

Abstract
As systems biology expands its multi-omic spectrum to increasing resolutions, distinguishing cells based on single-cell profiles becomes feasible. Unlike traditional bulk assays that average cellular responses and blur the distinct identities of responsive cells, single-cell technologies enable sensitive detection of small cellular changes and precise identification of those cells perturbed by toxicants. Among the suite of omic technologies that continue to expand and become affordable, single-cell RNA sequencing (scRNA-seq) is at the cutting edge and leading the way to transform systems toxicology. Single-cell systems toxicology can provide a wealth of information to elucidate cell-specific alterations and response trajectories, detect points-of-departure, map and develop dynamical models of toxicity pathways.
Keywords: single-cell RNA sequencing, transcriptomics, bulk assay, systems toxicology, computational toxicology, machine learning
Introduction:heterogeneity of single-cell responses and limitations of bulk assays
When the abundance of liver cytochrome P450 enzymes such as CYP1A1 and 1A2 was first examined some thirty years ago – through immunohistochemistry – in rats exposed to dioxin, phenobarbital or some other compounds, dose-dependent zonal induction of the enzymes dominated the observed patterns within the liver lobules [1,2]. In individual hepatocytes, these enzymes appeared to be induced in an all-or-none fashion; and as the dose of the test compounds increased, the zones of induced hepatocytes expanded either from the centrilobular to periportal regions or vice versa, depending on the specific enzymes and compounds examined. It was first thought that the location and thus the microenvironment of these hepatocytes may contribute to the heterogeneous responses between cells. However, cultured primary rat hepatocytes also responded in a heterogeneous fashion [3,4]. Of course, the use of primary cells did not rule out the possibility that these cells might have inherited their in vivo characteristics that underpin the heterogeneous responses observed in whole animals. Later studies using cell lines of hepatocytes, which eliminated any position-related variabilities, still showed binary yet asynchronous gene induction among cells, demonstrating that at least part of the heterogeneity resides inherently within an otherwise isogenic cell population [5,6]. Beyond liver enzyme induction, it is now widely recognized that cell-to-cell variability in basal states and in dynamical processes, such as proliferation, differentiation, apoptosis, and stress response, is a ubiquitous phenomenon even within a clonal population of cells and can be attributed to both extrinsic and intrinsic factors [7–12].
The ability to measure molecular changes in cells with high sensitivity and precision is essential for reliable determination of points-of-departure (PoDs) in biological responses. Its importance becomes even greater as toxicology and related fields in environmental health, in vitro toxicity testing and epidemiology move increasingly toward a multi-omic, systems biology approach, examining molecular alterations at the widest coverage achievable. Most bioassays, including microarray, RNA sequencing, and proteomic assays, were traditionally carried out in ensemble, at cell-population levels, despite the fact that the responses of individual cells are always heterogeneous and that the fraction of responsive cells often depends on the concentration of the perturbing agent. Cellular heterogeneity is masked in these bulk assays, in which cells, either as in a pure cell line or of mixed types from a tissue, are lysed in a single pool and the biomolecules released by all cells are quantified together to derive some mean values. This cell population-averaging approach is inherently insensitive to detect effects involving only a small fraction of the cell population. As illustrated in Fig. 1A, when assayed in aggregate where most cells are nonresponsive, large fold changes in gene expression in single, responsive cells are lost in the background, and the consequence of averaging is even worse for downregulated genes. For instance, changes in the levels of transcription factors (TFs), Pax5 and Blimp-1, which are down- and up-regulated respectively during antigen-stimulated B cell terminal differentiation, are much harder to detect in bulk lysis assays such as Western blot than in single-cell assays such as flow cytometry [13]. When tens of thousands of genes are measured in bulk assays such as in microarrays, low nominal fold changes can result in many false negatives given all the biological and technical variations and background noise. In contrast, measurements made at single-cell levels focused only on the responsive cells can be highly sensitive and specific, providing more reliable characterization of the responses at low doses and identification of PoDs.
Figure 1. Illustration of the biasing effects of bulk assays.
