

Abstract
Soybean is a major source of protein for human consumption and animal feed. Releasing new cultivars with high nutritional value is one of the major goals in soybean breeding. To achieve this goal, genome-wide association studies of seed amino acid contents were conducted based on 249 soybean accessions from China, US, Japan, and South Korea. The accessions were evaluated for 15 amino acids and genotyped by sequencing. Significant genetic variation was observed for amino acids among the accessions. Among the 231 single nucleotide polymorphisms (SNPs) significantly associated with variations in amino acid contents, fifteen SNPs localized near 14 candidate genes involving in amino acid metabolism. The amino acids were classified into two groups with five in one group and seven amino acids in the other. Correlation coefficients among the amino acids within each group were high and positive, but the correlation coefficients of amino acids between the two groups were negative. Twenty-five SNP markers associated with multiple amino acids can be used to simultaneously improve multi-amino acid concentration in soybean. Genomic selection analysis of amino acid concentration showed that selection efficiency of amino acids based on the markers significantly associated with all 15 amino acids was higher than that based on random markers or markers only associated with individual amino acid. The identified markers could facilitate selection of soybean varieties with improved seed quality.
Keywords: Glycine max, genome-wide association study, genomic selection, genotyping by sequencing, amino acid concentration, single nucleotide polymorphism
Introduction
Soybean [Glycine max (L.) Merr.] is a major source of protein for humans and livestock in the world. For the past several decades, soybean meal has been the leading protein feed source for the animal and poultry production operations because of its high concentration of protein. Poultry and livestock industries use about 68 and 77% of the soybean meal consumed in the European Union and United States, respectively1,2. A major function of proteins in nutrition is to supply adequate amounts of required amino acids (Friedman and Brandon, 2001). Thus, genetic improvement of amino acid composition and balance is an important goal in soybean breeding. Developing new molecular markers for marker assisted selection (MAS) and genomic selection (GS) of amino acid composition in soybean will help to achieve this goal.
Quantitative trait loci (QTL) mapping of amino acids have been reported in soybean. Panthee et al. (2006) identified 32 simple sequence repeat (SSR) markers associated with 16 amino acids in soybean seeds based on 101 F6-derived recombinant inbred lines (RIL) from a cross of N87-984-16 × TN93-99. Fallen et al. (2013) reported ten QTLs associated with 17 amino acids and three genomic regions on chromosome 13 (4.89, 21.51, 40.69 cM) controlled multiple amino acids in 282 F5:9 RILs derived from a cross of Essex × Williams 82. As a sole dietary source of protein, soybean is deficient in lysine (Lys), threonine (Thr), methionine (Met), and cysteine (Cys) for poultry and swine. Warrington et al. (2015) conducted QTL analysis for the four amino acids in the Benning × Danbaekkong soybean population with 98 SSRs and 323 single nucleotide polymorphism (SNP) markers, and detected two QTLs on chr 8 and 20 for Lys; three on chr 9, 17, and 20 for Thr; four on Chr 6, 9, 10, and 20 for Met; and one on chr 10 for Cys (Van Warrington, 2011; Warrington et al., 2015). Khandaker et al. (2015) analyzed MD96-5722” × “Spencer” RIL population and identified 13 QTLs associated with amino acids. However, reports of genetic diversity of amino acids and mapping of QTLs controlling amino acid in soybean germplasm are limited.
Because SSR, SNPs, and indels are abundant in plants and can be assayed with high-throughput technology, the markers have been widely used for genetic linkage mapping, association studies, diversity analysis, and tagging of genes controlling important traits (Liang et al., 2010; Lehne et al., 2011; Li et al., 2014; Shi et al., 2016; Taranto et al., 2016; Zatybekov et al., 2017; Qin et al., 2017a; Qin et al., 2017b; Chang et al., 2018). Genotyping by sequencing (GBS) takes advantage of the next-generation sequencing platforms and utilizes a highly-multiplexed system to assay DNA variants from reduced representation DNA libraries of plant materials (Elshire et al., 2011; Sonah et al., 2013). As a cost-effective technique, GBS has been successfully used in implementing genome wide association study (GWAS), genomic diversity study, genetic linkage analysis, molecular marker discovery and GS in plant breeding programs (Heslot et al., 2013; He et al., 2014; Qin et al., 2016; Shi et al., 2017).
With the decreased genotyping cost and improved statistical methods, GWAS and GS offer new approaches for genetic improvement of complex traits in crop species (Bernardo and Yu, 2007; Li et al., 2013; Morris et al., 2013; Yano et al., 2016; Zhang et al., 2017). GWAS is one of the powerful tools to overcome limitations in traditional QTL mapping (Luo et al., 2019). To date, it has been used to identify molecular markers for a broad range of complex traits in different plant species including Arabidopsis (Angelovici et al., 2017), wheat (Peng et al., 2018), maize (Li et al., 2013; Deng et al., 2017), rice (Huang et al., 2010; Yano et al., 2016), soybean (Fang et al., 2017); sorghum (Morris et al., 2013). In soybean research, GWAS were used in agronomic traits (Zatybekov et al., 2017; Chang et al., 2018), seed quality (Zhang et al., 2018), seed traits (Xia et al., 2018), phosphorus efficiency (Lü et al., 2018), disease resistance (Qin et al., 2017b; Hanson et al., 2018) etc. As soybean is globally cultivated primarily for its protein and oil, and soybean protein is a complete protein as it contains all the essential amino acid that are required for human health. Numerous studies have reported on the QTL mapping and GWAS for protein (Li et al., 2018; Li et al., 2019). GS is to select desired individual within a population based on genomic estimated breeding values (GEBVs) (Hayes et al., 2009), GS has been shown more efficient than the traditional MAS for the improvement of traits controlled by QTL with minor effects (Bernardo and Yu, 2007; Heffner et al., 2009; Shikha et al., 2017; Zhang et al., 2017). GS has been applied to various agronomic traits and disease resistance in maize (Bernardo, 1996; Piepho, 2009; Albrecht et al., 2011; Technow et al., 2013; Shikha et al., 2017), rice (Onogi et al., 2015; Spindel et al., 2015; Duhnen et al., 2017), soybean (Jarquin et al., 2016; Xavier et al., 2016), and wheat (Heffner et al., 2011; Rutkoski et al., 2011; Poland et al., 2012; Battenfield et al., 2016), etc. Previous studies reported the efficiency of GS prediction by cross-validation approach (Dawson et al., 2013; Michel et al., 2016) and suggested that the size of the training population was critical (Xavier et al., 2016). Zhang et al. (2018) conducted GWAS for seed composition, including protein, oil, fatty acids, and amino acids, using 313 diverse soybean germplasm accessions genotyped with a high-density SNP array of the Illumina Infinium SoySNP50K BeadChip (Song et al., 2013). After filtered, a total of 31,850 SNPs with minor allele frequency (MAF) ≥5% were used for GWAS in their analysis and 87 chromosomal regions were identified to be associated with seed composition, explaining 8–89% of genetic variances.
However, little GWAS and no GS for amino acid concentrations in soybean has been reported so far. The main objectives of this study were to (1) evaluate amino acid compositions in soybean germplasm from China, Korea, Japan and U.S. (2) identify SNP markers associated with amino acid concentrations of soybean via GWAS, and (3) explore efficiency of GS for amino acids in soybean breeding. The newly identified markers are anticipated to facilitate MAS and GS of nutritional traits in soybean, and the soybean accessions with high concentrations of amino acids will be potential parents for soybean breeding.
Materials and Methods
Panel for Genome-Wide Association Analysis and Genomic Selection
The panel with a total of 249 soybean accessions was chosen for this study (Supplementary Table 1). These accessions were collected from China, United States, South Korea, and Japan with 169 (67.9% out of 249), 75 (30.1%), 3 (1.2%), and 2 (0.8%) accessions, respectively (Supplementary Table 1).
DNA Extraction, GBS, and SNP Discovery
Genomic DNA was extracted from freeze-dried fresh leaves of soybean plants using the CTAB (hexadecyltrimethyl ammonium bromide) method (Kisha et al., 1997). DNA library was prepared using the fragment digested by restriction enzyme ApeKI following the GBS protocol described by Elshire et al. (2011) and DNA sequencing was performed using GBS method (Elshire et al., 2011; Sonah et al., 2013). The 90 bp pair-end sequencing was obtained from each soybean genotype at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China. The GBS dataset contained 3.26 M short-reads or 283.74 Mbp of sequence for each accession. The short reads were aligned to soybean whole genome sequence (Wm82.a1.v1)3,4 using SOAPaligner/soap2 and SOAPsnp v. 1.05 was used for SNP calling (Li et al., 2009; Li, 2011).
Approximately a half million SNPs were discovered from the 249 soybean germplasm accessions. SNPs were eliminated if MAF was less than 5%, or missing and ambiguous alleles larger than 15%. After filtering, 23,279 SNPs remained for genetic diversity and association analyses.
Amino Acid Content Determination and Phenotypic Data Analysis
Soybean germplasm was grown at three locations, Shijiazhuang (114°83′E, 38°03′N), Cangzhou (116°7′E, 38°03′N), and Handan (114°48′E, 36°62′N) in Hebei province in a randomized complete block design with three replications in June 2012. Each plot consisted of six rows with a row length of three meters and raw space of 50 cm in all trials. The density was 225,000 plants per ha. The soil at Shijiazhuang was cinnamon. The organic matter, available P and available K concentration were 1.74% 29.9 mg/kg, 94.3 mg/kg, respectively. The soil at Cangzhou was light loamy. The organic matter, available P and available K concentration were 1.0–1.2%, 15 mg/kg, and 100 mg/kg, respectively. The soil at Handan was fluviatile loamy and the organic matter, available P and available K concentration were 1.6%, 19.3 mg/kg, 156.2 mg/kg, respectively. The plots were irrigated once at seed-filling stage. Plants were harvested after 95% leaves had fallen off. Ten plants were randomly chosen from the middle of a plot for seed traits analysis.
A total of 15 amino acids, Ala, Arg, Asp, Glu, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Tyr, and Val in soybean seeds were measured by Biochrom 30 amino acid analyzer (Biochrom Ltd, Cambridge, UK) using the acid hydrolysis method (Davies and Thomas, 1973; Tsugita and Scheffler, 1982). Analysis was carried out by ion exchange chromatography under the experimental conditions recommended for protein hydrolysates. Each sample containing 0.1 g soybean seed powder was acid hydrolyzed with 10 ml of 6 N HCl at 110°C for 22 h in a 15 ml vacuum-sealed glass tube. The top hydrolysate in the tube was filtered into another 50 ml tube, and water was added to the tube. A total of 1 ml liquid from the 50 ml tube was transferred to a 1.5 ml tube and dried at 55°C, re-dissolved with 1 ml loading buffer and measured in the analyzer. The amino acid composition was calculated from the standard area obtained from the integrator and expressed as a percentage of the total weight.
