

Abstract
Since ancient times it is known that melancholia and sleep disturbances co-occur. The introduction of polysomnography into psychiatric research confirmed a disturbance of sleep continuity in patients with depression, revealing not only a decrease in Slow Wave Sleep, but also a disinhibition of REM (rapid eye movement) sleep, demonstrated as a shortening of REM latency, an increase of REM density, as well as total REM sleep time. Initial hopes that these abnormalities of REM sleep may serve as differential-diagnostic markers for subtypes of depression were not fulfilled. Almost all antidepressant agents suppress REM sleep and a time-and-dose–response relationship between total REM sleep suppression and therapeutic response to treatment seemed apparent. The so-called Cholinergic REM Induction Test revealed that REM sleep abnormalities can be mimicked by administration of cholinomimetic agents. Another important research avenue is the study of chrono-medical timing of sleep deprivation and light exposure for their positive effects on mood in depression. Present day research takes the view on insomnia, i.e., prolonged sleep latency, problems to maintain sleep, and early morning awakening, as a transdiagnostic symptom for many mental disorders, being most closely related to depression. Studying insomnia from different angles as a transdiagnostic phenotype has opened many new perspectives for research into mechanisms but also for clinical practice. Thus, the question is: can the early and adequate treatment of insomnia prevent depression? This article will link current understanding about sleep regulatory mechanisms with knowledge about changes in physiology due to depression. The review aims to draw the attention to current and future strategies in research and clinical practice to the benefits of sleep and depression therapeutics.
Subject terms: Predictive markers, Circadian rhythms and sleep
Introduction—the phenomenology of sleep in depression
A brief historical overview
Robert Burton, in his Anatomy of Melancholia [1] remarked that ancient Greek physicians were well aware of the fact that melancholic individuals complained of difficulties falling asleep, maintaining sleep or of waking up too early in the morning. Treatment of difficulties with sleep in antique times consisted of listening to calm music, reading, or the use of opium or alcohol [2]. Emil Kraepelin, the founder of modern psychiatry, observed that mental symptoms fall mainly into two groups and he created the illness categories [3] ‘manic-depression’’ and ‘dementia praecox’’, today clinically similar to ‘affective’’ and ‘psychotic’’ phenotypes. He postulated that a certain type of sleep difficulty co-occurred with a certain depressive subtype, i.e., sleep onset problems with “neurotic” and sleep maintenance/early morning awakenings with “endogenous” depression. Thus, the idea that the type of sleep disturbance in depressed individuals might support differential-diagnostic considerations was already born over a 100 years ago and experienced a renaissance in the 1970s with the discovery of shortened rapid eye movement (REM) sleep latencies in depressed individuals. Before then, pharmacological agents like bromides, paraldehyde, scopolamine, or barbiturates were used to treat insomnia accompanying depression until these were replaced by benzodiazepine hypnotics and sedating antidepressants in the 1950/60s. The incidence of depression is steadily rising globally and present-day epidemiological data reveal that depression is now the third leading contribution to global disease burden [4] and an estimated 25% of the population in industrialized countries will suffer from depression once in a lifetime [5]. This review seamlessly ties in with and extends an earlier line of research of using sleep as a window into the brain’s neurobiological links between sleep processes and depression. By telling the story of an early success, a subsequent stagnation phase and new enthusiasm based on the integration of emerging neurobiological knowledge into the mechanistic overlap of sleep and depression disturbances, this review is paving a way for the integration of this knowledge into clinical practice.
REM sleep and depression—early hopes
The discovery of phases of REM during sleep in 1953 [6] and the ensuing interest in sleep research led to the establishment of psychiatric sleep research utilizing polysomnography (PSG). Kupfer et al. from Pittsburgh [7, 8] were among the first to suggest that changes of REM sleep, i.e., shortened REM sleep latency (shorter interval between sleep onset and the first occurrence of REM sleep), increased total REM sleep duration and increased REM density (higher phasic eyes movements during REM sleep), are typical sleep characteristics of patients with primary vs. secondary depression (see Fig. 1 for illustration). Furthermore, polysomnographically measured sleep continuity was disturbed (i.e., long sleep latency and frequent awakenings throughout the sleep period) and Slow Wave Sleep (SWS) reduced. These findings were met with enthusiasm at the time and promoted the idea, later known as biomarkers [9], to identify functional subtypes within and across diagnostic categories by which treatment could be stratified and response predicted for each patient in order to achieve remission.
Fig. 1.

