

Abstract
Application of Buchwald-Hartwig catalysis for development of biologically relevant arylspirodiamine compounds is reported. This synthetic methodology requires no inert atmosphere and affords yields up to 93% in just 20 min. Linear and sterically hindered angular spirodiamines in salt and free-base form are coupled with electron-rich and -withdrawing aryl chlorides, demonstrating a broad scope and applicability of this protocol.
Keywords: Pd cross-coupling, C-N cross-coupling, Arylation, Spirodiamine, RuPhos
Graphical Abstract
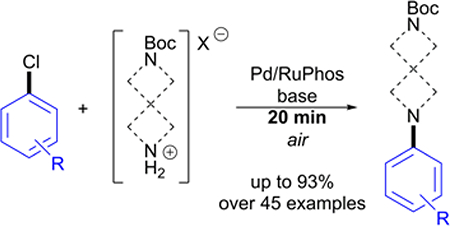
Introduction
Spirocylic scaffolds are structurally diverse compounds with broad applications throughout drug discovery,1 chiral ligand development,2 and organocatalysts for asymmetric synthesis.3 Reports containing these privileged structures have grown exponentially in the past 10 years alone due to the advantageous structural and pharmacological properties of these motiffs.4 The unique three-dimensional complexity of these compounds, measured by Fsp3 (fraction of sp3 carbon hybridization), correlates to increased physiochemical and biological properties, due to the enhanced selectivity of targeted proteins.5 Thus, spirodiamine cores, most notably arylspiroalkanes (Figure 1), are present in many biologically active compounds6 with reported antipsychotic,7 anti-insomnia,8 age-related macular degeneration (AMD),9 and anti-viral10 properties, among others.1d, 9, 11
Figure 1.

Arylspirodiamine scaffolds in compounds with pharmacological properties.
Despite the many examples of biologically active compounds containing arylspirodiamine scaffolds, reports illustrating the synthesis thereof are exceedingly rare. One attractive approach is the Hartwig-Buchwald amination, a powerful synthetic tool in C–N bond formation, thereby providing a direct approach to aryl amines.12 However, to our knowledge, the only report applying this catalysis to a broad scope of spirodiamine compounds was disclosed by Carreira and co-workers in 2008, which entailed anaerobic conditions and extended reaction times up to 46 h.13 A protocol that does not necessitate prolonged reactions times or an inert atmosphere would undoubtedly assist in furthering the development of arylspirodiamine stuctures.
Previous work by Buchwald and co-workers illustrated highly active Pd catalysts bearing biarylphosphine ligands in C–N cross-coupling of aryl halides and secondary amines.14 Inspired by these reports, we developed an aerobic piperazine arlyation protocol using precatalyst system Pd2(dba)3 and air- and moisture-stable biarylphosphine ligand RuPhos.15 This bench-top method affords excellent yields of the mono-arylated product in just 10 min and eliminates the need for an inert atmosphere and anhydrous solvents. More impressively, we found this Pd-ligand system to be highly efficient in coupling notoriously unreactive electron-rich aryl chlorides in short reaction times as well. Described herein is an extension of this methodology to include arylation of commercially available linear and angular spirodiamine salt and free-base compounds A-H (Figure 2).
Figure 2.

Spirodiamine compounds examined.
Currently, we have been examining A as a potential surrogate for piperazine due to the unique structural space the diamine can populate owing to the distinctive spirocyclic framework (Figure 3). Furthermore, pharmacokinetic properties, such as lipophilicity and metabolic stability of biologically relevant compounds can be advantageously modified by incorporation of A, along with analogous spiro[3.3]heptane structures, thus, providing an attractive alternative to piperazine, piperidines, and morpholine parent compounds.16 In order to develop a library of arylated spirodiamine A compounds in an efficient and practical time-scale, we applied modified reaction conditions from our previous Pd-catalyzed C–N cross-coupling report outlined in Scheme 1.
Figure 3.

Structural comparison of piperazine and 2,6-diazaspiro[3.3]heptane (A).
Scheme 1.

