

Abstract
Acyl phosphates of ATP (ATPAc) and related nucleotides have proven to be useful for the interrogation of known nucleotide binding sites via specific acylation of conserved lysines (K). In addition, occasional K acylations are identified in proteins without such known sites. Here we present a robust and specific acylation of procaspase-6 by ATPAc at K133 in Jurkat cell lysates. The K133 acylation is dependent on π–π stacking interactions between the adenine moiety of ATPAc and a conserved Y198–Y198 site formed at the homodimeric interface of procaspase-6. Significantly, the Y198A mutation in procaspase-6 abolishes K133 acylation but has no effect on the proteolytic activity of the mature, active caspase-6 Y198A variant. Additional in vitro studies show that ATP can inhibit the autoproteolytic activation of procaspase-6. These observations suggest that ATP, and possibly other nucleotides, may serve as the endogenous ligands for the allosteric site at the procaspase-6 dimer interface, a site that has persisted in its “orphan” status for more than a decade.
Graphical Abstract
Chemoproteomic analysis of nucleotide binding sites using acyl phosphates of nucleotide di- and triphosphates has become a broadly accepted method for identification of activesite lysines and profiling of inhibitors that compete with binding of the nucleotide probe (Figure 1A).1–3 The ATPAc and ADPAc probes provide broad coverage of protein kinases and other ATPase families, such as the heat shock proteins (HSP90s and HSP70s) and the AAA+ families.4 Incorporation of biotin, or an analogue thereof, into the acyl side chain of the probe permits selective capture of peptides containing acylated lysines. Exposure of cellular lysates to the nucleotide acyl phosphate probe causes transfer of the acyl moiety to the ε-amino group of closely positioned lysines within nucleotide binding sites. Weaker, second-order acylations of surface lysines rarely interfere in the analysis due to the “wash out” effect over many thousands of random peptides derived from the bulk of the proteome. The reaction is complete in 5–10 min and results in a covalent ε-amide on the modified lysine residues that is stable to enzymatic proteolysis. Following proteolysis, biotinylated peptides are captured and enriched on streptavidin beads and subjected to mass spectrometry analysis for identification and relative quantitation. Hundreds of ATP binding proteins can be simultaneously analyzed from a single proteomic sample based on detection of probe-labeled peptides (Figure S1).1–3
Figure 1.

Chemoproteomic identification of the acylated K133 peptide from procaspase-6. (a) ATPAc (Ac = desthiobiotinyl) along with the corresponding ADPAc, GTPAc, and GDPAc probes used in this study. Desthiobiotin shows superior properties for capture by and release from streptavidin beads compared to those of biotin on our chemoproteomic platform. For a full description of the platform, see Methods and Figure S1.(b) Identification of the acylated K133 peptide by LC–MS/MS spectra from a proteomic lysate of Jurkat cells treated with ATPAc. Peaks including the site of acylation (asterisk) are matched in agreement with a high cross-correlation score (Xcorr > 5.0). Translation of amino acid sequence using genomic database sequences confirmed the peptide was derived from CASP6. (c) Relative amounts of streptavidin-captured, acylated K133 peptide determined by LC–MS/MS using four related acyl phosphate probes. Percents are based on ATPAc capture defined as 100% under these experimental conditions.
For protein kinases, which contain highly conserved activesite lysines, robust acylation is clearly observed and more than 200 protein kinases are routinely identified from a single proteomic sample. In addition, acylation of lysines not directly in ATP binding sites has been observed in some kinases and HSP90s. For example, probe labeling of HSP90s has been shown to result from protein conformations where apparently distal lysines are brought into the proximity of the ATP binding site.4 In another example, acylation of p38α at K15, external to the ATP binding site, occurs only in the presence of MAPKAPK-2 and/or MAPKAPK-3, known binding partners of p38α.5 Protein–protein interactions orient K15 from p38α into the ATP binding site of the respective MAPKAPK binding partner, resulting in transacylation of K15. With few exceptions, nucleotide acyl phosphates affect acylation of lysines in a proximity-driven manner that requires nucleotide binding of the probe for efficient delivery of the acyl group. In this work, we present an example of an acylated lysine in procaspase-6, a protein not previously known to bind nucleotides.
MATERIALS AND METHODS
ATPAc-Labeled Sample Preparation.
Cell lysates were prepared by sonicating cell pellets in lysis buffer [50 mM HEPES (pH 7.5), 150 mM NaCl, 0.1% Triton X-100, and phosphatase inhibitors (Cocktail II, AG Scientific, catalog no. P-1518)]. Lysates were cleared by centrifugation, and MnCl2 was added to a final concentration of 20 mM. For small molecule studies, 5 μL of 100× compound or dimethyl sulfoxide (DMSO) was added to 445 μL of lysate; all samples were prepared in duplicate. Small molecules were incubated for 15 min prior to addition of 50 μL of a 10× desthiobiotin-nucleotide-acyl phosphate probe (ADP or ATP probe) for a final probe concentration of 20 μM. Probe labeling reactions were allowed to proceed for 15 min prior to further processing.
Targeted Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS) Analysis.
Samples were prepared for MS analysis as described previously6 with the only exception that Asp-N was used, in place of trypsin, to cleave intact proteins into the corresponding peptide complement. Briefly, probe-labeled lysates were denatured and reduced [6 M urea and 10 mM dithiothreitol (DTT) at 65 °C for 15 min], alkylated (40 mM iodoacetamide at 37 °C for 15 min), and gel filtered (Bio-Rad Econo-Pac 10G) into 2 M urea, 5 mM methionine, and 1 M Tris. The desalted protein mixture was digested with Asp-N (1 μg/mL) for 24 h at 37 °C, and desthiobiotinylated peptides were captured using 12.5 μL of high-capacity streptavidin resin (Thermo Scientific). Bead-bound peptides were washed extensively, and probe-labeled peptides were eluted with two 35 μL washes of a 50% CH3CN/water mixture with 0.1% trifluoroacetic acid.