Statistical analyses of the 15 amino acids were performed by JMP Genomics 7 (SAS Institute, Cary, NC, USA)5 (Sall et al., 2012). The mean, range, standard deviation (SD), standard error (SE) and coefficient of variation (CV) were estimated for each amino acid concentration using ‘Tabulate’; the distributions of amino acid concentrations were drawn using ‘Distribution’ in JMP Genomics 7.
Population Structure, Genetic Diversity, and Association Analysis
STRUCTURE, a program that uses Bayesian method to analyze multi-loci data in population genetics (Pritchard et al., 2000)6, was used to analyze population structure and to create Q-matrix for association analysis. We used the default parameters of STRUCTURE 2.0 software: Admixture Model; Allele Frequencies Correlated; and Compute Probability of the Data (Kaeuffer et al., 2007). The number of subpopulation (K) was assumed to be between 1 and 12. Thus, each K was run 10 times, the Markov Chain Monte Carlo (MCMC) length of the burn-in period was 20,000 and the number of MCMC iterations after the burn-in was 20,000. For each simulated K, the statistical value delta K was calculated using the formula described by Evanno et al. (2005). The optimal K was determined using STRUCTURE HARVESTER7 (Earl, 2012). After optimal K was determined, a Q-matrix was obtained and used in TASSEL 5 (Bradbury et al., 2007) for association analysis. Each soybean accession was then assigned to a cluster (Q) based on the probability that the genotype belonged to that cluster. The cut-off probability for the assignment to a cluster was 0.5. Based on the optimum K, a bar plot with ‘Sort by Q’ was obtained to visualize the population structure among the 249 accessions. Genetic diversity was also assessed and the phylogenic tree was drawn using MEGA 6 (Tamura et al., 2013) based on the Maximum Likelihood (ML) tree method (Shi et al., 2016) with the following parameters. Test of phylogeny: bootstrap method with No. of Bootstrap replications 500; Model/Method: General Time Reversible model, Rates among Sites: Gamma distributed with Invariant sites (G/I), Number of Discrete Gamma Categories: 6, Gaps/Missing Data Treatment: Use all sites, ML Heuristic Method: Subtree-Pruning-Regrafting-Ex-tensive (SPR level 5), Initial Tree for ML: Make initial tree automatically (Neighbor Joining), and Branch Swap Filter: Moderate. The population structure and the cluster information were imported to MEGA 6 for combined analysis of genetic diversity. For sub-tree of each Q (cluster), the shape of ‘Node/Subtree Marker’ and the ‘Branch Line’ was drawn using the same color scheme of the STRUCTURE analysis.
Association mapping for the 15 amino acids was conducted separately based on the mixed linear model (MLM-Q+K) in TASSEL 58 (Bradbury et al., 2007) The SNP markers were considered significantly associated with amino acids if logarithm of the odds (LOD) value ≥3.0 based on MLM-Q+K models.
Linkage Disequilibrium Analysis and SNP-Based Haplotype Blocks
TASSEL 5.0 (Bradbury et al., 2007) was used to calculate the linkage disequilibrium (LD) (r2) for all pairwise loci within a window of 1MB of each chromosome. Haplotype blocks (HAP) were constructed in Haploview (Barrett et al., 2004) with a cutoff of 1% (Contreras-Soto et al., 2017). The LD (r2) for all marker pairs was performed using the R script LDit9.
Candidate Gene Selection
Two databases including the annotations for genes at Soybase at www.soybase.org/dlpages/ 10 and the plant metabolic network (PMN) database11, were used for searching candidate genes related to amino acids in soybean.
Currently, three Williams 82 genome sequence assemblies are available at Soybase (Glyma1.1, and Glyma 2.0)10. However, we used Glyma1.1 as the reference because the SNP data were provided by Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, at the time, Glyma1.1 was the best assembly available. We downloaded gene annotation of Glyma1.1 from Soybase and the corresponding gene positions in the Glyma 2.0 were obtained from https://www.soybase.org/correspondence/index.php12. For each SNP significantly associated with amino acids, we searched candidate genes within 10 kb of the SNP position. We also downloaded gene annotation from PMN for candidate gene discovery, because the metabolic pathway in PMN is updated with newer version of the genome (Phytozome v12: Gmax_275_Wm82.a2.v1.protein.fa).
Genomic Selection
Method 1: Ridge Regression Best Linear Unbiased Prediction
Ridge regression best linear unbiased prediction (RR-BLUP) was used to predict genomic estimated breeding value (GEBV) in GS and performed in the rrBLUP package (Endelman, 2011) with the R software Version 3.5.0 (Thuiller et al., 2009). The rr-BLUP is an effective and accurate prediction method as demonstrated in a wide range of traits and crops (Heslot et al., 2012; Jarquín et al., 2014; Lipka et al., 2014; Zhang et al., 2016).
We used 4:1 size ratio of training set and validation set randomly selected from the 249 accessions, which is a four-fold cross-validation, and repeated 100 times. Each training population subset consisting of 199 accessions was randomly selected from the association panel, and the remaining 50 accessions as the validation set (Resende et al., 2012; Shikha et al., 2017).
Two sets of SNPs were used to predict GEBV for each amino acid concentration in each accession: (1) all 23,279 high quality SNPs from GBS, and (2) all 231 SNP markers associated with 15 amino acid concentrations with LOD ≥3.0 from GWAS. In addition, we predicted GEBV for each amino acid concentration based on the SNP markers associated with the amino acid.
The prediction accuracy was estimated using the average Pearson’s correlation coefficient (r) between the GEBVs and observed values for each amino acid concentration in the validation set (Zhang et al., 2010; Resende et al., 2012; Shikha et al., 2017). The training and validation sets were randomly created 100 times and the r value was estimated each time. The average r value was calculated for each amino acid. The r value indicates the prediction accuracy and the selection efficiency of GS.
Method 2: Genomic Best Linear Unbiased Prediction
GS was also performed with the genomic best linear unbiased prediction (gBLUP) and the method was extended to compressed best linear unbiased prediction (cBLUP) by using the Compressed Mixed Linear Model (CMLM) approach in GAPIT (Lipka et al., 2012; Tang et al., 2016; www.zzlab.net/GAPIT/gapit_help_document.pdf). In order to conduct a four-fold cross-validation for estimating prediction efficiency, we randomly selected 199 accessions as the training set and the remaining 50 accessions as the validation set to predict GEBV for each accession. GEBV was calculated using the cBLUP in GAPIT using the SNP markers which were associated with the 15 amino acid concentrations with LOD ≥3.0 from GWAS. The Pearson’s correlation coefficient (r) between GEBV and observed value of the amino acid concentrations in both training and validation sets were calculated based on the 249 accessions. A total of 100 replications were used to calculate the r values and the average r value for each amino acid was used as the indicator of prediction accuracy.
Results
Phenotypic Variation and Association of Amino Acids in Soybean Seeds
The concentration of 15 amino acids, Ala, Arg, Asp, Glu, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Tyr, and Val varied widely among the 249 accessions (Supplementary Table 2). Concentration distribution of all amino acids except for Val, Ile and Gly in the accessions was near normal, indicating the amino acids are complex traits (Supplementary Figure 1). Glu and Asp were the main components of soybean seeds, which consisted of 20.1% and 13.3% of the total 15 amino acids, respectively. Glu had the highest concentration (74.42 ppm) among the 15 amino acids, followed by Asp (49.15 ppm). Two to five times of difference were observed between the accessions with the lowest and the highest concentration of Arg, Gly, Ile, Leu, Pro, Thr, and Val (Supplementary Table 2). The large variations of the amino acids were also indicated by the high CV values (Supplementary Table 2).
Most of the correlation coefficients among the 15 amino acids were greater than the threshold of 0.124 at P = 0.05 significant level (Table 1). Significant and negative coefficients were also observed between Asp and Ile, Asp and Val, Ile and Gly, Ile and Ser, etc. (Table 1). Based on the correlation coefficient values, the 15 amino acids except for Arg, His, and Pro could be divided into two groups (Table 1). Group one consisted of five amino acids: Ala, Asp, Glu, Gly and Ser, their pairwise correlation coefficients were greater than 0.75 except for the pair between Glu and Gly (r = 0.6) (Table 1). Group two contained seven amino acids: Ile, Leu, Lys, Phe, Thr, Tye, and Val with r values greater than 0.48 for all pairs. However, most correlation coefficients of amino acids between the two groups were negative (Table 1). Since the content of amino acids within each group were all significantly and highly correlated, they could have practical application in breeding program, e.g. breeders don’t need to improve amino acid individually, they can simultaneously improve multiple amino acids within the same group.
Table 1.
Correlation coefficients among 15 amino acid concentrations in soybean seeds.
| Group 1 | Group 2 | Arg | His | Pro | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ala | Asp | Glu | Gly | Ser | Ile | Leu | Lys | Phe | Thr | Tyr | Val | |||||
| Group 1 | Ala | 1 | ||||||||||||||
| Asp | 0.849* | 1 | ||||||||||||||
| Glu | 0.752* | 0.763 | 1 | |||||||||||||
| Gly | 0.785 | 0.846 | 0.600 | 1 | ||||||||||||
| Ser | 0.797 | 0.927 | 0.786 | 0.759 | 1 | |||||||||||
| Group 2 | Ile | -0.392 | -0.627 | -0.262 | -0.713 | -0.627 | 1 | |||||||||
| Leu | 0.015 | -0.236 | 0.114 | -0.350 | -0.194 | 0.776 | 1 | |||||||||
| Lys | 0.117 | -0.020 | 0.219 | -0.229 | -0.018 | 0.651 | 0.682 | 1 | ||||||||
| Phe | 0.072 | -0.093 | 0.190 | -0.336 | -0.099 | 0.754 | 0.795 | 0.891 | 1 | |||||||
| Thr | -0.102 | -0.233 | 0.146 | -0.531 | -0.213 | 0.790 | 0.702 | 0.762 | 0.827 | 1 | ||||||
| Tyr | 0.268 | 0.086 | 0.324 | -0.154 | 0.117 | 0.573 | 0.806 | 0.795 | 0.830 | 0.701 | 1 | |||||
| Val | -0.274 | -0.491 | -0.191 | -0.557 | -0.496 | 0.850 | 0.626 | 0.628 | 0.724 | 0.615 | 0.481 | 1 | ||||
| Arg | 0.406 | 0.371 | 0.445 | 0.157 | 0.274 | 0.272 | 0.422 | 0.617 | 0.678 | 0.446 | 0.535 | 0.325 | 1 | |||
| His | 0.604 | 0.556 | 0.547 | 0.477 | 0.458 | 0.080 | 0.313 | 0.619 | 0.546 | 0.266 | 0.529 | 0.217 | 0.793 | 1 | ||
| Pro | 0.337 | 0.203 | 0.398 | 0.004 | 0.160 | 0.304 | 0.389 | 0.542 | 0.563 | 0.581 | 0.529 | 0.182 | 0.517 | 0.438 | 1 | |
*The significance threshold based on 249 samples: r = 0.124 at P = 0.05; r = 0.162 at P = 0.01; and r = 0.206 at P = 0.001. P < 0.00001 for those r values bolded.