Arylation of A with aryl chloridesa
aConditions: Pd2(dba)3 (1 mol %), RuPhos (2 mol %), aryl chloride (0.5 mmol), A (0.55 mmol), NaOt-Bu (3.0 equiv), and dioxane (1.5 mL), 20 min. Isolated yields. Reaction monitored by LCMS. b2.5 equiv of NaOt-Bu was used. cPd2(dba)3 (2 mol %), RuPhos (4 mol %), 1o (1.1 mmol), A (1.0 mmol), NaOt-Bu (3.0 equiv), and dioxane (3.0 mL), 20 min.
We began our investigation by examining 1a halide derivatives to determine if the outlined one-pot reaction conditions could be expanded beyond aryl chlorides. Amination of 1a substrates proceeded smoothly in each trial run, affording comparable 2a yields to previous Pd-catalyzed arylation report in which a higher catalyst loading, along with 21 h reaction time, were required.13 Sterically congested and electron-rich 1b was efficiently coupled with A to afford 2b at 74%. Reaction conditions were tolerable for trifluoromethoxy functional group, 1c, providing a synthetically useful 44% yield. Substrates 2-chloro and 2-bromo anisole were both examined, with 2-chloroanisol providing a higher 2d product yield. Impressive C–N cross-coupling activity was also observed with extremely electron-rich 1f, yielding 64% in just 20 min.
As expected, para-substituted electron-deficient aryl chlorides provided excellent yields including NO2 substituted 1i. However, a noticeable decrease in C–N cross-coupling activity was observed with ortho-substituted electron-withdrawing functional groups. For example, only 39% of the desired product 2j was afforded when compared to the higher yields obtained with substrates containing electron-donating substituents, traditionally less reactive substrates, in the ortho position (1a-b and 1d). We then examined if 1k-l would also result in a decrease in activity after affording excellent yields with analogous para-substituted aryl chlorides 1g-h. Indeed, reaction yields dropped to 74% and 76% for 2k and 2l, respectively, demonstrating a consistent trend in diminished cross-coupling activity with aryl chlorides containing ortho-substituted electron-withdrawing groups. Nonetheless, this method provides a more efficient route to 2l, compared to the 38% yield obtained in 24 h reaction time reported by Petrukhin and co-workers.9 Arylspirodiamine chlorides 2m-o accessed using reported conditions further demonstrates the synthetic versatility of this protocol with di- and tri-chloro aryl substrates. A higher yield of 2o was achieved by increasing the Pd/Ruphos loading to 2 mol% and 4 mol%, respectively, and using 1o in slight excess.
We next investigated if conditions would be tolerable with N-aryl chlorides due to the prevalence of nitrogen heterocycles biologically active compounds (Scheme 2). The coupling of 2-chloropyrazine was met with modest activity, yielding 62% of the desired product 4a. A comparable yield of 4b was obtained at a faster rate and lower catalyst loading than those previously reported by Carreira under anaerobic conditions.13 Electron-rich and –poor pyridine chlorides were also well tolerated, as good yields were afforded for both products, 4c-d. Screening continued with bicyclic N-aryl chlorides quinolone and quinoxaline, obtaining 4e-f in excellent yields. Efficient coupling continued with benzothiazole and benzooxazxole substrates, 3g-h, compounds with known anti-cancer17 and antipsychotic properties18, respectively. We briefly examined 5-membered heterocycle rings with the chloro and bromo variants of 3i, affording respectable yields for the aminated thiazole product.
Scheme 2.

Arylation of A with N-aryl chloridesa
aConditions: Pd2(dba)3 (1 mol %), RuPhos (2 mol %), aryl chloride (0.5 mmol), A (0.55 mmol), NaOt-Bu (3.0 equiv), and dioxane (1.5 mL), 20 min. Isolated yields. Reaction monitored by LCMS. b3.0 equiv of Cs2CO3 used instead of NaOt-Bu
With reports outlining synthetic methods of spirodiamine cores increasing over the past few years,1a, 4, 16, 19 we sought to apply this protocol to other structurally diverse spirodiamine compounds. We expanded the scope to include examples of C–N cross-coupling with B-H (outlined in Figure 2) and aryl chlorides 1a, 1e, and 1g (Scheme 3). These aryl substrates were selected to examine how sterics and electronics affect catalytic C–N coupling with linear and angular spirodiamine compounds B-H.
Scheme 3.

Arylation of B-Ha
aConditions: Pd2(dba)3 (1 mol %), RuPhos (2 mol %), aryl chloride (1.0 mmol), B-H (1.1 mmol), NaOt-Bu (3.0 equiv), and dioxane (3.0 mL), 20 min. Isolated yields. Reaction monitored by LCMS.
Excellent cross-coupling activity was observed with B and 1a yielding 6a at 87%. Electron-rich 1e proved to be an unfavorable coupling partner, affording 6b at 32% yield. Reaction conditions were more tolerable with 1g, as product yield of 6i occurred at 77%. High yields were obtained with 1a and 1g when coupled with B, however only modest reactivity was again observed with 1e yielding 6e at 54%. Sluggish reactivity was observed with D and aryl substrates 1a and 1e resulting in modest yields, although product yield improved slightly with 1g. Compound E, a common core in molecular scaffolds with applications in type 2 diabetes mellitus (T2DM) and obesity,20 was coupled with 1a and 1e affording good yields of 6j-k. Decreased activity, however, resulted with substrate 1e, yielding only 40% of desired product 6l.
Catalytic activity was considerably lower with sterically hindered F, affording low yields for 6m-o. We postulated the reduced product formation of 6m-o, compared to higher yields afforded with angular spirodiamine B, could be a result of increased steric congestion illustrated in Figure 4. A decrease in bond distance is observed when comparing the bond distance between carbon (C1) in F and aryl carbon (C2) in the coupled arene ring, compared to the analogous atoms of angular spirodiamine B. This increase in steric crowding could provide an unfavorable environment for the active catalyst, resulting in the poor yields observed for 6m-o.
Figure 4.