Whole Protein–Western Blot Analysis.
Probe labeling of intact caspase-6 was evaluated using a Western blot-coupled probe-based assay, as described previously.7 After the probe labeling reaction, samples were denatured, reduced, alkylated, and gel filtered as described above. However, samples were directly incubated with streptavidin beads to capture probe-labeled, intact proteins (with overnight incubation). Samples were washed extensively with phosphate-buffered saline to remove nonspecifically bound proteins. Probe-labeled proteins were then eluted with two 50 μL washes of reducing gel sample buffer, separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and detected by Western blotting for the target of interest.
Western Blotting.
Proteins were transferred to polyvinylidene fluoride membranes after SDS–PAGE using a Bio-Rad trans-blot turbo system. Membranes were blocked with 5% nonfat dry milk in TBS and 0.1% Tween for 1 h prior to incubation with appropriate caspase-6 antibodies (Cell Signaling, catalog nos. 9761 and 9762). Primary antibodies were utilized at dilutions of 1:1000. Secondary antibodies obtained from LiCor were used at dilutions of 1:20000 (IRDye IgG antibodies, catalog nos. 5470S, 5151S, 5257S, and 5366S). Images were both collected and analyzed using a LiCor Odyssey classic imaging system.
LC–MS/MS.
Samples were analyzed by LC–MS/MS as described previously.1,6,8 Agilent 1100 series micro-HPLC systems with custom packed C18 separation columns were coupled to Thermo LTQ ion trap mass spectrometers. Data were collected using custom target lists including the m/z value for the probe-labeled procaspase-6 peptide.1,6,8 Extracted ion chromatograms were generated using MS2 fragments resulting from the probe-labeled caspase-6 peptide, and integrated peak areas were used to determine the relative signal between treated samples and controls.
Generation of Caspase Variants.
The full-length wild-type (FL WT) caspase-6 used in this study was derived from a synthetic, Escherichia coli codon-optimized (His)6 C-terminally tagged caspase-6 gene (Celtek Bioscience) that was ligated into the NdeI/BamHI sites of the pET11a vector. Caspase-6 variants (C163S, Y198A, and C163S/Y198A) as well as the N-terminally (His)6 tagged FL caspase-6 were generated using Phusion site-directed mutagenesis (Thermo Scientific) in the FL WT caspase-6 construct. Fully cleaved and active caspase-6 was also used in the form of a constitutive two chain (CT), which was designed to independently express the large and small subunits of caspase-6 with the prodomain (residues 1–23) and linker (residues 180–193) removed.9
Caspase Protein Expression and Purification.
All caspase-6 constructs used in this study were transformed into the BL21(DE3) T7 express strain of E. coli (NEB). Overnight seed cultures were initially grown in 2×YT medium supplemented with 0.1 mg/mL ampicillin (Sigma) at 37 °C. Dense cultures were then diluted 1000-fold with 2×YT medium containing 0.1 mg/mL ampicillin at 37 °C until A600 reached 0.6. For the following caspase-6 variants [FL procaspase-6 C163S (active-site knockout), FL procaspase-6 C163S/Y198A, and FL Y198A], protein expression was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyr-anoside (IPTG) (GoldBio) at 20 °C for 18 h. FL N-terminally (His)6-tagged caspase-6 was induced by addition of 1 mM IPTG (GoldBio) at 37 °C for 10 min. Cells were centrifuged at 4700g for 10 min at 4 °C and stored at −80 °C until they were used. Frozen and thawed cells were lysed using a microfluidizer (Microfluidics, Inc.) in ice-cold lysis buffer [50 mM Tris (pH 8.5), 300 mM NaCl, 5% glycerol, and 50 mM imidazole] and centrifuged at 30600g for 1 h at 4 °C. The supernatant was loaded into a 5 mL HiTrap nickel-affinity column (GE Healthcare) and washed with lysis buffer until the absorbance returned to baseline. The protein was eluted with elution buffer [50 mM Tris (pH 8.5), 300 mM NaCl, 5% glycerol, and 250 mM imidazole] and diluted 5-fold with buffer A [20 mM Tris (pH 8.5) and 2 mM DTT] to reduce the salt concentration. This protein sample was then loaded into a 5 mL HiTrap Q HP column (GE Healthcare). The column was developed with a linear NaCl gradient, and the protein was eluted in 20 mM Tris (pH 8.5), 200 mM NaCl, and 2 mM DTT. This eluted protein was stored at −80 °C until it was used. The purified caspases were analyzed by SDS–PAGE to confirm their identity and purity.
Caspase Activity Assays.
