

Abstract
Background:
Targeting of drugs to the subcellular compartments represents one of the modern trends in molecular pharmacology. The approach for targeting mitochondria was developed nearly 50 years ago, but only in the last decade has it started to become widely used for delivering drugs. A number of pathologies are associated with mitochondrial dysfunction, including cardiovascular, neurological, inflammatory and metabolic conditions.
Objective:
This mini-review aims to highlight the role of mitochondria in pathophysiological conditions and diseases, to classify and summarize our knowledge about targeting mitochondria and to review the most important preclinical and clinical data relating to the antioxidant lipophilic cations MitoQ and SkQ1.
Methods:
This is a review of available information in the PubMed and Clinical Trials databases (US National Library of Medicine) with no limiting period.
Results and Conclusion:
Mitochondria play an important role in the pathogenesis of many diseases and possibly in aging. Both MitoQ and SkQ1 have shown many beneficial features in animal models and in a few completed clinical trials. More clinical trials and research efforts are needed to understand the signaling pathways influenced by these compounds. The antioxidant lipophilic cations have great potential for the treatment of a wide range of pathologies.
Keywords: Mitochondria, Targeted drug delivery, Uncouplers of oxidative phosphorylation, Reactive oxygen species, Antioxidants, Metabolic syndrome
1. INTRODUCTION
It has been more than a century since Paul Ehrlich postulated his famous idea of a ‘magic bullet’, but only recently has this concept become a reality. The main problems faced by the concept of targeted drug delivery consist of searching for a target for a particular disease, searching for a medicine and searching for an effective way of delivering drugs to the target.
Targeted drug delivery may take place on three levels: (i) first-order drug targeting to an organ or tissue, (ii) second-order drug targeting to specific cells and (iii) third-order drug targeting aimed at an intracellular compartment such as the nucleus, endoplasmic reticulum or mitochondria [1, 2].
Currently, mitochondria are recognized as an emerging pharmacological target, since this intracellular organelle has a number of vital functions and mitochondrial damage is crucial for the development of many diseases (see section 2.4 of the current review).
There is no universally accepted definition of a Mitochondria-Targeted Compound (MTC). We suggest defining an MTC as a substance that is selectively accumulated in mitochondria in amounts of at least 90% of the total substance added to the cells. The first MTC was synthesized in 1995 by Murphy and co-workers [3]. In the last decade, a whole set of MTCs has been synthesized and tested, both in vitro and in vivo. Many experimental data have been obtained confirming the efficacy of the MTCs in the prevention and treatment of a wide range of diseases. Moreover, a few dozens of clinical trials are either in an active state or have been completed.
The purpose of this review is to consider mitochondria as a promising target for treating a wide range of human diseases and pathological conditions and to review the principles of mitochondrially targeted drug delivery and the most significant results obtained with MTCs.
2. STRUCTURE AND FUNCTIONS OF MITOCHONDRIA
2.1. Structural Aspects
Two membrane layers that differ in both chemical and structural composition envelop mitochondria. Between the Outer Mitochondrial Membrane (OMM) and the Inner Mitochondrial Membrane (IMM), there is an intermembrane space, and the mitochondrial matrix is located inside the IMM. The components of the Electron Transport Chain (ETC) are located in the IMM (Fig. 1).
Fig. (1).
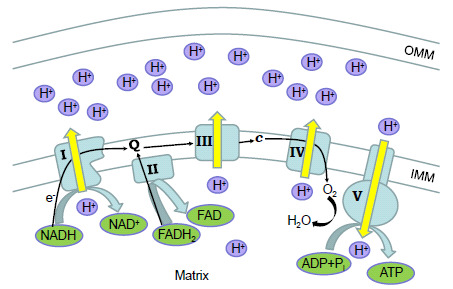
Scheme of mitochondrial electron transport chain (ETC). Electron flow (black arrows) from NADH or FADH2 substrates through complexes I - IV of ETC is accompanied by pumping of protons. This results in a proton gradient between the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM). This proton-motive force is used by ATP synthase (complex V) to make ATP from ADP and a phosphate (Pi). Abbreviations: Q - coenzyme Q10, c - cytochrome c.
Mitochondria have their own self-replicating genome in the form of circular mitochondrial DNA (mtDNA). Typically, one animal cell contains from 100 to 10, 000 mtDNA copies. Human mtDNA codes for mitochondrial transport RNA, ribosomal RNA and some hydrophobic ETC proteins. Mutations in either mtDNA or in nuclear DNA genes coding for mitochondrial proteins result in the development of inherited mitochondrial diseases [4].
The outer membrane is permeable to a wide range of small molecules. The permeability of the IMM is selective; special channels regulate the transport of molecules through the IMM. Thus, the electrochemical potential and the content of the mitochondrial matrix are significantly different from those of the cytosol.
2.2. Main Functions
Mitochondria are implicated in many vital processes in animal cells, including energy production, fatty-acid oxidation and the Tricarboxylic Acid (TCA) cycle, calcium signaling, permeability transition, apoptosis and heat production.
The main function of mitochondria is to produce Adenosine Triphosphate (ATP). In the cell, the necessary energy in the form of ATP is produced in two ways: in the cytosol as a product of glycolysis, and in the mitochondria as a product of oxidative phosphorylation (OXPHOS). The substrates, in the form of fatty acids and pyruvate, are oxidized via fatty acid β-oxidation and the TCA cycle respectively. The Nicotinamide Adenine Dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) produced by these reactions are used by the electron transport chain to generate ATP.
The mitochondrial electron transport chain consists of the enzyme complexes located in the IMM (Fig. 1). The electrons supplied by NADH are accepted by complex I and the electrons from FADH2 are transferred to complex II. Then, the electrons are transported by coenzyme Q to complex III and finally by cytochrome c through complex IV to oxygen molecules. The electron flow is accompanied by pumping of protons from the mitochondrial matrix to the intermembrane space. The resulting proton gradient generates ATP through complex V (ATP synthase).
In brown adipose tissue, mitochondria promote heat production via Uncoupling Protein 1 (UCP1) [5]. This protein induces mitochondrial uncoupling of oxidative phosphorylation and respiration via an increase in proton conductivity of the IMM. The energy of the protons is not used for ATP production but instead is dissipated in the form of thermal energy. In human adults, brown adipose tissue is found in small quantities only. However, UCP1-mediated heat production is of great importance in newborns, to avoid life-threatening hypothermia.
The main role in the storage of calcium cations in the cells is performed by endoplasmic reticulum, but mitochondria are also capable of storing calcium transiently [6]. The outer mitochondrial membrane is permeable to Ca2+ ions, which enter the mitochondrial matrix via the calcium uniporter located in the IMM. The release of calcium initiates calcium spikes or calcium waves, with a great impact on cellular signaling cascades (reviewed in [7]).
The Mitochondrial Permeability Transition (MPT) is an instant increase of the IMM permeability to molecules less than 1, 500 Da. MPT opening leads to depolarization, Ca2+ release and Reactive Oxygen Species (ROS) production, in response to certain stimuli, such as increased Ca2+, ER stress, fatty acids and inorganic phosphate [8]. Short-term openings probably fulfil physiological functions such as Ca2+ regulation and ROS production. Long-term MPT opening may result in detrimental effects including cell death [9]. Mitochondria form a central cellular hub regulating the intrinsic pathway of apoptosis. MPT opening leads to cytochrome c exit, caspase activation and apoptosis [10]. Even without the activation of caspases, permeabilization of the outer mitochondrial membrane may induce caspase-independent cell death [11].
2.3. Mitochondria and ROS
Oxidative phosphorylation always results in ROS production. The primary ROS is a superoxide anion (.O2-) produced by both complex I and complex III. Most superoxide anions are converted by superoxide dismutase (SOD) into hydrogen peroxide, which in turn is converted into water by catalase or by peroxidase enzymes. However, antioxidant enzymes are not able to inactivate all the ROS. Excessive amounts of superoxide anion may interact with other molecules, resulting in production of multiple secondary radicals including reactive nitrogen species and lipid radicals.
Mitochondrial ROS (mtROS) are also synthesized in a number of enzymatic reactions in mitochondria including those of glycerol-3-phosphate dehydrogenase, cytochrome P450, monoamine oxidase and cytochrome b5 reductase (reviewed in [12]).
It is widely accepted that excessive mtROS production is detrimental to the cells and surrounding tissues [13]. Many diseases and pathological conditions are now linked to mtROS (see the next section). However, besides their well-known destructive role, mtROS appear to be necessary for many physiological reactions, including apoptosis [14], activation of macrophages’ inflammasomes [15] and possible monoamine-induced calcium signaling in astrocytes [16].