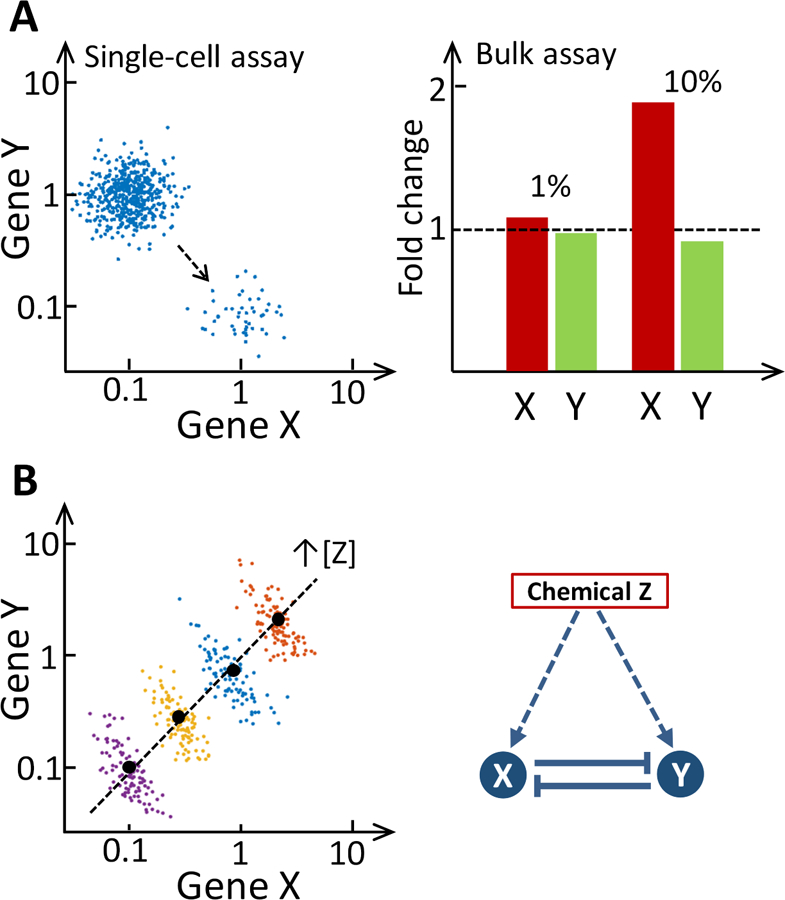
(A) Left panel: In a single-cell study, the small population of responsive cells and the magnitudes of gene expression changes in these cells can be easily distinguished from the nonresponsive population (each dot represents a cell). Right panel: Bulk assays averaging the responses of the total population disguise the magnitude of changes. For instance, if a perturbation renders 1% of the cell population responsive, in which the expression of gene X increases by 10-fold and that of gene Y decreases by 10-fold at single-cell level, then if measured with bulk assays, the expression of gene X will only increase by 9% ((10×1%+1×99%)/(1×100%) − 1 = 9%), and the expression of gene Y will only decrease by a meager 0.9% ((0.1×1%+1×99%)/(1×100%) − 1 = − 0.9%). When the responsive cells are 10% of the total population, gene X is upregulated by 90% ((10×10%+1×90%)/(1×100%) − 1 = 90%), but gene Y is downregulated by only 9% ((0.1×10%+1×90%)/(1×100%) − 1 = −9%) in bulk assays. (B) Right panel: Genes X and Y mutually repress each other but are commonly induced by chemical Z. Left panel: illustration of reversal of gene-gene correlation due to Simpson’s paradox. The true inverse correlation between the expression of genes X and Y can be revealed in single-cell studies (each color dot represents a cell) and clusters are obtained at different concentrations of chemical Z. However, with bulk assays which can only measure the averaged expression levels of genes X and Y (black dot in each cluster), the correlation between X and Y will appear positive when using the averages (dashed line).
Another problem with bulk assays is related to the so-called Simpson’s paradox, where correlations between biomolecular species observed within individual groups of cells may disappear or even reverse direction when the correlations are assessed across cell groups using the mean molecular abundance in each group [14]. These cell groups can either represent different cell types or they can be the same type of cells but are exposed to different doses of a chemical. As shown in Fig. 1B, if two genes mutually repress each other, within a population of cells under the same exposure condition, the expression of the two genes is inversely correlated because as one gene is transiently perturbed by noise, the expression of the other gene will likely move in the opposite direction. If a chemical dose-dependently upregulates both genes, the aforementioned inverse correlation still remains within each dose group. However, when using bulk assays to measure the average gene expression within each dose group and examining the correlation between the means across the dose groups, a completely opposite conclusion would be drawn: the two genes are co-expressed and thus positively correlated. As gene-gene relationships are often inferred through bulk assays conducted at different conditions such as different chemical doses, the resulting gene network can be wrong and misleading [15,16].
The sources and biological significance of cell-to-cell heterogeneity
As alluded to by the studies of enzyme induction in hepatocytes, cell-to-cell variability can result from both extrinsic and intrinsic factors. Cells of the same type residing in different locations within a tissue could be exposed to a different microscopic milieu, leading to different characteristics and functional variations. For instance, along the central-to-periportal axis of the liver lobule, there are concentration gradients for a number of extracellular factors, including oxygen, nutrients, hormones and morphogens, which may be partially responsible for the differential zonal behavior [17]. Within a tissue, extracellular factors may be necessary for initiating and maintaining distinct cell subtypes, but the molecular identities of cells may also be self-sustained through attractor states underpinned by feedback loops among TFs and/or epigenetic memory [18,19].