To measure caspase activity, 100 nM purified caspase-6 variants were assayed over 7 min at 37 °C in caspase-6 activity assay buffer (100 mM HEPES, 120 mM NaCl, 0.1% CHAPS, 10% sucrose, and 5 mM DTT). For substrate titration, a range of 0–500 μM fluorogenic substrate VEID-AMC [N-acetyl-Val-Glu-Ile-Asp-(7-amino-4-methyl-coumarin), Enzo Life Sciences Inc.] was used for caspase-6. Fluorescence kinetic measurements (λex of 365 nm and λem of 495 nm) were performed in three independent trials on three different days in 100 μL reaction mixtures in a 96-well format using a microplate reader (SpectraMax M5, Molecular Devices). Initial velocities versus substrate concentration were fit to a rectangular hyperbola using GraphPad Prism (GraphPad Software, San Diego, CA) to determine the kinetic parameters Km and kcat. Enzyme concentrations were determined by active-site titration with the quantitative covalent inhibitor VEID-CHO (N-acetyl-Val-Glu-Ile-Asp-aldehyde; Enzo Life Sciences Inc.) for caspase-6. Protein was added to inhibitor solvated in DMSO in 96-well V-bottom plates at room temperature for 1.5 h in caspase activity assay buffer. Aliquots (90 μL) were transferred in duplicate to black-well plates and assayed with a 50-fold molar excess of the substrate. The protein concentration was determined to be the lowest concentration at which full inhibition was observed and was thus used to calculate kcat.
Effect of ATP on the Activity of Cleaved, Active Caspase-6.
Cleaved and active caspase-6 ΔN D179CT (20 nM) was incubated with increasing concentrations of ATP (Fisher Scientific, catalog no. S25123) (0–40 mM, 1.5-fold dilution) in caspase-6 activity assay buffer [100 mM HEPES (pH 7.5), 10% sucrose, 0.1% CHAPS, 120 mM NaCl, and 5 mM DTT] at 37 °C for 1.5 h. The VEID-AMC fluorogenic substrate (Enzo Life Sciences Inc.) was then added to a final concentration of 60 μM. The final volume was 30 μL in a 384-well black flat-bottom plate (BD Falcon). Fluorescence kinetic measurements (λex of 365 nm and λem of 495 nm) were performed over 7 min, and the initial velocities were calculated. The data are presented as the percent activity of caspase-6 setting the buffer-only control as 100%. The data represent an average of two independent experiments (each run in duplicate), and the reported errors represent the standard error of the mean.
Effect of ATP on the Self-Activation of Procaspase-6.
The catalytically competent full-length version of procaspase-6, Caspase-6 FL N-terminally (His)6 (3 μM), was incubated with varying concentrations of ATP (0–60 mM) in caspase-6 assay buffer [100 mM HEPES (pH 7.5), 10% sucrose, 0.1% CHAPS, 120 mM NaCl, and 5 mM DTT] at 37 °C for 6 h. SDS loading buffer was added to the sample, and the sample was boiled for 10 min and analyzed by 16% SDS–PAGE. The gels were imaged using the ChemiDocTM MP imaging system (Bio-Rad), and band intensities were quantified using Image Lab software (Bio-Rad). The intensities of full-length procaspase-6 was set at 100%, and the extent of cleavage with varying ATP concentration was reported as the percent procaspase-6 remaining. The IC50 values were determined using a nonlinear curve-fitting algorithm (four-parameter equation) in GraphPad Prism (GraphPad Software). A time point experiment (0, 0.25, 1, 2, 4, 6, and 8 h) was also performed to assess the extent of procaspase-6 activation in the presence or absence of 20 mM ATP.
Molecular Modeling.
All computational procedures were carried out using Schrödinger’s Small-Molecule Drug Discovery suite (version 2016–1, Schrodinger, LLC, New York, NY), using the following applications: Maestro, Protein Preparation Wizard, Epik, Glide, Prime, and Desmond. An all-atom model for docking and molecular dynamics studies was generated using a crystal structure of procaspase-6 liganded at the homodimeric interface [Protein Data Bank (PDB) entry 4N6G] via a sequence of protonation state assignment (Epik), missing atom/loop reconstitution (Prime, OPLS3 force field), and constrained minimization (Prime, OPLS3 force field) within Maestro’s Protein Preparation Wizard tool. Docking (Glide) was initiated by centering the interfacial cavity into a 36 Å × 36 Å × 36 Å grid box and implementing a high-precision (XP) algorithm without any constraints with three-dimensional models of nucleotide conjugates in physiologically relevant protonation states. The resulting structures were further refined through restrained minimization (Prime, OPLS3 force field). The best scoring models served as starting points for molecular dynamics (MD) simulations, which were set up as NPT ensembles using neutralized systems (Na+ ions) with explicit water (SPC solvent model)) within a 10 Å buffer box and 200 mM NaCl or MgCl2 buffer salts (Desmond). The resulting ensembles were then subjected to unconstrained 1.2 ns MD simulations at 300 K and 1 atm. The quality of the simulation and the validity of the complex were evaluated by the root-mean-square deviation of the interacting proteins throughout the simulation. The resulting trajectories were analyzed using the Simulation Interaction Diagram tool within Desmond to reveal the relative persistence of molecular contacts.
RESULTS
Identification of Procaspase-6 as an ATP Binder in a Native Screen of Human Cell Lysates.
During proteomic analyses of Jurkat cell lysates with the ATPAc probe, we were struck by the robust acylation of a 15-amino acid peptide identified by LC–MS/MS after Asp-N proteolysis as DAK*IE-IQTLTGLFKG (the asterisk indicates the site of acylation), corresponding to amino acids 131–145 from the CASP6 gene (Figure 1B). Acylation of this peptide at K133 was inhibited by addition of ATP to the cellular lysate, suggesting that the acylation was not due to the second-order reaction of a surface lysine but rather was dependent on the ATP moiety in the probe.
Two other probe-related observations were intriguing. GTPAc was as effective as ATPAc in K133 acylation, and both ADPAc and GDPAc were significantly less effective in the acylation of K133 than the corresponding triphosphate probes (Figure 1C).