ROS are also produced from non-mitochondrial sources, including NADPH oxidase [17], myeloperoxidase [18], xanthine oxidase [19], monoamine oxidase [20] and uncoupled nitric oxide synthase [21]. NADPH oxidases are considered the major enzymatic source of ROS [22]. There are seven isoforms of NADPH oxidases, expressed in a variety of cells including phagocytes, vascular cells, smooth muscle cells and fibroblasts [23]. ROS produced by NADPH oxidases are necessary for proper functioning of innate immunity and signal transduction. However, excessive ROS production is involved in the development of many forms of the cardiovascular disease [22] and in many neurological diseases, including stroke, traumatic brain injury and neurodegenerative diseases [24]. Interestingly, though neutrophils and endothelial cells produce ROS mostly via NADPH oxidase and xanthine oxidase respectively, synthesis of mtROS is necessary for inflammatory signal transduction [25, 26]. In addition, secondary ROS production via NADPH oxidase was inhibited by quenching mtROS in an animal model of cisplatin-induced nephropathy [27]. Thus, mtROS may regulate ROS production by non-mitochondrial sources. The exact impact of mtROS in total ROS production in different cells is still not completely clear.
2.4. Mitochondria and Diseases
Mitochondrial dysfunction is typically characterized by a disturbance of the basic mitochondrial functions: Bioenergetic, antioxidant and regulatory. This dysfunction results in a decrease in ATP synthesis, dysregulated cell death processes and increased ROS production.
Proper mitochondrial functioning is crucial for every enucleated cell in a body. A number of diseases are characterized by dysfunction of muscular or neural systems or metabolic reactions. All these diseases and pathophysiological conditions are developed against a specific genetic background, together with environmental factors. Some examples of mitochondrial involvement in the development of these diseases are given below.
Our review does not focus on inherited mitochondrial diseases caused by mutations either in mtDNA or in nuclear DNA. The readers interested in this topic are directed to recent excellent reviews [4, 28, 29]. It should be noted that mitochondrial diseases affect cells in tissues with high energy demands: neural and muscular systems. These systems are also affected in all non-hereditary diseases accompanied or even caused, by dysfunctional mitochondria.
2.4.1. Metabolic Syndrome
Metabolic syndrome is a group of conditions combining hypertension, hyperglycemia, abdominal obesity and abnormal cholesterol or triglyceride levels. Metabolic syndrome greatly increases the risk of cardiovascular disease, stroke and type 2 diabetes (T2DM). There are numerous reports mentioning mitochondrial dysfunction and lower oxidative phosphorylation capacity in patients with T2DM compared with healthy individuals [30-32]. Both T2DM and obesity result in the accumulation of plasma acylcarnitines, probably due to reduction of β-oxidation of fatty acids inside mitochondria [33-35].
In skeletal muscle of both rodents and humans, a high-fat diet increases the H2O2-emitting potential of mitochondria, shifts the cellular redox environment to a more oxidized state and decreases the redox buffering capacity in the absence of any change in mitochondrial respiratory function. Furthermore, it was shown that attenuating mitochondrial H2O2 emission in mice muscles completely preserves insulin sensitivity, despite a high-fat diet [36].
2.4.2. Cardiovascular Diseases
The cardiovascular system strongly depends on mitochondrial function. Cardiomyocytes have very high mitochondrial content in order to produce the necessary ATP, and mitochondrial dysfunction inevitably leads to the development of cardiovascular diseases (for more details see [37]).
Alterations in mitochondrial calcium transport, which lead to ROS generation and MPT opening, also result in abnormalities of ATP production, thus leading to heart failure and ischemia-reperfusion injury (see the review in [38]). Mitochondrial dysfunction is also found in mice models of diabetic cardiomyopathy [39].
Peripheral arterial disease has a complex pathophysiology but is also shown to result from decreased mitochondrial respiratory capacity and oxidative stress (reviewed in [40]).
2.4.3. Neurodegenerative Disorders
There is now increasing evidence of mitochondrial dysfunction in Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Huntington’s disease, and amyotrophic lateral sclerosis (ALS).
The pathophysiology of AD remains unclear and the amyloid cascade hypothesis has dominated others to date. However, none of the drug candidates preventing accumulation of amyloid β-peptide has succeeded even in the delay of this disease. Thus, mitochondrial dysfunction was proposed as the primary event in the sporadic form of AD that causes amyloid β-peptide (Aβ) deposition, synaptic degeneration and formation of neurofibrillary tangles [41]. This hypothesis is supported by the evidence of mitochondrial dysfunction in AD patients [42] and in animal models [43, 44]. This approach also identifies new alternative therapeutic targets located in the mitochondria [45].
PD is characterized by resting tremor, bradykinesia and rigidity. These symptoms are developed due to loss of dopaminergic neurons in the substantia nigra. Patients with PD have a deficiency in the activity of mitochondrial electron transport chain complex I in a number of tissues, including the substantia nigra (reviewed in [46]). The primary role of mitochondrial dysfunction in PD was proved using transgenic mice lacking the mitochondrial transcription factor A gene. These animals slowly developed a typical Parkinsonian phenotype [47, 48].
Huntington’s disease is caused by an abnormal accumulation of CAG repeats in the huntingtin gene. Multiple mitochondrial abnormalities are found in the neurons of Huntington’s disease patients (reviewed in [49]). The main neuropathological features of Huntington’s disease are reproduced in animals treated with the mitochondrial toxin 3-nitropropionic acid [50].
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease affecting motor neurons in the brain and spinal cord. ALS etiology is still largely unknown, but in some familiar forms of ALS it occurs due to a mutation in the cytoplasmic Superoxide Dismutase (SOD1) gene. This mutated enzyme is also located in the mitochondria, causing alterations in axonal mitochondrial morphology and distribution [51]. Expressing mutant SOD1 targeted to mitochondria is sufficient to produce loss of motor neurons and an ALS phenotype [52].
2.4.4. Muscular Disorders
Duchenne muscular dystrophy is an X-linked recessive disease with a deleterious mutation in the dystrophin protein [53]. Duchenne dystrophy also has some features of mitochondrial dysfunction, including decreased ATP synthesis and MPT pore opening leading to the death of the myofibers [54-56].
Inclusion body myositis is a type of inflammatory myopathy that, like AD, is an amyloid disease characterized by inflammation and accumulation of Aβ oligomers and other protein aggregates in muscle fibers. Mitochondrial homeostasis is greatly affected in this disease (reviewed in [57]).
2.4.5. Inflammatory Diseases
Indeed, nearly all the diseases mentioned above are characterized by systemic chronic inflammatory reactions. In addition, mitochondrial dysfunction plays a significant role in the inflammatory response in acute human pathologies. Systemic Inflammatory Response Syndrome (SIRS) is a pathological state with a systemic immune reaction to severe damage, including ischemia, acute pancreatitis, trauma and sepsis [58]. SIRS is elicited by different factors including Damage-Associated Molecular Patterns (DAMPs), Pathogen-Associated Molecular Patterns (PAMPs) and cytokines [59]. Excessive inflammatory reactions may result in the development of fatal multiple organ failure. The important role of mitochondrial ROS in SIRS was reviewed recently [60].
Production of the pro-inflammatory cytokine IL-1β is regulated by NLRP3 inflammasome, which has been linked with various human auto-inflammatory and autoimmune diseases [61]. ROS-generating mitochondria greatly enhance the activity of the NLRP3 inflammasome complex [15].
Since mitochondria have a bacterial ancestry, it is not surprising that many mitochondrial components may function as DAMPs, promoting acute inflammatory reactions. Mitochondrial DAMPs elicit a strong immune response through interaction with the corresponding pattern-recognition receptors. It was recently shown that newly synthesized mtDNA enhances activation of the NLRP3 inflammasome [62]. Extracellular mtDNA also triggers inflammatory reactions in many cell types, including neutrophils [63, 64]. Cardiolipin, a phospholipid of the inner mitochondrial membrane, also functions as a DAMP [65]. Like bacteria, mitochondria start protein synthesis with the N-formylmethionine residue. These proteins also function as DAMPs, activating neutrophils and driving their chemotaxis [66].
2.4.6. Other Diseases
Many other diseases are interconnected with mitochondrial dysfunction. Due to space limitation, we direct readers to the recent review on this topic covering liver diseases, cancer, kidney diseases, eye diseases, pulmonary diseases (including chronic obstructive pulmonary disease) and drug-induced mitochondrial toxicity [67].
2.5. Mitochondria and Aging
A number of age-related processes are associated with mitochondrial dysfunction (reviewed in [68]) and most popular aging theories take this into account. The mitochondrial theory of aging suggests that the accumulation of damage to mitochondria and mtDNA promotes aging of humans and animals [69]. The theory supposes that there is a so-called vicious cycle: the accumulation of damage in mtDNA leads to the synthesis of nonfunctional proteins of the respiratory chain, which in turn increases the production of mitochondrial ROS, again damaging mtDNA. A number of facts confirm this theory: transgenic ‘mutator’ mice with defective mitochondrial DNA polymerase γ had accelerated accumulation of mutations in mtDNA, decreased lifespan and a senescent phenotype [70]. Later, Schrinner and colleagues showed that the overexpression of the mitochondria-targeted catalase increased lifespan in these mice [71]. Nowadays, however, this theory is seriously challenged by data showing that: (i) oxidative mtDNA damage during the lifetime does not result in accumulation of the mutations [72] and (ii) heterozygous ‘mutator’ mice have a higher mutational load in mtDNA than old wild-type mice, although their life spans are identical.