Based on 15 amino acid concentrations, we identified three accessions with the highest concentrations in each of the 15 amino acid concentrations. In addition, we ordered the 249 soybean accessions based on the concentration of each amino acid, and chosen 20 soybean accessions with at least one amino acid concentration topping three among the 249 soybean accessions. These 20 soybean accessions, Zhonghuang 10, Zhongzuo 983, 8588, Jian 31, Jidou 12, Zhengzhou 135, Wandou 15, Nanguanxiaopiqing, Lu 93748-1, Dabaipi, Bendidahuangdou, Jidou 12-3l, Lvrouheipidou, Xinliuqing, PI 547850, Zhongdou 33, Zheng 8516, Yudou 12, Huaheihu, and Lv 96150 would be good amino acid resources for improving amino acids concentration in soybean breeding programs (Supplementary Table 2 and Figure 1).
Figure 1.

The maximum likelihood tree of the 20 soybean germplasm accessions that ranked in the top three for at least one amino acid concentration among the 249 soybean accessions.
Association Mapping and SNP Marker Identification
The population structure of the 249 soybean accessions was initially inferred using STRUCTURE 2.3.4 (Pritchard et al., 2000) and the peak of delta K was observed at K = 6, indicating the presence of six sub-populations (clusters, Q1-Q6) (Figure 2A). In total, 51 of the 249 accessions were assigned to Q1 sub-population with 50 accessions from China; 65 assigned to Q2 with 42 from U.S., 21 from China and two from Korea; 55 assigned to Q3 with 54 cultivars from China; 42 assigned to Q4 with 27 cultivars from China and 12 accessions from U.S.; 21 assigned to Q5 with 16 from U.S.; and 15 to Q6 with all 15 from China (Figure 2B, and Supplementary Table 1). Phylogenetic analysis of the 249 soybean accessions using MEGA 6 also showed that the clustering of accessions was consistent with that inferred by STRUCTURE (Figure 2C).
Figure 2.

Structure analysis: (A) Delta K values for different numbers of populations (K) from the STRUCTURE analysis, x-axes shows different numbers of populations (K), y-axes shows Delta K values for different numbers of populations (K). (B) Classification of 249 accessions into six sub-populations using STRUCTURE version 2.3.4, where the x-axis shows accessions, and the y-axis shows the probability (from 0 to 1) of each accession belong to sub-population (Q = K) membership. The membership of each accessions belonging to sub-populations is indicated by different colors (Q1, red; Q2, green; Q3, blue; Q4, yellow; Q5, pink; and Q6: cyan). (C) Maximum Likelihood (ML) tree of the 249 accessions drawn in MEGA 6. The color code for each subpopulation is the same as that in the (B and C).
A total of 318 SNP markers consisted of 231 SNPs were associated with the 15 individual amino acid at LOD ≥3 (Supplementary Table 3 and Supplementary Figure 2). Because some SNPs were associated with two or more amino acids as pleiotropic association, the number of SNPs was only 231 (Table 2). Of the 318 SNPs, 11 were associated with Ala, 29 with Arg, 9 with Asp, 34 with Glu, 29 with Gly, 19 with His, 51 with Ile (Figure 3), 20 with Leu, 14 with Lys, 9 with Phe, 24 with Pro, 11 with Ser, 21 with Thr, 13 with Tyr, and 24 with Val (Supplementary Table 3 and Supplementary Figure 3).
Table 2.
List of SNP markers associated with each amino acid concentrations at LOD ≥ 3.0, respectively.
| SNP ID (chr_pos) | Trait | SNP ID (chr_pos) | Trait | SNP ID (chr_pos) | SNP ID (chr_pos) | Trait | |
|---|---|---|---|---|---|---|---|
| Gm01_33262451 | Ile | Gm07_4574178 | Ser | Gm13_39628016 | Ile | Gm16_6737312 | Thr |
| Gm01_45320366 | Ile | Gm07_5923593 | Arg | Gm13_39628019 | Ile | Gm16_780258 | Ile, Val |
| Gm01_53597652 | Gly | Gm08_14156183 | Ala, Glu | Gm13_39628049 | Ile | Gm17_14444779 | Pro |
| Gm01_571041 | His | Gm08_1969577 | Glu | Gm13_39628054 | Ile | Gm17_23967094 | Tyr |
| Gm01_571048 | His | Gm08_3446621 | Lys | Gm13_40242572 | Pro | Gm17_2459036 | Pro |
| Gm02_15368490 | Val | Gm08_43340095 | Lys | Gm13_40242573 | Pro | Gm17_2475262 | Pro |
| Gm02_45763574 | His | Gm08_45648867 | Asp | Gm13_40242709 | Pro | Gm17_2674908 | Pro |
| Gm02_47034495 | Ile, Thr, Val | Gm08_8091680 | Glu | Gm13_41203949 | His, Lys, Pro | Gm17_37708047 | His, Pro |
| Gm02_48215047 | Pro | Gm08_8480396 | Gly | Gm13_7762318 | Arg | Gm17_37708072 | His, Pro |
| Gm02_49856130 | Ile, Val | Gm08_8538031 | Gly | Gm14_28719225 | Gly | Gm17_37708077 | His, Pro |
| Gm02_50224425 | Arg | Gm09_43473530 | Ala | Gm14_42728555 | Glu | Gm17_37708117 | His, Pro |
| Gm02_50269310 | Arg | Gm09_43488824 | Ala, Asp | Gm14_42900467 | Glu | Gm17_37712338 | Arg, Pro |
| Gm02_5190606 | Ile | Gm10_12029489 | Ala | Gm14_43163207 | Glu | Gm18_1231280 | Ile |
| Gm02_6671113 | Gly | Gm10_35214322 | Val | Gm14_43163233 | Glu | Gm18_12797087 | Thr |
| Gm02_6721375 | Asp, Gly, Ile, Ser | Gm10_44070578 | Ile | Gm14_43163234 | Glu | Gm18_1449038 | Glu, Ser |
| Gm03_36272238 | Thr | Gm10_45237186 | Ile | Gm14_43163255 | Glu | Gm18_14877256 | Phe |
| Gm03_36417795 | Thr | Gm10_46037693 | Glu | Gm14_43163263 | Glu | Gm18_1564092 | Glu |
| Gm03_36465287 | Thr | Gm10_46037954 | Ala, Glu | Gm14_43163268 | Glu | Gm18_2026494 | Thr |
| Gm03_36530224 | Pro | Gm10_46045322 | Glu | Gm14_43163302 | Glu | Gm18_23446982 | Ile |
| Gm03_40600088 | Pro | Gm10_47770916 | Arg | Gm14_43163309 | Glu | Gm18_23680823 | Ile |
| Gm03_40600203 | Pro | Gm10_48103776 | Asp, Glu, His, Ser | Gm14_43163317 | Glu | Gm18_45637951 | Ile |
| Gm03_6537448 | Arg, His | Gm10_48367427 | Ser | Gm14_670550 | Arg | Gm18_54941806 | Leu |
| Gm04_29795804 | Gly, Ile, Thr | Gm10_4877563 | Arg | Gm14_670770 | Arg | Gm18_54941806 | Tyr |
| Gm04_3722529 | Pro | Gm10_4877661 | Arg | Gm15_42452169 | Lys, Phe, Val, Lys, Phe, Val | Gm18_55570016 | Arg |
| Gm04_43205897 | Gly | Gm10_50892012 | Glu | Gm15_46888773 | His | Gm18_57994827 | Arg |
| Gm04_43205900 | Gly | Gm10_50945017 | Glu | Gm15_6364620 | Gly | Gm18_57994865 | Arg |
| Gm04_43207187 | Gly, Ile, Thr, Phe | Gm10_50945124 | Glu | Gm15_6364624 | Gly | Gm18_58356668 | Gly |
| Gm04_43207248 | Gly, Ile, Phe, Thr | Gm10_6088950 | Arg | Gm15_6364658 | Gly | Gm18_61819070 | Leu |
| Gm04_43247307 | Gly | Gm10_6127825 | Arg | Gm15_6364660 | Gly | Gm18_61846089 | Leu |
| Gm04_43247365 | Gly | Gm10_6158335 | Arg | Gm15_6364671 | Gly | Gm18_61846097 | Leu |
| Gm04_45172948 | Ile | Gm11_17324386 | Leu | Gm16_19302037 | Ile | Gm18_61846199 | Leu |
| Gm05_1131617 | Thr, Tyr, Leu | Gm11_36252840 | Lys, Phe, Tyr | Gm16_19309923 | Ile, Val | Gm18_61846240 | Leu |
| Gm05_1364762 | Gly | Gm11_36391557 | Pro | Gm16_19310296 | Ile, Val | Gm18_61846255 | Leu |
| Gm05_1956615 | Glu, Ser | Gm11_38372080 | Ile | Gm16_19474288 | Ile | Gm18_61846357 | Leu |
| Gm05_21977894 | Ile, Val | Gm12_1283279 | Ile | Gm16_26668643 | Pro | Gm18_829983 | Leu |