Comparison of the polysomnographic (PSG) profile of a good sleeper (upper panel) and a patient hospitalized for severe depression according to DSM-IV criteria (lower panel). Both subjects have been free from intake of any psychotropic drug for at least 14 days. The y-axis lists arousal (micro-arousals), wake and sleep stages (REM, stage N1 to N3) and eye movements). The x-axis is the time axis. Sleep in depression is characterized by alterations of sleep continuity (prolonged sleep onset and sleep maintenance problems), a decrement of SWS (Slow Wave Sleep, also measurable as a decline in Delta-Power) and a disinhibition of REM sleep: this encompasses shortening of REM latency, prolongation of the first REM period and increase of REM density. Original data from Freiburg sleep lab, hitherto unpublished
REM sleep and depression—disappointed expectations
The question of the differential-diagnostic value of REM sleep abnormalities puzzled psychiatric sleep research for almost two decades [10]. It was thought that REM sleep abnormalities might be more typical for the ‘primary/melancholic/endogenous’’ subtype compared to the ‘secondary/neurotic/non-melancholic’’ subtype. With increasing research efforts, however, it turned out that especially age, and to a certain extent sex, has a very strong effect on REM sleep latency, whereby REM sleep latency decreases with age. Revisiting the primary/secondary dichotomy and REM latency, carefully controlling for age and severity of depression, Thase et al. [11] concluded that initial positive findings may have represented epiphenomena of sample differences with respect to age or severity of depression. Additional research into other mental disorders (especially schizophrenia, borderline personality disorder and alcohol dependency) revealed that patients with these diagnostic entities also display some degree of REM sleep alterations [12, 13], further weakening the assumptions of a high specificity of REM sleep abnormalities for depression.
Antidepressants and sleep in depression
An important impetus for the field of sleep research in depression was the observation that almost all antidepressants influence sleep, notably by strongly suppressing REM sleep [14], whereby the extent of REM sleep suppression covaried with clinical ratings indicating therapeutic efficacy of antidepressants response [10]. Thus, REM suppression appeared as a promising early predictor of subsequent treatment response. Identifying such early predictor that could function as a biomarker would be of high clinical value given the long latency to response and the limited response rate of ~60% of depressed patients. Insofar, the suppression of REM sleep seemed to constitute a window to the brain reflecting therapeutic efficacy of antidepressant substances. Table 1 summarizes the effects of antidepressants on different aspects of polysomnographically recorded sleep [14].
Table 1.
Impact of antidepressants on PSG recorded sleep (according to Riemann and Nissen, 2012 [14])
| Types of antidepressantsa | Sleep continuity | Slow wave sleep | REM sleep |
|---|---|---|---|
| Nonspecific monoamine reuptake inhibitors (TCAs) | |||
| Amitriptyline | ↑ | ↔SWS % | ↓ REM %, ↑ REM latency |
| Doxepin | ↑ | ↔SWS % | ↓ REM %, ↑ REM latency |
| Clomipramine | ↑ | ↑ | ↓ REM % |
| Desipramine | ↑ | ↑ | ↓ REM % |
| Nortriptyline | ? | ↑ | ↓ REM % |
| Imipramine | ? | ? | ↓ REM % |
| Selective serotonine reuptake inhibitors (SSRIs) | |||
| Citalopram | ? | ? | ↓ |
| Fluvoxamine | ↓ | ? | ↓ |
| Fluoxetine | ↓ | ? | ↓ |
| Paroxetine | ↓ | ? | ↓ |
| Noradrenaline reuptake inhibitors (NRIs) | |||
| Maprotiline | ↑ | ? | ↓ |
| Viloxazine | ↓ | ↓ SWS % | ↓ |
| Norepinephrine-dopamine reuptake inhibitors (NDRIs) | |||
| Bupropion | ? | ↓ | ↑ |
| Serotonine-noradrenaline reuptake inhibitors (SNRIs) | |||
| Venlafaxine | ↓ | ? | ↓ REM % |
| Monoamine oxidase inhibitors (MAOis) | |||
| Moclobemide | ↓ | ? | ↓ REM % |
| Phenelzine | ↓ | ? | ↓ REM % |
| Other mechanisms of action | |||
| Trimipramine | ↑ | ↔SWS % | ↔REM % |
| Mirtazapine | ↑ | ↔SWS % | ↔REM % |
| Trazodon | ↑ | ↑ SWS % | ↔REM % (↑ to ↓, individual studies) |
aReported effects are based on preponderance of evidence from published studies (see text for details). Many effects are inconsistent between individual studies. “↑” Indicates increase from pre-treatment baseline; “↓” indicates decrease from pre-treatment baseline; “ ↔ ” indicates no change from pre-treatment baseline
Data in the table demonstrate that most antidepressants suppress REM sleep, with only a few exceptions such as trimipramine, trazodone and mirtazapine. Many of these substances lead to an enhanced SWS and improved sleep continuity but it is important to note that certain drug classes, especially SSRIs (Selective Serotonin Reuptake Inhibitors), may induce a deterioration of sleep continuity, which is clinically expressed as increased insomnia complaints. This heterogeneity posed a challenge to the initial hope of using PSG to predict treatment response and it has been given up by and large [14]—partly due to failures to replicate the predictive value of REM sleep suppression for later therapeutic response, partly due to the inherent costs and clinical impracticability of using PSG as a drug monitoring tool. Today, miniaturized EEG monitors and refined semi-automatic algorithms for sleep analyses might allow the introduction of sleep EEG into the clinics [15]. An intrinsic value of this line of research, however, is that it is now widely acknowledged that drug wash-out periods of at least 7–14 days (for fluoxetine even longer) are needed to obtain valid sleep data. This, however, renders PSG research into depression complicated, because today most patients with severe depression who come into contact with a research facility are pre-medicated and drug-withdrawal would not be consistent with standard guidelines and considered unethical.