Another theory describes aging as a chronic low-grade inflammation causing cumulative damage to organic compounds via ROS [73]. Aging is accompanied by increased mitochondrial ROS production in the heart [74] and in vessels [75]. The ROS are able to activate redox-sensitive pathways, including transcription factor NF-κB, and can also stimulate secretion of pro-inflammatory cytokines, leading to the development of vascular inflammation and/or promoting atherosclerosis [76]. Both mitochondrial dysfunction and mitochondrial DAMPs are suspected sources of chronic inflammation in aging [77].
Apart from these theories, there are many experimental data confirming the key role of mitochondria in aging. For instance, two recent papers published in the journal Cell have shown that mitochondrial stress early in nematode development causes chromatin rearrangement and a subsequent increase in the life span of Caenorhabditis elegans [78, 79]. Thus, mitochondria also appear to be an attractive target for aging retardation.
3. HOW TO TARGET MITOCHONDRIA
3.1. Targeting Principles
It is likely that mitochondria originated via endosymbiosis with ancient prokaryotic organisms such as proteobacteria [80]. Thus, among all other organelles, mitochondria still have some unique features that can be used for targeting (Fig. 2): high transmembrane potential (ΔΨm) across the inner mitochondrial membrane, unique phospholipid composition in the IMM and a special protein import machinery.
Fig. (2).
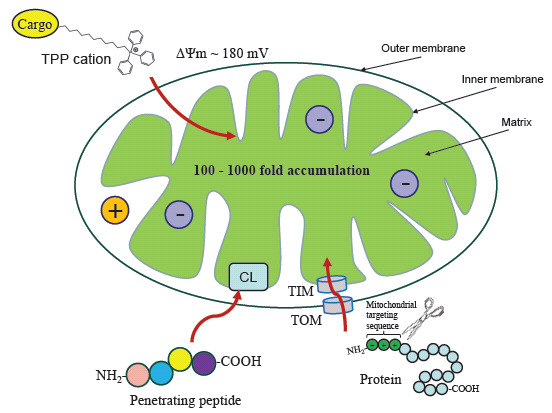
Three main types of mitochondrial targeting. Mitochondrial uptake of lipophilic cations such as triphenylphosphonium (TPP) occurs due to the transmembrane potential (ΔΨm), and the cations are accumulated at the inner mitochondrial membrane. Peptides penetrating mitochondria and binding to the mitochondrial phospholipid cardiolipin (CL). Cellular proteins with N-terminal mitochondrial targeting sequences are imported into mitochondria via TOM and TIM channels.
Firstly, mitochondria are the only intercellular organelles with a negative charge inside. The ΔΨm between the matrix and the intermembrane space is around 180 mV. Mitochondria use this potential as a proton-motive force to drive ATP synthesis, and ΔΨm is a key parameter that indicates the bioenergetic competence of mitochondria [81]. The ΔΨm is exploited for mitochondrial targeting by the use of positively charged ions (cations) that are attracted to the mitochondria via an electrostatic force.
Secondly, the IMM has a unique structure and lipid composition, which can also be used for the targeted delivery of drugs to the mitochondria. The phospholipid cardiolipin is found almost exclusively in the IMM, where it provides structural support for respiratory chain complexes and also plays a key role in apoptosis [82].
While establishing endosymbiotic relationships between the primary eukaryotic cell and mitochondria, the latter lost most of their genetic material and were forced to use proteins encoded in the nuclear genome. Thus, mitochondria have their own protein import machinery that recognizes proteins with a special amino acid sequence [83] (Fig. 2).
Thus, drug delivery to mammalian mitochondria is carried out using one of these approaches, or a combination thereof. The main groups of MTCs exploiting various targeting principles are listed below.
3.2. Lipophilic Cation Moieties
Mitochondria-penetrating cations were discovered about half a century ago [84]. These cations should possess the following properties, allowing them to penetrate through the cellular and mitochondrial membranes of the animal cell. Firstly, they must be to an optimal degree lipophilic. If lipophilicity is less than a certain limit, then the molecules cannot penetrate biological membranes. If lipophilicity is excessively high, then the compounds will accumulate mainly in the cell membrane. Secondly, the positive charge of lipophilic cations should be delocalized, i.e., the charge should be distributed between at least three atoms but not within the single internuclear region between two adjacent atoms.
The most widely used lipophilic cation is triphenylphosphonium (TPP) (Fig. 3). Normally, the drug of interest is linked to the TPP cation via a carbon aliphatic linker. The idea of using lipophilic cations as the carriers to mitochondria was first implemented by Murphy and co-workers [3] to synthesize thiobutyltriphenylphosphonium bromide. Later, the thiobutyl group was replaced by the natural antioxidant α-tocopherol (MitoVitE) [85]. Nowadays, many dozens of TPP-based MTCs have been synthesized [86] and characterized in various in vitro and in vivo studies.
Fig. (3).
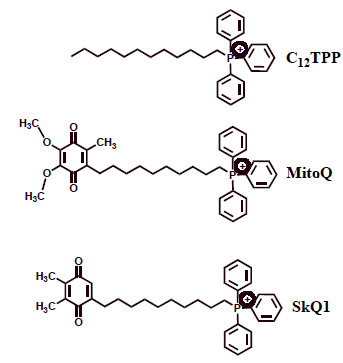
Chemical structures of the lipophilic cations triphenylphosphonium (TPP), MitoQ and SkQ1. MitoQ and SkQ1 are shown in the oxidized (quinone) forms.
There are also less common lipophilic systems besides TPP, such as rhodamine, quaternary ammonium salts, pyridinium, cyanine and berberine derivatives. Dequalinium (DQA) is another lipophilic molecule with two cations. DQA has been used for a long time as a topical antimicrobial agent. DQA can form liposome-like aggregates called DQAsomes that are able to deliver different molecules to mitochondria [87]. Unfortunately, the usefulness of this promising delivery system is limited by low transfection efficiency [88].
3.3. Cardiolipin Targeting (Penetrating Peptides)
Penetrating peptides are small, cell-permeable, mitochondria-targeted, antioxidant peptides that can protect mitochondria from oxidative damage [89]. The exact mechanism of the peptides’ uptake into mitochondria is still unclear, but most probably it does not rely on mitochondrial potential [89] but depends on direct interaction with cardiolipin in the IMM [90].
These peptides (also sometimes called Szeto-Schiller peptides) carry aromatic residues providing necessary hydrophobicity and basic amino acids providing positive charge, at physiological pH values [89]. The peptides have antioxidant properties attributed to the tyrosine or dimethyltyrosine residue. Tyrosine reacts with ROS forming relatively inert tyrosyl radicals, which either create dityrosine or react with superoxide to form tyrosine hydroperoxide [91]. Dimethyltyrosine acts in the same way but appears to be more effective than tyrosine. These peptides efficiently neutralize ROS in mitochondria and prevent the oxidation of cardiolipin, thus inhibiting mitochondrial apoptosis associated with oxidative stress [89].
The first of the Szeto-Schiller peptides, SS-31, is currently in multiple clinical trials, mostly targeted at cardiovascular, age-related and mitochondrial diseases (please refer to the informative reviews in [92, 93]).
3.4. Mitochondrial Targeting Signal Peptides
More than 1, 000 mitochondria proteins are encoded in the nuclear genome. Mitochondrial protein import machinery is naturally used by cells for the delivery of nuclear-encoded mitochondrial proteins. These proteins normally have a cleavable 20-40-amino acid N-terminal targeting sequence. Though not conserved, this sequence is typically charged positively and contains amphiphilic α-helices [94]. TOM importing complex is located on the OMM and is responsible for the translocation of the imported proteins to the TIM complex located at the IMM. After passing through the IMM, the targeting sequence is cleaved by mitochondrial peptidases and the imported protein is folded into mature conformation by the mitochondrial chaperones (reviewed in [95]).
Most of the researchers’ efforts were aimed at using a mitochondrial targeting sequence for DNA or gene delivery into the mitochondria as a possible way to cure mitochondrial diseases [96, 97]. This approach is also used to study the specific role of mitochondria in the development of some diseases, e.g., the role of mitochondrial accumulation of Aβ peptide in the alteration of mitochondrial function and cell death [98].
However, no potential drug with mitochondrial targeting signal peptides has yet been tested, since this approach has many practical limitations: (i) large molecules can hardly be transported, (ii) efficient targeting peptides are quite long, (iii) the peptides have low hydrophilicity and cellular permeability and (iv) the price for chemical synthesis is very high. These drawbacks severely limit the use of MTCs based on the mitochondrial targeting signal peptides.
4. MTCS IN PRECLINICAL AND CLINICAL TRIALS
4.1. Main Types of MTCs and New Drug Discovery
The vast majority of synthesized MTCs fall into one of the following categories:
antioxidants - lower mitochondrial ROS production
uncouplers of oxidative phosphorylation and respiration - lower ΔΨm and ATP production
poisons - mitotoxic and cytotoxic compounds inducing cell death, mainly apoptosis
probes and sensors - for detection of reactive oxygen, nitrogen and sulfur species.
