

Abstract
cAMP acts as a second messenger in many cellular processes. Three protein types mainly mediate cAMP-induced effects: PKA, exchange protein directly activated by cAMP (Epac), and cyclic nucleotide–modulated channels (cyclic nucleotide–gated or hyperpolarization-activated and cyclic nucleotide–modulated (HCN) channels). Discrimination among these cAMP signaling pathways requires specific targeting of only one protein. Previously, cAMP modifications at position N6 of the adenine ring (PKA) and position 2′-OH of the ribose (Epac) have been used to produce target-selective compounds. However, cyclic nucleotide–modulated ion channels were usually outside of the scope of these previous studies. These channels are widely distributed, so possible channel cross-activation by PKA- or Epac-selective agonists warrants serious consideration. Here we demonstrate the agonistic effects of three PKA-selective cAMP derivatives, N6-phenyladenosine-3′,5′-cyclic monophosphate (N6-Phe-cAMP), N6-benzyladenosine-3′,5′-cyclic monophosphate (N6-Bn-cAMP), and N6-benzoyl-adenosine-3′,5′-cyclic monophosphate (N6-Bnz-cAMP), on murine HCN2 pacemaker channels. Electrophysiological characterization in Xenopus oocytes revealed that these derivatives differ in apparent affinities depending on the modification type but that their efficacy and effects on HCN2 activation kinetics are similar to those of cAMP. Docking experiments suggested a pivotal role of Arg-635 at the entrance of the binding pocket in HCN2, either causing stabilizing cation–π interactions with the aromatic ring in N6-Phe-cAMP or N6-Bn-cAMP or a steric clash with the aromatic ring in N6-Bnz-cAMP. A reduced apparent affinity of N6-Phe-cAMP toward the variants R635A and R635E strengthened that notion. We conclude that some PKA activators also effectively activate HCN2 channels. Hence, when studying PKA-mediated cAMP signaling with cAMP derivatives in a native environment, activation of HCN channels should be considered.
Keywords: cAMP, PKA, cell signaling, second messenger, protein targeting, cyclic nucleotide binding domain, HCN channels, ligand selectivity, off-target effect, target specificity
Introduction
cAMP, the first identified second messenger (1), plays a key role in living organisms ranging from Dictyostelium to Homo sapiens. It is involved in a wide variety of cellular processes, including proliferation, differentiation, secretion, migration, pacemaking, sensation, and apoptosis. The concentration of cytosolic cAMP is controlled by the action of adenylyl cyclases, catalyzing the conversion of ATP to cAMP, and cyclic nucleotide phosphodiesterases, catalyzing the conversion of cAMP to 5′-AMP. In mammalian cells, four main types of proteins respond to cAMP: PKA, exchange protein directly activated by cAMP (Epac1 and Epac2),3 CN-modulated ion channels (cyclic nucleotide–gated channels and hyperpolarization-activated and cyclic-nucleotide modulated (HCN) channels) (2) and the relatively new class of Popeye domain–containing proteins (3).
HCN channels, as one type of CN-modulated ion channels, are activated by hyperpolarizing membrane potentials as a primary stimulus but are secondarily stimulated by direct binding of cyclic nucleotides, mainly cAMP, to intracellular binding sites (4–10). Structurally, HCN channels belong to the superfamily of tetrameric voltage-gated ion channels (11). In these channels, each subunit contains a voltage sensor domain and a pore domain. In contrast to most other members of this superfamily, HCN channel subunits additionally contain a cyclic nucleotide–binding domain (CNBD) in the C terminus, which is connected to the membrane portion by a so-called C-linker (12). It has been shown that the unoccupied CNBD together with the C-linker has an inhibitory effect on channel gating, which is relieved by CN binding (13). Such a relieving effect is reflected by a shift of the steady-state activation curve to more depolarized voltages, thereby increasing the maximum current amplitude, accelerating activation and decelerating deactivation (14, 15).
The CNBD is a highly conserved structure (16). Combined data from PKA, Epac, and ion channel studies illustrate that CNBDs use a common mechanism to bind and sense cAMP (17). This similarity in the cAMP-binding mechanism of all cAMP-responsive proteins makes it challenging to target only one type of protein to discriminate between the different cAMP signaling pathways. However, discrimination between two proteins, PKA and Epac, could be realized and has been used widely (e.g. (18–22). Although Epac tolerates 2′-OH modifications, modifications at the N6 position of the purine ring are not accepted by this protein. However, such N6-modified derivatives are often potent agonists for PKA and can be used to exclude Epac activation (23). Notably, fully functional ion channels were often outside the scope when studying the selectivity of cAMP derivatives.
Besides a systematic study performed by Ng et al. (24) comparing binding and gating parameters for different cyclic nucleotides in whole HCN channels, the effect of purine ring modifications at cAMP on functional channels have so far not been tested systematically. To contribute to the urgent problem of developing cAMP analogs that can discriminate between the different cAMP-binding proteins, here we tested three N6-modified cAMP derivatives: N6-phenyladenosine-3′,5′-cyclic monophosphate (N6-Phe-cAMP), N6-benzyladenosine-3′,5′-cyclic monophosphate (N6-Bn-cAMP), and N6-benzoyladenosine-3′,5′-cyclic monophosphate (N6-Bnz-cAMP), known to be activators of PKA, on functional HCN2 channels. We show that all of them are HCN2 channel activators with efficacies similar to cAMP but with different apparent affinities. Docking and mutagenesis experiments revealed the specific interactions between molecule and binding pocket that underlie those differences.
Results
Effect of the native ligand cAMP on HCN2 channels in oocyte macropatches
HCN2 channels produce slowly activating inward currents in response to hyperpolarizing voltage jumps. It has been shown that the cyclic nucleotide–binding domain has an inhibitory effect on channel gating that is relieved by cAMP binding (13). Such a relieving effect is reflected by an increase in current amplitude at a given voltage, by a shift of the voltage of half-maximum activation, V1/2, to more depolarized values, and by an acceleration of activation kinetics and a deceleration of deactivation kinetics (14). This could also be observed here for mHCN2 channels expressed in Xenopus laevis oocytes (Fig. 1).
Figure 1.