The cholinergic REM induction test
An experimental avenue towards a mechanistic understanding of REM sleep abnormalities in depression constituted the Cholinergic REM Induction Test (CRIT). This strategy, developed by Gillin and Sitaram [16], was based on the reciprocal interaction model of Non-REM/REM sleep regulation [17, 18] positing that REM sleep onset can be advanced in animals and healthy volunteers by cholinergic stimulation [19] because REM sleep is largely governed by cholinergic neurons in the brain stem (see Fig. 2).
Fig. 2.

a, b The Hobson/McCarley Model and the regulation of Non-REM and REM sleep in good sleepers and patients with depression. Experiments by Hobson and McCarley in cats especially in the brain stem were able to show that manipulation of cholinergic or aminergic cell groups is able to change sleep. In the case of good sleepers (a) the aminergic systems (dorsal raphe/locus coeruleus) are active during Non-REM sleep and cease their activity around the onset of REM sleep, where cholinergic activity becomes dominant. The reciprocal interaction between the aminergic and the cholinergic system determines the Non-REM-REM cycle. In depression (b), an overactive cholinergic system or a weakened aminergic system leads to an earlier onset of REM sleep. Based on refs [10, 18], permission granted
Berger et al. [20] showed that such a cholinergic stimulus elicited an even more-pronounced response in patients with depression compared to good sleepers and patients with other mental disorders. Other studies showed that this early onset of REM sleep may also be provoked by cholinomimetic agents in individuals at high risk for depression [21], who later displayed a higher rate of episodes of depression over a follow-up period of >10 years [22]. Thus, it was demonstrated that early onset of REM sleep after cholinergic stimulation is not only a state and trait marker, but also a vulnerability marker for depression. Given that drug wash-out periods of at least 7–14 days are necessary to obtain valid CRIT data, it is understandable that, apart from its scientific value, the CRIT never saw widespread clinical application for determining risk profiles.
Biological timing and sleep deprivation in depression
Wehr et al. [23] published ground-breaking work on the likely involvement of the biological time keeping system in the pathogenesis of affective disorders, high-lighting that an advance of the sleep period by 6 h normalized the REM sleep phases and induced a longer-lasting remission of depressive symptoms. The authors inferred from their longitudinal observations that REM sleep rhythmicity and their underlying biochemical rhythms must have altered internal phase-relationships that are associated with certain psychopathological phenotypes. Similar remissions of depressive symptoms were also achieved in patients undergoing complete wake therapy (full night of sleep deprivation), that was first tested by Pflug and Toelle [24]. These results were better understood with the two-process model of sleep regulation proposed by Borbély et al. [25–27], in which a homeostatic sleep process (S) and a circadian process (C), interact in a threshold- and time-dependent manner (see Fig. 3), thereby incorporating that appropriate timing of sleep with respect to the internal clock is crucial for stable mood as described in the ‘internal coincidence’’ model [28, 29].
Fig. 3.