It should be mentioned that applying any of the MTCs nearly always has multiple effects, such as alterations in the redox status, ETC activity, ATP synthesis, etc. This occurs due to the complex interconnected processes existing within mitochondria. For example, at certain concentrations, uncouplers of oxidative phosphorylation and respiration also lower ROS production in mitochondria [99]. High doses of the antioxidants may exert pro-oxidant effects [100] and inhibit ETC [101].
Mitochondria-targeted antioxidants represent the major part of all tested MTCs because of the important role of mtROS in many pathophysiological processes (see above). Most of the in vivo results and all the clinical trial results are obtained using either the lipophilic TPP antioxidants MitoQ and SkQ1 (Fig. 3) or the cell-penetrating tetrapepride SS-31
(also known as elamipretide, MTP-131). Below, we review the most important preclinical data and clinical trials with MitoQ and SkQ1 (Table 1).
Table 1. Mitochondria-targeted compounds used in clinical studies.
| Molecule Name (Type) | Targeted Disease | Study Title | Clinical Trial Identifier1 | Status | Main Outcome(s) / References |
|---|---|---|---|---|---|
| MitoQ | Multiple Sclerosis Fatigue |
MitoQ for Fatigue in Multiple Sclerosis | NCT03166800 | Recruiting | ND |
| MitoQ | Aging | The Efficacy of Oral Mitoquinone (MitoQ) Supplementation for Improving Physiological in Middle-aged and Older Adults | NCT02597023 | Completed | Improved vascular function [102] |
| MitoQ | Parkinson’s Disease | A Trial of MitoQ for the Treatment of People with Parkinson’s Disease | NCT00329056 | Completed | No effect on the PD progression [103] |
| MitoQ | Diastolic Dysfunction | MitoQ Supplementation and Cardiovascular Function in Healthy Men and Women | NCT03586414 | Recruiting | ND |
| MitoQ | Chronic Hepatitis C | Trial of MitoQ for Raised Liver Enzymes Due to Hepatitis C | NCT00433108 | Completed | Significantly decreased plasma alanine and aspartate aminotransferase [104] |
| MitoQ | Peripheral Arterial Disease | Impacts of Mitochondrial-Targeted Antioxidant on Peripheral Artery Disease Patients | NCT03506633 | Not yet recruiting | ND |
| MitoQ | Non-Alcoholic Fatty Liver Disease |
A Study to Compare MitoQ and Placebo to Treat Non-Alcoholic Fatty Liver Disease (NAFLD) | NCT01167088 | Terminated | Terminated due to poor participant recruitment |
| MitoQ | Alzheimer’s Disease, Early Onset |
Effects of Mitochondrial-Targeted Antioxidant on Alzheimer’s Disease | NCT03514875 | Not yet recruiting | ND |
| MitoQ | Chronic Kidney Disease | Mitochondrial Oxidative Stress and Vascular Health in Chronic Kidney Disease | NCT02364648 | Recruiting | ND |
| MitoQ | Chronic Obstructive Pulmonary Disease, Pulmonary Artery Hypertension, Heart Failure, Hypertension |
Vascular Function in Health and Disease | NCT02966665 | Recruiting | ND |
| MitoQ | Hypogonadism | Cardiovascular Outcomes of Low Testosterone | NCT02758431 | Recruiting | ND |
| SkQ1 | Keratoconjunctivitis Sicca | A Clinical Study to Assess the Safety and Efficacy of an Ophthalmic Solution (SkQ1) in the Treatment of Dry Eye Syndrome | NCT02121301 | Completed | Significant improvements in dry eye signs and symptoms [105] |
1 - as indicated at www.clinicaltrials.gov.
Development of new mitochondria-targeted antioxidants is an urgent task in modern pharmacology. Antioxidant capacities of ‘classical’ antioxidants like glutathione, ascorbate or N-acetylcysteine may easily be evaluated using established assays to determine total antioxidant capacities both in vitro and in vivo [106]. Other types of antioxidant, (e.g., MitoQ and SkQ1) have the ability to quench radical reactions inside mitochondria and to be subsequently recharged by ETC. There is no established method of quantifying the potency or potential of a new drug candidate of this type. We can suggest the following steps. The pro-oxidant and antioxidant activities of the drug candidate are determined in aqueous solutions, planar bilayer phospholipid membranes, isolated mitochondria and cell cultures. Measurement of lipid peroxidation is of particular importance in these systems [107]. The main assays on the isolated mitochondria include measurements of transmembrane mitochondrial potential, oxygen consumption, ATP synthesis, the ability of the drug to be reduced by ETC and overall OXPHOS efficiency. The in vitro assays include cell survival assays under oxidative stress, (e.g., treatment with hydrogen peroxide). The MTCs with good characteristics proceed to further in vivo tests.
4.2. MitoQ and SkQ1
MitoQ and SkQ1 are the derivatives of ubiquinone and plastoquinone respectively (Fig. 3) attached via the C10 hydrophobic linker to the TPP cation. Importantly, both compounds may not only be oxidized by ROS but also reduced afterwards by the mitochondrial ETC. This ability makes these antioxidants ‘rechargeable’ in contrast to many other mitochondria-targeted antioxidants [108].
SkQ1 has lower pro-oxidant activity than MitoQ [100]. Thus, SkQ1 exerts antioxidant effects at very low concentrations. The range of working ‘antioxidant’ activity of SkQ1 is also larger than that of MitoQ, providing a potential advantage for clinical SkQ1 applications [100].
Interestingly, SkQ1 also possesses the ability to bind to mitochondrial cardiolipin and prevent its oxidation, just as the penetrating peptide SS-31 does [109]. MitoQ also binds to the cardiolipin, though with lower affinity than SkQ1 [109]. Nevertheless, unlike SS-31, MitoQ and SkQ1 do rely on ΔΨm to penetrate mitochondria.
Another important feature of both MitoQ and SkQ1 is the uncoupling activity. It was shown that TPP-based cations may be paired with fatty acids and these pairs cycle across the IMM increasing the transmembrane proton-conducting activity [110]. This feature may be used to treat a variety of conditions associated with mitochondrial hyperpolarization.
There are many other data concerning the in vitro properties of MitoQ and SkQ1 (see a recent review [111]). The main properties are decreased ROS levels, prevention of lipid peroxidation, decreased protein oxidation and prevention of cell apoptosis and necrosis. It should be mentioned that these compounds are active at nanomolar concentrations [111].
Mitochondria-targeted TPP-based compounds are inevitably lipophilic. SkQ1 and MitoQ have octanol-PBS partition coefficients of 13, 000:1 and 3, 000:1 respectively [100, 112]. It should be noted that TPP-based compounds are ‘membranophilic’ rather than lipophilic, since they tend to accumulate at the layer between water and octanol and at the lipid bilayer membranes. This may produce biased results in different experimental settings. The water solubility of these compounds was determined as ~ 1.1 µM. At higher concentrations they start to form micelles [100]. Orally consumed SkQ1 and MitoQ are subjected to first-pass metabolism in the liver and intestinal wall [113], thus reducing their quantities. The oral bioavailability of MitoQ was determined as ~ 10%, and the major metabolites in urine are glucuronides and sulfates of the reduced hydroquinone form, along with demethylated compounds [114]. Nevertheless, there is a possibility that cardiac, hepatic and/or renal failure may strongly affect the pharmacokinetics and the in vivo effectiveness of mitochondria-targeted lipophilic drugs.
Significant doses of TPP-based compounds could be fed safely to mice over long periods, coming to steady-state distributions within the heart, brain, liver and muscle [115]. Animal studies with tritiated compounds indicate that the half-life of TPP-based antioxidants is about 1.5 days [115]. In phase I trials, MitoQ showed satisfactory pharmacokinetic behavior with oral dosing at 80 mg (1 mg/kg), resulting in a plasma Cmax of ∼ 33 ng/ml and Tmax of ∼ 1 hour (h) [114].
MitoQ and SkQ1 have both shown many beneficial features in animal models, including neuroprotection and protection against ischemia-reperfusion injury, kidney diseases, metabolic syndrome and obesity, wound healing, SIRS, cardiovascular pathologies, eye diseases, arthritis and many age-related pathologies (reviewed in [111, 116-118]). Importantly, these compounds also exhibited anti-aging effects. For example, SkQ1 greatly improved the health and life span of the PolG ‘mutator’ mice [119] and MitoQ extended the life span of a transgenic C. elegans model of Alzheimer’s disease [120].
Based on impressive results, clinical trials began a decade ago (Table 1). Below, we review the results of clinical trials and the possible molecular mechanisms underlying the observed effects.
MitoQ is now available as a dietary supplement that is well tolerated and safety-approved when administered orally for three weeks to healthy young adults without adverse effects [121].
