

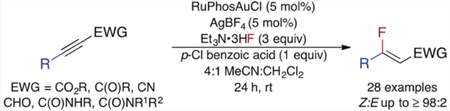
Abstract
The gold(I)-catalyzed, stereoselective hydrofluorination of electron-deficient alkynes with triethylamine trihydrogen fluoride (Et3N·3HF) is described. Fluorinated α,β-unsaturated aldehydes, amides, esters, ketones, and nitriles were isolated in moderate to good yields as single diastereomers. In all but four cases, the (Z)-vinyl fluorides were initially formed in ≥97% diastereoselectivity. This work constitutes the first catalytic example of the diastereoselective preparation of a variety of β-alkyl, β-fluoro Michael acceptors from alkynes. Additionally, the described work expands access to β-aryl, β-fluoro Michael acceptors to the synthesis of β-fluoro-α,β-unsaturated amides and nitriles. The monofluoroalkenes formed through this strategy were readily transformed into other fluorine-containing compounds, and the developed method was applied to the synthesis of a fluorinated analogue of Exoderil, a topical antimycotic.
Keywords: monofluoroalkenes, michael acceptors, hydrofluorination, gold catalysis, fluorine
Graphical Abstract
New routes toward the selective fluorination of small molecules have been targeted in recent years due to the differences in the physical and biological properties between fluorinated compounds and those of their nonfluorinated analogues.1 A fluorinated motif of particular interest is the monofluoroalkene. Monofluoroalkenes are isosteric with peptide bonds, and several bioactive compounds containing this motif have been reported.2 Although several synthetic protocols exist to access α-fluoro, α,β-unsaturated carbonyl compounds—the Horner–Wadsworth–Emmons reaction,3 the Julia Olefination,4 the Peterson Olefination,5 and the Reformatsky reaction6—the stereoselective synthesis of β-fluoro, α,β-unsaturated carbonyl compounds has proven to be a challenge, especially if β-alkyl substituents are desired.7 Previous methods to access (Z)-β-fluoro-α,β-unsaturated carbonyl compounds are limited by the formation of products with low diastereoselectivities or yields,8 the requirement for prefunctionalized starting materials,9 and narrow functional group tolerance.9b,c,10 Because of these limitations, a stereoselective and functional-group-tolerant method to access (Z)-β-alkyl, β-fluoro-α,β-unsaturated carbonyl compounds would be highly desirable.
The hydrofluorination of electron-deficient alkynes is perhaps the most direct method to generate (Z)-β-fluoro α,β-unsaturated carbonyl compounds from commercially available starting materials. Although some electron-deficient alkynes can undergo hydrofluorination in the absence of a catalyst, the diastereoselectivities of these reactions are generally moderate, especially for β-alkyl substrates.8a,b,10 Traditional chromatographic techniques often fail to separate (E) and (Z) isomers of monofluoroalkenes; therefore, it is essential that the desired monofluoroalkenes are synthesized with high diastereomeric ratios.11
Since Sadighi’s seminal report of the gold-catalyzed hydrofluorination of internal alkynes in 2007, other research groups have expanded the use of coinage metals for alkyne hydrofluorination.12 Both Jiang, with excess AgF (Scheme 1a), and Nolan, with a catalytic amount of gold (Scheme 1b), prepared β-aryl, β-fluoro-α,β-unsaturated esters or ketones from electron-deficient, unsymmetrical alkynes.12c,e However, neither procedure reported the synthesis of β-alkyl, β-fluoro Michael acceptors or utilized alternative electron-withdrawing groups such as nitriles or amides. Alternative conditions were described by Hammond and Xu for the gold-catalyzed hydrofluorination of alkynes with a new DMPU/HF fluorinating reagent, but this procedure did not expand access to (Z)-β-fluoro-α,β-unsaturated carbonyl compounds.12d The first gold-catalyzed synthesis of a β-alkyl-β-fluoro Michael acceptor was demonstrated by Hammond and Xu in 2017 (Scheme 1c).12f
Scheme 1.