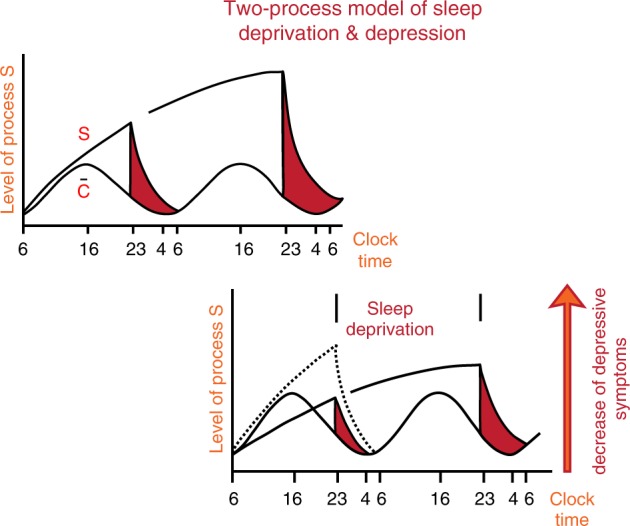
The Two-Process-Model is based on two components or factors, i.e., process C and process S. Process C reflects circadian rhythmicity, which is under control of the light–dark cycle. Process S reflects sleep or sleep pressure and can be measured as delta waves during night sleep. The interaction between C and S describes or determines the sleep-wake cycle in good sleepers (upper left panel). In contrast, in depression (lower right panel), process S is deficient, as reflected by low levels of slow wave sleep/delta power in the spectral analysis of the sleep EEG. Sleep deprivation may act antidepressive because it enhances process S, thus leading to an increased amount of slow wave sleep after sleep deprivation. Personal permission by A. Borbely
Germain and Kupfer [30] summarized further chronobiologically inspired experiments based on circadian/ sleep hypotheses of depression, including the circadian phase-shift effects of light [31] or the social rhythm model [32]. At present, the most comprehensive, decisive review on sleep deprivation therapy explaining its antidepressant effects on the level of molecular circadian regulation was compiled by Bunney and Bunney [33]. Clinically, therapeutic sleep deprivation (SD) is a rapid acting treatment for a subset of patients with major depression. Within 24 h it provokes a transient, but strong decrease in depressive symptoms in those who respond [34, 35]. Unfortunately, the effect is not long lasting and a relapse into depression occurs after the next night of sleep. Even brief daytime sleep periods after SD can reverse the therapeutic effect [36]. Strategies to enhance or prolong the antidepressive effect are concomitant pharmacotherapy, light therapy or sleep phase advance, if correctly timed and any deviation from the schedule avoided to prevent a rebound into depressive symptomatology. Paradigms of SD in depression include total sleep deprivation, partial sleep deprivation (mostly concerning the second half of the night), selective REM sleep deprivation and selective slow wave sleep (SWS) deprivation [37]. Clinical predictors for positive SD response include the melancholic subtype of depression, a high level of vigilance, the propensity to produce diurnal mood variations and short REM latency [38, 39]. From a scientific point of view, SD presents a unique paradigm to study the neurobiology of major depression within a short period of time and without pharmacological interference because patients can be investigated in a depressed and non-depressed/euthymic state. Unfortunately, up to now, the neurobiological basis of the immediate antidepressant response to SD and its rapid drop with more sleep is not sufficiently understood. The rapid antidepressive mechanisms probably is quite different from conventional treatments (pharmacological/psychotherapy). Therefore, elucidating the neuronal mechanisms of SD has become crucially important for novel perspectives on antidepressive treatment.
We recently integrated the synaptic plasticity hypothesis of major depression [40, 41] and the synaptic homeostasis hypothesis of sleep-wake regulation [42, 43] into a synaptic plasticity model of sleep wake regulation. The model posits, in brief, that sleep deprivation, through an increase of overall synaptic strength, shifts patients with major depression into a more favorable window of synaptic plasticity and network function [44]. While certainly oversimplified, the model allows for testing specific predictions, such as proposed antidepressive properties of selective slow wave sleep suppression, for instance through auditory closed-loop stimulation [45, 46] and translation into animal models of depression for further deciphering the neural mechanisms of rapid treatment response.
Sleep and depression: neuroimaging results
Advances in human brain imaging techniques, particularly [18F]2-fluoro-2-deoxy-d-glucose PET (Positron Emission Tomography) studies, have led to new insights into changes in brain metabolism during the sleep-wake cycle in healthy humans and those with depression, which are not accessible to surface electrophysiology. The integration of these studies suggests that functional neuroanatomic correlates can be assigned to characteristic PSG alterations in depression, of which three main components can be distinguished. First, persistent hyperactivity in the basic ascending arousal system throughout the sleep-wake cycle in depression might be implicated in the experience of hyperarousal during waking and disturbed sleep continuity [47]. Second, persistent hyperactivity in the ventral emotional system that includes the amygdala and the ventral anterior cingulate cortex (ACC) might relate to disturbances in affect, including depressed mood, and to the characteristic enhancement of REM sleep in depression [48]. Third, hypoactivity in the dorsal executive system throughout the sleep-wake cycle that includes the dorsolateral prefrontal cortex (DLPFC) might be implicated in the attenuation of executive functions and reduced slow-wave sleep in depression [49]. It is of note that these observations do not directly inform about the direction of causality. Interventional studies, such as non-invasive brain stimulation studies, would be needed to test whether the described alterations of sleep in depression can be corrected and whether this kind of non-invasive stimulation might exert therapeutic effects.