Cell-to-cell variations also exist in a clonal population of cells, as a result of stochastic gene expression [20]. This so-called gene expression noise can arise intrinsically from the randomness of biochemical reactions in the promotor, transcriptional, translational, and degradation events involving molecular entities present at low abundance. While stochasticity may be deleterious, it has also been shown to play important roles in determining cellular fates in development, pattern formation, dose response, and information transmission [11,21]. As a result of gene expression noise, two identical daughter cells may diverge in their gene expression levels over time and differ significantly after several rounds of cell division [22]. In this way, the cellular phenotype and response profile at the single-cell level can be transient as the gene expression states in cells fluctuate. This transient nature of the cellular phenotype is best demonstrated by cytotoxicity studies. When a clonal population of cells are exposed to an anti-tumor drug at a particular concentration, a fraction of cells are killed. After the drug is removed and the surviving cells are allowed to recover for several cell generations, a new population emerges. However, when the new population is exposed to the same drug at the same concentration as before, a similar fraction of cells are killed, as observed in the original population [23]. These experiments demonstrated that cellular response to chemical perturbation is not necessarily a fixed property for single cells, and stochastic gene expression can reconstitute surviving cells into a population whose distribution of heterogeneity resembles that of the original population.
Single-cell technologies
Traditional experimental approaches at single-cell resolutions include immunocytochemistry and in situ hybridization for within-tissue studies, flow cytometry for dissociated cells, and live cell imaging for real-time observations. While providing valuable information of single-cell behaviors, these techniques are limited in coverage and throughput and can be multiplexed to simultaneously monitor only a small number of proteins or mRNAs. Recent improvements in real-time quantitative reverse transcription PCR and mass cytometry allow measuring up to tens of genes and proteins, but still a far cry from a full omic-wide coverage [24,25]. In the past few years, rapid technological advancements have been made in nearly all omic-level single-cell assays. While affordable single-cell proteomic [26], metabolomic [27], and epigenomic [28] assays are still on the horizon, single-cell RNA sequencing (scRNA-seq) has evolved and matured to a state where this technology is economically accessible. Since its introduction in 2009 [29], this technology has quickly undergone multiple iterations of improvement, in areas including cell capturing, barcoding, reverse transcription, amplification, and sequencing, which lead to enhanced data quality, reduced batch effect, increased throughput and reduced cost. single-cell RNA sequencing (scRNA-seq) has been applied to study a number of organs and tissues including embryo, brain, bone marrow, liver, kidney, and blood. Embracing the promise of scRNA-seq, initiatives such as the Human Cell Atlas have been launched aiming to map every cell in the human body [30]. At the same time, an expanding suite of bioinformatic and machine-learning tools have flourished to handle and analyze the unique, high-dimensional datasets generated with scRNA-seq and help to extract biologically meaningful information as much as possible.
The rest of the article focuses on the potential applications of scRNA-seq in toxicology, a discipline yet to reap benefits from this new technology. Besides technical maturity and affordability, a transcriptomic focus is also justified by the general conclusion drawn from a number of functional toxicogenomic studies in recent years [31–33]. Most of these studies demonstrated that the benchmark chemical dose and time at which transcriptional alterations begin to appear in the most sensitive intracellular molecular pathways may also demarcate the PoD for an adverse apical-endpoint outcome. Recently the National Center for Computational Toxicology at the United States Environmental Protection Agency (USEPA) has prioritized high-throughput transcriptomics as one of the top-tier screening assays in their next-generation blueprint for chemical testing [34]. Embracing the potentials brought by single-cell technologies, the US National Academies’ Standing Committee on Use of Emerging Science for Environmental Health Decisions convened, in March, 2019, a workshop on the Promise of Single Cell and Single Molecule Analysis Tools to Advance Environmental Health Research. It is recognized that the molecular signatures of individual cells accessible through scRNA-seq and other single-cell omic technologies will bring unprecedented resolution and innovation to examining biological systems and their perturbations by chemicals.
Cell type identification and cell type-specific responses
One of the most common and useful applications of scRNA-seq is clustering cells in a biological sample, using a variety of unsupervised machine-learning algorithms, into distinct groups based on select features of the high-dimensional mRNA data [35,36]. Clustering is often conducted on reduced dimensions, achieved through dimension reduction algorithms, such as principle component analysis (PCA), a traditional linear approach that captures the global relationship between distinct clusters, and t-distributed stochastic neighbor-embedding (t-SNE), a more recent nonlinear approach that aims to preserve the local relationship between similar cells (Fig. 2A) [37]. While PCA and t-SNE can be applied in tandem to produce better spatially-resolved clusters, a novel nonlinear algorithm published in 2018, uniform manifold approximation and projection (UMAP), claimed to preserve the global structure and the local structure at nearly ten times the computational speed as t-SNE can deliver and has been quickly adopted by the scRNA-seq community [38]. Cell types and subtypes of the clusters, visualized on these low-dimension maps, can be identified based on domain knowledge of specific marker genes; however, novel cell types or subtypes, existing as rare physiological populations or emerging because of chemical perturbations, can also be discovered with this approach. A number of recent scRNA-seq studies of the liver, pancreas, brain, kidney, and so on confirmed that cell types, traditionally defined based on morphology, function, and biomarkers, are only crude classifications of a continuous spectrum of otherwise distinct subtypes, many of which were not previously known or appreciated [39–42]. In addition, by following up with single-molecule RNA fluorescence in situ hybridization or intersectional genetic approaches to microscopically locate the expression of landmark genes, identified cell types or subtypes can be mapped back to their anatomical positions in intact organs [41,43].