| Gm05_36368612 | Tyr | Gm12_1966701 | Leu, Val | Gm16_26668804 | Pro | Gm18_849773 | Leu |
| Gm06_14669414 | Lys | Gm12_2246393 | Ile, Leu, Phe, Thr, Val | Gm16_26760058 | Pro | Gm18_8944865 | Lys, Thr |
| Gm06_1655912 | Arg, Tyr | Gm12_2246402 | Thr | Gm16_27656811 | Ile, Leu | Gm19_14283927 | Ile, Val |
| Gm06_20941559 | Glu | Gm12_2246405 | Thr | Gm16_27675722 | Ile, Leu | Gm19_34599708 | Ala |
| Gm06_289575 | Gly | Gm12_2246408 | Thr | Gm16_28109123 | Ile | Gm19_35491961 | Ile, Val |
| Gm06_399885 | Ala, Asp, Gly | Gm12_2246409 | Thr | Gm16_30033799 | Val | Gm19_35491974 | Ile, Val |
| Gm06_46691924 | Gly | Gm12_37250318 | Ile, Leu | Gm16_31565242 | Lys, Val | Gm19_35491994 | Ile, Val |
| Gm06_48160139 | His | Gm12_37253606 | Ile | Gm16_31565425 | Lys, Val | Gm19_35491998 | Ile, Val |
| Gm06_48405502 | Gly | Gm12_37699937 | Pro | Gm16_32344691 | Arg | Gm19_35492018 | Ile, Val |
| Gm06_49021688 | Gly | Gm12_37700016 | Pro | Gm16_32636611 | Arg | Gm19_35492028 | Ile, Val |
| Gm06_582930 | Gly | Gm12_4525326 | Arg | Gm16_32891444 | Arg | Gm19_35492061 | Ile, Val |
| Gm07_16345870 | Phe | Gm12_4525341 | Arg | Gm16_33487136 | Arg, His | Gm19_35492063 | Ile, Lys, Val |
| Gm07_3374472 | Gly | Gm12_9802063 | Ile | Gm16_33595082 | Arg, His | Gm19_36853376 | Ala, Ser |
| Gm07_3374492 | Gly | Gm13_17646967 | Asp, Ile, Ser | Gm16_33670373 | Asp | Gm19_36856526 | Ala, Glu, Ser |
| Gm07_36388230 | Asp | Gm13_21744787 | Asp, Glu, Ser | Gm16_33761779 | Arg, His | Gm19_38354186 | Tyr |
| Gm07_36390103 | Glu | Gm13_21758530 | Ala, Asp | Gm16_33853366 | Arg, HIs | Gm19_41048945 | Glu |
| Gm07_36524487 | Glu | Gm13_22508206 | Arg | Gm16_35244130 | His | Gm20_31240721 | Leu, Tyr |
| Gm07_36542987 | Glu | Gm13_38830655 | Lys | Gm16_35747794 | Ser | Gm20_31240801 | Tyr |
| Gm07_36543902 | Glu | Gm13_39627980 | Ile | Gm16_36927834 | Tyr | Gm20_31387086 | His |
| Gm07_36633143 | Glu | Gm13_39627983 | Ile | Gm16_36927871 | Tyr | Gm20_35630363 | Leu |
| Gm07_36633260 | Glu | Gm13_39627986 | Ile | Gm16_6737154 | His, Lys, Thr | Gm20_42531505 | Gly, Ile, Leu, Tyr, Phe, Thr |
| Gm07_3811476 | Arg | Gm13_39628010 | Ile | Gm16_6737218 | Thr | Gm20_42569717 | Lys, Tyr |
| Gm07_39077446 | Ala | Gm13_39628014 | Ile | Gm16_6737289 | Thr |
Figure 3.

The QQ plot between the expected LOD (-log(P-value)) value and the estimated LOD (log(P-value)) value of amino acid Ile based on 23,279 SNPs as an example (all 15 QQ-plot for the 15 amino acids showed in Supplementary Figure 3).
The total number of haplotype blocks was 3,458 based on 23,279 SNPs, the 231 SNPs were positioned in 85 of these haplotype blocks (Supplementary Table 3). Many haplotype blocks contained more than two SNP markers. For example, Gm12_4525341 and Gm12_4525326 were in the same haplotype block and associated with Arg; Gm06_289575, Gm06_399885, and Gm06_582930 were in the same haplotype block on Chr 6 and were associated with Gly (Supplementary Table 3).
The number of the haplotype blocks varied among chromosomes, e.g. 12 of the 85 haplotype blocks were on Chr 16; 11 haplotype blocks on Chr 18; 1 on Chrs 6 and 9. Twenty of the 85 haplotype blocks had significant association with more than one amino acids, e.g. Gm20_42531505 on the Chr. 20_Block 2 was significantly associated with Thr, Gly, Ile, Tyr, Leu, Phe; Two SNP markers, Gm04_43207248 and Gm04_43207187 in the Chr. 4_Block 3, were significantly associated with Ile, Phe, Gly and Thr; and two markers, Gm15_42452169 and Gm15_42452285 in the Chr. 15_Block 2 associated with Val, Phe and Lys (Supplementary Table 4).
Based on phenotypic patterns of the amino acid concentration among accessions, the 15 amino acids could be divided into two groups which were showed in phenotypic variance section. SNP markers associated with amino acids in each group were also found. Twenty-five SNP markers were associated with five amino acids, Ala, Asp, Glu, Gly, and Ser in group one (Table 3), and 28 SNP markers with seven amino acids, Ile, Leu, Lys, Phe, Thr, Tyr, and Val in group two (Table 4). The SNP markers in each group can be used to simultaneously select multiple amino acids within the group. Such as Gm10_48103776 was associated with five amino acids, Ala, Asp, Glu, Gly, and Ser in group one with LOD values of 2.93, 3.15, 3.51, 2.35, and 3.60, respectively (Table 3) and it can be used to simultaneously select soybean lines with higher contents of the five amino acids in soybean breeding progress. For group two, such as Gm20_42531505 was associated with seven amino acids, Ile, Leu, Lys, Phe, Thr, Tye, and Val with LOD values of 3.53, 4.55, 2.89, 4.79, 5.04, 3.87, and 2.10, respectively (Table 4), indicating that it can be used to simultaneously select the soybean lines with higher contents of seven amino acids. Meanwhile, both phenotypic and genetic data supported there were two groups of amino acids existed in soybean.
Table 3.
Twenty-five SNP markers associated with five amino acids of group one, simultaneously.
| SNP ID | Chr | Position (bp) | Ala | Asp | Glu | Gly | Ser |
|---|---|---|---|---|---|---|---|
| Gm02_6671113 | 2 | 6671113 | 2.19* | 2.83 | 0.68 | 3.46 | 2.48 |
| Gm02_6721375 | 2 | 6721375 | 2.85 | 5.41 | 0.83 | 3.36 | 4.63 |
| Gm06_399885 | 6 | 399885 | 3.14 | 2.58 | 1.86 | 4.15 | 2.35 |
| Gm07_36388230 | 7 | 36388230 | 2.38 | 3.06 | 2.13 | 2.29 | 2.95 |
| Gm07_36542987 | 7 | 36542987 | 1.18 | 2.36 | 3.68 | 2.20 | 2.01 |
| Gm07_36633143 | 7 | 36633143 | 2.06 | 2.13 | 3.26 | 0.83 | 2.15 |
| Gm07_36633260 | 7 | 36633260 | 2.06 | 2.13 | 3.26 | 0.83 | 2.15 |
| Gm10_46037693 | 10 | 46037693 | 2.88 | 2.10 | 3.84 | 2.10 | 2.35 |
| Gm10_46037954 | 10 | 46037954 | 3.07 | 2.38 | 3.86 | 2.70 | 2.44 |
| Gm10_48103776 | 10 | 48103776 | 2.93 | 3.15 | 3.51 | 2.35 | 3.60 |
| Gm13_21744787 | 13 | 21744787 | 2.31 | 3.05 | 3.03 | 2.66 | 3.27 |
| Gm13_21758530 | 13 | 21758530 | 3.12 | 3.18 | 2.92 | 2.04 | 2.97 |
| Gm14_43163207 | 14 | 43163207 | 2.10 | 2.46 | 3.83 | 0.90 | 2.17 |
| Gm14_43163233 | 14 | 43163233 | 2.10 | 2.46 | 3.83 | 0.90 | 2.17 |
| Gm14_43163234 | 14 | 43163234 | 2.58 | 2.54 | 4.37 | 1.33 | 2.57 |
| Gm14_43163255 | 14 | 43163255 | 2.58 | 2.54 | 4.37 | 1.33 | 2.57 |
| Gm14_43163263 | 14 | 43163263 | 2.10 | 2.46 | 3.83 | 0.90 | 2.17 |
| Gm14_43163268 | 14 | 43163268 | 2.58 | 2.54 | 4.37 | 1.33 | 2.57 |
| Gm14_43163302 | 14 | 43163302 | 2.10 | 2.46 | 3.83 | 0.90 | 2.17 |
| Gm14_43163309 | 14 | 43163309 | 2.10 | 2.46 | 3.83 | 0.90 | 2.17 |
| Gm14_43163317 | 14 | 43163317 | 2.58 | 2.54 | 4.37 | 1.33 | 2.57 |
| Gm16_35747794 | 16 | 35747794 | 2.78 | 2.22 | 1.46 | 2.40 | 3.02 |
| Gm18_1449038 | 18 | 1449038 | 2.34 | 2.93 | 3.22 | 2.74 | 3.39 |
| Gm19_36853376 | 19 | 36853376 | 3.07 | 2.37 | 2.51 | 1.17 | 3.52 |
| Gm19_36856526 | 19 | 36856526 | 3.04 | 2.42 | 3.64 | 1.97 | 3.54 |
*LOD (-log(P-value)) from MLM of Tassel.
Table 4.