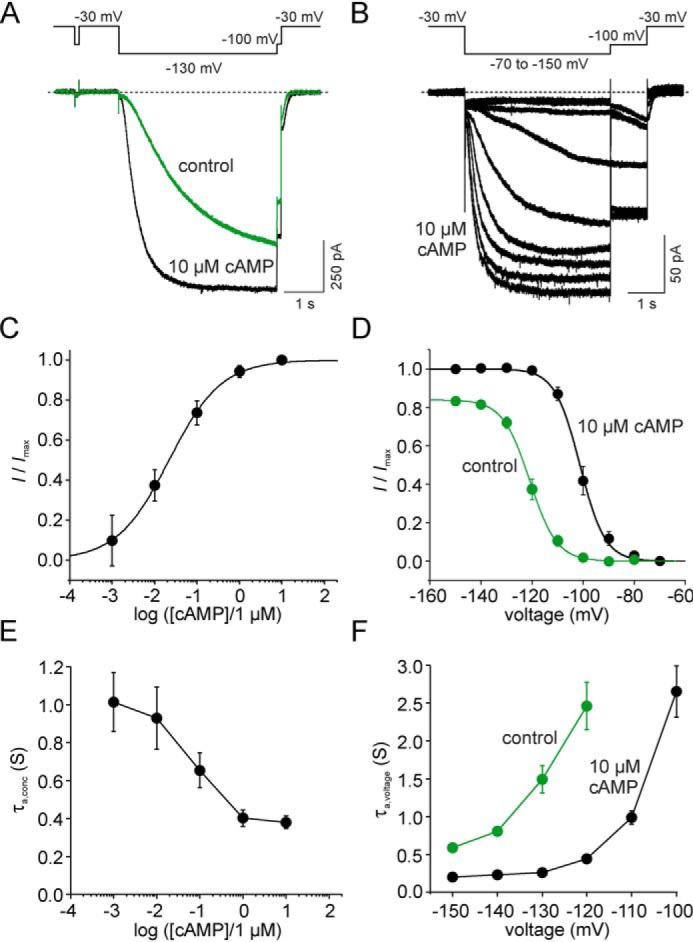
cAMP effects on steady-state and nonsteady state parameters of mHCN2 channel activation. A, protocol and representative current traces to study concentration-dependent gating at a fixed command voltage. Tail currents were obtained from a −100 mV pulse following an activating −130 mV pulse. B, protocol and representative current traces (exemplary of a saturating concentration of 10 μm cAMP) to study voltage-dependent gating at a fixed agonist concentration. A voltage family from −70 mV to −150 mV was applied with 10-mV increments. Tail currents were obtained from a −100 mV pulse following the variable test pulse. C, concentration–response relationship for cAMP. Mean values for I/Imax were obtained from six to 13 recordings and plotted against the cAMP concentration. The Hill equation (Equation 1) was approximated to the data, yielding EC50 (21.3 ± 3.3 nm) and H (1.1 ± 0.2), respectively. D, steady-state activation relationship at zero and saturating [cAMP]. Mean values for relative current amplitudes (I/Imax) for zero cAMP (n = 14) and for saturating cAMP of 10 μm (n = 13) were plotted against the command voltage. The Boltzmann equation was approximated to the data, yielding V1/2 = −117.8 ± 1.5 mV and a slope of zδ = 4.2 ± 0.3 for zero and V1/2 = −97.9 ± 1.9 mV and a slope of zδ = 4.2 ± 0.3 for 10 μm cAMP. E, activation kinetics at different cAMP concentrations. Activation time constants were obtained from approximating a monoexponential function (Equation 3) to the current time courses, yielding τa,conc. Mean values were obtained from three to seven recordings. The protocol used is shown in A. F, activation kinetics at different command voltages at zero and saturating [cAMP]. Activation time constants were obtained from approximating a monoexponential function (Equation 3) to the current time courses, yielding τa,voltage. Mean values were obtained from 14 recordings for zero and 13 recordings for 10 μm cAMP. The protocol used is shown in B.
Fig. 1, A and B, shows representative current traces and the respective protocols used to determine channel activation at varying agonist concentrations at a given command voltage of −130 mV (Fig. 1A) and to determine channel activation at varying voltages at zero or saturating agonist concentrations (Fig. 1B), respectively. The cAMP concentration required to cause EC50, a measure of the apparent affinity of the agonist to the receptor, was determined by approximating the Hill equation (Equation 1) to relative current amplitudes plotted against the cAMP concentration (Fig. 1C). Current amplitudes were obtained from tail currents at −100 mV following an activating pulse of −130 mV. The EC50 value was found to be 21.3 ± 3.3 nm, and the Hill coefficient was 1.1 ± 0.2. From this, 10 μm was defined as the saturating concentration for all experiments.
To estimate V1/2, relative current amplitudes were plotted versus the command voltage. The Boltzmann equation was fitted to the data points of individual recordings, yielding V1/2 under control conditions of −117.8 ± 1.5 mV and after application of 10 μm cAMP of −97.9 ± 1.9 mV. Thus, the steady-state activation was shifted by 19.9 ± 1.2 mV to more depolarized values because of cAMP binding (Fig. 1D). The slopes of the curves were similar with and without cAMP (4.2 ± 0.2 and 4.2 ± 0.3 mV/e -fold change, respectively).
The time constant of activation, given as τa,conc, decreased with increasing cAMP concentrations (Fig. 1E). Furthermore, the time constant of activation decreased with increasing hyperpolarizing voltages under control conditions without cAMP as well as after application of 10 μm cAMP (Fig. 1F).
All three tested cAMP derivatives were able to promote HCN2 channel activation
We tested three cAMP derivatives, used as PKA activators, for their ability to activate HCN2 channels: N6-Phe-cAMP, N6-Bn-cAMP, and N6-Bnz-cAMP (Fig. 2A). In these derivatives, a phenyl group, a benzyl group, or a benzoyl group, respectively, is attached to the amino group in position 6 of the adenine moiety. All three N6-modified derivatives have been shown to be site-selective for site A of both PKA type I and II (27–29).
Figure 2.

Structure and apparent affinities of the three tested derivatives. A, molecular formulas for three N6-modified derivatives known to activate PKA. B, representative current responses before and after application of the respective ligand. Black traces represent recordings in the absence of the ligand, and colored traces represent recordings during application of a saturating concentration of the respective ligand. Each ligand turned out to be an activator of heterologously expressed HCN2 channels. C, concentration–response relationships for the three derivatives (colored symbols and fits) compared with mHCN2 WT data (black symbols and fits). The Hill equation (Equation 1) was approximated to the data to obtain EC50 and H. Error bars indicate S.E. D, box plot of EC50 values obtained from C. Filled circles indicate individual recordings. Error bars indicate S.D. E, box plot of Hill coefficients obtained from C. Filled circles indicate individual recordings. Error bars indicate S.D.