Figure 2. Illustration of dimension reduction, clustering and pseudotime trajectory of scRNA-seq data.
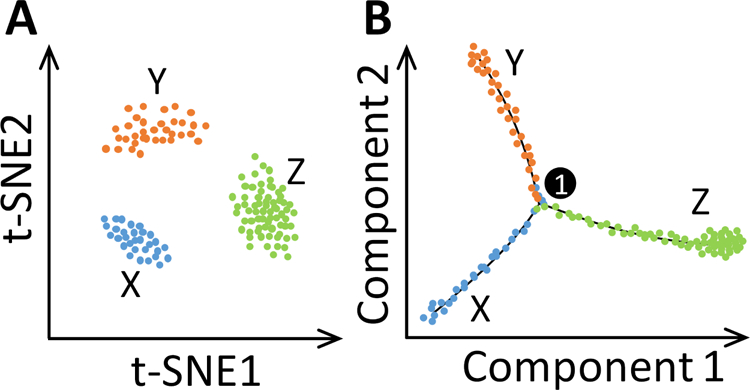
(A) Visualization and clustering of cells in a 2-D t-SNE map. Cluster X represents a precursor cell type, and clusters Y and Z represent the two cell types of sub-lineages of X. (B) Pseudotime trajectories showing ordering of cells based on their developmental pseudotime and bifurcation of precursor X into sub-lineages Y and Z. #1 in circle denotes the first bifurcation point (and the only one in this illustration). Each color dot represents a cell.
A recent single-cell study by Halpern et al. revealed characteristic division of labor among hepatocytes by showing that mouse hepatocytes can be clustered into 9 zonal layers within a liver lobule, each with a distinct transcriptomic profile [41]. While some genes were expressed monotonically across the zonal layers, many others were found to be nonmonotonic. Conceivably, toxicological studies of chemical perturbation followed by scRNA-seq can characterize differential activation of hepatocytes in different lobular zones by different chemicals, doses, and exposure durations. Such studies would help to better understand hepatic metabolism of chemicals and their potential hepatotoxicity, and provide new insights into organ-level responses. A recent scRNA-seq study of the kidney showed the traditionally defined glomerular and tubular cells and their subtypes, but also identified a novel cell type in the collecting duct that occupies a developmentally transitional state between principal and intercalated cells [40]. Future scRNA-seq studies of chemically induced perturbation can help to pinpoint the cellular targets of renal toxic chemicals under complex exposure conditions. One such scenario is cadmium accumulation in diabetic individuals, where cadmium selectively damages proximal tubular epithelia whereas high blood glucose damages glomerular endothelia and podocytes. Recent scRNA-seq studies of developing midbrains in mice and humans have revealed multiple dopaminergic (DA) lineage cells: radial glial-like cells, neuroprogenitors, medial neuroblasts, DA neuroblasts, and various immature and mature DA subtypes [39]. In the adult brain, as many as 5 DA neuron subtypes were identified with distinct but overlapping axonal projections and anatomical locations in the midbrain. Given the developmental neural toxicity of many insecticides in this brain region, it will be intriguing to investigate whether these compounds alter the DA neuron subtypes and their developmental trajectories to cause adverse neurobehavioral outcomes. A recent scRNA-seq study of asbestos-induced lung fibrosis in mice was able to identify, among tissue-resident interstitial macrophages, alveolar macrophages, and monocyte-derived alveolar macrophages present in the fibrotic niche, that only the latter subpopulation is causally associated with fibrosis, a result that is consistent with human pulmonary fibrosis studies [44]. In a study on the effects of tobacco-smoking, scRNA-seq of human peripheral blood mononuclear cells (PBMCs) revealed that natural killer-like CD8+ T lymphocytes, which are a rare subtype of T cells in nonsmokers, increase about 4 times, constituting nearly 9% of PBMCs in smokers [45].
Pseudotime, developmental trajectories, and cell state transition
One type of information that can be extracted from scRNA-seq data is “pseudotime”, which provides a timestamp to each cell in a heterogeneous population to specify where the cell resides along the biological trajectory following a physiological stimulation or perturbation. The most straightforward application of pseudotime is to order cells during development. Progression of individual cells differentiating along a lineage path is asynchronous, and pseudotime analysis takes advantage of this phenomenon to assign developmental timestamps to cells based on their transcriptomic similarity [46]. While pseudotime allows ordering of identified cell types according to their maturity in the lineage, pseudotime-based trajectory inference, using algorithms such as Monocle and Wishbone, takes it one step further to also delineate the bifurcation point where two sub-lineages branch out from common precursor cells (Fig. 2B) [47,48]. scRNA-seq studies have now revealed developmental trajectories in a variety of tissues, such as the brain, kidney, and immune system [39,48,49]. A potential application in toxicology is to identify possible alterations in developmental trajectories of cells perturbed by chemicals capable of producing developmental toxicity. Pseudotime analysis can also be applied to order cells according to their phase in the cell cycle or other biological rhythms, eliminating the need to experimentally synchronize these cells to maximize the ability to detect molecular changes obscured otherwise [50].
