [Google Scholar]
- 39.Thomas E, Chiquet M, Sander B, Zschiesche E, Flochlay AS. Field efficacy and safety of fluralaner solution for administration in drinking water for the treatment of poultry red mite (Dermanyssus gallinae) infestations in commercial flocks in Europe. Parasit Vectors. 2017;10:457. doi: 10.1186/s13071-017-2390-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisbroth SH. The differentiation of Dermanyssus gallinae from Ornithonyssus sylviarum. Avian Dis. 1960;4:133–137. doi: 10.2307/1587499. [DOI] [Google Scholar]
- 41.Moss WW. An illustrated key to the species of the acarine genus Dermanyssus (Mesostigmata: Laelapoidea: Dermanyssidae) J Med Entomol. 1968;5:67–84. doi: 10.1093/jmedent/5.1.67. [DOI] [PubMed] [Google Scholar]
- 42.Dowling APG, Bauchan GR, Ochoa R, Beard JJ. Scanning electron microscopy vouchers and genomic data from an individual specimen: maximizing the utility of delicate and rare specimens. Acarologia. 2010;50:479–485. doi: 10.1051/acarologia/20101983. [DOI] [Google Scholar]
- 43.Li JB, Li YX, Sun JT, Xue XF, Xu XN, Hong X. COI barcoding as a molecular assay for the identification of phytoseiid mites. Syst Appl Acarol. 2012;17:397–406. [Google Scholar]
- 44.The European Bioinformatics Institute (EMBL-EBI). http://www.ebi.ac.uk/Tools/msa/clustalw2/. Accessed 20 Jan 2019.
- 45.Primer-BLAST. https://www.ncbi.nlm.nih.gov/tools/primer-blast/. Accessed 20 Jan 2019.
- 46.Navajas M, Gutierrez J, Bonato O, Bolland HR, Mapangoudivassa S. Intraspecific diversity of the cassava green mite Mononycellus progresivus (Acari, Tetranychidae) using comparisons of mitochondrial and nuclear ribosomal DNA-sequences and cross-breeding. Exp Appl Acarol. 1994;18:351–360. doi: 10.1007/BF00116316. [DOI] [PubMed] [Google Scholar]
- 47.Ros VID, Breeuwer JAJ. Spider mite (Acari: Tetranychidae) mitochondrial COI phylogeny reviewed: host plant relationships, phylogeography, reproductive parasites and barcoding. Exp Appl Acarol. 2007;42:239–262. doi: 10.1007/s10493-007-9092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar Sudhir, Stecher Glen, Tamura Koichiro. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bandelt H, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 51.PopART (Population Analysis with Reticulate Trees). http://popart.otago.ac.nz. Accessed 2 Mar 2019.
- 52.ExPASy:SIB Bioinformatics Resource Portal. https://web.expasy.org/translate/. Accessed 12 Mar 2019. [DOI] [PMC free article] [PubMed]
- 53.Rozas J, Sanchez-Delbarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by coalescent and other methods. Bioinform. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 54.Lynch M, Crease TJ. The analysis of population survey data on DNA sequence variation. Mol Biol Evol. 1990;7:377–394. doi: 10.1093/oxfordjournals.molbev.a040607. [DOI] [PubMed] [Google Scholar]
- 55.Excoffier L, Laval G, Schneider S. ARLEQUIN, version 3.0: an integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:47–50. doi: 10.1177/117693430500100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hall RD, Gross WB, Turner EC. Preliminary observations on northern fowl mite infestations on estrogenized roosters and in relation to initial egg production in hens. Poult Sci. 1978;57:1088–1090. doi: 10.3382/ps.0571088. [DOI] [PubMed] [Google Scholar]
- 59.Arthur FH, Axtell RC. Northern fowl mite population development on laying hens caged at three colony sizes. Poult Sci. 1983;62:424–427. doi: 10.3382/ps.0620424. [DOI] [PubMed] [Google Scholar]
- 60.Hebert PDN, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit I divergences among closely related species. Proc R Soc Lond. 2003;270:S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jansson DS, Otman F, Lundqvist L, Höglund J, Engström A, Chirico J. Northern fowl mite (Ornithonyssus sylviarum) in Sweden. Med Vet Entomol. 2014;28:443–446. doi: 10.1111/mve.12053. [DOI] [PubMed] [Google Scholar]
- 62.Slatkin M, Hudson RR. Pairwise comparisons of mitochondrial-DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dabert M, Coulson SJ, Gwiazdowicz DJ, Moe B, Hanssen SA, Biersma EM, Pilskog HE, Dabert J. Differences in speciation progress in feather mites (Analgoidea) inhabiting the same host: the case of Zachvatkinia and Alloptes living on arctic and long-tailed skuas. Exp Appl Acarol. 2015;65:163–179. doi: 10.1007/s10493-014-9856-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- 65.Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae) Exp Appl Acarol. 1992;15:27–40. doi: 10.1007/BF01193965. [DOI] [PubMed] [Google Scholar]
- 66.Sargison ND, Shahzad K, Mazeri S, Chaudhry U. A high throughput deep amplicon sequencing method to show the emergence and spread of Calicophoron daubneyi rumen fluke infection in United Kingdom cattle herds. Vet Parasitol. 2019;268:9–15. doi: 10.1016/j.vetpar.2019.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included within the article.