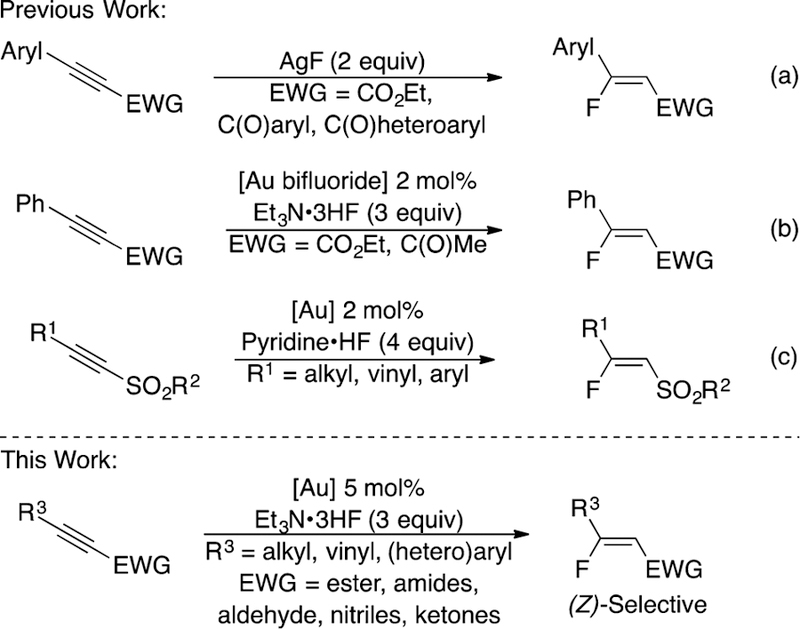
Generation of β-Fluoro Michael Acceptors from Alkynes with Coinage Metals
Although β-alkyl-β-fluorovinylsulfones could be accessed in a (Z)-selective manner, alkynes that did not bear a sulfonyl group—such as aroyl and phosphonyl—failed to undergo hydrofluorination. Despite these advances in alkyne hydrofluorination by coinage metals, a general procedure to synthesize a variety of (Z)-β-alkyl, β-fluoro Michael acceptors from electron-deficient alkynes is still an unsolved challenge.
Herein, we report a method for the preparation of a diverse array of β-alkyl, β-fluoro Michael acceptors from the gold-catalyzed hydrofluorination of electron-deficient alkynes. In addition to forming β-fluoro-α,β-unsaturated esters and ketones, this method is the first gold-catalyzed procedure to generate β-fluoro-α,β-unsaturated amides, nitriles, and aldehydes. A variety of β-alkyl as well as β-aryl substituents were tolerated; notably, 3° alkyl, alkenyl, and o-tolyl. Furthermore, we demonstrate that the monofluoroalkene products are synthetically versatile fluorinated building blocks.
The hydrofluorination of ethyl 2-butynoate (1a) with Et3N· 3HF to form ethyl (Z)-3-fluorobut-2-enoate (1b) was selected as a model reaction. Monofluoroalkene 1b formed in moderate yields and low stereoselectivities under conditions similar to those reported by Sadighi (see Table 1, entry 1).12a Reactions employing AgBF4 as the silver salt afforded alkene 1b in greater chemical yield compared to reactions conducted in the presence of other silver salts (entry 1 and 2, see the Supporting Information for further details). Upon switching from gold catalysts bearing NHC-ligands to gold catalysts bearing phosphine ligands, modest improvements in both yield and stereoselectivity were observed (entries 3 and 4). Unfortunately, reactions conducted with several triaryl or trialkyl phosphine gold(I) complexes as catalysts generated a purple hue after several hours in the presence of Et3N·3HF, which has been reported by others as a visual indication of catalyst decomposition.13
Table 1.
Effect of the Reaction Conditions on the Hydrofluorination of 1a
 | ||||
|---|---|---|---|---|
| entry | L | solvent | additive | yield [%] (Z:E)b |
| 1 | IPr | CH2CI2 | KHSO4 | 50 (66:34) |
| 2c | IPr | CH2CI2 | KHSO4 | 43 (70:30) |
| 3 | PPh3 | CH2CI2 | KHSO4 | 55 (60:40) |
| 4 | PCy3 | CH2CI2 | KHSO4 | 64 (75:25) |
| 5 | CyJohnPhos | CH2CI2 | KHSO4 | 84 (55:45) |
| 6 | RuPhos | CH2CI2 | KHSO4 | 57 (56:44) |
| 7d | RuPhos | CH2CI2 | KHSO4 | 62 (97:3) |
| 8e | RuPhos | CH2CI2 | p-CI BAe | 66 (97:3) |
| 9 | RuPhos | CH3CN | p-CI BA | 70 (97:3) |
| 10 | RuPhos | 1:4 CH2CI2:CH3CN | p-CI BA | 76 (96:4) |
| 11f | RuPhos | 1:4 CH2CI2:CH3CN | p-CI BA | 71 (96:4) |
| 12 | RuPhos | 1:4 CH2CI2:CH3CN | none | 65 (96:4) |
| 13g | RuPhos | 1:4 CH2CI2:CH3CN | p-CI BA | 80 (96:4) |
General reaction conditions: 0.2 mmol 1a, plastic vial.