Comparison of C–C bond distances of sipirodiamine F and B.21
Excellent reactivity was observed with G, yielding >80% for both 6p and 6q, and a respectable 67% for 6r. Reaction conditions were also tolerable for H, a common core found in biologically active compounds with application in thrombotic disease, pain and inflammation, and inhibition of GPIIb-IIIa.21
Conclusion
In summary, Pd/Ruphos catalyst system has been shown to be highly active in arylation of linear and angular spirodiamines in salt and free-base form. This extension of our previous work is a rare example of C–N bond formation that does not require an inert atmosphere or extended reaction times. Finally, reactions with activated and deactivated aryl chlorides were afforded at moderate to excellent yield at a constant catalyst loading in just 20 min.
Supplementary Material
Acknowledgments
We are grateful for the financial support of National Institute on Drug Abuse [(R01 DA29840-07 R.H.M.) and (R01 DA23957-06 R.R. Luedtke)].
Footnotes
Supplementary Material
Electronic Supplementary Information (ESI) available: Experimental procedure, NMR and mass spectral data of the isolated product (PDF). See DOI: 10.1039/x0xx00000x.
References and notes
- 1.(a) Grygorenko OO, Radchenko DS, Volochnyuk DM, Tolmachev AA and Komarov IV, Chem. Rev, 2011, 111, 5506. [DOI] [PubMed] [Google Scholar]; (b) Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, et al. , J. Med. Chem, 2005, 48, 3474. [DOI] [PubMed] [Google Scholar]; (c) Smith LK and Baxendale IR, Org. Biomol. Chem, 2015, 13, 9907. [DOI] [PubMed] [Google Scholar]; (d) Zheng Y, Tice CM and Singh SB, Bioorg. Med. Chem. Lett, 2014, 24, 3673. [DOI] [PubMed] [Google Scholar]
- 2.Ding K, Han Z and Wang Z, Chem.-Asian J, 2009, 4, 32. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ćwiek R, Niedziejko P and Kałuża Z, J. Org. Chem, 2014, 79, 1222. [DOI] [PubMed] [Google Scholar]; (b) Planas L. c., Pérard-Viret J and Royer J, Tetrahedron: Asymmetry, 2004, 15, 2399 [Google Scholar]; (c) Jiang M, Zhu S-F, Yang Y, Gong L-Z, Zhou X-G, et al. , Tetrahedron: Asymmetry, 2006, 17, 384 [Google Scholar]; (d) Sala X, García Suárez EJ, Freixa Z, Benet-Buchholz J and van Leeuwen PWNM, Eur. J. Org. Chem, 2008, 2008, 6197. [Google Scholar]
- 4.Carreira EM and Fessard TC, Chem. Rev, 2014, 114, 8257. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lovering F, MedChemComm, 2013, 4, 515 [Google Scholar]; (b) Lovering F, Bikker J and Humblet C, J. Med. Chem, 2009, 52, 6752. [DOI] [PubMed] [Google Scholar]
- 6.Marson CM, Chem. Soc. Rev, 2011, 40, 5514. [DOI] [PubMed] [Google Scholar]
- 7.Zheng LT, Hwang J, Ock J, Lee MG, Lee W-H, et al. , J. Neurochem, 2008, 107, 1225. [DOI] [PubMed] [Google Scholar]
- 8.Betschart C, Hintermann S, Behnke D, Cotesta S, Fendt M, et al. , J. Med. Chem, 2013, 56, 7590. [DOI] [PubMed] [Google Scholar]
- 9.Cioffi CL, Dobri N, Freeman EE, Conlon MP, Chen P, et al. , J. Med. Chem, 2014, 57, 7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong H, Foulk M, Aschenbrenner L, Fan J, Tiong-Yip C-L, et al. , Bioorg. Med. Chem. Lett, 2013, 23, 6789. [DOI] [PubMed] [Google Scholar]