Another double-blinded and placebo-controlled study tested the efficacy of MitoQ against impaired vascular function in healthy older adults aged 60-79. After six weeks of administration of MitoQ, these individuals had improved endothelial function at ~ 42%, measured as brachial artery flow-mediated dilation. They also had reduced levels of plasma oxidized low-density lipoproteins, a marker of oxidative stress [102]. These results confirmed previously obtained preclinical data showing that four weeks of oral supplementation of MitoQ in old mice completely restored endothelium-dependent dilation by improving nitric oxide (NO) bioavailability [122]. Endothelial dysfunction is thought to be caused by reduced NO bioavailability due to increased oxidative stress [123, 124]. The mitochondria-derived superoxide anion reacts with NO to form peroxynitrite, which in turn can irreversibly damage tetrahydrobiopterin (a cofactor for endothelial NO synthase). Thus, NO synthase produces more additional superoxide, rather than NO [123]. SkQ1 also showed angioprotective and anti-inflammatory activity, lowering expression of NO synthase and interleukin-6 in lipopolysaccharide-treated mice [125].
SkQ1 decreased age-related heart hypertrophy in mice [126] and also prevented age-related inflammatory activation of endothelium in the aortas of old mice via NF-κB-dependent reduced expression of cell adhesion molecules (ICAM-1) [26, 127]. Probably, the complex interplay between mtROS and endothelium influences many signaling pathways that require further investigation.
MitoQ demonstrated promising results in a cell model of Parkinson’s disease [128]. However, in human clinical trials, MitoQ had no effect on the PD progression [103]. Probably, lack of MitoQ efficacy could be due to insufficient brain penetration or the severity of neuronal damage and dopaminergic deficiency at the time the treatment was applied [103].
Hepatitis C virus (HCV) infection induces mitochondrial oxidative stress leading to cell death and tissue fibrosis in liver [129]. Randomized placebo-controlled clinical trials of MitoQ in patients with chronic HCV infection did not result in a decrease of viral load but significantly decreased plasma alanine transaminase and aspartate aminotransferase levels, thus indicating reduced liver damage [104]. In agreement with these results, both MitoQ and SkQ1 also protected against liver damage in various animal models [130, 131].
Early animal studies of SkQ1 revealed its high efficacy against a wide range of eye diseases, such as dry eye syndrome, retinopathy, glaucoma, uveitis and conjunctivitis [100, 132-134]. The first human clinical trials of the SkQ1-based Visomitin eye drops for treatment of dry eye syndrome were successfully carried out in 2011. Visomitin greatly reduced hyperemia and edema of the conjunctiva and healed corneal microerosions [135]. In another international, multicenter, randomized, double-masked, placebo-controlled clinical study of Visomitin, the eye drops increased tear film stability and reduced corneal damage in patients with dry eye syndrome [136]. Phase 2 US clinical trials of Visomitin eye drops for safety and efficacy confirmed significant improvements of the signs and symptoms of dry eye [105]. The underlying mechanism of SkQ1 therapeutic action is likely to involve protection of the cardiolipin against oxidation [100]. In a rabbit model of dry eye disease, SkQ1 also stimulated the activity of the antioxidant enzymes (glutathione peroxidase and glutathione reductase) and lowered pro-inflammatory cytokine production in tears [137]. Nevertheless, most of the involved signaling pathways affected by SkQ1 probably remain unstudied.
CONCLUSION
Mitochondria are crucial cellular organelles, and mitochondrial dysfunction, accompanied by excessive mtROS production, decreased ATP synthesis and increased cell death, plays an important role in the pathogenesis of many diseases. Most of the prominent results in the treatment of these diseases were obtained with the lipophilic antioxidant cations MitoQ and SkQ1, or the penetrating peptide SS-31. These antioxidants were also investigated in multiple clinical trials completed up to phase 2. Further studies are needed to elucidate the signaling pathways influenced by these compounds, including their long-term effects on gene expression, proteomics, metabolomics and epigenetics. Such studies will also help in understanding the pathophysiology of mitochondria-related diseases.
The prospects for new drug discoveries for treating mitochondrial dysfunction will mostly depend on increasing the bioavailability of the membranophilic drugs. Unfortunately, current pharmaceutical technologies do not offer a ready solution, though the use of microemulsions seems to be very promising [138]. Another problem is that existing MTCs are not targeted to certain organs or tissues and their distribution in the body relies only on their physico-chemical properties. There is a need for the combination of both first- and second-order drug targeting with MTCs.
Acknowledgements
Declared none.
LIST OF ABBREVIATIONS
- AD
Alzheimer’s Disease
- ALS
Amyotrophic Lateral Sclerosis
- Aβ
Amyloid β Peptide
- ATP
Adenosine Triphosphate
- DAMPs
Damage-associated Molecular Patterns
- DQA
Dequalinium
- FADH2
Flavin Adenine Dinucleotide
- HCV
Hepatitis C Virus
- ETC
Electron Transport Chain
- ICAM-1
Intercellular Adhesion Molecule 1
- IMM
Inner Mitochondrial Membrane
- MPT
Mitochondrial Permeability Transition
- MTC
Mitochondria-targeted Compound
- mtDNA
Mitochondrial DNA
- mtROS
Mitochondrial Reactive Oxygen Species
- NADH
Nicotinamide Adenine Dinucleotide
- NO
Nitric Oxide
- OMM
Outer Mitochondrial Membrane
- OXPHOS
Oxidative Phosphorylation
- PAMPs
Pathogen-associated Molecular Patterns
- PD
Parkinson’s Disease
- ROS
Reactive Oxygen Species
- SIRS
Systemic Inflammatory Response Syndrome
- SOD
Superoxide Dismutase
- T2DM
Type 2 Diabetes Mellitus
- TCA
Tricarboxylic Acid Cycle
- TPP
Triphenylphosphonium Cation
- UCP1
Uncoupling Protein 1
- ΔΨm
Transmembrane Potential
Consent for Publication
Not applicable.
Funding
The work of the authors was supported by grants from the Russian Science Foundation (No.14-24-00107) (sections about mitochondria-targeted compounds) and from the Russian Foundation for Basic Research (No. 18-04-01110).
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Davis S.S. Biomedical applications of nanotechnology--implications for drug targeting and gene therapy. Trends Biotechnol. 1997;15(6):217–224. doi: 10.1016/S0167-7799(97)01036-6. [DOI] [PubMed] [Google Scholar]
- 2.Keservani R.K., Sharma A.K., Kesharwani R.K. CRC Press. 2017. Drug Delivery Approaches and Nanosystems, Volume 2: Drug Targeting Aspects of Nanotechnology. [Google Scholar]
- 3.Burns R.J., Smith R.A., Murphy M.P. Synthesis and characterization of thiobutyltriphenylphosphonium bromide, a novel thiol reagent targeted to the mitochondrial matrix. Arch. Biochem. Biophys. 1995;322(1):60–68. doi: 10.1006/abbi.1995.1436. [DOI] [PubMed] [Google Scholar]
- 4.Wallace D.C., Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholls D.G., Bernson V.S., Heaton G.M. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl. 1978;32:89–93. doi: 10.1007/978-3-0348-5559-4_9. [DOI] [PubMed] [Google Scholar]
- 6.Bagur R., Hajnóczky G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell. 2017;66(6):780–788. doi: 10.1016/j.molcel.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clapham D.E. Calcium signaling. Cell. 2007;131(6):1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 8.Lemasters J.J. Molecular Pathology. Elsevier; 2018. Molecular Mechanisms of Cell Death. pp. 1–24. [Google Scholar]
- 9.Kim J-S., He L., Lemasters J.J. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003;304(3):463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 10.Kalkavan H., Green D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. doi: 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chautan M., Chazal G., Cecconi F., Gruss P., Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr. Biol. 1999;9(17):967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- 12.Angelova P.R., Abramov A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016;100:81–85. doi: 10.1016/j.freeradbiomed.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial ROS-induced ROS release: an update and review. Biochim. Biophys. Acta (BBA)-. Bioenergetics. 2006;1757(5-6):509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 14.Orrenius S., Gogvadze V., Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 15.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 16.Angelova P.R., Kasymov V., Christie I., Sheikhbahaei S., Turovsky E., Marina N., Korsak A., Zwicker J., Teschemacher A.G., Ackland G.L., Funk G.D., Kasparov S., Abramov A.Y., Gourine A. V Functional Oxygen Sensitivity of Astrocytes. J. Neurosci. 2015;35(29):10460–10473. doi: 10.1523/JNEUROSCI.0045-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedard K., Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 18.Klebanoff S.J. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 2005;77(5):598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 19.Kellogg E.W., Fridovich I. Superoxide, hydrogen peroxide, and singlet oxygen in lipid peroxidation by a xanthine oxidase system. J. Biol. Chem. 1975;250(22):8812–8817. [PubMed] [Google Scholar]