A related application of scRNA-seq is to predict critical cellular state transitions that may underpin the PoD to an adverse health outcome such as cancer. Such critical state transitions, often irreversible, can be driven by environmental toxicants accumulating in the human body. Stable cell types normally correspond to stable cellular steady states (attractors) regulated by relevant TF networks [18]. When sufficiently perturbed, the TF network, operating as a nonlinear dynamical system, may switch to an alternative attractor state corresponding to an adverse phenotype, i.e., disease or toxicity. To predict how close the biological system is to the tipping point, it is possible to exploit the critical-slowing-down phenomenon commonly observed in nonlinear dynamical systems operating near bifurcation (tipping) points [51]. In an scRNA-seq study, this phenomenon is embodied in changes in three statistical metrics of the TF network genes as the system approaches a tipping point: (i) an increase in the average of variations of the expression levels of each TF across cells (<SDTF>, Fig. 3A and 3B, left panels); (ii) an increase in the average of pair-wise, TF-TF Pearson correlations across cells (<PCCTF>, Fig. 3A and 3B, left panels); and (iii) a decrease in the average of pair-wise, cell-cell Pearson correlations across TFs (<PCCC>, Fig. 3A and 3B, right panels). A composite index derived from these three statistics, CI = <SDTF><PCCTF>/<PCCC>, is expected to rise and peak when the network is approaching the tipping point and decrease past the tipping point (Fig. 3C) [52,53]. Such composite indices may be used as novel biomarkers to predict the PoD to adverse outcomes [54].
Figure 3. Illustration of changes in gene expression statistics in single cells when an underlying TF network is perturbed (e.g. by a chemical) to move closer to a bifurcation (tipping) point that demarcates the separation of a healthy attractor state from an adverse attractor state.
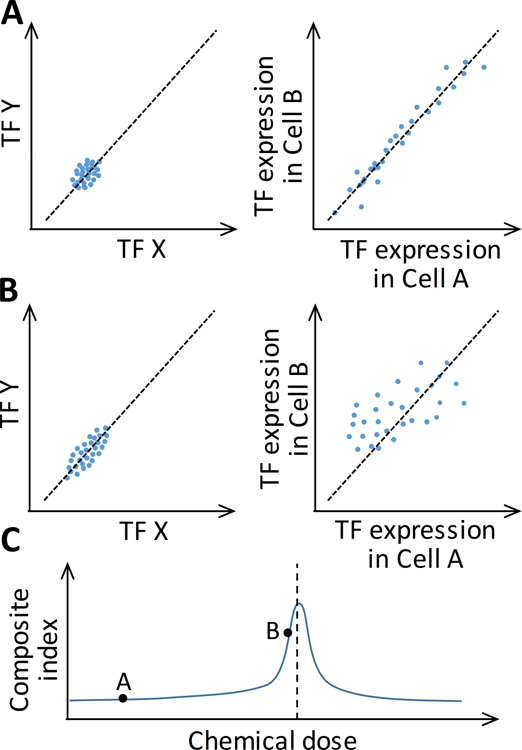
(A) Cells are at a state far away from the bifurcation point and (B) at a state near the bifurcation point. Left panels: the expression variations (<SDTF>) of a pair of TFs (X and Y) in the network and their correlations (<PCCTF>) increase from (A) to (B); each dot represents a cell. Right panel: the similarity between a pair of cells, as defined by the correlation (<PCCC>) of expression levels of all TFs in the network, decreases from (A) to (B); each dot represents a TF in the network. (C) As chemical dose increases to push the TF network closer to the bifurcation (tipping) point (vertical dashed line), the composite index (<SDTF><PCCTF>/<PCCC>) increases, peaks, and then declines. Black dots on curve correspond to states A and B.
Dynamical network modeling
The ability to simulate and predict the dynamical response of a biological system composed of diverse cells to chemical perturbations is one of the goals of systems toxicology. This bottom-up systems biology endeavor can be greatly aided by the top-down, single-cell omic approach, especially scRNA-seq, in several ways. First, scRNA-seq data can help identify and map the gene regulatory network (GRN) underlying the dynamic cellular response to chemicals. A variety of in silico network inference methods have been developed in the past based on bulk microarray or RNA-seq data, ranging from simple Pearson correlations to more complex Bayesian networks, ordinary differential equation frameworks, and mutual information correlations [55]. As discussed previously, approaches using bulk assay data suffer from Simpson’s paradox to various extents. In contrast, single-cell data provide much richer and reliable information regarding the relationship between genes within the same cells. With many recently developed algorithms such as SCODE and SCENIC, more reliable GRN structures can be reverse engineered and reconstructed based on single-cell data to feed dynamical modeling [56,57]. Second, although the counts of unique molecular identifiers (UMI) for each cell can be noisy, depending on the cDNA labeling efficiency, sequencing depth and many other technical variations in the scRNA-seq workflow, they nevertheless provide extremely valuable information on the abundance (copy numbers) of each mRNA species for modeling single-cell GRNs. In particular, stochastic models of GRNs that simulate the abundance of mRNAs and proteins as discrete, integral numbers and explicitly consider noise in gene expression, will become more feasible. Finally, the temporal order of transcriptional events, based on pseudotime trajectories, provides essential information on the dynamics of the underlying GRN and can be used to guide the calibration of the dynamical models.