Twenty-eight SNP markers associated with seven amino acids of group two, simultaneously.
| SNP ID | Chr. | Position (bp) | Ile | Leu | Lys | Phe | Thr | Tyr | Val |
|---|---|---|---|---|---|---|---|---|---|
| Gm03_36417795 | 3 | 36417795 | 1.95* | 1.34 | 2.06 | 2.91 | 3.38 | 2.44 | 2.44 |
| Gm03_36465287 | 3 | 36465287 | 2.98 | 1.95 | 2.46 | 2.85 | 3.68 | 2.68 | 2.34 |
| Gm04_43207187 | 4 | 43207187 | 3.39 | 1.95 | 2.52 | 3.57 | 3.17 | 2.46 | 2.39 |
| Gm04_43207248 | 4 | 43207248 | 3.39 | 1.95 | 2.52 | 3.57 | 3.17 | 2.46 | 2.39 |
| Gm04_45172948 | 4 | 45172948 | 3.11 | 2.29 | 2.11 | 2.15 | 2.06 | 1.42 | 1.95 |
| Gm05_1131617 | 5 | 1131617 | 2.74 | 3.69 | 2.54 | 2.51 | 3.32 | 3.24 | 1.15 |
| Gm05_1364762 | 5 | 1364762 | 2.70 | 1.78 | 2.52 | 2.13 | 2.78 | 2.46 | 1.72 |
| Gm05_21977894 | 5 | 21977894 | 3.59 | 1.72 | 2.67 | 2.18 | 2.64 | 2.60 | 3.28 |
| Gm08_3446621 | 8 | 3446621 | 2.39 | 0.90 | 3.99 | 2.73 | 2.05 | 1.30 | 2.66 |
| Gm11_36252840 | 11 | 36252840 | 1.13 | 2.48 | 3.94 | 3.48 | 2.62 | 4.30 | 1.26 |
| Gm12_1966701 | 12 | 1966701 | 2.95 | 3.24 | 1.70 | 2.20 | 1.51 | 2.38 | 3.13 |
| Gm12_2246393 | 12 | 2246393 | 3.40 | 3.02 | 2.36 | 3.27 | 4.13 | 2.64 | 3.39 |
| Gm12_9802063 | 12 | 9802063 | 3.32 | 2.04 | 1.81 | 2.38 | 2.64 | 1.38 | 2.12 |
| Gm15_42452169 | 15 | 42452169 | 2.19 | 1.55 | 3.80 | 3.35 | 2.06 | 1.87 | 3.79 |
| Gm16_6737218 | 16 | 6737218 | 2.58 | 1.11 | 2.89 | 2.24 | 3.59 | 1.90 | 2.19 |
| Gm16_27675722 | 16 | 27675722 | 3.03 | 3.63 | 1.62 | 2.39 | 2.30 | 2.12 | 1.35 |
| Gm18_1231280 | 18 | 1231280 | 3.17 | 0.91 | 2.15 | 2.81 | 2.35 | 1.54 | 2.34 |
| Gm18_14877256 | 18 | 14877256 | 2.86 | 2.18 | 2.67 | 3.09 | 2.27 | 1.92 | 1.60 |
| Gm18_54941806 | 18 | 54941806 | 2.45 | 3.91 | 2.47 | 2.54 | 2.27 | 3.60 | 1.52 |
| Gm19_35491974 | 19 | 35491974 | 4.20 | 0.59 | 2.48 | 2.38 | 2.90 | 0.95 | 4.17 |
| Gm19_35491994 | 19 | 35491994 | 4.03 | 0.64 | 2.51 | 2.26 | 2.61 | 0.93 | 3.99 |
| Gm19_35491998 | 19 | 35491998 | 4.19 | 0.62 | 2.51 | 2.35 | 2.82 | 0.88 | 4.04 |
| Gm19_35492018 | 19 | 35492018 | 4.19 | 0.62 | 2.51 | 2.35 | 2.82 | 0.88 | 4.04 |
| Gm19_35492028 | 19 | 35492028 | 4.19 | 0.62 | 2.51 | 2.35 | 2.82 | 0.88 | 4.04 |
| Gm19_35492061 | 19 | 35492061 | 4.30 | 0.64 | 2.71 | 2.50 | 2.94 | 0.97 | 4.23 |
| Gm19_35492063 | 19 | 35492063 | 4.15 | 0.68 | 3.22 | 2.81 | 2.98 | 1.32 | 3.35 |
| Gm20_42531505 | 20 | 42531505 | 3.53 | 4.55 | 2.89 | 4.79 | 5.04 | 3.87 | 2.10 |
| Gm20_42569717 | 20 | 4256971 | 1.64 | 2.09 | 3.43 | 2.92 | 2.47 | 3.00 | 1.42 |
*LOD (-LOG(P-value)).
Candidate Gene Selection
The linkage disequilibrium (LD) of soybean genome was analyzed, the average distance of markers at half of the maximum LD decay rate was about 200kb. Considering the LD decay value may vary from genomic region to region, we used the 10kb windows as previously reported (Xie et al., 2018). We identified 704 genes with all or partial sequence within the 10 kb windows that flanked each of the 217 out of 231 unique SNPs associated with one or more amino acids (Supplementary Table 5) and the other 14 SNPs did not have any candidate genes at the 10 kb windows on the chromosomes.
Based on gene annotations of the soybean whole genome assembly Gmax_275_Wm82.a2.v1 from Soybase and PMN (Phytozome v12: Gmax_275_Wm82.a2.v1.protein.fa), we found that 15 SNPs were in 14 genes related to amino acid metabolism in gene ontology annotation terms (Supplementary Table 6), e.g. in the region flanking the SNP Gm03_36417795, there was a candidate gene “Glyma03g28476 (Glyma 1.1)/Glyma.03g129100 (Glyma 2.0)” encoding for pyrroline-5-carboxylate reductase (Delauney and Verma, 1990)13 (Supplementary Table 6). This enzyme catalyzes the last step of L-proline biosynthesis through the L-glutamate degradation pathway. In the region flanking the SNP Gm03_36465287, there was a gene Glyma03g28530 (Glyma 1.1)/Glyma.03g129700 (Glyma 2.0) encoding β L-selenocystathionase, a key enzyme catalyzing L-homocysteine and L-cysteine interconversion. L-homocysteine and L-cysteine interconversion is an intermediate step for conversion between methionine and cysteine (McCluskey et al., 1986)14 (Supplementary Table 6).
Genomic Selection for Amino Acid Concentration Based on RR-BLUP in rrBLUP
Based on RR-BLUP in rrBLUP, the GEBV of each amino acid was estimated using three different sets of SNPs, i.e. 23,279 SNPs, 231 SNP markers associated with 15 amino acid, and SNP markers associated with an individual amino acid.
The correlation coefficients between GEBV and observed value varied among amino acids based on all 23,279 SNPs (column-2 in Table 5), the r value was 0.61 for Arg; 0.50 for Phe; between 0.35 and 0.50 for His, Lys, Thr and Tyr; between 0.25 and 0.35 for Ala, Glu, Ile, Leu, Pro, and Val; and less than 0.25 for Asp, Gly, and Ser. The r values for most amino acids were less than 0.5, suggesting GS prediction accuracy for most amino acids was low based on genome-wide random SNPs.
Table 5.
The averaged correlation coefficient (r) among 15 amino acids between the observed values (each amino acid content) and the GEBVs predicted from (1) all 23,279 SNPs, (2) the 231 SNP markers, and (3) only the associated SNP markers with the specific amino acid content using RR-BLUP in rrBLUP software, and from (4) the 231 SNP markers in reference set (training set) and inference set (validation set) using CBLUPin Gapit.
| RR-BLUP in rrBLUP | CBLUP in Gapit | ||||
|---|---|---|---|---|---|
| 23279 SNPs in 249 accessions | 231 SNPs associated with amino acids | Associated SNPs* | 231 SNPs in training set | 231 SNPs in validation set | |
| Ala | 0.30 | 0.45 | 0.33 | 0.76 | 0.52 |
| Arg | 0.61 | 0.68 | 0.61 | 0.80 | 0.59 |
| Asp | 0.22 | 0.53 | 0.41 | 0.77 | 0.57 |
| Glu | 0.31 | 0.48 | 0.48 | 0.74 | 0.52 |
| Gly | 0.23 | 0.56 | 0.35 | 0.79 | 0.60 |
| His | 0.46 | 0.57 | 0.46 | 0.76 | 0.55 |
| Ile | 0.25 | 0.61 | 0.53 | 0.80 | 0.61 |
| Leu | 0.30 | 0.53 | 0.49 | 0.77 | 0.53 |
| Lys | 0.42 | 0.61 | 0.54 | 0.82 | 0.59 |
| Phe | 0.50 | 0.68 | 0.35 | 0.84 | 0.68 |
| Pro | 0.26 | 0.49 | 0.34 | 0.76 | 0.46 |
| Ser | 0.18 | 0.47 | 0.42 | 0.73 | 0.51 |
| Thr | 0.39 | 0.63 | 0.50 | 0.85 | 0.63 |
| Tyr | 0.36 | 0.54 | 0.44 | 0.81 | 0.57 |
| Val | 0.25 | 0.52 | 0.53 | 0.75 | 0.54 |
| Average | 0.34 | 0.56 | 0.45 | 0.78 | 0.56 |
*Associated SNPs signifies that the average correlation coefficient (r) for each amino acid in column-4 was calculated with the SNP markers only associated with the individual amino acid to predict the GEBV for each amino acid, such as for r = 0.33 for Ala, which was calculated from 11 associated SNPs.
The correlation coefficients between GEBV and observed value of the 15 amino acids were equal or higher from 231 SNPs than those from the 23,279 SNPs (column-3 vs column-2 in Table 5). The r value was larger than 0.6 for Arg, Ile, Lys, Phe, and Thr, and between 0.5 and 0.6 for Asp, Gly, His, Leu, Tyr, and Val, indicating that associated markers were more efficient to predict amino acids for soybean lines than all the SNPs (Figure 4 and column-3 in Table 5).
Figure 4.

The correlation coefficient (r) among 15 amino acids between the observed values (each amino acid concentration) and the GEBVs predicted from the 231 SNP markers using RR-BLUP in rrBLUP software.
Of the 231 SNPs, a total of 171, 42, 12, 4, 1 and 1 SNPs were associated with only one, two, three, four, five, and six amino acids, respectively. A total of 11, 29, 9, 34, 29, 19, 51, 20, 14, 9, 24, 11, 21, 13, and 24 SNP markers were associated with Ala, Arg, Asp, Glu, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Tyr, and Val, respectively (Supplementary Table 3). We used the SNP markers only associated with individual amino acid to predict the GEBV for each amino acid, the r values for the 14 amino acids were higher than those from the 23,279 SNPs except for Phe, but equal or lower than those from the 231 SNP markers except for Val (Table 5).
T-test was conducted to compare the r values from the 231 SNPs and from the all 23,279 SNPs and found that the r value from the 231 SNPs in column-3 for each amino acids was significantly higher than that in column-2 from all SNPs with P = 0.01 level in Table 5, indicating that using the associated SNPs had better prediction for GS than using all randomly SNPs (Table 5).