Yields and Z:E ratios were determined by 19F NMR spectroscopy with 2,4-dinitrofluorobenzene as an internal standard.
5 mol % AgSbF6.
4 h.
p-chlorobenzoic acid.
10 mol % p-CI BA.
3.0 equiv Et3N·3HF.
Cationic-gold(I) complexes with dialkylbiarylphosphine ligands are known to be more stable toward decomposition pathways than cationic gold(I) complexes triaryl or trialkyl phosphines.14 Upon switching the gold catalyst to CyJohnPhosAuCl, monofluoroalkene 1b was generated in 84% yield. However, the stereoselectivity of the reaction conducted with CyJohnPhosAuCl decreased relative to the stereoselectivity of the reaction conducted with Cy3PAuCl as the catalyst (entry 3 and 4). Examination of a variety of dialkylbiaryl phosphinegold(I) complexes revealed that only reactions with RuPhos as the ligand afforded the greatest Z:E selectivity of 1b (entries 6 and 7). For instance, in the presence of CyJohnPhos the yield of 1b after 4 h was 85% but with a Z:E of 77:23.
In addition to the ligand effect on the reaction, both the solvent and additive were found to influence the yield and stereoselectivity of the hydrofluorination of alkynoate 1a. Switching from potassium bisulfate to p-chlorobenzoic acid (p-Cl BA), a more soluble acid coadditive, resulted in a modest improvement in the yield of monofluoroalkene 1b (entry 8). Reactions conducted with RuPhosAuCl and CH3CN as the solvent afforded the hydrofluorination product in a further improved yield while maintaining the Z-selectivity observed at shorter reaction times (entry 8 and 9). The change in solvent also ensured that the Z:E ratio did not decrease over time, permitting easier reaction monitoring as alkene isomerization was largely suppressed. Ultimately, reactions conducted in a solvent mixture of CH3CN:CH2Cl2 maintained the high stereoselectivity of the hydrofluorination of alkyne 1a while affording alkene 1b in an improved yield (entry 9 and 10). The beneficial improvement in the yield of 1b was observed with as little as 10 mol % p-Cl BA (entry 11 and 12). Other acid additives were examined, but benzoic acid derivatives appeared to provide an optimal pKa range (see Supporting Information, Table S4). Increasing the equivalents of Et3N·3HF did not have a significant influence on the reaction (entry 13); however, reactions with Et3N·2HF, Et3N·HF, and pyridine·HF (70% HF) failed to generate alkene 1b (See Supporting Information).
Having identified suitable reactions conditions for the hydrofluorination of alkyne 1a, we investigated the hydrofluorination of β-alkyl alkynoates, alkynones, alkynamides, and alkynenitriles (Table 2). Methyl, 1° alkyl, 2° alkyl, and vinyl β-substituted alkynoates underwent hydrofluorination in the presence of Et3N·3HF in a Z-selective manner in good yields. Notably, the final products were all isolated as a single diastereomer after standard silica gel column chromatography. Importantly, these results highlight this operationally simple, one-step route to β-alkyl, β-fluoro Michael acceptors from alkynes. The hydrofluorination reaction was also shown to be scalable, as fluoroalkenes 3b and 5b were both prepared on a gram scale in good yield and with excellent Z-selectivity. For substrate 6a with a bulky β-substituents, a higher reaction temperature was required to obtain the product in moderate yield (6b). The hydrofluorination of alkynoates bearing β-vinyl substituents provided straightforward access to fluorinated dienes 7b and 8b. Hydrofluorination of the γ,δ-alkene of either 7a or 8a was not detected by 19F NMR spectroscopy. The reaction conditions for the hydrofluorination of β-alkyl alkynoates were also suitable for the hydrofluorination of β-alkyl (hetero)aryl alkynones 9b and 10b. Although methyl ketone 11a proved to be a challenging substrate, 11b was isolated in good yield with only a trace amount of the E-isomer. Both 2° and 3° β-alkyl alkynamides (12−14a) as well as alkynonitrile derivative 15a underwent hydrofluorination to provide 12−15b in moderate yields. Dec-2-ynal was the only substrate that did not undergo hydrofluorination in a diastereoselective manner under the standard conditions in Table 2 (72%, Z:E = 51:49). However, conducting the reaction at 5 °C did afford a Z:E ratio of >98:2 and 22% yield after 24 h. Unfortunately, after 96 h at 5 °C, the yield increased to 51% but the Z:E ratio decreased to 70:30.