Challenges in single-cell systems toxicology
Despite a variety of application potentials, single-cell technologies, as they are applied to toxicology, face a number of technical and computational challenges. Toxicological studies often involve comparing biological samples across different chemical exposure conditions such as doses and durations. The accuracy of comparison is subject to not only traditional technical variations associated with animal treatment, sample collection and handling, but also single-cell technology-specific variations including tissue and cell dissociation, library preparation and downstream processing. So far in nearly all scRNA-seq studies, tissue samples collected under different conditions are processed separately through the scRNA-seq workflow. The associated batch effects can affect the quantifications of cell numbers of identified cell types and pseudotime trajectories perturbed by chemical exposures. To reduce batch effects, efforts to multiplex through sample-specific cell tagging are underway so that samples obtained from different conditions can be pooled and processed together and subsequently identified bioinformatically [58].
Another challenge lies in the computational analysis tasks including cell clustering across conditions. For a single normal condition, cells can be grouped into physiological cell types based on differentially expressed marker genes. But when cells are exposed to chemicals, they also tend to cluster based on the differential transcriptional responses to different exposure conditions, which will interfere with clustering cells into physiological cell types. Therefore, clustering cells appropriately and comparing them under complex conditions can be challenging. To facilitate comparison, the most recent iteration of the single-cell computational tool Seurat uses canonical correlation analysis to first identify physiological cell type clusters across conditions and then examine cell type-specific responses [59]. While such approach mitigates the issue of cross-condition comparison, it does not completely eliminate it, and better algorithms need to be developed. Furthermore, with single-cell studies, we are comparing not only means and variances between samples, but also distributions of cells, cell types/subtypes, and genome-wide RNA levels. Establishing the ground truth of these metrics for normal tissues and being able to tell the adverse from the normality can pose challenges.
Concluding remarks
Systems toxicology is at a great time of rapid biotechnological innovation. Single-cell technologies are providing an expanding, high-resolution view of the cellular perturbations of biological organisms by the internal and external environment. As single-cell omic technologies improve and costs decline, the coming deluge of high-dimensional datasets will challenge not only our computational resources, but also our intellectual abilities to make best use of them. As the field simultaneously develops novel computational analysis tools, the information content that can be extracted from single-cell datasets will expand and improve our understanding of the physiological and toxicological processes constantly playing out in the cells of our body. Challenges aside, as with any nascent technology, single-cell omics opens new avenues for toxicological research and brings higher precision to the risk assessment of adverse outcomes from chemical exposures [60,61].
Acknowledgements
The work was supported by NIEHS Superfund Research grant P42ES04911 and Pilot Grant through NIEHS HERCULES grant P30ES019776.
Abbreviations:
- GRN
gene regulatory network
- PCA
principle component analysis
- PoD
point-of-departure
- scRNA-seq
single-cell RNA sequencing
- TF
transcription factor
- t-SNE
t-distributed stochastic neighbor embedding
- UMAP
uniform manifold approximation and projection
- UMI
unique molecular identifiers
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest
None. Mention of products and trade names does not indicate endorsement by the federal government. Conclusions drawn in this study neither constitute nor necessarily reflect U.S. EPA policy.
References
* of special interest
* * of outstanding interest
- *1.Bars RG, Elcombe CR: Dose-dependent acinar induction of cytochromes P450 in rat liver. Evidence for a differential mechanism of induction of P450IA1 by beta-naphthoflavone and dioxin. Biochem J 1991, 277 (Pt 2):577–580.Along with *2, one of the first studies showing zonal induction of CYP enzymes in liver lobules by toxicants.