Genomic Selection for Amino Acid Concentration Based on CMLM in GAPIT
Based on cBLUP method using CMLM in GAPIT, the average r was estimated (Table 5 and Figure 5). The average correlation coefficient in the training set was greater than 0.7 and was higher than those in validation set. The average values in validation set were greater than 0.5 for amino acids except for Pro.
Figure 5.
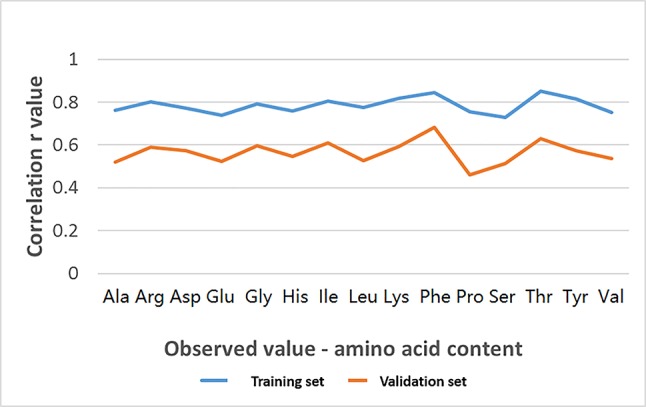
The average correlation coefficient (r) among 15 amino acids between the observed values (each amino acid concentration) and the GEBVs predicted in both training set and validation set from the 231 SNP markers using cBLUP method in GAPIT.
Two comparisons were tested to validate the stability of GS using different estimate methods and approaches: (1) RR-BLUP in rrBLUP vs cBLUP in Gapit, and (2) self-validation (training set by itself) vs cross-validation (training set). For the first comparison, the 15 r values in column-3 (“231 SNPs in 249 accessions”) was compared to those in column-6 (“231 SNPs in validation set”) in Table 5 and we found a strong association between the average r values from RR-BLUP in rrBLUP and from cBLUP in Gapit (r = 0.85) based on the 231 associated SNPs. For the second comparison, the 15 r values in column-5 (“231 SNPs in training set”) was compared to those in column-6 (“231 SNPs in validation set”) in Table 5 and we found a strong association between the average r values from cBLUP in Gapit (r = 0.84) based on the 231 associated SNPs. The strong association with high r value >0.8 between different methods and approach showed that we can use the 231 SNPs to select high amino acid content in soybean through GS.
Discussion
Application of Marker-Assisted Selection to Genetic Improvement of Soybean
Previous studies using bi-parental segregating populations have identified QTLs controlling 15 amino acids in soybean seeds (Panthee et al., 2006; Fallen et al., 2013; Khandaker et al., 2015; Warrington et al., 2015). The QTL were associated with 84 molecular markers on 14 chromosomes (Supplementary Table 7). In this study, we identified 231 unique SNP markers significantly associated with 15 amino acids (Supplementary Table 3). Eight SNPs were in the same regions of SSR markers that were associated with amino acid concentrations reported by Panthee et al. (2006), e.g. the SNP marker, Gm07_4574178 (located at 4.5 Mb on chr 7) associated with Ser was near the SSR marker, Satt 567 (located at 63,663 bp on chr 7), Gm19_41048945 at 41 Mb on chr 19 for Glu was near Satt076 at 374,148 bp of chr 19; Gm02_15368490 at 15,368,490 bp on chr 2 for Val near Satt537; Gm01_45320366 at 45,320,366 bp on chr 1 for Ile near Satt203; Gm19_35491961 at 35,491,961 bp on chr 19 for Ile near Satt313; Gm02_50269310 at 50,269,310 bp on chr 2 for Arg also near Satt274 and Satt196; and Gm09_43488824 at 43,488,824 bp on chr 9 for Asp near Satt196 (Panthee et al., 2006). Two SNP markers, Gm09_43488824 at 43,488,824 bp on chr 9 for Asp and Gm10_48103776 at 48,103,776 bp on chr 10 for His were close to the regions controlling the two amino acids reported by Fallen et al. (2013) (Supplementary Table 7). In addition, Gm09_43488824 at 43,488,824 bp on chr 9 associated with Asp was in the regions reported by Panthee et al. (2006) and Fallen et al. (2013).
As GWAS for amino acid concentrations in soybean, Zhang et al. (2018) reported that 54 SNPs, as 92 markers were associated with 18 amino acids; 38 of the 54 SNPs associated with only one amino acid; and 11 SNPs associated with 2 to 12 amino acids. The SNP markers for each amino acid were located at one chromosome such as Pro or Ser, nine chromosomes such as Arg or Asp, up to 11 chromosomes such as Try (Supplementary Table 7). Comparisons with the SNP markers associated with amino acids reported by Zhang et al. (2018), most of SNP markers were located at different regions of soybean chromosomes. However, there were four regions similar to our results: (1) 3.71–3.82 Mb of chr 7 for Arg; (2) 33.85–35.73 Mb of chr 16 for Arg; (3) 16.28–17.65 Mb of chr13 for Asp; and (4) 8.27–9.33 Mb of Chr 8 for Gly. From our study, the SNP marker Gm07_3811476 was associated with Arg at 3,811,476 bp on chr 7, which was near with around 98 kb to the SNP markers ss715597475 at 3,713,267 bp on chr7 for Arg reported by Zhang et al. (2018). Another SNP, Gm16_33853366 close to ss715624781 with 1.87 M distance on chr 16 was also associated with Arg; Gm16_33853366 was at 33,853,366 bp and ss715624781 at 35,721,993 bp on chr 16. For Asp, the Gm13_17646967 at 17,646,967 bp was close to ss715616790 at 16,286,313 bp with a distance 1,360,654 bp on chr 13. The SNP markers, Gm08_8480396 and Gm08_8538031 associated with Gly from this study were close to the two SNP markers, ss715602750 and ss715602851 with Gly (Zhang et al., 2018) and the four markers are located at a region with one Mb distance on chr 8. Thus, the four regions were validated to be associated with one of the amino acid, Arg, Asp, or Gly.
These SNPs identified for 15 amino acids in this study can be used as molecular markers to select lines with high amino acids content through marker-assisted selection (MAS). PCR-based KASP SNP genotyping can be used in soybean breeding program to select high amino acids through MAS. Targeted region sequencing such as tGBS (targeted genotyping-by-sequencing) (Simko et al., 2018) can also be used for MAS and GS based on the sequences flanking these SNPs (Ott et al., 2017).
From this study, 14 candidate genes were found to be related to amino acid metabolism based on gene annotations from Soybase and PMN with gene ontology annotation terms using the DNA sequences in the 15 regions with the 15 SNPs in column-B of Supplementary Table 7 significantly associated with amino acids (Supplementary Table 6). Our further research will develop the molecular markers such as PCR-based assays or targeted region sequencing to validate these candidate genes in our association panel and others. Gene-silence through CRISPR/Cas9 may be used as an approach to validate these candidate genes.
Genomic Selection
Prediction accuracy is the main parameter to measure the performance of GS (Jarquin et al., 2016; Zhang et al., 2016; Duhnen et al., 2017). Prediction accuracy is affected by several factors including GS models, marker density, level of LD, QTL number, the population size specially the training population size, relationship between training population and validation population, and trait heritability (Jarquin et al., 2016).
Zhang et al. (2016) estimated prediction accuracy (r value) of seed size based on 309 soybean accessions and reported r = 0.85 when 2000 SNPs or 31,045 SNPs were included, r = 0.8 when 1000 SNPs or 500 SNPs were used. They also identified 48 SNPs on 12 chromosomes associated with seed size based on GWAS. The r value ranged from 0.64 to 0.74 when 5, 10, and 15 of the 48 SNP markers were used, which were 25% higher than those calculated from the same number of randomly selected SNPs. Our results showed that the highest r value (0.56) was obtained based on the model including 231 SNPs signific antly associated with one or multiple amino acids, followed by the model including SNPs significantly associated with individual amino acid (r = 0.45), and the least was the model including all SNPs (r = 0.34). A t-test showed r values were significantly different among the sets.
We also estimated the GEBV and r values using the cBLUP in GAPIT. Based on the set of 231 SNPs, the correlation coefficient was greater than 0.7 in the training population and greater than 0.5 in validation population. The high correlation between the reference and inference (0.84) based on 15 amino acids, further confirmed the reliability of the GS. A high correlation (0.85) of the prediction accuracy between rrBLUP and GAPIT based on 231 SNPs, indicated that both RR-BLUP in rrBLUP or cBLUP in GAPIT were consistent.
Conclusion
In this study, soybean accessions with high concentrations of amino acids in seeds, and molecular markers associated with individual and groups of amino acids were identified. These soybean accessions with high amino acid concentrations could be used as parents in soybean breeding programs. The SNP markers strongly associated with the concentrations of the amino acids could be used to improve the nutritional quality of soybean through marker-assisted selection. In addition, fourteen candidate genes that were related to amino acid metabolism were also identified. These candidate genes will lead to a better understanding of the molecular mechanisms that control amino acids metabolism in soybean seeds. Genomic selection analysis of amino acid concentration showed that the selection efficiency of amino acids based on the markers significantly associated with 15 amino acids was higher than that based on genome-wide random markers or markers only associated with an individual amino acid. These results suggest that including a set of markers significantly associated with multiple amino acids in genomic selection is likely to help breeders to efficiently select soybean varieties with improved amino acid content.
Data Availability Statement
SNP data can be found in the ENA using accession number PRJEB34546 (www.ebi.ac.uk/ena/data/view/PRJEB34546).
Ethics Statement
All data and materials are not related to human and animals. This research is not related to any plant specimens to be deposited as vouchers or any other association for this section.
Author Contributions
JQ, AS, YC, FW, and WR carried out phenotyping and genotyping. AS, JQ, SL, and QS analyzed the data. JQ composed the draft of the manuscript. MZ and CY directed and managed this research. AS and QJS reviewed and edited the manuscript. All authors have read, made corrections, and approved the final manuscript.