The immediate protein biosynthesized from the gene, CASP6, is procaspase-6, the inactive dimeric zymogen of active caspase-6. This homodimer is composed of a pro-domain (residues 1–23), a large subunit (residues 24–179), an intersubunit linker (residues 180–193), and a small subunit (residues 194–293) for each monomer. The zymogen dimer requires activation by cleavage of the pro-domain at D23 and of a linker region at D179–D193, yielding a dimer of heterodimers that constitutes active caspase-6. This maturation of procaspase-6, removal of the active prodomain and the intersubunit linker, results in structural–dynamic changes.9,10 Because both procaspase-6 and active caspase-6 contain amino acids 131–145, the zymogen/enzyme selectivity of ATPAc for acylation of K133 remained to be determined. Moreover, the mechanism and specificity for both ATPAc recognition and acylation were unresolved because no ATP binding site had been reported for CASP6. With these questions in hand, we investigated further.
Procaspase-6 Is the Target of K133 Acylation.
To determine whether procaspase-6 and/or active caspase-6 generates the observed probe-labeled peptide, we evaluated cellular lysates from resting cells and apoptotic cells induced by anti-Fas antibody 4C3.11 Nearly complete apoptosis-induced cleavage of procaspase-6 to active caspase-6 was confirmed by immunoblotting (Figure 2A). When these samples were analyzed for K133 acylation, the amount was proportional to the amount of procaspase-6 in the lysates (Figure 2B), suggesting that ATPAc exclusively recognizes procaspase-6 for acylation of K133.
Figure 2.

(a and b) ATPAc labeling of resting and apoptotic Jurkat cell lysates and (c) inhibition of labeling in resting cells by addition of nucleotides. (a) Protein immunoblots of procaspase-6 and caspase-6 in lysates from resting and apoptotic Jurkat cells (induced by anti-FAS antibody 4C3). (b) Both lysates were treated with ATPAc and subjected to our chemoproteomic platform, and the acylated K133 peptides were identified and quantified. The resting lysate containing predominantly procaspase-6 was defined as 100% acylation. (c) Lysates from resting Jurkat cells were individually pretreated (15 min.) with the stated nucleotides (5 mM) and then subjected to the described procedures to quantify the inhibition of formation of the acylated K133 peptide.
Inhibition of K133 Acylation by Nucleotides.
Addition of exogenous nucleotides to cellular lysates has been shown to inhibit ATPAc acylation of active-site lysines in a variety of protein kinases and ATPases and serves as a corroborating test for binding specificity.1,6,12 Thus, several nucleotides were tested for their inhibitory effect on the K133 acylation of procaspase-6 in Jurkat lysates (Figure 2C). As expected, ATP and GTP inhibited K133 acylation by the corresponding acyl phosphates. The IC50 for inhibition by ATP was determined by Western blotting and by LC–MS/MS. The results of both procedures were similar and in the range of 1–3 mM in cell lysates. Both ADP and GDP showed similar IC50 values for inhibition of K133 acylation, suggesting that the corresponding ADPAc and GDPAc probes should be capable of binding to procaspase-6. Thus, the reduced level of acylation observed with diphosphate probes (Figure 1C) is possibly due to the shortened “reach” of the terminal acyl β-phosphate compared to the corresponding terminal acyl γ-phosphate of the triphosphate probes, preventing efficient transfer of an acyl group to K133. The observation that multiple nucleotides inhibited transfer of an acyl group to K133 with roughly similar potency suggested a relative lack of nucleic acid base specificity in the binding interaction (Figure 2C). In a related immunoblot experiment, dATP showed no significant inhibition of probe acylation at ≤100 mM, suggesting a role for the 2′-OH in ATP binding (Figure S2A). In addition, the enhancement of acylation by divalent metal ions like Mn2+, seen in protein kinases, was also observed for K133 acylation (Figure S2B).
A Role for Y198 in Nucleotide Binding.
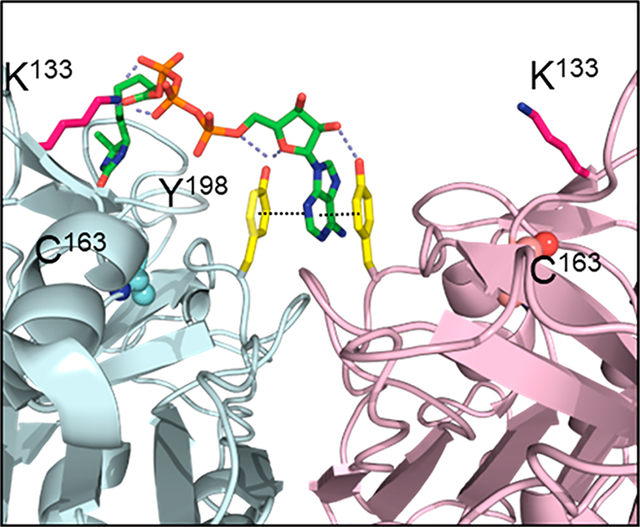
The recent identification of a small molecule binding site at the procaspase-6 dimer interface provided important clues for elucidating the mechanism of K133 acylation by ATPAc.13 Crystallographic studies determined that a hydrophobic crevice extends along the dimer interface of procaspase-6 (Figure 3A), similar to those observed in the other executioner caspase-3 and −7.14–18 Upon closer inspection of the dimer interface, the K133 pair is found to be symmetrically positioned near the edge of the crevice, close (~14 Å) to a π–π stacking motif formed by the Y198 pair at the hydrophobic interface (Figure 3A, inset).
Figure 3.