Table 2.
Scope of β-Alkyl, β-Fluoro Michael Acceptors
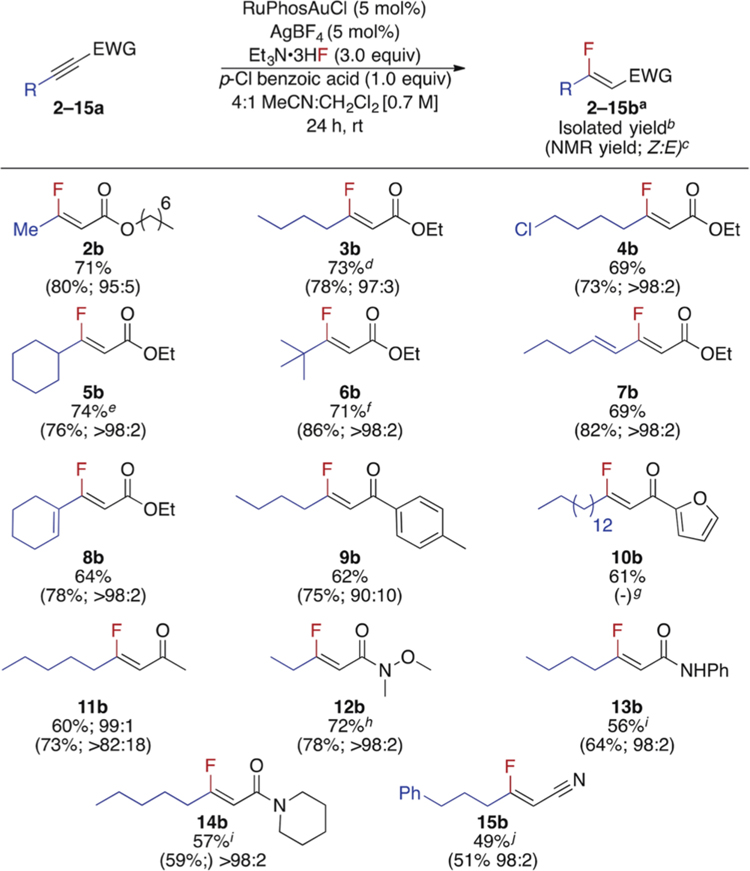 |
Standard reaction conditions: 0.5 mmol 2–15a, 3.0 equiv Et3N·3HF, 1.0 equiv p-CI BA, 5 mol % RuPhosAuCl, 5 mol % AgBF4, 4:1 MeCN:CH2CI2 [0.7M], rt, 24 h.
2-15b isolated as a single isomer except 11b.
Detemined by 19F NMR spectroscopy with PhF as an internal standard.
6.0 mmol scale.
5.0 mmol scale.
55 °C ginsoluble product.
1.25 M, 4.0 equiv Et3N·HF.
1.25 M, 4.0 equiv Et3N·3HF.
1.43 M, 4.5 equiv Et3N·3HF, 50 °C.
To showcase the generality of this method, the hydrofluorination reactions of a variety of electron deficient alkynes bearing β-aryl substituents were also explored (Table 3). Generally, the yields of β-aryl-monofluoroalkenes 16–30b were comparable to those of their β-alkyl-analogues 2–15b. In contrast to previous procedures, even a monofluoroalkene bearing an ortho-substituted aryl group (19b) was generated in modest yield.12e Compared with the esters and ketones, even the more electrophilic 2-phenylpropiolaldehyde afforded 27b in a Z-selective manner. Moreover, both β-aryl alkynonitriles and alkynamides were suitable substrates, generating otherwise difficult to access fluorinated motifs (28–30b). Finally, this methodology was found to be complementary to that reported by Hammond and Xu (See Supporting Information, Table S5)12f
Table 3.