To test whether the three cAMP derivatives are able to modulate HCN2 channels, channel activation was monitored at zero cAMP and in the presence of different agonist concentrations. Representative current recordings for zero cAMP and saturation are shown in Fig. 2B. All three derivatives caused an increase in current amplitude and acceleration of the activation kinetics, as known for cAMP (Fig. 1).
cAMP modifications at position N6 affected the apparent affinity of the agonist for HCN2
To study the concentration dependence in more detail, concentration–response relationships were recorded for each derivative (Fig. 2C) and compared with the relationship for cAMP. For all cases, the Hill equation (Equation 1) was approximated to the relative currents of each individual recording. The results for EC50 and the Hill coefficient, H, are summarized in Fig. 2, D and E, respectively. The nature of the N6 modification had a major influence on the apparent affinity (EC50,Phe = 3.02 ± 0.83 nm, EC50,Bn = 5.12 ± 1.03 nm, EC50,Bnz = 374 ± 91.3 nm) (Fig. 2D). Adding a phenyl or benzyl group to position N6 shifted the EC50 value to lower concentrations compared with native cAMP, causing an apparent affinity one order of magnitude higher than for cAMP. However, adding a benzoyl group shifted the EC50 value to higher concentrations, indicating an apparent affinity one order of magnitude lower than for cAMP (Fig. 2D). The Hill coefficients for the derivatives were not significantly different from that for cAMP: HPhe = 1.4 ± 0.3, HBn = 1.2 ± 0.3, and HBnz = 0.9 ± 0.2 (Student's t test, p = 0.05) (Fig. 2E).
None of the cAMP modifications tested changed the efficacy
We used two measures to compare the efficacy of the cAMP derivatives with that of native cAMP: the maximal agonist-induced voltage shift (ΔV1/2,max) and the maximal fractional increase in tail current amplitude (Imax,agonist/Imax,cAMP) observed with saturating agonist concentrations (Fig. 3) (30).
Figure 3.

Efficacy of the three tested derivatives. A, steady-state activation relationships at zero and saturating agonist concentrations. The graphs show the relationships of the tested derivatives at saturating concentrations and in the absence of any ligand. Except for N6-Bnz-cAMP, for which 100 μm was used as a saturating concentration, the concentration was 10 μm for saturation. I/Imax are mean values obtained from three to six recordings. The Boltzmann equation was approximated to the relative current values to yield V1/2. The results of the fits were as follows: −97.5 ± 2.0 mV for N6-Phe-cAMP, −104.9 ± 2.7 mV for N6-Bn-cAMP, and −98.3 ± 1.4 mV for N6-Bnz-cAMP. The slopes were 3.4 ± 0.2, 4.1 ± 0.2, and 4.4 ± 0.3, respectively. B and C, comparison of the efficiency of the three derivatives compared with cAMP in two measures. B, box plots of Imax/Imax,cAMP. Filled circles indicate individual recordings. Error bars indicate S.D. C, box plots of ΔV1/2. Filled circles indicate individual recordings. Error bars indicate S.D.
To yield ΔV1/2,max, V1/2 values before and after application of saturating agonist concentrations were estimated by fitting the Boltzmann equation to steady-state activation relationships (Fig. 3A). There was no difference between ΔV1/2,max of cAMP and either one of the tested derivatives (16.1 ± 2.3 mV for cAMP, 18.3 ± 1.4 mV for N6-Phe-cAMP, 18.6 ± 2.4 mV for N6-Bn-cAMP, and 22.3 ± 1.1 mV for N6-Bnz-cAMP) (Fig. 3B).
To yield the maximal fractional increase in tail current amplitude, we determined the tail current amplitudes at a test pulse of −100 mV, following a hyperpolarizing pulse of −130 mV, for each cAMP derivative at saturating concentrations and related that to the maximum tail current amplitude of 10 μm cAMP in the same patch. All derivatives caused a fractional current of around 1.0, suggesting that they all cause a similar current increase as cAMP (Fig. 3C). The values were 1.02 ± 0.14 for N6-Phe-cAMP, 0.98 ± 0.05 for N6-Bn-cAMP, and 1.00 ± 0.02 for N6-Bnz-cAMP (Fig. 3B). Thus, both measures led us conclude that the modifications performed in the tested cAMP derivatives did not affect the efficacy of the agonists.
None of the cAMP modifications tested changed activation kinetics
Fig. 4 summarizes the results regarding activation kinetics upon hyperpolarizing voltage jumps. First, we studied the concentration dependence of these kinetics. For this purpose, we applied the agonists at different concentrations, covering a wide range, and recorded the currents at a nearly saturating voltage of −130 mV. Kinetics were quantified by τa,conc obtained from approximating a monoexponential equation (Equation 3) to the current time courses. The values were plotted against the normalized concentration, which is the ratio of applied concentration and apparent affinity [agonist]/EC50 (Fig. 4A). Such normalization was required because large differences in the EC50 values for the derivatives and cAMP did not allow comparison at absolute concentrations. As a result, the concentration dependence of τa,conc for derivatives could be superimposed with that of cAMP.
Figure 4.
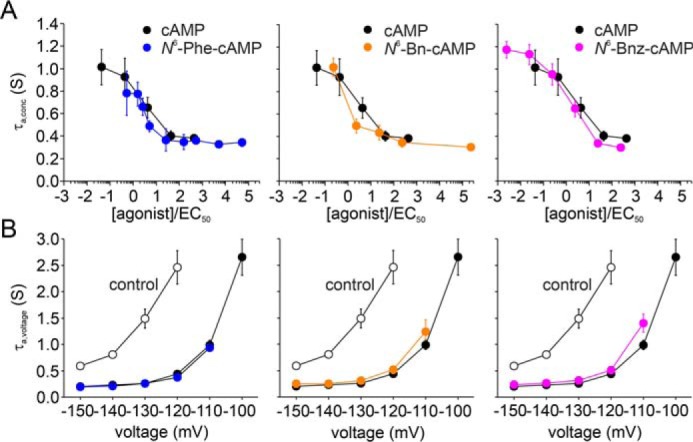
Activation kinetics for the tested cAMP derivatives. A, activation kinetics in dependence on agonist concentration at a given command voltage of −130 mV. τa,conc values, obtained from approximating a monoexponential equation (Equation 3) to the current time courses, are plotted against the cAMP concentration. Mean values were obtained from three to nine recordings. Black symbols in each plot illustrate the case for cAMP. The protocol used is shown in Fig. 1A. B, activation kinetics in dependence on command voltage at a saturating agonist concentration. τa,voltage values were plotted against the command voltage. Open symbols represent recordings in the absence of ligands and filled symbols in the presence of saturating agonist concentrations. Mean values were obtained from three to 19 recordings. The protocol used is shown in Fig. 1B.
Second, we studied the voltage dependence of activation kinetics. For this purpose, we applied a family of hyperpolarizing voltages at saturating agonist concentrations and in the absence of agonists, respectively. The results are shown in Fig. 4B. Analogous to the concentration dependence, the voltage dependence of τa,voltage could be superimposed with that of cAMP. From these results, we conclude that all derivatives had a similar accelerating effect as the native ligand.