Structural models of the ATP binding site at the dimer interface of procaspase-6. (a) Structure of the procaspase-6 dimer highlighting the hydrophobic cavity (yellow) at the dimer interface. This cavity is observed in the executioner caspase-3, −6, and −712,13 as well as caspase-1.11,12 Tyrosine 198 (yellow sticks) has been identified as a site that is critical for ATP binding (inset). (b) The adenine ring in the acyl phosphate probe (green sticks) is predicted to bind between the two Y198 residues at the dimer interface. This allows covalent modification of K133, which is located at the edge of the cavity ~18 Å from the adenine-Y198 pair. (c) The crystal structure of compound 3 (brown sticks), identified by SPR as a caspase-6 inhibitor, can be similarly observed sandwiched between the Y198 (yellow) pair of the procaspase-6 dimer.13 (d) The modeled interaction between the acyl phosphate probe and procaspase-6 shows excellent shape complementarity.
The Y198 pair in procaspase-6 was identified recently as the binding site for aromatic heterocyclic compounds using surface plasmon resonance (SPR).13 X-ray crystallography established that compound 3 (KD = 1.8 μM), an isoquinolinone with an exoamino group (3-aminoisoquinolin-1-ol), was sandwiched between both faces by π–π interactions with the Y198 pair (Figure 3B). Because the Y198 stack is disrupted by structural changes in the active caspase-6, structural analogues of compound 3 were selective for the zymogen and were proposed to be allosteric inhibitors of caspase-6 activation (Figure S3). To date, no endogenous allosteric molecules that access the Y198 pair have been identified.
Molecular Modeling.
The position of the K133 pair relative to the Y198 stack suggested that the stacking-capable, electron-deficient adenine ring of ATPAc could bind in the Y198 interface forming a π–π interaction sandwich similar to that reported with compound 3.13 Extension of the triphosphate linker could favorably position the acyl phosphate moiety in the proximity of the ε-amino group of K133 for efficient acyl transfer (Figure 3C). In fact, the fully extended form of ATP presents the acyl carbonyl up to 14 Å from the anomeric carbon (C-1′) of the pentose, which matches well the distance between the α-carbons of Y198 and K133.
Docking studies of the acyl phosphate probes using the crystal structure of procaspase-6 in a complex with compound 3 (PDB entry 4N6G) supported this hypothesis and provided an explanation for the apparent preference for ATP over ADP in the acyl phosphate reaction. In the optimal docking arrangements, the adenine heterocycle forms a π–π–π sandwich with the Y198 pair that is stabilized by H-bonding between the proximal (to the acyl group) Y198 hydroxyl and the endocyclic oxygen of the ribose ring. Importantly, H-bonding is also predicted to occur between the distal Y198 hydroxyl and the 2′-OH of ribose (Figure 3C, inset). The absence of this contact in dATP could explain its significantly weaker binding to the Y198 pair. This arrangement also features stacking of both Y198 hydroxyphenyls with the pyrimidine portion of the purine system. The phosphates projecting from the ribose are guided via a network of H-bond donors (N125, G124, and H126) to project the acyl group toward K133. The appended desthiobiotin moiety is predicted to make several favorable hydrophobic and H-bonding contacts with a solvent-excluding cavity, formed by a combination of functionally diverse residues (Y128, E123, R164, and G165) ~18 Å from the adenine-Y198 pair (Figures S4–S6). The remote position of this secondary subsite requires a relatively extended conformation from the probe, which cannot be attained by the ADPAc probe. Regardless of the desthiobiotin accommodation, the flexible ammonium from K133 can form solvent-exposed H-bonds with the carbonyl oxygen of the acyl phosphate in both ATP- and ADP-derived probes, preventing a conclusive accounting of experimentally observed trends via analysis of the static docking models.
Relatively short (1.2 ns) molecular dynamics (MD) simulations of the ATPAc and ADPAc complexes provided a compelling rationale for their distinct reactivities (Figure S7 and S8). The simulations supported the persistent engagement by the surface cavity (E123, Y128, R164, and G165) of the desthiobiotin fragment within the ATP-derived probe. No such persistent contacts were seen for the ADP-desthiobiotin probe. The MD runs also confirmed the persistence of the H-bonding arrangement between at least one of the Y198 hydroxyls and the ribose ring. Importantly, only in the case of ATPAc is the acyl phosphate group positioned for a substantial fraction of the simulation period (19%) in an H-bonding arrangement with the carbonyl of the acyl group and a bond rotation away from the required reactive arrangement, commonly associated with the Burgi–Dunitz angle (~107°) between nucleophilic (amine) and electrophilic (carbonyl) reactivity centers.19 In contrast, with the bound ADPAc, the ε-ammonium group of K133 cannot reach this arrangement without severely distorting the adenine-Y198 pair sandwich. Finally, a follow-up MD simulation with explicit magnesium ions (i.e., 150 mM MgCl2) highlights putative roles that divalent oxophilic metal centers, commonly chelated by polyphosphate nucleotides, and terminal γ-phosphate could play in this reaction: Lewis acid and general base, respectively. Both elements combine to transform the solvent-exposed and, therefore, protonated lysine into a nucleophile while enhancing the electrophilicity of the acyl group and the leaving group capacity of triphosphate. Further analysis of the whole-proteome reactivity supports the critical requirement for divalent metal ions for the acylation reaction (Figure S7).
Chemoproteomic Analysis of Procaspase-6 Variants.