Scope of β-aryl, β-fluoro Michael acceptors
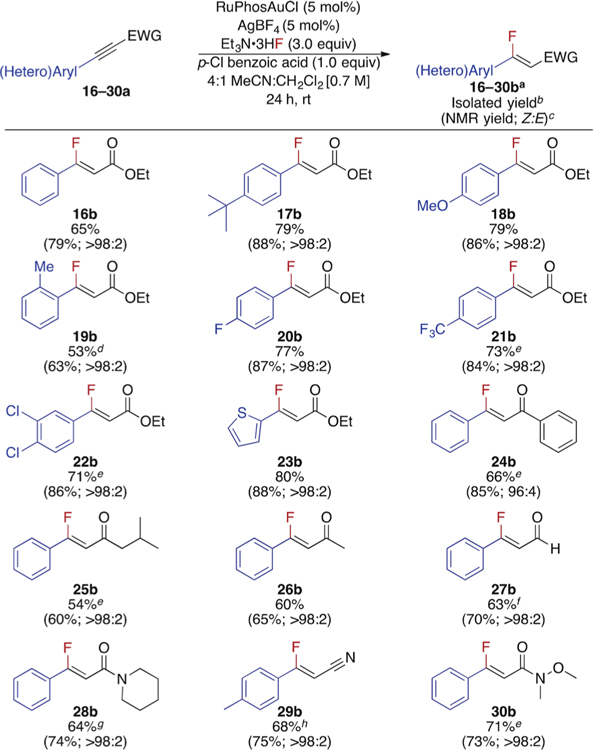 |
Standard reaction conditions: 0.5 mmol 16-30a, 3.0 equiv Et3N· 3HF, 1.0 equiv p-CI BA, 5 mol % RuPhosAuCl, 5 mol % AgBF4, 4:1 MeCN:CH2CI2 [0.7M], rt, 24 h.
16-30b isolated as a single isomer.
Determined by 19F NMR spectroscopy with PhF as an internal standard.
1.43 M, 45 °C.
45 °C.
4.5 mmol.
1.25 M, 55 °C, 4.0 equiv Et3N·3HF.
45 °C, 48 h, 4.0 equiv Et3N·3HF.
The monofluoroalkenes generated from our catalytic process underwent a series of transformations demonstrating that β-fluoro Michael acceptors are valuable fluorinated building blocks (Scheme 2). For example, ester 3b was reduced in the presence of DIBAL–H to yield the fluorinated allylic alcohol 1c in high yield.15 Aldehyde 27b underwent Wittig olefination in modest yield to afford a 1-fluoro-2,4-diene 2c.16 In the presence of a suitable 1,3-ylide, ester 5b underwent a regioselective [3 + 2] cycloaddition to generate a pyrrolidine with a quaternary fluorine center (3c).17 Finally, amide 31b was reduced in the presence of Meerwein’s salt to furnish a fluorine-containing analogue of Exoderil 4c.18
Scheme 2.