Funding
The authors would like to thank Prof. Lijuan Qiu (Chinese Academy of Agricultural Sciences) for providing seeds of 249 soybean accessions. This study was supported by the Hebei Province Natural Science Foundation for Distinguished Young Scholars (C2014301035), National Natural Science Foundation of China (31100880), and Key Project of the Natural Science Foundation of Hebei Province (C2012301020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
http://www.soystats.com, accessed on August 10, 2019
https://www.fediol.eu, accessed on August 10, 2019
https://www.jmp.com/en_us/software/genomics-data-analysis-software.html; accessed on August 10, 2019
https://web.stanford.edu/group/pritchardlab/structure_software/release_versions/v2.3.4/html/structure.html, accessed on August 10, 2019
https://github.com/rossibarra/r_buffet/blob/master/LDit.r, verified on May 10, 2018
https://www.soybase.org/dlpages/; accessed on August 10, 2019
https://www.plantcyc.org/; accessed on August 10, 2019
https://www.soybase.org/correspondence/index.php12, accessed on August 10, 2019
https://link.springer.com/article/10.1007/BF00259392, accessed on August 10, 2019
https://doi.org/10.1016/0031-9422(86)80067-X, accessed on August 10, 2019
Abbreviations
SNP Single nucleotide polymorphism; GWAS Genome-wide association study; AA Amino acids; Ala Alanine; Arg Arginine; Asp Aspartic acid; Glu Glutamic acid; Gly Glycine; His Histidine; Ile Isoleucine; Leu Leucine; Lys Lysine; Phe Phenylalanine; Pro Proline; Ser Serine; Thr Threonine; Tyr Tyrosine; Val Valine; SSR Simple sequence repeat; MAS Marker-assisted selection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01445/full#supplementary-material
References
- Albrecht T., Wimmer V., Auinger H.-J., Erbe M., Knaak C., Ouzunova M., et al. (2011). Genome-based prediction of testcross values in maize. Theor. Appl. Genet. 123, 339. 10.1007/s00122-011-1587-7 [DOI] [PubMed] [Google Scholar]
- Angelovici R., Batushansky A., Deason N., Gonzalez-Jorge S., Gore M. A., Fait A., et al. (2017). Network-guided GWAS improves identification of genes affecting free amino acids. Plant Physiol. 173, 872–886. 10.1104/pp.16.01287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J. C., Fry B., Maller J., Daly M. J. (2004). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. 10.1093/bioinformatics/bth457 [DOI] [PubMed] [Google Scholar]
- Battenfield S. D., Guzmán C., Gaynor R. C., Singh R. P., Peña R. J., Dreisigacker S., et al. (2016). Genomic selection for processing and end-use quality traits in the CIMMYT spring bread wheat breeding program. Plant Genome 9, 22. 10.3835/plantgenome2016.01.0005 [DOI] [PubMed] [Google Scholar]
- Bernardo R. (1996). Best linear unbiased prediction of maize single-cross performance. Crop Sci. 36, 50–56. 10.2135/cropsci1996.0011183X003600010009x [DOI] [PubMed] [Google Scholar]
- Bernardo R., Yu J. (2007). Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 47, 1082–1090. 10.2135/cropsci2006.11.0690 [DOI] [Google Scholar]
- Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y., Buckler E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. 10.1093/bioinformatics/btm308 [DOI] [PubMed] [Google Scholar]
- Chang F., Guo C., Sun F., Zhang J., Wang Z., Kong J., et al. (2018). Genome-wide association studies for dynamic plant height and number of nodes on the Main stem in summer sowing soybeans. Front. Plant Sci. 9, 1184. 10.3389/fpls.2018.01184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Soto R. I., Mora F., de Oliveira M. A., Higashi W., Scapim C. A., Schuster I. (2017). A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PloS One 12, e0171105. 10.1371/journal.pone.0171105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M. G., Thomas A. J. (1973). An investigation of hydrolytic techniques for the amino acid analysis of foodstuffs. J. Sci. Food Agric. 24, 1525–1540. 10.1002/jsfa.2740241208 [DOI] [PubMed] [Google Scholar]
- Dawson J. C., Endelman J. B., Heslot N., Crossa J., Poland J., Dreisigacker S., et al. (2013). The use of unbalanced historical data for genomic selection in an international wheat breeding program. Field Crops Res. 154, 12–22. 10.1016/j.fcr.2013.07.020 [DOI] [Google Scholar]
- Delauney A. J., Verma D. P. S. (1990). A soybean gene encoding Δ1-pyrroline-5-carboxylate reductase was isolated by functional complementation in Escherichia coli and is found to be osmoregulated. MGG 221, 299–305. 10.1007/BF00259392 [DOI] [PubMed] [Google Scholar]
- Deng M., Li D., Luo J., Xiao Y., Liu H., Pan Q., et al. (2017). The genetic architecture of amino acids dissection by association and linkage analysis in maize. Plant Biotechnol. J. 15, 1250–1263. 10.1111/pbi.12712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhnen A., Gras A., Teyssèdre S., Romestant M., Claustres B., Daydé J., et al. (2017). Genomic Selection for Yield and Seed Protein Content in Soybean: A Study of Breeding Program Data and Assessment of Prediction Accuracy. Crop Sci. 57, 1325–1337. 10.2135/cropsci2016.06.0496 [DOI] [Google Scholar]
- Earl D. A. (2012). Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Elshire R. J., Glaubitz J. C., Sun Q., Poland J. A., Kawamoto K., Buckler E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One 6, e19379. 10.1371/journal.pone.0019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endelman J. B. (2011). Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4, 250–255. 10.3835/plantgenome2011.08.0024 [DOI] [Google Scholar]
- Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Fallen B. D., Hatcher C. N., Allen F. L., Kopsell D. A., Saxton A. M., Chen P., et al. and (2013). Soybean seed amino acid content QTL detected using the Universal Soy Linkage Panel 1.0 with 1,536 SNPs. 10.5147/jpgs.2013.0089 [DOI]
- Fang C., Ma Y., Wu S., Liu Z., Wang Z., Yang R., et al. (2017). Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 18, 161. 10.1186/s13059-017-1289-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M., Brandon D. L. (2001). Nutritional and health benefits of soy proteins. J. Agric. Food Chem. 49, 1069–1086. 10.1021/jf0009246 [DOI] [PubMed] [Google Scholar]
- Hanson A. A., Lorenz A. J., Hesler L. S., Bhusal S. J., Bansal R., Michel A. P., et al. (2018). Genome-wide association mapping of host-plant resistance to soybean aphid. Plant Genome. 10.3835/plantgenome2018.02.0011 [DOI] [PubMed]
- Hayes B. J., Bowman P. J., Chamberlain A., Goddard M. (2009). Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 92, 433–443. 10.3168/jds.2008-1646 [DOI] [PubMed] [Google Scholar]
- He J., Zhao X., Laroche A., Lu Z.-X., Liu H., Li Z. (2014). Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5, 484. 10.3389/fpls.2014.00484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner E. L., Jannink J.-L., Iwata H., Souza E., Sorrells M. E. (2011). Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop Sci. 51, 2597–2606. 10.2135/cropsci2011.05.0253 [DOI] [Google Scholar]
- Heffner E. L., Sorrells M. E., Jannink J.-L. (2009). Genomic selection for crop improvement. Crop Sci. 49, 1–12. 10.2135/cropsci2008.08.0512 [DOI] [Google Scholar]
- Heslot N., Rutkoski J., Poland J., Jannink J.-L., Sorrells M. E. (2013). Impact of marker ascertainment bias on genomic selection accuracy and estimates of genetic diversity. PloS One 8, e74612. 10.1371/journal.pone.0074612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslot N., Yang H.-P., Sorrells M. E., Jannink J.-L. (2012). Genomic selection in plant breeding: a comparison of models. Crop Sci. 52, 146–160. 10.2135/cropsci2011.06.0297 [DOI] [Google Scholar]
- Huang X., Sang T., Zhao Q., Feng Q., Zhao Y., Li C., et al. (2010). Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961. 10.1038/ng.695 [DOI] [PubMed] [Google Scholar]
- Jarquín D., Kocak K., Posadas L., Hyma K., Jedlicka J., Graef G., et al. (2014). Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genomics 15, 740. 10.1186/1471-2164-15-740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarquin D., Specht J., Lorenz A. (2016). Prospects of genomic prediction in the USDA Soybean Germplasm Collection: Historical data creates robust models for enhancing selection of accessions. G3: Genes Genomes Genet. 6, 2329–2341. 10.1534/g3.116.031443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer R., Réale D., Coltman D., Pontier D. (2007). Detecting population structure using STRUCTURE software: effect of background linkage disequilibrium. Heredity 99, 374. 10.1038/sj.hdy.6801010 [DOI] [PubMed] [Google Scholar]
- Khandaker L., Akond M., Liu S., Kantartzi S. K., Meksem K., Bellaloui N., et al. (2015). Mapping of QTL associated with seed amino acids content in "MD96-5722" by "Spencer" RIL population of soybean using SNP markers. Food Nutr. Sci. 6, 974. 10.4236/fns.2015.611101 [DOI] [Google Scholar]
- Kisha T., Sneller C., Diers B. (1997). Relationship between genetic distance among parents and genetic variance in populations of soybean. Crop Sci. 37, 1317–1325. 10.2135/cropsci1997.0011183X003700040048x [DOI] [Google Scholar]
- Lehne B., Lewis C. M., Schlitt T. (2011). From SNPs to genes: disease association at the gene level. PloS One 6, e20133. 10.1371/journal.pone.0020133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Zhao X., Han Y., Li W., Xie F. (2019). Genome-wide association mapping for seed protein and oil contents using a large panel of soybean accessions. Genomics 111, 90–95. 10.1016/j.ygeno.2018.01.004 [DOI] [PubMed] [Google Scholar]
- Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Peng Z., Yang X., Wang W., Fu J., Wang J., et al. (2013). Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43. 10.1038/ng.2484 [DOI] [PubMed] [Google Scholar]
- Li P., Guo M., Wang C., Liu X., Zou Q. (2014). An overview of SNP interactions in genome-wide association studies. Briefings Funct. Genomics 14, 143–155. 10.1093/bfgp/elu036 [DOI] [PubMed] [Google Scholar]
- Li Y.-H., Reif J. C., Hong H.-L., Li H.-H., Liu Z.-X., Ma Y.-S., et al. (2018). Genome-wide association mapping of QTL underlying seed oil and protein contents of a diverse panel of soybean accessions. Plant Sci. 266, 95–101. 10.1016/j.plantsci.2017.04.013 [DOI] [PubMed] [Google Scholar]
- Liang H.-Z., Yu Y.-L., Wang S.-F., Yun L., Wang T.-F., Wei Y.-L., et al. (2010). QTL mapping of isoflavone, oil and protein contents in soybean (Glycine max L. Merr.). Agric. Sci. China 9, 1108–1116. 10.1016/S1671-2927(09)60197-8 [DOI] [Google Scholar]