To evaluate the role of Y198 in nucleotide binding and zymogen-related processes, several mutants of full-length procaspase-6 were constructed, expressed in E. coli, and analyzed for K133 acylation (Figure 4). Under the expression conditions tested, wild-type procaspase-6 was fully processed to mature active caspase-6 through self-activation prior to analysis, resulting in little acylation of K133 (0.9%, data not shown). In contrast, the active-site knockout C163S variant, which is enzymatically incompetent for self-activation due to replacement of the catalytic cysteine, persisted as the full-length zymogen and demonstrated robust acylation of K133 (defined as 100%). The absence of wild-type zymogen and the persistence of the corresponding C163S variant are consistent with earlier observations of procaspase-6 self-activation, a noncanonical route for caspase activation.20,21 Because procaspase-6 has a loop 2 (L2) that is longer than those of procaspase-3 and −7, the scissile peptide bond between Asp-193 and Ala-194 within the intersubunit linker, a contiguous part of L2, sits close to the active-site Cys-163 and can then be primed for hydrolysis by intramolecular self-cleavage. In E. coli lysates, the inability to isolate wild-type procaspase-6 is consistent with this self-activation, while the C163S procaspase-6 stability confirms that there is no caspase-like activity in the E. coli lysate to efficiently process the variant zymogen. Unlike E. coli, mammalian cells such as Jurkat and HEK293 produce stable wild-type procaspase-6, suggesting the presence of a mammalian factor(s) that prevents autoactivation, such as the presence of zinc22 or endogenous inhibitors of apoptosis.21,23
Figure 4.

ATPAc labeling of procaspase-6 variants. The amount of intact procaspase-6 was determined by immunoblotting and compared to the amount of ATPAc probe labeling detected. Strong probe labeling was observed for the C163S variant compared to that of the C163S/Y198A variant, even though the proenzyme was present in similar amounts for both variants.
Several Y198A variants were generated to directly assess the effect of Y198 on ATPAc. The Y198A variant was weakly acylated by ATPAc (1.9%). However, we observed that the Y198A zymogen was also processed to the mature Y198A caspase-6 via the self-activation pathway (confirmed by immunoblotting), suggesting that loss of acylation might be due to the loss of procaspase-6 protein. In fact, isolation of Y198A procaspase-6 with the active-site cysteine intact has proven to be intractable in E. coli. Removal of the Y198 pair has no apparent deleterious effect on self-activation, and this variant is as active as WT caspase-6 in substrate-based assays (Table 1 and Figure S9). In contrast, the Y198A/C163S variant yielded full-length procaspase-6 with a dramatically reduced capacity for K133 acylation by ATPAc (1.7%). These findings strongly suggest that Y198 π–π stacking in procaspase-6 is the primary factor for ATPAc binding to procaspase-6 and, thereby, K133 acylation.
Table 1.
Enzymatic Parameters for Caspase-6 Isoforms
| caspase-6 isoform | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| WT | 56.1 ± 6.2 | 2.98 ± 0.10 | 53202 |
| Y198A | 64.4 ± 3.5 | 2.84 ± 0.04 | 44064 |
Procaspase-6 Self-Processing in Vitro Is Inhibited by ATP.
Given that ATPAc robustly labels procaspase-6 and the site of binding has been identified, it seemed plausible that ATP would impact some aspect of procaspase-6 function or self-processing, considering the well-characterized changes in intracellular ATP concentration during apoptosis.24–29 While we routinely use a C-terminal His6 tag to isolate active caspase-6, this construct was not appropriate for isolating procapase-6 because it repeatedly resulted in predominantly active (cleaved) caspase-6. Using a new N-terminal His6 construct, we were able to isolate a nearly homogeneous preparation of procaspase-6 that enabled subsequent analyses regarding the impact of ATP on procaspase-6 self-processing in vitro. Procaspase-6 was incubated for ≤8 h in the presence or absence of 20 mM ATP. At this concentration of ATP, cleavage of procaspase-6 was significantly impeded (Figure 5A). Dose–response studies (Figure 5B) demonstrated an IC50 of 19 ± 0.8 mM for ATP-dependent inhibition of procaspase-6 self-processing (Figure 5C). Conversely, the activity of mature caspase-6 is not affected by increasing concentrations of ATP (Figure 5D). From these analyses, it is clear that procaspase-6 is sensitive to the presence of ATP, which inhibits self-processing, but that the activity of mature caspase-6 is not sensitive to ATP.
Figure 5.

ATP attenuates procaspase-6 autoactivation but not caspase-6 activity. (a) Self-proteolysis of procaspase-6 [caspase-6 FL N-terminally (His)6] in the absence and presence of 20 mM ATP at 37 °C for different time intervals (0, 0.25, 1, 2, 4, 6, and 8 h). An increasing incubation time promotes self-activation of procaspase-6, generating the large and small subunits of active caspase-6 (FL, full-length; ΔN, without the pro-domain; Lg+L, large subunit attached to the linker; Lg, large subunit; and Sm, small subunit). After 6 h, the pro-domain is fully cleaved in the absence of ATP but remains intact in the presence of ATP. (b) Self-proteolysis of procaspase-6 [FL N-terminally (His)6] after incubation for 6 h at 37 °C in the presence of various ATP concentrations (0, 2.5, 3.75, 5, 7.5, 10, 15, 20, 25, 30, 35, 40, and 60 mM). (c) Quantification of remaining band intensities to determine the potency for ATP-mediated inhibition of procaspase-6 self-activation (IC50 = 19 ± 0.8 mM). (d) Proteolytic activity of active caspase-6 measured using the fluorogenic VEID-AMC substrate in the presence of increasing concentrations of ATP.