- *2.Tritscher AM, Goldstein JA, Portier CJ, McCoy Z, Clark GC, Lucier GW: Dose-response relationships for chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in a rat tumor promotion model: quantification and immunolocalization of CYP1A1 and CYP1A2 in the liver. Cancer Res 1992, 52:3436–3442. [PubMed] [Google Scholar]
- 3.Bars RG, Mitchell AM, Wolf CR, Elcombe CR: Induction of cytochrome P-450 in cultured rat hepatocytes. The heterogeneous localization of specific isoenzymes using immunocytochemistry. Biochem J 1989, 262:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.French CT, Hanneman WH, Chubb LS, Billings RE, Andersen ME: Induction of CYP1A1 in primary rat hepatocytes by 3,3’,4,4’,5-pentachlorobiphenyl: evidence for a switch circuit element. Toxicol Sci 2004, 78:276–286. [DOI] [PubMed] [Google Scholar]
- 5.Broccardo CJ, Billings RE, Chubb LS, Andersen ME, Hanneman WH: Single cell analysis of switch-like induction of CYP1A1 in liver cell lines. Toxicol Sci 2004, 78:287–294. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman TE, Acerbo ER, Carranza KF, Gilberto VS, Wallis LE, Hanneman WH: Ultrasensitivity dynamics of diverse aryl hydrocarbon receptor modulators in a hepatoma cell line. Arch Toxicol 2018. [DOI] [PubMed]
- 7.Andersen ME, Dennison JE, Thomas RS, Conolly RB: New directions in incidence-dose modeling. Trends Biotechnol 2005, 23:122–127. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell S, Roy K, Zangle TA, Hoffmann A: Nongenetic origins of cell-to-cell variability in B lymphocyte proliferation. Proc Natl Acad Sci U S A 2018, 115:E2888–E2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Bhattacharya S, Kline DE, Crawford RB, Conolly RB, Thomas RS, Kaminski NE, Andersen ME: Stochastic modeling of B lymphocyte terminal differentiation and its suppression by dioxin. BMC Syst Biol 2010, 4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, Yarnitzky T, Liron Y, Polak P, Lahav G, et al. : Oscillations and variability in the p53 system. Mol Syst Biol 2006, 2:2006 0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Symmons O, Raj A: What’s Luck Got to Do with It: Single Cells, Multiple Fates, and Biological Nondeterminism. Mol Cell 2016, 62:788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK: Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu H, Crawford RB, Kaplan BL, Kaminski NE: 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated disruption of the CD40 ligand-induced activation of primary human B cells. Toxicol Appl Pharmacol 2011, 255:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson EH: The Interpretation of Interaction in Contingency Tables. Journal of the Royal Statistical Society Series B-Statistical Methodology 1951, 13:238–241. [Google Scholar]
- 15.Petri T, Altmann S, Geistlinger L, Zimmer R, Kuffner R: Addressing false discoveries in network inference. Bioinformatics 2015, 31:2836–2843. [DOI] [PubMed] [Google Scholar]
- 16.Freitas AA: Investigating the role of Simpson’s paradox in the analysis of top-ranked features in high-dimensional bioinformatics datasets. Brief Bioinform 2019. [DOI] [PubMed]
- 17.Gebhardt R, Matz-Soja M: Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J Gastroenterol 2014, 20:8491–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang S, Eichler G, Bar-Yam Y, Ingber DE: Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett 2005, 94:128701. [DOI] [PubMed] [Google Scholar]
- 19.Berry S, Dean C, Howard M: Slow Chromatin Dynamics Allow Polycomb Target Genes to Filter Fluctuations in Transcription Factor Activity. Cell Syst 2017, 4:445–457 e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raj A, van Oudenaarden A: Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 2008, 135:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levchenko A, Nemenman I: Cellular noise and information transmission. Curr Opin Biotechnol 2014, 28:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigal A, Milo R, Cohen A, Geva-Zatorsky N, Klein Y, Liron Y, Rosenfeld N, Danon T, Perzov N, Alon U: Variability and memory of protein levels in human cells. Nature 2006, 444:643–646. [DOI] [PubMed] [Google Scholar]
- 23.Flusberg DA, Roux J, Spencer SL, Sorger PK: Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol Biol Cell 2013, 24:2186–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brodin P: The biology of the cell - insights from mass cytometry. FEBS J 2018. [DOI] [PubMed]
- 25.Stahlberg A, Bengtsson M: Single-cell gene expression profiling using reverse transcription quantitative real-time PCR. Methods 2010, 50:282–288. [DOI] [PubMed] [Google Scholar]
- 26.Specht H, Slavov N: Transformative Opportunities for Single-Cell Proteomics. J Proteome Res 2018, 17:2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emara S, Amer S, Ali A, Abouleila Y, Oga A, Masujima T: Single-Cell Metabolomics. Adv Exp Med Biol 2017, 965:323–343. [DOI] [PubMed] [Google Scholar]
- 28.Shema E, Bernstein BE, Buenrostro JD: Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet 2019, 51:19–25. [DOI] [PubMed] [Google Scholar]
- 29.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. : mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009, 6:377–382. [DOI] [PubMed] [Google Scholar]
- 30.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA: The Human Cell Atlas: from vision to reality. Nature 2017, 550:451–453. [DOI] [PubMed] [Google Scholar]
- 31.Farmahin R, Williams A, Kuo B, Chepelev NL, Thomas RS, Barton-Maclaren TS, Curran IH, Nong A, Wade MG, Yauk CL: Recommended approaches in the application of toxicogenomics to derive points of departure for chemical risk assessment. Arch Toxicol 2017, 91:2045–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas RS, Wesselkamper SC, Wang NC, Zhao QJ, Petersen DD, Lambert JC, Cote I, Yang L, Healy E, Black MB, et al. : Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol Sci 2013, 134:180–194. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q, Bhattacharya S, Pi J, Clewell RA, Carmichael PL, Andersen ME: Adaptive Posttranslational Control in Cellular Stress Response Pathways and Its Relationship to Toxicity Testing and Safety Assessment. Toxicol Sci 2015, 147:302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, Frithsen JB, Grulke CM, Gwinn MR, Harrill JA, et al. : The next generation blueprint of computational toxicology at the U.S. Environmental Protection Agency. Toxicol Sci 2019. [DOI] [PMC free article] [PubMed]
- 35.Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR, et al. : SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 2017, 14:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menon V: Clustering single cells: a review of approaches on high-and low-depth single-cell RNA-seq data. Brief Funct Genomics 2018. [DOI] [PMC free article] [PubMed]
- **37.van der Maaten L, Hinton G: Visualizing Data using t-SNE. Journal of Machine Learning Research 2008, 9:2579–2605.This is the original article reporting the algorithm of t-SNE for nonlinear high-dimension reduction.