Diversification of Fluorinated Michael Acceptors
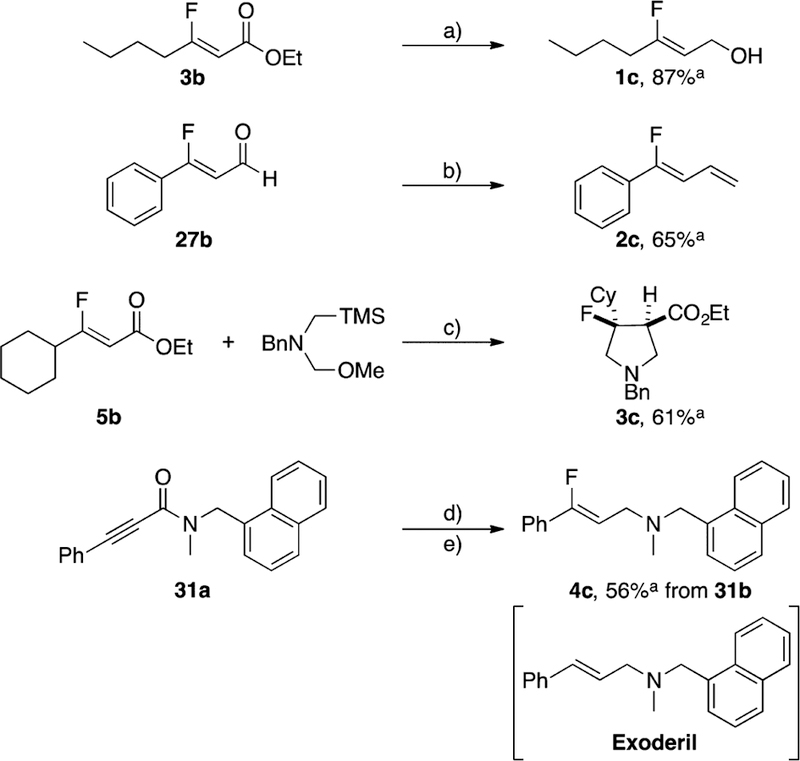
aYield given is for isolated product at specified scale. (a) 3b (2.32 mmol), DIBAL–H, CH2CI2, 0 °C. (b) nBuLi, Ph3PMeBr, 27b (0.3 mmol), THF, 0 °C. (c) 5b (0.3 mmol), ylide, TFA, 0 °C–rt. (d) 31a (1.96 mmol), 5 mol % RuPhosAuCI, 5 mol % AgBF4, 1.0 equiv p-CI BA, 4.0 equiv Et3N·3HF, 45 °C, 48 h, 4:1 MeCN:CH2CI2[1.25 M]. (e) Me3OBF4, 2,6-DI-tBu-pyridine, 31b (0.54 mmol), CH2CI2, rt; NaBH4, MeOH, −10 °C.
In conclusion, we have developed a stereoselective hydrofluorination of electron-deficient alkynes catalyzed by a RuPhos-ligated gold(I) complex. For the first time, direct access to a variety of (Z)-β-alkyl, β-fluoro Michael acceptors was achieved. In addition, (Z)-β-aryl, β-fluoro α,β-unsaturated amides and nitriles were conveniently accessed with the disclosed method. The synthetic potential of the resulting monofluoroalkene was demonstrated with various transformations of the products without the loss of the newly installed fluorine atom, and with the synthesis of a fluorinated analogue of Exoderil.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the National Institute of General Medical Sciences (R35 GM118190) for financial support of this work. T.J.O. thanks the NSF (DGE 1752814) for a predoctoral fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b01341.
Experimental details and compound characterization data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Bizet V; Besset T; Ma J-A; Cahard D Recent Progress in Asymmetric Fluorination and Trifluoromethylation Reactions. Curr. Top. Med. Chem 2014, 14, 901–940 [DOI] [PubMed] [Google Scholar]; (b) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330 [DOI] [PubMed] [Google Scholar]; (c) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369 [DOI] [PubMed] [Google Scholar]; (d) Muller K; Faeh C; Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886 [DOI] [PubMed] [Google Scholar]; (e) Yamazaki T; Taguchi T; Ojima I Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons, Ltd, Chichester, U.K., 2009; pp 3–624. [Google Scholar]
- (2).(a) Landelle G; Bergeron M; Turcotte-Savard MO; Paquin JF Synthetic Approaches to Monofluoroalkenes. Chem. Soc. Rev 2011, 40, 2867–2908. [DOI] [PubMed] [Google Scholar]; (b) Yanai H; Taguchi T Synthetic Methods for Fluorinated Olefins. Eur. J. Org. Chem 2011, 2011, 5939–5954. [Google Scholar]
- (3).Machleidt H; Wessendorf R Carbonyl-Fluorolefinierungen. Justus Liebigs Ann. Chem 1964, 674, 1–10. [Google Scholar]
- (4).Zajc B; Kake S Exceptionally Mild, High-Yield Synthesis of α-Fluoro Acrylates. Org. Lett 2006, 8, 4457–4460. [DOI] [PubMed] [Google Scholar]
- (5).Welch JT; Herbert RW The Stereoselective Construction of (Z)-3-Aryl-2-fluoroalkenoates. J. Org. Chem 1990, 55 (16), 4782–4784. [Google Scholar]
- (6).Barma DK; Kundu A; Zhang H; Mioskowski C; Falck JR (Z)-α-Haloacrylates: An Exceptionally Stereoselective Preparation via Cr(II)-Mediated Olefination of Aldehydes with Trihaloacetates. J. Am. Chem. Soc 2003, 125, 3218–3219. [DOI] [PubMed] [Google Scholar]