- 20.Pizzinat N., Copin N., Vindis C., Parini A., Cambon C. Reactive oxygen species production by monoamine oxidases in intact cells. Naunyn Schmiedebergs Arch. Pharmacol. 1999;359(5):428–431. doi: 10.1007/pl00005371. [DOI] [PubMed] [Google Scholar]
- 21.Montezano A.C., Touyz R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012;110(1):87–94. doi: 10.1111/j.1742-7843.2011.00785.x. [DOI] [PubMed] [Google Scholar]
- 22.Sahoo S., Meijles D.N., Pagano P.J. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin. Sci. (Lond.) 2016;130(5):317–335. doi: 10.1042/CS20150087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nauseef W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008;283(25):16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rastogi R., Geng X., Li F., Ding Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016;10:301. doi: 10.3389/fncel.2016.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vorobjeva N., Prikhodko A., Galkin I., Pletjushkina O., Zinovkin R., Sud’ina G., Chernyak B., Pinegin B. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur. J. Cell Biol. 2017;96(3):254–265. doi: 10.1016/j.ejcb.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Zinovkin R.A., Romaschenko V.P., Galkin I.I., Zakharova V.V., Pletjushkina O.Y., Chernyak B.V., Popova E.N. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging (Albany NY) 2014;6(8):661–674. doi: 10.18632/aging.100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukhopadhyay P., Horváth B., Zsengellér Z., Zielonka J., Tanchian G., Holovac E., Kechrid M., Patel V., Stillman I.E., Parikh S.M., Joseph J., Kalyanaraman B., Pacher P. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012;52(2):497–506. doi: 10.1016/j.freeradbiomed.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuppen H.A.L., Blakely E.L., Turnbull D.M., Taylor R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta Bioenerg. 2010;1797(2):113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Lightowlers R.N., Taylor R.W., Turnbull D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science. 2015;349(6255):1494–1499. doi: 10.1126/science.aac7516. [DOI] [PubMed] [Google Scholar]
- 30.Phielix E., Schrauwen-Hinderling V.B., Mensink M., Lenaers E., Meex R., Hoeks J., Kooi M.E., Moonen-Kornips E., Sels J-P., Hesselink M.K.C., Schrauwen P. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57(11):2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelley D.E., He J., Menshikova E.V., Ritov V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 32.Ritov V.B., Menshikova E.V., Azuma K., Wood R., Toledo F.G.S., Goodpaster B.H., Ruderman N.B., Kelley D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010;298(1):E49–E58. doi: 10.1152/ajpendo.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mihalik S.J., Goodpaster B.H., Kelley D.E., Chace D.H., Vockley J., Toledo F.G.S., DeLany J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010;18(9):1695–1700. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newgard C.B., An J., Bain J.R., Muehlbauer M.J., Stevens R.D., Lien L.F., Haqq A.M., Shah S.H., Arlotto M., Slentz C.A., Rochon J., Gallup D., Ilkayeva O., Wenner B.R., Yancy W.S., Eisenson H., Musante G., Surwit R.S., Millington D.S., Butler M.D., Svetkey L.P. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams S.H., Hoppel C.L., Lok K.H., Zhao L., Wong S.W., Minkler P.E., Hwang D.H., Newman J.W., Garvey W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009;139(6):1073–1081. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson E.J., Lustig M.E., Boyle K.E., Woodlief T.L., Kane D.A., Lin C-T., Price J.W., Kang L., Rabinovitch P.S., Szeto H.H., Houmard J.A., Cortright R.N., Wasserman D.H., Neufer P.D. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown D.A., Perry J.B., Allen M.E., Sabbah H.N., Stauffer B.L., Shaikh S.R., Cleland J.G.F., Colucci W.S., Butler J., Voors A.A., Anker S.D., Pitt B., Pieske B., Filippatos G., Greene S.J., Gheorghiade M. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017;14(4):238–250. doi: 10.1038/nrcardio.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halestrap A.P., Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta. 2009;1787(11):1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 39.Boudina S., Sena S., O’Neill B.T., Tathireddy P., Young M.E., Abel E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 40.Rontoyanni V.G., Nunez Lopez O., Fankhauser G.T., Cheema Z.F., Rasmussen B.B., Porter C. Mitochondrial Bioenergetics in the Metabolic Myopathy Accompanying Peripheral Artery Disease. Front. Physiol. 2017;8:141. doi: 10.3389/fphys.2017.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swerdlow R.H., Burns J.M., Khan S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010;20(Suppl. 2):S265–S279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Onyango I.G., Dennis J., Khan S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016;7(2):201–214. doi: 10.14336/AD.2015.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lustbader J.W., Cirilli M., Lin C., Xu H.W., Takuma K., Wang N., Caspersen C., Chen X., Pollak S., Chaney M., Trinchese F., Liu S., Gunn-Moore F., Lue L-F., Walker D.G., Kuppusamy P., Zewier Z.L., Arancio O., Stern D., Yan S.S., Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 44.Manczak M., Anekonda T.S., Henson E., Park B.S., Quinn J., Reddy P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006;15(9):1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 45.Moreira P.I., Zhu X., Wang X., Lee H-G., Nunomura A., Petersen R.B., Perry G., Smith M.A. Mitochondria: A therapeutic target in neurodegeneration. Biochim. Biophys. Acta. 2010;1802(1):212–220. doi: 10.1016/j.bbadis.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beal M.F. Mitochondria and neurodegeneration. Novartis Found. Symp. 2007;287:183–192. doi: 10.1002/9780470725207.ch13. [DOI] [PubMed] [Google Scholar]
- 47.Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A., Hoffer B., Cullheim S., Mohammed A.H., Olson L., Larsson N-G. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA. 2007;104(4):1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galter D., Pernold K., Yoshitake T., Lindqvist E., Hoffer B., Kehr J., Larsson N-G., Olson L. MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson’s disease. Genes Brain Behav. 2010;9(2):173–181. doi: 10.1111/j.1601-183X.2009.00542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carmo C., Naia L., Lopes C., Rego A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018;1049:59–83. doi: 10.1007/978-3-319-71779-1_3. [DOI] [PubMed] [Google Scholar]
- 50.Beal M.F., Brouillet E., Jenkins B.G., Ferrante R.J., Kowall N.W., Miller J.M., Storey E., Srivastava R., Rosen B.R., Hyman B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993;13(10):4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi P., Gal J., Kwinter D.M., Liu X., Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2010;1802(1):45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Igoudjil A., Magrané J., Fischer L.R., Kim H.J., Hervias I., Dumont M., Cortez C., Glass J.D., Starkov A.A., Manfredi G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci. 2011;31(44):15826–15837. doi: 10.1523/JNEUROSCI.1965-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffman E.P., Brown R.H., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 54.Sperl W., Skladal D., Gnaiger E., Wyss M., Mayr U., Hager J., Gellerich F.N. High resolution respirometry of permeabilized skeletal muscle fibers in the diagnosis of neuromuscular disorders. Mol. Cell. Biochem. 1997;174(1-2):71–78. [PubMed] [Google Scholar]
- 55.Vila M.C., Rayavarapu S., Hogarth M.W., Van der Meulen J.H., Horn A., Defour A., Takeda S., Brown K.J., Hathout Y., Nagaraju K., Jaiswal J.K. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017;24(2):330–342. doi: 10.1038/cdd.2016.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly-Worden M., Thomas E. Mitochondrial Dysfunction in Duchenne Muscular Dystrophy. Open J. Endocr. Metab. Dis. 2014;4(8):211–218. [Google Scholar]
- 57.Rygiel K.A., Miller J., Grady J.P., Rocha M.C., Taylor R.W., Turnbull D.M. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 2015;41(3):288–303. doi: 10.1111/nan.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balk R.A. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. 2014;5(1):20–26. doi: 10.4161/viru.27135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rangel-Frausto M.S., Pittet D., Costigan M., Hwang T., Davis C.S., Wenzel R.P. The natural history of the systemic inflammatory response syndrome (SIRS): A prospective study. JAMA. 1995;273(2):117–123. [PubMed] [Google Scholar]
- 60.Zakharova V.V., Pletjushkina O.Y., Zinovkin R.A., Popova E.N., Chernyak B. V Mitochondria-Targeted Antioxidants and Uncouplers of Oxidative Phosphorylation in Treatment of the Systemic Inflammatory Response Syndrome (SIRS). J. Cell. Physiol. 2017;232(5):904–912. doi: 10.1002/jcp.25626. [DOI] [PubMed] [Google Scholar]