Molecular docking suggests a pivotal role of Arg-635 for determining the binding behavior of N6-modified derivatives
To determine the structural basis of the higher apparent affinities of N6-Phe-cAMP and N6-Bn-cAMP and the lower apparent affinity of N6-Bnz-cAMP, we predicted and compared the binding modes of cAMP, N6-Phe-cAMP, N6-Bn-cAMP, and N6-Bnz-cAMP in mHCN2J by molecular docking (Fig. 5). The selected docking protocol was able to reproduce the crystallographically determined binding mode of cAMP with a heavy-atom RMSD of 0.60 Å (Fig. 5A), indicating that the binding modes of the N6-substituted derivatives can be reliably predicted. The predicted binding modes of the cAMP moiety of N6-Phe-cAMP and N6-Bn-cAMP deviate only marginally from the native binding mode of cAMP (heavy-atom RMSDs of 0.73 Å and 0.91 Å, respectively; Fig. 5, B and C). Docking scores (XP GScores) for all three compounds were in a similar range (cAMP, −13.53 kcal mol−1; N6-Phe-cAMP, −10.20 kcal mol−1; N6-Bn-cAMP, −11.54 kcal mol−1).
Figure 5.

Predicted binding modes of the tested cAMP derivatives. A–D, predicted binding modes of cAMP (A, gray; crystallographic pose (PDB code 1Q5O (25) shown in gold), N6-Phe-cAMP (B, blue), N6-Bn-cAMP (C, orange), and N6-Bnz-cAMP (D, magenta). In A–C, hydrogen bonds to and from protein side chains are depicted as dashed yellow lines, hydrogen bonds to and from the protein main chain as dashed green lines, and cation–π interactions as dashed blue lines. E, predicted binding mode of the cAMP fragment in N6-Bn-cAMP extended with a benzoyl group as found in N6-Bnz-cAMP. The steric clash with Arg-635 resulting from this extension is highlighted (red box), explaining the inverted binding mode of N6-Bnz-cAMP shown in D.
Compared with cAMP, the predicted binding modes of N6-Phe-cAMP and N6-Bn-cAMP reveal stabilizing cation–π and/or π–π stacking interactions between the additional phenyl ring and the side chain of Arg-635 (Fig. 5, A–C), which can explain the higher apparent affinities of these two derivatives. This interaction may be more stable in the case of N6-Phe-cAMP because of the more restricted conformational freedom of the N6-ring bonds compared with the less restricted N6-benzyl carbon bond in N6-Bn-cAMP, which can explain the higher apparent affinity of N6-Phe-cAMP compared with N6-Bn-cAMP.
In contrast to the N6-Phe and N6-Bn derivatives, the orientation of the cAMP moiety in the predicted binding mode of N6-Bnz-cAMP did not match the crystallographic pose (RMSD of 8.66 Å) but was instead inverted (Fig. 5D) and associated with a markedly lower docking score (−6.11 kcal mol−1). To determine why N6-Bnz-cAMP cannot adopt the binding mode favorable for cAMP and the other N6-substituted derivatives, we used the predicted binding mode of N6-Bn-cAMP as a template and replaced the benzyl moiety with a benzoyl moiety. Because of the planarity of the amide group in N6-Bnz-cAMP, the phenyl ring is then forced into an orientation in which it inevitably clashes with Arg-635 (Fig. 5E), resulting in a strongly disfavorable interaction.
Substituting Arg-635 at the entrance of the binding pocket prevented the higher apparent affinity of N6-Phe-cAMP
The arginine at position 635 was substituted with either alanine for neutralization (R635A) or glutamate for charge reversal (R635E). If Arg-635 is indeed involved in the higher apparent affinity of N6-Phe-cAMP and N6-Bn-cAMP, then the mutations should cause an apparent affinity that resembles that of cAMP to WT channels. To test this, the concentration-activation relationship was tested for N6-Phe-cAMP in both R635A and R635E. Both constructs, R635A and R635E, formed functional channels and expressed in high densities in Xenopus oocytes. First, we tested whether those constructs are still reactive to 10 μm cAMP. As for WT channels, we found a substantial current increase and an acceleration of the activation speed (Fig. 6A). When studying the concentration-response relationships for N6-Phe-cAMP, we found, as expected, that the EC50 values for both constructs were increased (Fig. 6, B and C), 27.7 ± 5.5 nm for R635A and 33.9 ± 5.3 nm for R635E. They are not different from the EC50 values found for cAMP (21.3 ± 3.3 nm) in WT channels.
Figure 6.
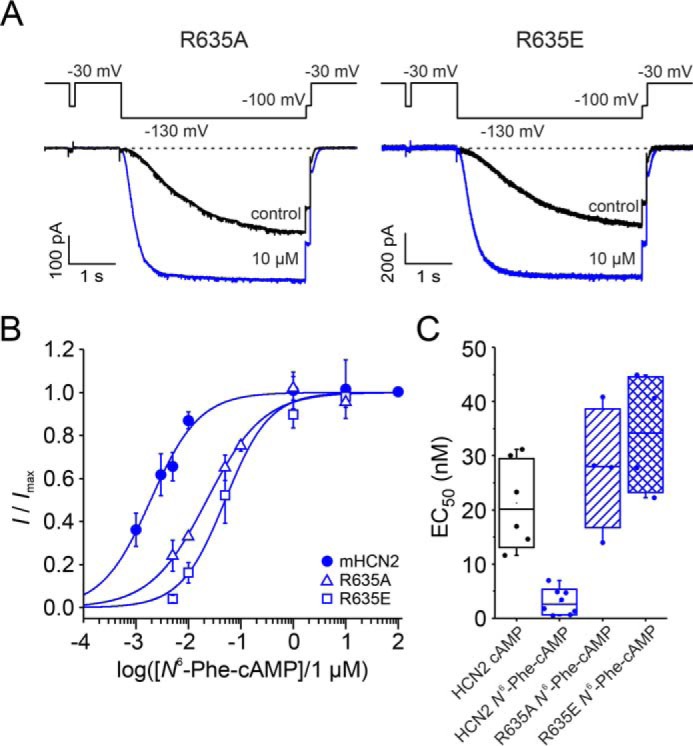
Response to N6-Phe-cAMP after charge neutralization or reversal at position Arg-635 in the CNBD. A, representative current traces for R635A and R635E at zero and saturating cAMP. B, concentration–activation relationships for mHCN2, R635A, and R635E. The Hill equation (Equation 1) was approximated to the relative currents to obtain EC50 and the Hill coefficient. C, box plot of EC50 values. Filled circles indicate individual recordings. Error bars indicate S.D. EC50 values obtained with N6-Phe-cAMP for R635A and R635E, respectively, are not different from the EC50 value obtained for cAMP and mHCN2 (Student's t test).
Discussion
PKA agonists activate HCN2 channels
Here we performed a comparative study of three cAMP derivatives, N6-Phe-cAMP, N6-Bn-cAMP, and N6-Bnz-cAMP, earlier described as activators of PKA, to investigate their effects on HCN2 channels. In the last decades, these derivatives have been shown to be useful tools when discrimination between activation of PKA and Epac was required (18–22). Interestingly, all three tested derivatives turned out to also be activators for HCN2 channels heterologously expressed in Xenopus laevis oocytes by promoting HCN2 channel gating in the presence of a primary hyperpolarizing voltage stimulus.