- (7).(a) Yanai H; Taguchi T Synthetic Methods for Fluorinated Olefins. Eur. J. Org. Chem 2011, 2011 (30), 5939–5954. [Google Scholar]; (b) Champagne PA; Desroches J; Hamel JD; Vandamme M; Paquin JF Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]; (c) Drouin M; Hamel J-D; Paquin J-F Synthesis of Monofluoroalkenes: A Leap Forward. Synthesis 2018, 50, 881–955. [Google Scholar]
- (8).(a) Albert P; Cousseau J Tetrabutylammonium and Polymer-supported Dihydrogentrifluoride: New Hydrofluorinating Reagents for Electrophilic Alkynes. J. Chem. Soc., Chem. Commun 1985, 961–962.; (b) Gorgues A; Stéphan D; Cousseau J Mono-hydrofluorination of Electrophilic Alkynes by the Liquid Biphasic CsF-H2O-DMF System (DMF = N,N-dimethylformamide). J. Chem. Soc., Chem. Commun 1989, 1493–1494.; (c) Sano K; Fukuhara T; Hara S Regioselective Synthesis of β-fluoro-α,β-unsaturated Ketones by the Reaction of β-diketones with DFMBA. J. Fluorine Chem 2009, 130, 708–713. [Google Scholar]
- (9).(a) Zhang J; Liu L; Duan J; Gu L; Chen B; Sun T; Gong, Stereoselective One-Pot Sequential Dehydrochlorination/trans-Hydrofluorination Reaction of β-Chloro-α,β-unsaturated Aldehydes or Ketones: Facile Access to (Z)-β-Fluoro-β-arylenals/β-Fluoro-β-arylenones. Adv. Synth. Catal 2017, 359, 4348–4358. [Google Scholar]; (b) Yoshida M; Kawakami K; Hara S An Efficient Stereoselective Synthesis of (E)-β-Fluoroalkenyliodonium Salts. Synthesis 2004, 2004, 2821–2824. [Google Scholar]; (c) Yoshida M; Komata A; Hara S Stereoselective Synthesis of Fluoroalkenes via (Z)-2-Fluoroalkenyliodonium Salts. Tetrahedron 2006, 62, 8636–8645. [Google Scholar]
- (10).(a) McElroy KT; Purrington ST; Bumgardner CL; Burgess JP Lack of Polymerization of Fluorinated Acrylates. J. Fluorine Chem 1999, 95, 117–120. [Google Scholar]; (b) Krishnan G; Sampson P Synthesis of β-Fluoro-α,β-unsaturated Esters and Nitriles via a Fluoro-Pummerer Rearrangement. Tetrahedron Lett 1990, 31, 5609–5612. [Google Scholar]; (c) Patrick TB; Neumann J; Tatro A Cycloaddition Reactions of ethyl (E)- and (Z)-3-fluoropropenoate. J. Fluorine Chem 2011, 132, 779–782. [Google Scholar]
- (11).(a) Zhao Y; Jiang F; Hu J Spontaneous Resolution of Julia-Kocienski Intermediates Facilitates Phase Separation to Produce Z- and E-monofluoroalkenes. J. Am. Chem. Soc 2015, 137, 5199–5203. [DOI] [PubMed] [Google Scholar]; (b) Thornbury RT; Toste FD Palladium-Catalyzed Defluorinative Coupling of 1-Aryl-2,2-Difluoroalkenes and Boronic Acids: Stereo-selective Synthesis of Monofluorostilbenes. Angew. Chem., Int. Ed 2016, 55, 11629–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sommer H; Fürstner A Stereospecific Synthesis of Fluoroalkenes by Silver-Mediated Fluorination of Functionalized Alkenylstannanes. Chem. - Eur. J 2017, 23, 558–562. [DOI] [PubMed] [Google Scholar]; (d) Hu J; Han X; Yuan Y; Shi Z Stereoselective Synthesis of Z Fluoroalkenes through Copper-Catalyzed Hydrodefluorination of gem-Difluoroalkenes with Water. Angew. Chem., Int. Ed 2017, 56, 13342–13346. [DOI] [PubMed] [Google Scholar]