- 61.Jo E-K., Kim J.K., Shin D-M., Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016;13(2):148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong Z., Liang S., Sanchez-Lopez E., He F., Shalapour S., Lin X., Wong J., Ding S., Seki E., Schnabl B., Hevener A.L., Greenberg H.B., Kisseleva T., Karin M. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prikhodko A.S., Vitushkina M.V., Zinovkina L.A., Popova E.N., Zinovkin R.A. Priming of human neutrophils is necessary for their activation by extracellular DNA. Biochem. 2016;81(6):609–614. doi: 10.1134/S0006297916060079. [DOI] [PubMed] [Google Scholar]
- 65.Chakraborty K., Raundhal M., Chen B.B., Morse C., Tyurina Y.Y., Khare A., Oriss T.B., Huff R., Lee J.S., St Croix C.M., Watkins S., Mallampalli R.K., Kagan V.E., Ray A., Ray P. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 2017;8:13944. doi: 10.1038/ncomms13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raoof M., Zhang Q., Itagaki K., Hauser C.J. Mitochondrial Peptides Are Potent Immune Activators That Activate Human Neutrophils Via FPR-1. J. Trauma Inj. Infect. Crit. Care. 2010;68(6):1328–1334. doi: 10.1097/TA.0b013e3181dcd28d. [DOI] [PubMed] [Google Scholar]
- 67.Oliveira P.J., editor. Mitochondrial Biology and Experimental Therapeutics. Cham: Springer; 2018. [Google Scholar]
- 68.Sun N., Youle R.J., Finkel T. The Mitochondrial Basis of Aging. Mol. Cell. 2016;61(5):654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harman D. The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 1972;20(4):145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 70.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly-Y M., Gidlöf S., Oldfors A., Wibom R., Törnell J., Jacobs H.T., Larsson N-G. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 71.Schriner S.E., Linford N.J., Martin G.M., Treuting P., Ogburn C.E., Emond M., Coskun P.E., Ladiges W., Wolf N., Van Remmen H., Wallace D.C., Rabinovitch P.S. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 72.Kennedy S.R., Salk J.J., Schmitt M.W., Loeb L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9(9):e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chung H.Y., Cesari M., Anton S., Marzetti E., Giovannini S., Seo A.Y., Carter C., Yu B.P., Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009;8(1):18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Judge S., Jang Y.M., Smith A., Hagen T., Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19(3):419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 75.Ungvari Z., Orosz Z., Labinskyy N., Rivera A., Xiangmin Z., Smith K., Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2007;293(1):H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 76.Dai D-F., Rabinovitch P.S., Ungvari Z. Mitochondria and cardiovascular aging. Circ. Res. 2012;110(8):1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanada F., Taniyama Y., Muratsu J., Otsu R., Shimizu H., Rakugi H., Morishita R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018;5:12. doi: 10.3389/fcvm.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S.D., Quiros P.M., Steffen K.K., Williams E.G., Mouchiroud L., Tronnes S.U., Murillo V., Wolff S.C., Shaw R.J., Auwerx J., Dillin A. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165(5):1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tian Y., Garcia G., Bian Q., Steffen K.K., Joe L., Wolff S., Meyer B.J., Dillin A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR mt. Cell. 2016;165(5):1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gray M.W., Burger G., Lang B.F. Mitochondrial evolution. Science. 1999;283(5407):1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 81.Adam-Vizi V., Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006;27(12):639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 82.Kagan V.E., Borisenko G.G., Tyurina Y.Y., Tyurin V.A., Jiang J., Potapovich A.I., Kini V., Amoscato A.A., Fujii Y. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004;37(12):1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 83.Pfanner N., Geissler A. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2001;2(5):339–349. doi: 10.1038/35073006. [DOI] [PubMed] [Google Scholar]
- 84.Liberman E.A., Topaly V.P., Tsofina L.M., Jasaitis A.A., Skulachev V.P. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature. 1969;222(5198):1076–1078. doi: 10.1038/2221076a0. [DOI] [PubMed] [Google Scholar]
- 85.Smith R.A., Porteous C.M., Coulter C.V., Murphy M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999;263(3):709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 86.Amorim R., Benfeito S., Teixeira J., Cagide F., Oliveira P.J., Borges F. In: Mitochondrial Biology and Experimental Therapeutics. Oliveira P.J., editor. Cham: Springer; 2018. pp. 333–358. [Google Scholar]
- 87.Weissig V. In: Mitochondrial Medicine. Weissig V., Edeas M., editors. Vol. II. Humana Press; 2015. pp. 1–11. [Google Scholar]
- 88.D’Souza G.G.M., Boddapati S.V., Weissig V. Mitochondrial leader sequence-plasmid DNA conjugates delivered into mammalian cells by DQAsomes co-localize with mitochondria. Mitochondrion. 2005;5(5):352–358. doi: 10.1016/j.mito.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 89.Zhao K., Zhao G-M., Wu D., Soong Y., Birk A.V., Schiller P.W., Szeto H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004;279(33):34682–34990. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 90.Birk A.V., Liu S., Soong Y., Mills W., Singh P., Warren J.D., Seshan S.V., Pardee J.D., Szeto H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013;24(8):1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szeto H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006;8(2):E277–E283. doi: 10.1007/BF02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cerrato C.P., Langel Ü. Mitochondrial Biology and Experimental Therapeutics. Cham: Springer International Publishing; 2018. Cell-Penetrating Peptides Targeting Mitochondria. pp. 593–611. [Google Scholar]
- 93.Szeto H.H., Birk A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014;96(6):672–683. doi: 10.1038/clpt.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heijne G. von Mitochondrial targeting sequences may form amphiphilic helices. EMBO J. 1986;5(6):1335–1342. doi: 10.1002/j.1460-2075.1986.tb04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neupert W., Herrmann J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 96.Flierl A., Jackson C., Cottrell B., Murdock D., Seibel P., Wallace D.C. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol. Ther. 2003;7(4):550–557. doi: 10.1016/s1525-0016(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 97.Yu H., Koilkonda R.D., Chou T-H., Porciatti V., Ozdemir S.S., Chiodo V., Boye S.L., Boye S.E., Hauswirth W.W., Lewin A.S., Guy J. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA. 2012;109(20):E1238–E1247. doi: 10.1073/pnas.1119577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cha M-Y., Han S-H., Son S.M., Hong H-S., Choi Y-J., Byun J., Mook-Jung I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One. 2012;7(4):e34929. doi: 10.1371/journal.pone.0034929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 100.Antonenko Y.N., Avetisyan A.V., Bakeeva L.E., Chernyak B.V., Chertkov V.A., Domnina L.V., Ivanova O.Y., Izyumov D.S., Khailova L.S., Klishin S.S., Korshunova G.A., Lyamzaev K.G., Muntyan M.S., Nepryakhina O.K., Pashkovskaya A.A., Pletjushkina O.Y., Pustovidko A.V., Roginsky V.A., Rokitskaya T.I., Ruuge E.K., Saprunova V.B., Severina I.I., Simonyan R.A., Skulachev I.V., Skulachev M.V., Sumbatyan N.V., Sviryaeva I.V., Tashlitsky V.N., Vassiliev J.M., Vyssokikh M.Y., Yaguzhinsky L.S., Zamyatnin A.A., Skulachev V.P. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry (Mosc.) 2008;73(12):1273–1287. doi: 10.1134/s0006297908120018. [DOI] [PubMed] [Google Scholar]
- 101.Fink B.D., Herlein J.A., Yorek M.A., Fenner A.M., Kerns R.J., Sivitz W.I. Bioenergetic effects of mitochondrial-targeted coenzyme Q analogs in endothelial cells. J. Pharmacol. Exp. Ther. 2012;342(3):709–719. doi: 10.1124/jpet.112.195586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rossman M.J., Santos-Parker J.R., Steward C.A.C., Bispham N.Z., Cuevas L.M., Rosenberg H.L., Woodward K.A., Chonchol M., Gioscia-Ryan R.A., Murphy M.P., Seals D.R. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertens. (Dallas, Tex. 1979) 2018;71(6):1056–1063. doi: 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O’Sullivan J.D., Fung V., Smith R.A.J., Murphy M.P., Taylor K.M. Protect Study Group A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010;25(11):1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 104.Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M., Frampton C.M., Taylor K.M., Smith R.A.J., Murphy M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30(7):1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 105.Petrov A., Perekhvatova N., Skulachev M., Stein L., Ousler G. SkQ1 Ophthalmic Solution for Dry Eye Treatment: Results of a Phase 2 Safety and Efficacy Clinical Study in the Environment and During Challenge in the Controlled Adverse Environment Model. Adv. Ther. 2016;33(1):96–115. doi: 10.1007/s12325-015-0274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sies H. Total antioxidant capacity: Appraisal of a concept. J. Nutr. 2007;137(6):1493–1495. doi: 10.1093/jn/137.6.1493. [DOI] [PubMed] [Google Scholar]