To study the agonistic effect in more detail, we determined the concentration–activation relationship and quantified EC50 as a measure of the apparent affinity and the Hill coefficient of activation. The apparent affinity was considerably affected by the type of N6 modification: Substituting one of the N6-bound hydrogens with a phenyl or benzyl group dramatically increased the apparent affinity (seven times and four times higher than cAMP, respectively), whereas a benzoyl ring decreased it (18 times lower than cAMP). However, because EC50 is a function of both binding affinity and efficacy, this parameter alone does not allow any interpretation of the mechanisms behind the agonist-dependent differences. The Hill coefficients for all agonists were similar to the Hill coefficient for cAMP. Even though this parameter is often used to describe the magnitude of cooperativity in ligand binding to allosteric systems, we hesitate to interpret those numbers in detail because, when derived from electrophysiological measurements, the Hill coefficient is just an empirical description without physical meaning because it depends on both binding and gating (31).
To address the question of why the tested agonists show those clear differences in their apparent affinities, we determined the efficacy, the ability of a ligand to elicit a response upon binding to a receptor (32), compared with unmodified cAMP. Two parameters were quantified: the maximum extent of shifting the steady-state activation to more depolarized voltages, ΔV1/2, and the ability of increasing the current in channels maximally activated by voltage. For both parameters, none of the three agonists differed from cAMP, indicating a similar efficacy of the native ligand and the three derivatives. Following a definition formulated by Colquhoun (31), according to which efficacy is determined by the sum of transduction events that follow the initial binding reaction, in HCN2 channels, those transduction events are similar whether matter they bind N6-Phe-cAMP, N6-Bn-cAMP, N6-Bnz-cAMP, or the native ligand cAMP. Provided that cAMP is a full agonist in HCN2 channels (33), those data further suggest consideration of all three N6-modified derivatives also as full agonists.
In addition to those steady-state parameters, we quantified the activation kinetics of the macroscopic current time course following a hyperpolarizing voltage jump. The kinetics of a macroscopic current reflect the kinetics of the underlying channels and are determined by the rate at which a new equilibrium occurs after a perturbation. We compared the effect of the N6-modified derivatives under two conditions: at varying voltages in the presence of a saturating agonist concentration and at varying concentrations at a saturating voltage. For both conditions, the activation time constants for each of the three derivatives were not different from the activation time constants for cAMP. This supports the interpretation of efficacy that the transduction events that follow the initial binding reaction are similar for all tested agonists. Together, those data led us to suggest that the N6-modified derivatives tested here are differently potent in HCN2 channels because they differ in their ability to bind to the HCN2 binding sites.
Arginine 635 in the CNBD affects apparent affinities
We performed docking experiments to identify molecular interactions between N6 derivatives and binding site residues that might underlie the observed differences in apparent affinities. The binding modes of N6-Phe-cAMP and N6-Bn-cAMP are stabilized by a cation–π and/or π–π stacking interaction between the additional phenyl moiety and the side chain of Arg-635. The higher apparent affinity of N6-Phe-cAMP compared with N6-Bn-cAMP is likely explained by the more restricted conformational freedom of the amino-phenyl side chain in the former case, which leads to a reduced loss in configurational entropy upon binding. In contrast, the benzoyl moiety in N6-Bnz-cAMP enforces coplanarity of the two aryl systems, which would lead to a clash between the phenyl ring and Arg-635 if the cAMP moiety of N6-Bnz-cAMP adopted the usual binding mode in the cAMP binding site. Based on these results, we hypothesized that the cation–π and/or π–π stacking interaction between the phenyl ring and Arg-635 is the main cause of the differential apparent affinities of N6-Phe-cAMP and N6-Bn-cAMP versus N6-Bnz-cAMP.
To test this hypothesis, we either neutralized or reversed the charge at position 635 by replacing the arginine with either alanine (R635A) or glutamate (R635E). Neutralization resulted in an apparent affinity of N6-Phe-cAMP similar to the apparent affinity of cAMP for mHCN2 WT channels. From this result, we suggest that now N6-Phe-cAMP interacts with R635A in a similar manner as cAMP with the mHCN2 WT. Reversing the charge by replacing the arginine with a glutamate led to an apparent affinity of N6-Phe-cAMP slightly lower than that of cAMP for mHCN2 WT channels. This might be caused by a repulsive force between the aromatic ring of N6-Phe-cAMP and the negative side chain of the newly introduced glutamate. Together, these data support the hypothesis resulting from the docking experiments, proposing a stabilizing or destabilizing interaction between N6 modifications and Arg-635. A role of Arg-635 for binding and selectivity was discussed earlier by Zhou and Siegelbaum (30). They described this residue as being involved in discriminating between cAMP and cGMP without contributing to efficacy.
Usability of N6 modifications in cAMP to discriminate between PKA and HCN channels
What can we learn from the results presented here regarding discrimination between PKA and HCN channel activation? To compare the apparent affinities of the derivatives at HCN2 channels with that at PKA, we plotted relative EC50 values for PKAI and PKAII obtained from the literature (EC50/EC50, cAMP) versus relative EC50 values for HCN2 obtained here (Fig. 7) (29, 34–36).
Figure 7.

Comparison of relative apparent affinity for PKA and HCN2. A, apparent affinities for cAMP derivatives were related to apparent affinities for unmodified cAMP. Among the tested derivatives, the best discrimination can be realized with N6-Bnz-cAMP. Data for PKA were obtained from Refs. 29, 34–36. Filled symbols represent PKAI and empty symbols PKAII. B, sequence alignment comparing the distal parts of the C-helix sequences of mouse and human HCN isoforms with those from A and B sites in regulatory subunits of human PKAI and II. Alignments and structure predictions (α-helix C marked in green) are shown as proposed by Berman et al. (39), who used a structure and transformation method (SAT). Prediction of the HCN C-helix follows a suggestion from Lee and MacKinnon (40). The asterisks mark Arg-635 mutated herein. Positively charged residues are shown in red and negatively charged in blue.
The very high apparent affinity of N6-Phe-cAMP as well as the very low one of N6-Bnz-cAMP for HCN2 channels brings the N6 position into focus for the search for discriminating agonists. However, in the case of N6-Phe-cAMP, the apparent affinity for PKA I and PKAII has also been shown to be relatively high, with a factor of ∼2.4 and ∼2.8 higher for PKAI and PKAII, respectively, compared with cAMP (29). Thus, the discrimination between HCN and PKA is not very strong.
N6-Bn-cAMP was also very potent for HCN2 channels, whereas it was in most cases less potent than cAMP in PKAI (29, 34, 36). The differences are not very high; however, a benzyl ring in the N6 position could be at least one of several modifications in the cAMP molecule to yield discrimination between HCN2 and PKAI. A discrimination to PKAII is questionable because the apparent affinity has been shown to be either unchanged (29) or two times better than for cAMP (36).