DISCUSSION
The dimer interfaces of the executioner caspase-3, −6, and −7 have received attention as possible sites of allosteric regulation and activation of these key apoptotic enzymes.30 The discovery of the hydrophobic cavity at the interface of caspase-3 and −7 led to the identification of small drug-like aromatic hetero-cycles by screening a library of 10000 thiol-containing compounds against accessible cysteines in the cavity.14,15 The compounds inhibited native caspase activity by reorienting a side chain at the dimer interface, which blocked the correct conformation of the active-site loops, thus preventing substrate binding at the active site. These compounds appeared to function by trapping the active caspase into a zymogen-like and, therefore, inactive structure. The identification of allosteric inhibitors of procaspase-6 using SPR marked a significant departure from the caspase-3 and −7 studies in that it focused on compounds that could specifically prevent zymogen activation.13 The choice of SPR as the binding readout was necessary because it provided a direct measurement of the binding to the inactive C163S zymogen in place of an activity assay. However, the in vitro conditions required to measure binding by SPR may not translate to cellular conditions. For instance, the KD for compound 3 in the SPR assay (~1.8 μM) was not predictive of its binding (IC50 > 1 mM) observed in cellular lysates using ATPAc. This is not surprising because the relatively pristine, nonphysiological conditions of the SPR assay do not reflect the myriad of factors that might attenuate binding of compound 3 in a whole cellular lysate.
The Y198 pair appears to be structurally unique to procaspase-6. The Y223 pair in caspase-7 was shown to interact with the inhibitors mentioned above in an extended arrangement not comparable to the π–π stack reported here. The fact that Y198 is needed for ATP binding supports the notion that ATP and perhaps other nucleotides or aromatic intracellular compounds bind at the dimer interface allosteric site and modulate caspase-6 function.
The key features mediating the ATP–procaspase-6 interaction, Y198, K133, and associated protein motifs, are completely conserved across all primates and highly conserved across all mammalian orthologs with few exceptions, the most notable being a Y198H single-point mutation in a number of aquatic and southeast Asian mammals and, remarkably, a Y198S single-point mutation found in virtually all members of the cat family (Felidae). Caspase-6 orthologs identified in birds, lizards, modern amphibians, and teleosts also show high levels of conservation for Y198 and K133.
Our suggestion that ATP is an endogenous ligand for dimeric procaspase-6 is strengthened by the generally accepted range of intracellular ATP concentrations of roughly 1–10 mM (average of ~3.2 mM), by far the most abundant nucleotide.31 The use of low levels (micromolar) of the irreversible acyl phosphate probe and short reaction times (minutes) in our chemoproteomic platform results in a low extent of acylation across the hundreds of susceptible active-site lysines in the whole cell lysate. This yields IC50’s for competitive inhibition of acylation by nucleotides that are close approximations of the Ki’s under these conditions.1,2 Thus, ATP appears to be unique among the nucleotides tested in that its binding constant (~2 mM) with procaspase-6 lies within range of its intracellular concentration.
In vitro inhibition of procaspase-6 self-activation by ATP (Figure 5) is complicated by intervening enzymatic processing, relative enzyme concentrations, and modifications in procaspase-6 that alter autoactivation. The relationship between the Ki for binding of ATP to the dimer interface and the IC50 for the inhibition of procaspase-6 self-processing is, therefore, correspondingly complex, leading to IC50’s that could be considerably larger than the Ki for ATP binding. However, the 19 mM ATP IC50 for procaspase-6 self-processing is still reasonably close to the physiological ATP concentration range. We are exploring other in vitro conditions to better define the mechanisms for this discrepancy.
In summary, our chemoproteomic analysis and supporting studies presented here suggest that ATP is a bona fide endogenous procaspase-6 ligand and potentially an in vivo inhibitor of self-processing at concentrations approximating cellular levels. Our findings should prompt further investigations to identify additional native ligands for the procaspase-6 dimer interface, the biological outcomes of ligand binding, and the regulation of other procaspases and active caspases through similar mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge Jane Wu, Senait Alemayehu, and Maria Sykes for maintenance and operation of LC–MS/MS and sample processing equipment.
Funding
This work was supported by the National Institutes of Health (GM80532 to J.A.H.). In addition, I.S. was supported in part by the UMass Chemistry-Biology Interface Training Program (National Research Service Award T32 GM 08515 from the National Institutes of Health).
ABBREVIATIONS
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- ATPAc
desthiobiotin acyl phosphate of ATP
- ADPAc
desthiobiotin acyl phosphate of ADP
- GTPAc
desthiobiotin acyl phosphate of GTP
- GDPAc
desthiobiotin acyl phosphate of GDP
- ATP
adenosine triphosphate
- ADP
adenine diphosphate
- GTP
guanosine triphosphate
- GDP
guanosine diphosphate
- CTP
cytidine triphosphate
- UTP
uridine triphosphate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00290.
Procedures for probe labeling, LC–MS/MS analyses, generation of caspase-6 variants, recombinant enzyme purification, and enzymology; data illustrating caspase enzymology and nucleotide inhibition; and computational procedures for molecular modeling, including molecular dynamics interaction maps (PDF)
Accession Codes
UniProtKB: CASP6, P55212; CASP7, P55210; CASP3, P42574; p38-α, Q16539; MAPKAPK2, P49137; MAPKAPK3, Q16644.
The authors declare no competing financial interest.