- 38.McInnes L, Healy J, Melville J: UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, 1802.03426v2.
- 39.La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, Borm LE, Stott SRW, Toledo EM, Villaescusa JC, et al. : Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 2016, 167:566–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Susztak K: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018, 360:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **41.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, et al. : Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542:352–356.The first paper using scRNA-seq to cluster and locate lobular hepatocytes into 9 different functional layers.
- 42.Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, et al. : A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst 2016, 3:385–394 e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, Awatramani R: Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci 2018, 21:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **44.Joshi N, Watanabe S, Verma R, Jablonski RP, Chen C-I, Cheresh P, Reyfman PA, McQuattie-Pimentel AC, Sichizya L, Flozak AS, et al. : Single-cell RNA-seq reveals spatially restricted multicellular fibrotic niches during lung fibrosis. bioRxiv 2019:569855.Likely the first pubished scRNA-seq study in toxicology.
- 45.Martos SN, Campbell MR, Iannone MA, Bell DA: Single Cell RNA sequencing Reveals Natural Kill-Like, Altered Effector CD8+ T Lymphocytes in Smokers. In 58th Annual Meeting of Society of Toxicology Edited by Baltimore, MD; March 10–14, 2019 Abstract #2986. [Google Scholar]
- 46.Rostom R, Svensson V, Teichmann SA, Kar G: Computational approaches for interpreting scRNA-seq data. FEBS Lett 2017, 591:2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **47.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014, 32:381–386.This is the orginal article reporting the popular Monocle algorithm for psuedotime trajectory analysis.
- 48.Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, Choi K, Bendall S, Friedman N, Pe’er D: Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat Biotechnol 2016, 34:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindstrom NO, De Sena Brandine G, Tran T, Ransick A, Suh G, Guo J, Kim AD, Parvez RK, Ruffins SW, Rutledge EA, et al. : Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev Cell 2018, 45:651–660 e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Z, Lou H, Xie K, Wang H, Chen N, Aparicio OM, Zhang MQ, Jiang R, Chen T: Reconstructing cell cycle pseudo time-series via single-cell transcriptome data. Nat Commun 2017, 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheffer M, Bascompte J, Brock WA, Brovkin V, Carpenter SR, Dakos V, Held H, van Nes EH, Rietkerk M, Sugihara G: Early-warning signals for critical transitions. Nature 2009, 461:53–59. [DOI] [PubMed] [Google Scholar]
- *52.Mojtahedi M, Skupin A, Zhou J, Castano IG, Leong-Quong RY, Chang H, Trachana K, Giuliani A, Huang S: Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol 2016, 14:e2000640.Along with *53, this paper showed that critical state transition underpinning cell fate switching and thus tipping point can be predicted by using single-cell transcriptional network data.
- *53.Richard A, Boullu L, Herbach U, Bonnafoux A, Morin V, Vallin E, Guillemin A, Papili Gao N, Gunawan R, Cosette J, et al. : Single-Cell-Based Analysis Highlights a Surge in Cell-to-Cell Molecular Variability Preceding Irreversible Commitment in a Differentiation Process. PLoS Biol 2016, 14:e1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Q, Li J, Middleton A, Bhattacharya S, Conolly RB: Bridging the Data Gap From in vitro Toxicity Testing to Chemical Safety Assessment Through Computational Modeling. Front Public Health 2018, 6:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G: Revealing strengths and weaknesses of methods for gene network inference. Proc Natl Acad Sci U S A 2010, 107:6286–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, Hayashi T, Nikaido I: SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33:2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. : SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017, 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gehring J, Park JH, Chen S, Thomson M, Pachter L: Highly Multiplexed Single-Cell RNA-seq for Defining Cell Population and Transcriptional Spaces. bioRxiv 2018:315333.
- **59.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018, 36:411–420.This paper provides a streamlined computational algorithm for integrating scRNA-seq data across different experimental conditions, which can be applied directly in toxicological settings.
- 60.Zhang B, Huang K, Zhu L, Luo Y, Xu W: Precision toxicology based on single cell sequencing: an evolving trend in toxicological evaluations and mechanism exploration. Arch Toxicol 2017, 91:2539–2549. [DOI] [PubMed] [Google Scholar]
- 61.Wang W, Gao D, Wang X: Can single-cell RNA sequencing crack the mystery of cells? Cell Biol Toxicol 2018, 34:1–6. [DOI] [PubMed] [Google Scholar]