- (12).(a) Akana JA; Bhattacharyya KX; Muller P; Sadighi JP Reversible C–F Bond Formation and the Au-Catalyzed Hydrofluorination of Alkynes. J. Am. Chem. Soc 2007, 129, 7736–7737. [DOI] [PubMed] [Google Scholar]; (b) Gorske BC; Mbofana CT; Miller SJ Regio- and Stereoselective Synthesis of Fluoroalkenes by Directed Au(I) Catalysis. Org. Lett 2009, 11, 4318–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li Y; Liu X; Ma D; Liu B; Jiang H Silver-Assisted Difunctionalization of Terminal Alkynes: Highly Regio- and Stereoselective Synthesis of Bromofluoroalkenes. Adv. Synth. Catal 2012, 354, 2683–2688. [Google Scholar]; (d) Okoromoba OE; Han J; Hammond GB; Xu B Designer HF-Based Fluorination Reagent: Highly Regioselective Synthesis of Fluoroalkenes and gem-Difluoromethylene Compounds from Alkynes. J. Am. Chem. Soc 2014, 136, 14381–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nahra F; Patrick SR; Bello D; Brill M; Obled A; Cordes DB; Slawin AM; O’Hagan D; Nolan SP Hydrofluorination of Alkynes Catalysed by Gold Bifluorides. ChemCatChem 2015, 7, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zeng X; Liu S; Hammond GB; Xu B Divergent Regio- and Stereoselective Gold-catalyzed Synthesis of α-Fluorosulfones and β-Fluorovinylsulfones from Alkynylsulfones. Chem. - Eur. J 2017, 23, 11977–11981 [DOI] [PubMed] [Google Scholar]; (g) For reviews of gold catalysis and fluorine:. Miro J; del Pozo C Fluorine and Gold: A Fruitful Partnership. Chem. Rev 2016, 116, 11924–11966. [DOI] [PubMed] [Google Scholar]; (h) Hopkinson MN; Gee AD; Gouverneur V Gold Catalysis and Fluorine. Isr. J. Chem 2010, 50, 675–690. [Google Scholar]
- (13).Bartolomé C; Ramiro Z; Peñas-Defrutos MN; Espinet P Some Singular Features of Gold Catalysis: Protection of Gold(I) Catalysts by Substoichiometric Agents and Associated Phenomena. ACS Catal 2016, 6, 6537–6545. [Google Scholar]
- (14).(a) Malhotra D; Hammond GB; Xu B Ligand Design in Gold Catalysis and Chemistry of Gold-Oxonium Intermediates. Top. Curr. Chem 2014, 357, 1–24. [DOI] [PubMed] [Google Scholar]; (b) Gorin DJ; Sherry BD; Toste FD Ligand Effects in Homogeneous Au Catalysis. Chem. Rev 2008, 108, 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Konno T; Ikemoto A; Ishihara T A New Entry to the Construction of a Quaternary Carbon Center having a Fluorine Atom-S(N)2’ Reaction of γ-Fluoroallylic Alcohol Derivatives with Various Cyanocuprates. Org. Biomol. Chem 2012, 10, 8154–8163. [DOI] [PubMed] [Google Scholar]
- (16).Zhu S; Guo Z; Huang Z; Jiang H Bioinspired Intramolecular Diels-Alder Reaction: A Rapid Access to the Highly-Strained Cyclopropane-Fused Polycyclic Skeleton. Chem. - Eur. J 2014, 20, 2425–2430. [DOI] [PubMed] [Google Scholar]
- (17).(a) Mason JM; Murkin AS; Li L; Schramm VL; Gainsford GJ; Skelton BW A β-fluoroamine Inhibitor of Purine Nucleoside Phosphorylase. J. Med. Chem 2008, 51, 588020135884. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McAlpine I; Tran-Dube M; Wang F; Scales S; Matthews J; Collins MR; Nair SK; Nguyen M; Bian J; Alsina LM; Sun J; Zhong J; Warmus JS; O’Neill BT Synthesis of Small 3-Fluoro- and 3,3-Difluoropyrrolidines Using Azomethine Ylide Chemistry. J. Org. Chem 2015, 80, 7266–7274. [DOI] [PubMed] [Google Scholar]
- (18).Deiters A; Chen K; Eary CT; Martin SF Biomimetic Entry to the Sarpagan Family of Indole Alkaloids: Total Synthesis of (+)-Geissoschizine and (+)-N-Μethylvellosimine. J. Am. Chem. Soc 2003, 125, 4541–4550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.