REFERENCES
- (1).Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, and Kozarich JW (2007) Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–8. [DOI] [PubMed] [Google Scholar]
- (2).Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, and Kozarich JW (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Adachi J, Kishida M, Watanabe S, Hashimoto Y, Fukamizu K, and Tomonaga T (2014) Proteome-wide discovery of unknown ATP-binding proteins and kinase inhibitor target proteins using an ATP probe. J. Proteome Res 13, 5461–5470. [DOI] [PubMed] [Google Scholar]
- (4).Nordin BE, Liu Y, Aban A, Brown HE, Wu J, Hainley AK, Rosenblum JS, Nomanbhoy TK, and Kozarich JW (2015) ATP Acyl Phosphate Reactivity Reveals Native Conformations of Hsp90 Paralogs and Inhibitor Target Engagement. Biochemistry 54, 3024–36. [DOI] [PubMed] [Google Scholar]
- (5).Okerberg ES, Brown HE, Minimo L, Alemayehu S, Rosenblum J, Patricelli M, Nomanbhoy T, and Kozarich JW (2014) Monitoring native p38??:M K2/3 complexes via trans delivery of an ATP acyl phosphate probe. J. Am. Chem. Soc 136, 4664–4669. [DOI] [PubMed] [Google Scholar]
- (6).Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee J-D, Gray NS, and Kozarich JW (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Okerberg E, Shih A, Brown H, Alemayehu S, Aban A, and Nomanbhoy TK (2013) Profiling native kinases by immune-assisted activity-based profiling. Curr. Protoc. Chem. Biol, 213. [DOI] [PubMed] [Google Scholar]
- (8).Okerberg ES, Wu J, Zhang B, Samii B, Blackford K, Winn DT, Shreder KR, Burbaum JJ, and Patricelli MP (2005) High-resolution functional proteomics by active-site peptide profiling. Proc. Natl. Acad. Sci. U. S. A 102, 4996–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vaidya S, Velazquez-Delgado EM, Abbruzzese G, and Hardy JA (2011) Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol 406, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vaidya S, and Hardy JA (2011) Caspase-6 latent state stability relies on helical propensity. Biochemistry 50, 3282–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Parrish AB, Freel CD, and Kornbluth S (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harbor Perspect. Biol 5, a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Adachi J, Kishida M, Watanabe S, Hashimoto Y, Fukamizu K, and Tomonaga T (2015) Data for proteomic analysis of ATP-binding proteins and kinase inhibitor target proteins using an ATP probe. Data Br 5, 726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Murray J, Giannetti AM, Steffek M, Gibbons P, Hearn BR, Cohen F, Tam C, Pozniak C, Bravo B, Lewcock J, Jaishankar P, Ly CQ, Zhao X, Tang Y, Chugha P, Arkin MR, Flygare J, and Renslo AR (2014) Tailoring small molecules for an allosteric site on procaspase-6. ChemMedChem 9, 73–77. [DOI] [PubMed] [Google Scholar]
- (14).Hardy JA, Lam J, Nguyen JT, O’Brien T, and Wells JA (2004) Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U. S. A 101, 12461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hardy JA, and Wells JA (2009) Dissecting an allosteric switch in caspase-7 using chemical and mutational probes. J. Biol. Chem 284, 26063–26069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Scheer JM, Romanowski MJ, and Wells JA (2006) A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. U. S. A 103, 7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Datta D, Scheer JM, Romanowski MJ, and Wells JA (2008) An Allosteric Circuit in Caspase-1. J. Mol. Biol 381, 1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, and Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666. [DOI] [PubMed] [Google Scholar]
- (19).Burgi HB, Dunitz JD, Lehn JM, and Wipff G (1974) Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 30, 1563–1572. [Google Scholar]
- (20).Wang XJ, Cao Q, Liu X, Wang KT, Mi W, Zhang Y, Li LF, LeBlanc AC, and Su XD (2010) Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep 11, 841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Klaiman G, Champagne N, and LeBlanc AC (2009) Self-activation ofCaspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim. Biophys. Acta, Mol. Cell Res 1793, 592. [DOI] [PubMed] [Google Scholar]
- (22).Velázquez-Delgado EM, and Hardy JA (2012) Zinc-mediated allosteric inhibition of caspase-6. J. Biol. Chem 287, 36000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pop C, and Salvesen GS (2009) Human caspases: Activation, specificity, and regulation. J. Biol. Chem 284, 21777–21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Feldenberg LR, Thevananther S, del Rio M, de Leon M, and Devarajan P (1999) Partial ATP depletion induces Fas-and caspase-mediated apoptosis in MDCK cells. Am. J. Physiol 276, F837. [DOI] [PubMed] [Google Scholar]
- (25).Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, and Okada Y (2005) Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ 12, 1390–7. [DOI] [PubMed] [Google Scholar]
- (26).Hu Y, Benedict MA, Ding L, Nú Ñ, and Ez G (1999) Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 18, 3586–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ferrari D, Stepczynska A, Los M, Wesselborg S, and Schulze-Osthoff K (1998) Differential Regulation and ATP Requirement for Caspase-8 and Caspase-3 Activation during CD95-and Anticancer Drug—induced Apoptosis. J. Exp. Med 188, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chandra D, Bratton SB, Person MD, Tian Y, Martin AG, Ayres M, Fearnhead HO, Gandhi V, and Tang DG (2006) Intracellular Nucleotides Act as Critical Prosurvival Factors by Binding to Cytochrome C and Inhibiting Apoptosome. Cell 125, 1333–1346. [DOI] [PubMed] [Google Scholar]
- (29).Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, and Ravichandran KS (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dagbay K, Eron SJ, Serrano BP, Velázquez-Delgado EM, Zhao Y, Lin D, Vaidya S, and Hardy JA (2014) A multipronged approach for compiling a global map of allosteric regulation in the apoptotic caspases. Methods Enzymol 544, 215–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Traut TW (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.