- Lipka A. E., Lu F., Cherney J. H., Buckler E. S., Casler M. D., Costich D. E. (2014). Accelerating the switchgrass (Panicum virgatum L.) breeding cycle using genomic selection approaches. PloS One 9, e112227. 10.1371/journal.pone.0112227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka A. E., Tian F., Wang Q., Peiffer J., Li M., Bradbury P. J., et al. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399. 10.1093/bioinformatics/bts444 [DOI] [PubMed] [Google Scholar]
- Lü H., Yang Y., Li H., Liu Q., Zhang J., Yin J., et al. (2018). Genome-wide association studies of photosynthetic traits related to phosphorus efficiency in soybean. Front. Plant Sci. 9, 1226. 10.3389/fpls.2018.01584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Tomasi P., Fahlgren N., Abdel-Haleem H. (2019). Genome-wide association study (GWAS) of leaf cuticular wax components in Camelina sativa identifies genetic loci related to intracellular wax transport. BMC Plant Biol. 19, 187. 10.1186/s12870-019-1776-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey T. J., Scarf A. R., Anderson J. W. (1986). Enzyme catalysed α,β-elimination of selenocystathionine and selenocystine and their sulphur isologues by plant extracts. Phytochemistry 25, 2063–2068. 10.1016/0031-9422(86)80067-X [DOI] [Google Scholar]
- Michel S., Ametz C., Gungor H., Epure D., Grausgruber H., Löschenberger F., et al. (2016). Genomic selection across multiple breeding cycles in applied bread wheat breeding. Theor. Appl. Genet. 129, 1179–1189. 10.1007/s00122-016-2694-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. P., Ramu P., Deshpande S. P., Hash C. T., Shah T., Upadhyaya H. D., et al. (2013). Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. 110, 453–458. 10.1073/pnas.1215985110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onogi A., Ideta O., Inoshita Y., Ebana K., Yoshioka T., Yamasaki M., et al. (2015). Exploring the areas of applicability of whole-genome prediction methods for Asian rice (Oryza sativa L.). Theor. Appl. Genet. 128, 41–53. 10.1007/s00122-014-2411-y [DOI] [PubMed] [Google Scholar]
- Ott A., Liu S., Schnable J. C., Yeh C., Wang K. S., Schnable P. S. (2017). tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 1 45 (21), e178. 10.1093/nar/gkx853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panthee D., Pantalone V., Saxton A., West D., Sams C. (2006). Genomic regions associated with amino acid composition in soybean. Mol. Breed. 17, 79–89. 10.1007/s11032-005-2519-5 [DOI] [Google Scholar]
- Peng Y., Liu H., Chen J., Shi T., Zhang C., Sun D., et al. (2018). Genome-wide association studies of free amino acid levels by six multi-locus models in bread wheat. Front. Plant Sci. 9, 1196. 10.3389/fpls.2018.01196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho H.-P. (2009). Ridge regression and extensions for genomewide selection in maize. Crop Sci. 49, 1165–1176. 10.2135/cropsci2008.10.0595 [DOI] [Google Scholar]
- Poland J., Endelman J., Dawson J., Rutkoski J., Wu S., Manes Y., et al. (2012). Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 5, 103–113. 10.3835/plantgenome2012.06.0006 [DOI] [Google Scholar]
- Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Shi A., Mou B., Grusak M. A., Weng Y., Ravelombola W., et al. (2017. a). Genetic diversity and association mapping of mineral element concentrations in spinach leaves. BMC Genomics 18, 941. 10.1186/s12864-017-4297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Shi A., Xiong H., Mou B., Motes D., Lu W., et al. (2016). Population structure analysis and association mapping of seed antioxidant content in USDA cowpea (Vigna unguiculata L. Walp.) core collection using SNPs. Can. J. Plant Sci. 96, 1026–1036. 10.1139/CJPS-2016-0090 [DOI] [Google Scholar]
- Qin J., Song Q., Shi A., Li S., Zhang M., Zhang B. (2017. b). Genome-wide association mapping of resistance to Phytophthora sojae in a soybean [Glycine max (L.) Merr.] germplasm panel from maturity groups IV and V. PloS One 12, e0184613. 10.1371/journal.pone.0184613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende M. D., Resende M. F., Sansaloni C. P., Petroli C. D., Missiaggia A. A., Aguiar A. M., et al. (2012). Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 194, 116–128. 10.1111/j.1469-8137.2011.04038.x [DOI] [PubMed] [Google Scholar]
- Rutkoski J. E., Heffner E. L., Sorrells M. E. (2011). Genomic selection for durable stem rust resistance in wheat. Euphytica 179, 161–173. 10.1007/s10681-010-0301-1 [DOI] [Google Scholar]
- Sall J., Lehman A., Stephens M. L., Creighton L., (2012). JMP start statistics: a guide to statistics and data analysis using JMP. Sas Institute.
- Shi A., Buckley B., Mou B., Motes D., Morris J. B., Ma J., et al. (2016). Association analysis of cowpea bacterial blight resistance in USDA cowpea germplasm. Euphytica 208, 143–155. 10.1007/s10681-015-1610-1 [DOI] [Google Scholar]
- Shi A., Qin J., Mou B., Correll J., Weng Y., Brenner D., et al. (2017). Genetic diversity and population structure analysis of spinach by single-nucleotide polymorphisms identified through genotyping-by-sequencing. PloS One 12, e0188745. 10.1371/journal.pone.0188745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikha M., Kanika A., Rao A. R., Mallikarjuna M. G., Gupta H. S., Nepolean T. (2017). Genomic selection for drought tolerance using genome-wide SNPs in maize. Front. Plant Sci. 8, 550. 10.3389/fpls.2017.00550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simko I., Hayes R. J., Truco M.-J., Michelmore R. W., Antonise R., Massoudi M. (2018). Molecular markers reliably predict post-harvest deterioration of fresh-cut lettuce in modified atmosphere packaging. Horticulture Res. 5, 21. 10.1038/s41438-018-0022-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonah H., Bastien M., Iquira E., Tardivel A., Légaré G., Boyle B., et al. (2013). An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PloS One 8, e54603. 10.1371/journal.pone.0054603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q. J., Hyten D. L., Jia G. F., Quigley C. V., Fickus E. W., Nelson R. L., et al. (2013). Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PloS One 8, e54985. 10.1371/journal.pone.0054985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindel J., Begum H., Akdemir D., Virk P., Collard B., Redona E., et al. (2015). Genomic selection and association mapping in rice (Oryza sativa): effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PloS Genet. 11, e1004982. 10.1371/journal.pgen.1004982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Liu X., Wang J., Li M., Wang Q., Tian F., et al. (2016). GAPIT version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9, 9. 10.3835/plantgenome2015.11.0120 [DOI] [PubMed] [Google Scholar]
- Taranto F., D'agostino N., Greco B., Cardi T., Tripodi P. (2016). Genome-wide SNP discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genomics 17, 943. 10.1186/s12864-016-3297-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Technow F., Bürger A., Melchinger A. E. (2013). Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3: Genes Genomes Genet. 3, 197–203. 10.1534/g3.112.004630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuiller W., Lafourcade B., Engler R., Araújo M. B. (2009). BIOMOD-a platform for ensemble forecasting of species distributions. Ecography 32, 369–373. 10.1111/j.1600-0587.2008.05742.x [DOI] [Google Scholar]
- Tsugita A., Scheffler J. J. (1982). A rapid method for acid hydrolysis of protein with a mixture of trifluoroacetic acid and hydrochloric acid. Eur. J. Biochem. 124, 585–588. 10.1111/j.1432-1033.1982.tb06634.x [DOI] [PubMed] [Google Scholar]
- Van Warrington C. (2011). QTL mapping and optimum resource allocation for enhancing amino acid content in soybean. Athens, Georgia, USA: University of Georgia. [Google Scholar]
- Warrington C., Abdel-Haleem H., Hyten D., Cregan P., Orf J., Killam A., et al. (2015). QTL for seed protein and amino acids in the Benning×, Athens, Georgia, USA Danbaekkong soybean population. Theor. Appl. Genet. 128, 839–850. 10.1007/s00122-015-2474-4 [DOI] [PubMed] [Google Scholar]
- Xavier A., Muir W. M., Rainey K. M. (2016). Assessing predictive properties of genome-wide selection in soybeans. G3: Genes Genomes Genet. 6, 2611–2616. 10.1534/g3.116.032268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Wang Y., Li Y., Wu H., Hu B., Zheng J., et al. (2018). Genotyping of soybean cultivars with medium-density array reveals the population structure and QTNs underlying maturity and seed traits. Front. Plant Sci. 9, 610. 10.3389/fpls.2018.00610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D., Dai Z., Yang Z., Sun J., Zhao D., Yang X., et al. (2018). Genome-wide association study identifying candidate genes influencing important agronomic traits of flax (Linum usitatissimum L.) using SLAF-seq. Front. Plant Sci. 8, 2232. 10.3389/fpls.2017.02232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K., Yamamoto E., Aya K., Takeuchi H., Lo P.-C., Hu L., et al. (2016). Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927. 10.1038/ng.3596 [DOI] [PubMed] [Google Scholar]
- Zatybekov A., Abugalieva S., Didorenko S., Gerasimova Y., Sidorik I., Anuarbek S., et al. (2017). GWAS of agronomic traits in soybean collection included in breeding pool in Kazakhstan. BMC Plant Biol. 17, 179. 10.1186/s12870-017-1125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Wang X., Lu Y., Bhusal S. J., Song Q., Cregan P. B., et al. (2018). Genome-wide scan for seed composition provides insights into soybean quality improvement and the impacts of domestication and breeding. Mol. Plant 11, 460–472. 10.1016/j.molp.2017.12.016 [DOI] [PubMed] [Google Scholar]
- Zhang X., Pérez-Rodríguez P., Burgueño J., Olsen M., Buckler E., Atlin G., et al. (2017). Rapid cycling genomic selection in a multiparental tropical maize population. G3: Genes Genomes Genet. 7, 2315–2326. 10.1534/g3.117.043141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Sallam A., Gao L., Kantarski T., Poland J., Dehaan L. R., et al. (2016). Establishment and optimization of genomic selection to accelerate the domestication and improvement of intermediate wheatgrass. Plant Genome 9, 18. 10.3835/plantgenome2015.07.0059 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Liu J., Ding X., Bijma P., De Koning D.-J., Zhang Q. (2010). Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PloS One 5, e12648. 10.1371/journal.pone.0012648 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SNP data can be found in the ENA using accession number PRJEB34546 (www.ebi.ac.uk/ena/data/view/PRJEB34546).