- 11.Gadekar PK, Roychowdhury A, Kharkar PS, Khedkar VM, Arkile M, et al. , Eur. J. Med. Chem, 2016, 122, 475. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yang BH and Buchwald SL, J. Organomet. Chem, 1999, 576, 125 [Google Scholar]; (b) Wolfe JP, Wagaw S, Marcoux J-F and Buchwald SL, Acc. Chem. Res, 1998, 31, 805 [Google Scholar]; (c) Hartwig JF, Angew. Chem., Int. Ed, 1998, 37, 2046. [DOI] [PubMed] [Google Scholar]; (d) Surry DS and Buchwald SL, Angew. Chem., Int. Ed, 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Buchwald SL, Mauger C, Mignani G and Scholz U, Adv. Synth. Catal, 2006, 348, 23 [Google Scholar]; (f) Johansson Seechurn CCC, DeAngelis A and Colacot TJ, New Trends in Cross-Coupling: Theory and Applications, The Royal Society of Chemistry, 2015. [Google Scholar]
- 13.Burkhard J and Carreira EM, Org. Lett, 2008, 10, 3525. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fors BP and Buchwald SL, J. Am. Chem. Soc, 2010, 132, 15914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fors BP, Davis NR and Buchwald SL, J. Am. Chem. Soc, 2009, 131, 5766. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Biscoe MR, Fors BP and Buchwald SL, J. Am. Chem. Soc, 2008, 130, 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reilly SW and Mach RH, Org. Lett, 2016, 18, 5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Burkhard JA, Wagner B, Fischer H, Schuler F, Müller K, et al. , Angew. Chem., Int. Ed, 2010, 49, 3524. [DOI] [PubMed] [Google Scholar]; (b) Burkhard JA, Guérot C, Knust H and Carreira EM, Org. Lett, 2012, 14, 66. [DOI] [PubMed] [Google Scholar]; (c) Burkhard JA, Guérot C, Knust H, Rogers-Evans M and Carreira EM, Org. Lett, 2010, 12, 1944. [DOI] [PubMed] [Google Scholar]
- 17.(a) Enise Ece G, Ebru B, Irem D, Rengul C-A and Mine Y, Anti-Cancer Agents Med. Chem, 2015, 15, 382 [Google Scholar]; (b) Gurdal EE, Durmaz I, Cetin-Atalay R and Yarim M, J. Enzyme Inhib. Med. Chem, 2015, 30, 649. [DOI] [PubMed] [Google Scholar]
- 18.Huang L, Zhang W, Zhang X, Yin L, Chen B, et al. , Bioorg. Med. Chem. Lett, 2015, 25, 5299. [DOI] [PubMed] [Google Scholar]
- 19.(a) Smith AC, Cabral S, Kung DW, Rose CR, Southers JA, et al. , J. Org. Chem, 2016, 81, 3509. [DOI] [PubMed] [Google Scholar]; (b) Weinberg K, Stoit A, Kruse CG, Haddow MF and Gallagher T, Tetrahedron, 2013, 69, 4694 [Google Scholar]; (c) Orain D, Hintermann S, Pudelko M, Carballa D and Jedrzejczak A, Synlett, 2015, 26, 1815. [Google Scholar]
- 20.(a) Bhattacharya SK, Andrews K, Beveridge R, Cameron KO, Chen C, et al. , ACS Med. Chem. Lett, 2014, 5, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McClure KF, Jackson M, Cameron KO, Kung DW, Perry DA, et al. , Bioorg. Med. Chem. Lett, 2013, 23, 5410. [DOI] [PubMed] [Google Scholar]; (c) Kung DW, Coffey SB, Jones RM, Cabral S, Jiao W, et al. , Bioorg. Med. Chem. Lett, 2012, 22, 4281. [DOI] [PubMed] [Google Scholar]
- 21.(a) Mehrotra MM, Heath JA, Rose JW, Smyth MS, Seroogy J, et al. , Bioorg. Med. Chem. Lett, 2002, 12, 1103. [DOI] [PubMed] [Google Scholar]; (b) Smyth MS, Rose J, Mehrotra MM, Heath J, Ruhter G, et al. , Bioorg. Med. Chem. Lett, 2001, 11, 1289. [DOI] [PubMed] [Google Scholar]; (c) Pandey A, Seroogy J, Volkots D, Smyth MS, Rose J, et al. , Bioorg. Med. Chem. Lett, 2001, 11, 1293. [DOI] [PubMed] [Google Scholar]; (d) Hawkinson JE, Szoke BG, Garofalo AW, Hom DS, Zhang H, et al. , J. Pharmacol. Exp. Ther, 2007, 322, 619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.