The derivative with the best discrimination ability found here is N6-Bnz-cAMP. It is a potent agonist of PKAI, with a similar apparent affinity as cAMP (29, 35), but a relatively poor agonist of HCN2, with an apparent affinity 18 times worse than cAMP. This is a very important finding because N6-Bnz-cAMP is a well-established tool for studying cAMP-mediated signal transduction in a wide range of cellular processes (e.g. 23, 37, 38). Discrimination between PKAII and HCN2 is expected to be less strong because N6-Bnz-cAMP is two times less potent to PKAII than cAMP (29, 35).
The C-helix of CNBDs (green bars in Fig. 7) acts as a hydrophobic lid covering the base moiety of the cyclic nucleotide and thereby capping the binding pocket (39). Is the positively charged Arg-635 at the C-terminal end of this C-helix a unique feature of HCN channels and can it be used for further discrimination approaches? The alignment in Fig. 7 shows that, with the exception of mHCN4, Arg-635 is highly conserved in mouse and human HCN channel isoforms, which allows speculation regarding an important role in ligand binding. A comparison with A and B sites of PKAI and PKAII shows that there is no such arginine at the C-terminal ends of the PKA C-helices. However, there is an interesting pattern of charged amino acids right before R635 that reads negative–positive–X–negative–positive and can be found in most HCN channels. This pattern is also present in A sites of PKA RI and, slightly modified, in PKA RII, although not as part of the C-helix sequence but in the consecutive loop directly behind. Such a pattern could not be found in any B sites of PKA presented here. Another pattern in HCN CNBDs is a strong positive spot at the N-terminal part of the C-helix followed by negative residues two positions C-terminal from that. Again this pattern can be found in A sites of PKA RI and additionally in RII but not in B sites. Those differences might be responsible for N6-Phe-cAMP, N6-Bn-cAMP, and N6-Bnz-cAMP preferring A sites over B sites (34, 41).
Because the C-helix is the most variable structure of CNBDs (39), residues in that helix are potential key players in determining protein-specific ligand selectivity. After comparing sequences of HCN CNBDs with A sites and B sites, we speculate that there is a greater similarity between binding to HCN CNBDs and A sites than between HCN CNBDs and B sites. Thus, to identify further cAMP derivatives with a higher degree of discrimination between HCN and PKA than shown here, we propose to focus on the unique characteristics of binding to B sites.
In summary, our analysis shows that possible cross-activation of CN-modulated ion channels should be taken into account when CN-dependent cellular processes are investigated with N6-modified derivatives. To overcome this problem, we further suggest that, even with a missing all-or-nothing principle in protein activation by cyclic nucleotides, carefully performed concentration–activation relationships can be used to identify a concentration range in which selective protein activation is possible.
Experimental procedures
X. laevis oocytes as a heterologous expression system
Surgical removal of oocytes was performed under anesthesia (0.3% tricaine, MS-222, Pharmaq Ltd., Fordingbridge, UK) from adult females of the South African claw frog X. laevis. The oocytes were treated with collagenase A (3 mg/ml, Roche) for 105 min in Ca2+-free Barth's solution containing 82.5 mm NaCl, 2 mm KCl, 1 mm MgCl2, and 5 mm Hepes (pH 7.5). After this procedure, oocytes of stages IV and V were manually dissected and injected with cRNA encoding either mHCN2 channels of Mus musculus (NM_008226) or the mHCN2 mutants R635A and R635E, respectively. After injection with cRNA, the oocytes were cultured at 18 °C for 2–6 days in Barth's solution containing 84 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 0.82 mm MgSO4, 0.41 mm CaCl2, 0.33 mm Ca(NO3)2, and 7.5 mm Tris (pH 7.4). The procedures had approval from the authorized animal ethical committee of the Friedrich Schiller University Jena. The methods were carried out in accordance with the approved guidelines. Oocytes harvested in our own laboratory were complemented with ready-to-use oocytes purchased from Ecocyte Bioscience (Dortmund, Germany).
Molecular biology
The mouse HCN2 (accession number NM_008226) channel and the modified subunit variants were subcloned in front of the T7 promoter of pGEM-HCN2. The point mutations R635E and R635A were introduced by overlapping PCR using the following primers: PCR 1 forward, 5′-TCATACTCGCGCGCGGCCCCAAGGTTTC-3′ (restriction site MauBI, outer primer); PCR 1 reverse, 5′-CTTCTTGCCTATCTCATCTAGCCGGTCAATAGC-3′ (R635E) or 5′-CTTCTTGCCTATGGCATCTAGCCGGTCAATAGC-3′ (R635A); PCR 2 forward, 5′-GCTATTGACCGGCTAGATGAGATAGGCAAGAAG-3′ (R635E) or 5′-GCTATTGACCGGCTAGATGCCATAGGCAAGAAG-3′ (R635A); PCR 2 reverse, 5′-AGCAGGGTTGGTCTAGAGTCACAAGTTGGAAGAG-3′ (restriction site XbaI, outer primer). In a third PCR, the PCR products of PCR 1 and 2 were used as a template using the outer primers. The resulting fragment was subcloned into the pGEM-HCN2 subunit. Correctness of the sequences was confirmed by restriction analysis and sequencing (Microsynth SEQLAB). cRNAs were prepared using the mMESSAGE mMACHINE T7 Kit (Ambion).
Electrophysiological experiments
Macroscopic currents were recorded using the patch clamp technique in the inside-out configuration. All measurements were started after a delay of 3.5 min to minimize rundown phenomena. Patch pipettes were pulled from quartz tubing whose outer and inner diameters were 1.0 and 0.7 mm (Vitrocom), respectively, using a laser puller (P-2000, Sutter Instrument). The pipette resistance was 1.2–2.1 megaohm. The bath solution contained 100 mm KCl, 10 mm EGTA, and 10 mm Hepes (pH 7.2), and the pipette solution contained 120 mm KCl, 10 mm Hepes, and 1.0 mm CaCl2 (pH 7.2). For parts of the experiments, different concentrations of cAMP, N6-Phe-cAMP, N6-Bn-cAMP, or N6-Bnz-cAMP (BIOLOG LSI, Bremen, Germany) were applied with the bath solution. A saturating concentration of 10 μm cAMP was applied to each patch to define the maximum current amplitude. An HEKA EPC 10 USB amplifier (Harvard Apparatus) was used for current recording. Pulsing and data recording were controlled by the Patchmaster software (Harvard Apparatus). The sampling rate was 5 kHz. The holding potential was generally −30 mV.