- 107.Gasparovic A.C., Jaganjac M., Mihaljevic B., Sunjic S.B., Zarkovic N. Assays for the measurement of lipid peroxidation. Methods Mol. Biol. 2013;965:283–296. doi: 10.1007/978-1-62703-239-1_19. [DOI] [PubMed] [Google Scholar]
- 108.Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276(7):4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 109.Skulachev V.P., Antonenko Y.N., Cherepanov D.A., Chernyak B.V., Izyumov D.S., Khailova L.S., Klishin S.S., Korshunova G.A., Lyamzaev K.G., Pletjushkina O.Y., Roginsky V.A., Rokitskaya T.I., Severin F.F., Severina I.I., Simonyan R.A., Skulachev M.V., Sumbatyan N.V., Sukhanova E.I., Tashlitsky V.N., Trendeleva T.A., Vyssokikh M.Y., Zvyagilskaya R.A. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim. Biophys. Acta Bioenerg. 2010;1797(6-7):878–889. doi: 10.1016/j.bbabio.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 110.Severin F.F., Severina I.I., Antonenko Y.N., Rokitskaya T.I., Cherepanov D.A., Mokhova E.N., Vyssokikh M.Y., Pustovidko A.V., Markova O.V., Yaguzhinsky L.S., Korshunova G.A., Sumbatyan N.V., Skulachev M.V., Skulachev V.P. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc. Natl. Acad. Sci. USA. 2010;107(2):663–668. doi: 10.1073/pnas.0910216107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feniouk B.A., Skulachev V.P. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017;10(1):41–48. doi: 10.2174/1874609809666160921113706. [DOI] [PubMed] [Google Scholar]
- 112.Asin-Cayuela J., Manas A-R.B., James A.M., Smith R.A.J., Murphy M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571(1-3):9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 113.Pond S.M., Tozer T.N. First-pass elimination. Basic concepts and clinical consequences. Clin. Pharmacokinet. •••;9(1):1–25. doi: 10.2165/00003088-198409010-00001. [DOI] [PubMed] [Google Scholar]
- 114.Murphy M.P., Smith R.A.J. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 115.Smith R.A.J., Porteous C.M., Gane A.M., Murphy M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA. 2003;100(9):5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kezic A., Spasojevic I., Lezaic V., Bajcetic M. Mitochondria-Targeted Antioxidants: Future Perspectives in Kidney Ischemia Reperfusion Injury. Oxid. Med. Cell. Longev. 2016;2016:1–12. doi: 10.1155/2016/2950503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Silva F.S.G., Simoes R.F., Couto R., Oliveira P.J. Targeting Mitochondria in Cardiovascular Diseases. Curr. Pharm. Des. 2016;22(37):5698–5717. doi: 10.2174/1381612822666160822150243. [DOI] [PubMed] [Google Scholar]
- 118.Braakhuis A.J., Nagulan R., Somerville V. The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2018;2018:8575263. doi: 10.1155/2018/8575263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shabalina I.G., Vyssokikh M.Y., Gibanova N., Csikasz R.I., Edgar D., Hallden-Waldemarson A., Rozhdestvenskaya Z., Bakeeva L.E., Vays V.B., Pustovidko A.V., Skulachev M.V., Cannon B., Skulachev V.P., Nedergaard J. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging (Albany NY) 2017;9(2):315–339. doi: 10.18632/aging.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ng L.F., Gruber J., Cheah I.K., Goo C.K., Cheong W.F., Shui G., Sit K.P., Wenk M.R., Halliwell B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014;71:390–401. doi: 10.1016/j.freeradbiomed.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 121.Shill D.D., Southern W.M., Willingham T.B., Lansford K.A., McCully K.K., Jenkins N.T. Mitochondria-specific antioxidant supplementation does not influence endurance exercise training-induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J. Physiol. 2016;594(23):7005–7014. doi: 10.1113/JP272491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gioscia-Ryan R.A., LaRocca T.J., Sindler A.L., Zigler M.C., Murphy M.P., Seals D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014;592(12):2549–2561. doi: 10.1113/jphysiol.2013.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van der Loo B., Labugger R., Skepper J.N., Bachschmid M., Kilo J., Powell J.M., Palacios-Callender M., Erusalimsky J.D., Quaschning T., Malinski T., Gygi D., Ullrich V., Lüscher T.F. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000;192(12):1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bachschmid M.M., Schildknecht S., Matsui R., Zee R., Haeussler D., Cohen R.A., Pimental D., Loo B. van der Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann. Med. 2013;45(1):17–36. doi: 10.3109/07853890.2011.645498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Weidinger A., Müllebner A., Paier-Pourani J., Banerjee A., Miller I., Lauterböck L., Duvigneau J.C., Skulachev V.P., Redl H., Kozlov A.V. Vicious inducible nitric oxide synthase-mitochondrial reactive oxygen species cycle accelerates inflammatory response and causes liver injury in rats. Antioxid. Redox Signal. 2015;22(7):572–586. doi: 10.1089/ars.2014.5996. [DOI] [PubMed] [Google Scholar]
- 126.Manskikh V.N., Gancharova O.S., Nikiforova A.I., Krasilshchikova M.S., Shabalina I.G., Egorov M.V., Karger E.M., Milanovsky G.E., Galkin I.I., Skulachev V.P., Zinovkin R.A. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histol. Histopathol. 2015;30(3):353–360. doi: 10.14670/HH-30.353. [DOI] [PubMed] [Google Scholar]
- 127.Galkin I.I., Pletjushkina O.Y., Zinovkin R.A., Zakharova V.V., Birjukov I.S., Chernyak B.V., Popova E.N. Mitochondria-targeted antioxidants prevent TNFα-induced endothelial cell damage. Biochemistry (Mosc.) 2014;79(2):124–130. doi: 10.1134/S0006297914020059. [DOI] [PubMed] [Google Scholar]
- 128.Mao P., Manczak M., Shirendeb U.P., Reddy P.H. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta. 2013;1832(12):2322–2331. doi: 10.1016/j.bbadis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Korenaga M., Wang T., Li Y., Showalter L.A., Chan T., Sun J., Weinman S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005;280(45):37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 130.Chacko B.K., Srivastava A., Johnson M.S., Benavides G.A., Chang M.J., Ye Y., Jhala N., Murphy M.P., Kalyanaraman B., Darley-Usmar V.M. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology. 2011;54(1):153–163. doi: 10.1002/hep.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lukashev A.N., Skulachev M.V., Ostapenko V., Savchenko A.Y., Pavshintsev V.V., Skulachev V.P. Advances in development of rechargeable mitochondrial antioxidants. Prog. Mol. Biol. Transl. Sci. 2014;127:251–265. doi: 10.1016/B978-0-12-394625-6.00010-6. [DOI] [PubMed] [Google Scholar]
- 132.Skulachev V.P., Anisimov V.N., Antonenko Y.N., Bakeeva L.E., Chernyak B.V., Erichev V.P., Filenko O.F., Kalinina N.I., Kapelko V.I., Kolosova N.G., Kopnin B.P., Korshunova G.A., Lichinitser M.R., Obukhova L.A., Pasyukova E.G., Pisarenko O.I., Roginsky V.A., Ruuge E.K., Senin I.I., Severina I.I., Skulachev M.V., Spivak I.M., Tashlitsky V.N., Tkachuk V.A., Vyssokikh M.Y., Yaguzhinsky L.S., Zorov D.B. An attempt to prevent senescence: a mitochondrial approach. Biochim. Biophys. Acta. 2009;1787(5):437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 133.Markovets A.M., Fursova A.Z., Kolosova N.G. Therapeutic action of the mitochondria-targeted antioxidant SkQ1 on retinopathy in OXYS rats linked with improvement of VEGF and PEDF gene expression. PLoS One. 2011;6(7):e21682. doi: 10.1371/journal.pone.0021682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iomdina E.N., Khoroshilova-Maslova I.P., Robustova O.V., Averina O.A., Kovaleva N.A., Aliev G., Reddy V.P., Zamyatnin A.A., Skulachev M.V., Senin I.I., Skulachev V.P. Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Front. Biosci. Landmark Ed. 2015;20(1):892–901. doi: 10.2741/4343. [DOI] [PubMed] [Google Scholar]
- 135.Yani E.V., Katargina L.A., Chesnokova N.B., Beznos O.V., Savchenko A.Y., Vygodin V.A., Gudkova E.Y., Zamyatnin J.A.A., Skulachev M.V. The first experience of using the drug Vizomitin in the treatment of dry eyes. Per. Med. 2012;4(59):134–137. [Google Scholar]
- 136.Brzheskiy V.V., Efimova E.L., Vorontsova T.N., Alekseev V.N., Gusarevich O.G., Shaidurova K.N., Ryabtseva A.A., Andryukhina O.M., Kamenskikh T.G., Sumarokova E.S., Miljudin E.S., Egorov E.A., Lebedev O.I., Surov A.V., Korol A.R., Nasinnyk I.O., Bezditko P.A., Muzhychuk O.P., Vygodin V.A., Yani E.V., Savchenko A.Y., Karger E.M., Fedorkin O.N., Mironov A.N., Ostapenko V., Popeko N.A., Skulachev V.P., Skulachev M.V. Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 2015;32(12):1263–1279. doi: 10.1007/s12325-015-0273-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zernii E.Y., Gancharova O.S., Baksheeva V.E., Golovastova M.O., Kabanova E.I., Savchenko M.S., Tiulina V.V., Sotnikova L.F., Zamyatnin A.A., Philippov P.P., Senin I.I. Mitochondria-Targeted Antioxidant SkQ1 Prevents Anesthesia-Induced Dry Eye Syndrome. Oxid. Med. Cell. Longev. 2017;2017:1–17. doi: 10.1155/2017/9281519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Callender S.P., Mathews J.A., Kobernyk K., Wettig S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017;526(1-2):425–442. doi: 10.1016/j.ijpharm.2017.05.005. [DOI] [PubMed] [Google Scholar]