Quantification and statistical analysis
Concentration–activation relationships were analyzed by approximating the Hill equation to each individual recording using the OriginPro 9.0G software.
| (Eq. 1) |
I is the actual current amplitude at a given agonist concentration, Imax the maximal current amplitude at a saturating concentration of 10 μm cAMP, EC50 the concentration of half-maximum activation, and H the Hill coefficient. Current amplitudes were generally obtained from tail currents at a −100 mV pulse following an activating −130 mV pulse and corrected for leak currents obtained from a short −100 mV pulse at the beginning of each trace. Values for EC50 and H were yielded for each individual recording and averaged. Steady-state activation curves were analyzed by fitting the Boltzmann equation to each individual recording using the OriginPro 9.0G software.
| (Eq. 2) |
V1/2 is the voltage of half-maximum activation and zδ the effective gating charge. F, R, and T are the Faraday constant, the molar gas constant, and the temperature in Kelvin, respectively. I is the actual current amplitude and Imax the maximum current amplitude at the saturating hyperpolarizing voltage of −150 mV specified for each patch.
The time courses of current activation were fitted with a single exponential starting after an initial delay.
| (Eq. 3) |
A is the amplitude, t the time, and τ the time constant for activation.
Experimental data are given as mean ± S.E. Statistical analysis was performed by an unpaired Student's t test. A value of p < 0.05 was accepted as statistically significant.
Molecular modeling and docking experiments
Three-dimensional structures of the anionic species of cAMP, N6-Phe-cAMP, N6-Bn-cAMP, and N6-Bnz-cAMP were generated in Maestro (Schrödinger, LLC) and prepared using the LigPrep workflow (Schrödinger, LLC). One monomer (chain A) of the crystal structure of cAMP-bound mHCN2J (25) was prepared using the Protein Preparation Wizard in Maestro; the program was evoked to cap the termini with acetyl and N-methyl amide moieties and to convert selenomethionine residues back to methionine residues. The potential grid for docking was centered on the cocrystallized cAMP, and the ligand length was set to 16 Å or less. The dimensions of the inner box that restricts the region in which the diameter midpoint of each ligand can be located were set to 10 × 10 × 10 Å. No constraints, rotatable OH- and SH-groups, or excluded volumes were defined. Molecular docking was performed with Glide (Schrödinger, LLC) using the Extra Precision (26) mode with default options.
Author contributions
T. L., M. B., F. Spiegelhalter, and J. K. data curation; T. L., M. B., F. Spiegelhalter, and J. K. formal analysis; T. L., M. B., F. Spiegelhalter, F. Schwede, T. S., H. G., and J. K. methodology; T. L., M. B., F. Spiegelhalter, F. Schwede, T. S., and H. G. writing-review and editing; M. B. and J. K. validation; F. Schwede, H. G., and J. K. conceptualization; F. Schwede and T. S. resources; H. G. and J. K. supervision; H. G. and J. K. funding acquisition; J. K. writing-original draft; J. K. project administration.
Acknowledgments
We thank Sandra Bernhardt, Uta Enke, Andrea Kolchmeier, Claudia Ranke, and Karin Schoknecht for excellent technical assistance. We are grateful for computational support and infrastructure provided by the Zentrum für Informations- und Medientechnologie at the Heinrich Heine University Düsseldorf and the computing time provided by the John von Neumann Institute for Computing (to H. G.) on the supercomputer JUWELS at the Jülich Supercomputing Center (user IDs HKF7 and HDD17).
This work was supported by grant DFG Research Unit 2518 DynIon of the Deutsche Forschungsgemeinschaft (projects P2 (KU 3092/2–1) and P7 (GO 1367/2–1)). F. S. is Head of Research and Development of the BIOLOG Life Science Institute, which sells the cAMP analogs that were used in this study.
- Epac
- exchange protein directly activated by cAMP
- CN
- cyclic nucleotide
- HCN
- hyperpolarization-activated and cyclic nucleotide–modulated
- CNBD
- cyclic nucleotide–binding domain
- N6-Phe-cAMP
- N6-phenyladenosine-3′,5′-cyclic monophosphate
- N6-Bn-cAMP
- N6-benzyladenosine-3′,5′-cyclic monophosphate
- N6-Bnz-cAMP
- N6-benzoyl-adenosine-3′,5′-cyclic monophosphate
- RMSD
- root mean square deviation.
References
- 1. Sutherland E. W., and Rall T. W. (1958) Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 232, 1077–1091 [PubMed] [Google Scholar]
- 2. Beavo J. A., and Brunton L. L. (2002) Cyclic nucleotide research: still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3, 710–718 10.1038/nrm911 [DOI] [PubMed] [Google Scholar]
- 3. Schindler R. F., and Brand T. (2016) The Popeye domain containing protein family: a novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 120, 28–36 10.1016/j.pbiomolbio.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Craven K. B., and Zagotta W. N. (2006) CNG and HCN channels: two peas, one pod. Annu. Rev. Physiol. 68, 375–401 10.1146/annurev.physiol.68.040104.134728 [DOI] [PubMed] [Google Scholar]
- 5. DiFrancesco D. (1986) Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature 324, 470–473 10.1038/324470a0 [DOI] [PubMed] [Google Scholar]
- 6. DiFrancesco D. (1999) Dual allosteric modulation of pacemaker (f) channels by cAMP and voltage in rabbit SA node. J. Physiol. 515, 367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robinson R. B., and Siegelbaum S. A. (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65, 453–480 10.1146/annurev.physiol.65.092101.142734 [DOI] [PubMed] [Google Scholar]
- 8. Santoro B., and Tibbs G. R. (1999) The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann. N.Y. Acad. Sci. 868, 741–764 10.1111/j.1749-6632.1999.tb11353.x [DOI] [PubMed] [Google Scholar]
- 9. Wang J., Chen S., Nolan M. F., and Siegelbaum S. A. (2002) Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron 36, 451–461 10.1016/S0896-6273(02)00968-6 [DOI] [PubMed] [Google Scholar]
- 10. Wang J., Chen S., and Siegelbaum S. A. (2001) Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J. Gen. Physiol. 118, 237–250 10.1085/jgp.118.3.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clapham D. E. (1998) Not so funny anymore: pacing channels are cloned. Neuron 21, 5–7 10.1016/S0896-6273(00)80508-5 [DOI] [PubMed] [Google Scholar]
- 12. Santoro B., Grant S. G., Bartsch D., and Kandel E. R. (1997) Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. U.S.A. 94, 14815–14820 10.1073/pnas.94.26.14815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wainger B. J., DeGennaro M., Santoro B., Siegelbaum S. A., and Tibbs G. R. (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810 10.1038/35081088 [DOI] [PubMed] [Google Scholar]
- 14. DiFrancesco D., and Tortora P. (1991) Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351, 145–147 10.1038/351145a0 [DOI] [PubMed] [Google Scholar]
- 15. Wicks N. L., Wong T., Sun J., Madden Z., and Young E. C. (2011) Cytoplasmic cAMP-sensing domain of hyperpolarization-activated cation (HCN) channels uses two structurally distinct mechanisms to regulate voltage gating. Proc. Natl. Acad. Sci. U.S.A. 108, 609–614 10.1073/pnas.1012750108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shabb J. B., and Corbin J. D. (1992) Cyclic nucleotide-binding domains in proteins having diverse functions. J. Biol. Chem. 267, 5723–5726 [PubMed] [Google Scholar]
- 17. Rehmann H., Wittinghofer A., and Bos J. L. (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 10.1038/nrm2082 [DOI] [PubMed] [Google Scholar]
- 18. Chen L., Wang P., Andrade C. F., Zhao I. Y., Dubé P. E., Brubaker P. L., Liu M., and Jin T. (2005) PKA independent and cell type specific activation of the expression of caudal homeobox gene Cdx-2 by cyclic AMP. FEBS J. 272, 2746–2759 10.1111/j.1742-4658.2005.04694.x [DOI] [PubMed] [Google Scholar]
- 19. Hewer R. C., Sala-Newby G. B., Wu Y. J., Newby A. C., and Bond M. (2011) PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell Cardiol. 50, 87–98 10.1016/j.yjmcc.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang G., Joseph J. W., Chepurny O. G., Monaco M., Wheeler M. B., Bos J. L., Schwede F., Genieser H. G., and Holz G. G. (2003) Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic β-cells. J. Biol. Chem. 278, 8279–8285 10.1074/jbc.M211682200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rangarajan S., Enserink J. M., Kuiperij H. B., de Rooij J., Price L. S., Schwede F., and Bos J. L. (2003) Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the β2-adrenergic receptor. J. Cell Biol. 160, 487–493 10.1083/jcb.200209105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shimomura H., Imai A., and Nashida T. (2004) Evidence for the involvement of cAMP-GEF (Epac) pathway in amylase release from the rat parotid gland. Arch. Biochem. Biophys. 431, 124–128 10.1016/j.abb.2004.07.021 [DOI] [PubMed] [Google Scholar]
- 23. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., and Døskeland S. O. (2003) cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402 10.1074/jbc.M302179200 [DOI] [PubMed] [Google Scholar]
- 24. Ng L. C. T., Putrenko I., Baronas V., Van Petegem F., and Accili E. A. (2016) Cyclic purine and pyrimidine nucleotides bind to the HCN2 ion channel and variably promote C-terminal domain interactions and opening. Structure 24, 1629–1642 10.1016/j.str.2016.06.024 [DOI] [PubMed] [Google Scholar]
- 25. Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., and Gouaux E. (2003) Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425, 200–205 10.1038/nature01922 [DOI] [PubMed] [Google Scholar]
- 26. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., and Mainz D. T. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- 27. Døskeland S. O., Ogreid D., Ekanger R., Sturm P. A., Miller J. P., and Suva R. H. (1983) Mapping of the two intrachain cyclic nucleotide binding sites of adenosine cyclic 3′,5′-phosphate dependent protein kinase I. Biochemistry 22, 1094–1101 10.1021/bi00274a016 [DOI] [PubMed] [Google Scholar]
- 28. Ogreid D., Ekanger R., Suva R. H., Miller J. P., and Døskeland S. O. (1989) Comparison of the two classes of binding sites (A and B) of type I and type II cyclic-AMP-dependent protein kinases by using cyclic nucleotide analogs. Eur. J. Biochem. 181, 19–31 10.1111/j.1432-1033.1989.tb14689.x [DOI] [PubMed] [Google Scholar]
- 29. Ogreid D., Ekanger R., Suva R. H., Miller J. P., Sturm P., Corbin J. D., and Døskeland S. O. (1985) Activation of protein kinase isozymes by cyclic nucleotide analogs used singly or in combination: principles for optimizing the isozyme specificity of analog combinations. Eur. J. Biochem. 150, 219–227 10.1111/j.1432-1033.1985.tb09010.x [DOI] [PubMed] [Google Scholar]
- 30. Zhou L., and Siegelbaum S. A. (2007) Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 15, 655–670 10.1016/j.str.2007.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stephenson R. P. (1956) A modification of receptor theory. Br. J. Pharmacol. Chemother. 11, 379–393 10.1111/j.1476-5381.1956.tb00006.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flynn G. E., Black K. D., Islas L. D., Sankaran B., and Zagotta W. N. (2007) Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure 15, 671–682 10.1016/j.str.2007.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zorn M., Fladmark K. E., Ogreid D., Jastorff B., Døskeland S. O., and Dostmann W. R. (1995) Ala335 is essential for high-affinity cAMP-binding of both sites A and B of cAMP-dependent protein kinase type I. FEBS Lett. 362, 291–294 10.1016/0014-5793(95)00261-7 [DOI] [PubMed] [Google Scholar]
- 35. Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X. B., Christensen A. E., Schwede F., Genieser H. G., Bos J. L., Doskeland S. O., Beavo J. A., and Butt E. (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278 10.1038/nmeth0408-277 [DOI] [PubMed] [Google Scholar]
- 36. Chepurny O. G., Bertinetti D., Diskar M., Leech C. A., Afshari P., Tsalkova T., Cheng X., Schwede F., Genieser H. G., Herberg F. W., and Holz G. G. (2013) Stimulation of proglucagon gene expression by human GPR119 in enteroendocrine L-cell line GLUTag. Mol. Endocrinol. 27, 1267–1282 10.1210/me.2013-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lo K. W., Ashe K. M., Kan H. M., Lee D. A., and Laurencin C. T. (2011) Activation of cyclic amp/protein kinase: a signaling pathway enhances osteoblast cell adhesion on biomaterials for regenerative engineering. J. Orthop. Res. 29, 602–608 10.1002/jor.21276 [DOI] [PubMed] [Google Scholar]
- 38. Monje P. V., Athauda G., and Wood P. M. (2008) Protein kinase A-mediated gating of neuregulin-dependent ErbB2-ErbB3 activation underlies the synergistic action of cAMP on Schwann cell proliferation. J. Biol. Chem. 283, 34087–34100 10.1074/jbc.M802318200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berman H. M., Ten Eyck L. F., Goodsell D. S., Haste N. M., Kornev A., and Taylor S. S. (2005) The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. U.S.A. 102, 45–50 10.1073/pnas.0408579102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee C. H., and MacKinnon R. (2017) Structures of the human HCN1 hyperpolarization-activated channel. Cell 168, 111–120.e11 10.1016/j.cell.2016.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huq I., Dostmann W. R., and Ogreid D. (1996) Isoleucine 368 is involved in low-affinity binding of N6-modified cAMP analogues to site B of the regulatory subunit of cAMP-dependent protein kinase I. Biochem. J. 316, 337–343 10.1042/bj3160337 [DOI] [PMC free article] [PubMed] [Google Scholar]