Interim summary
Sleep and circadian rhythm research in psychiatry had temporarily lost much of its momentum over the disappointment of the unspecificity of REM sleep alterations for depression [12, 13], as well as due to the introduction of more easy-to-apply neuroimaging methods in psychiatry, and the rise of SSRIs as mainstay treatment for affective disorders. Promising strategies such as the cholinergic REM induction test or the application of PET to study sleep in depression provided highly interesting and exciting results, but were not followed-up because of their immense economic and time-consuming costs that precluded the introduction of these techniques into clinical routine. The fact that practically all available antidepressive medications have more or less strong effects on the physiology of sleep created another roadblock. Today, researchers are confronted with mainly pre-medicated patients for whom drug discontinuation (with a wash-out periods of at least 7–14 days) for research purposes would be considered unethical. A further big hurdle for sleep research (but indeed probably for all types of biological psychiatric research) constituted the fact that a categorical approach to nosology prevailed, hampering any biologically meaningful investigation into psychophysiological underlying mechanisms across diagnostic boundaries. This obstacle has finally been overcome by overwhelming evidence of transdiagnostic and dimensional constellations in pathophysiological traits. The transdiagnostic role of insomnia for many mental disorders, and especially considering its strong association with depression, either as a symptom, syndrome or a distinct diagnostic entity in itself, has become a viable strategy to investigate the sleep—depression relationship. It is our hope that this paradigm change in research may also be clinically more fruitful with respect to aspects of detection, diagnosis and treatment of depression.
The relationship between insomnia and depression
The previous chapters on the phenomenology of sleep and depression have exemplified that disturbances of sleep continuity ubiquitously accompany affective disorders. Whereas the low specificity of insomnia symptoms rendered the study of insomnia unattractive for years, today symptoms of insomnia are seen as a critical feature of depression and a better understanding of the processes involved should contribute to the refinement of pathophysiological concepts and therapeutic approaches for depressive disorders.
What is insomnia?
DSM-5 has given up the distinction between primary and secondary insomnia and now includes the diagnostic category “insomnia disorder” [50]. According to these criteria insomnia is defined as the experience of problems to fall asleep, to maintain sleep or to suffer from early morning awakening. These sleep symptoms have to be coupled with daytime impairments, like decreased attention or problems in concentration. These symptoms have to occur at least three times a week over a period of at least 3 months in order to be diagnosed as insomnia disorder. If the insomniac symptoms are clearly due to another medical/ mental disorder/sleep disorder or substance use, they are not diagnosed separately. However, in most cases, insomnia may have occurred before the medical or mental disorder, or may persist beyond the medical or mental disorder even when the other disorder has been treated successfully. In these cases, diagnostic comorbidity is preferred to single diagnoses. Insomnia as a symptom occurs very frequently (>50% of the population in a year) and in many cases will disappear, for example after the cessation of an acute stressor [51, 52]. It is assumed that ~10% of the population in industrialized countries suffer from chronic insomnia [53]. Recent treatment guidelines [54–56] have summarized that insomnia is associated with high costs for the health care system and also for a high degree of suffering for afflicted individuals. Treatment constitutes of benzodiazepines, benzodiazepine receptor agonists, ramelteon, suvorexant and other substances, which are administered off-label like sedating antidepressants—an interesting development further underlining the close relationships between insomnia and depression. As of now, the first-line treatment of insomnia according to current guidelines is cognitive-behavioral therapy for insomnia (CBT-I) [54–56]. Importantly, the diagnosis of insomnia—as with all other mental disorders—is based on the subjective experience of the afflicted individual and not on PSG defined criteria.
Insomnia and hyperarousal
The idea that insomnia might be due to “hyperarousal” is a very old one and can frequently be found already in the medical literature of the nineteenth century (“overexcitation of the nerves”) or even earlier. We have documented [51, 52, 57, 58] that the hyperarousal hypothesis offers an explanation into the origin of insomnia. The literature looking at psychophysiological variables of insomnia can be summarized as follows: parameters of the autonomous nervous system, the endocrine system, the neuroinflammatory system and neurophysiological indices clearly show that patients who suffer from insomnia show increased indices of hyperarousal, as reflected by altered heart rate, increased cortisol output, and an increase in fast waves as documented by increased amounts of beta waves measured by the sleep EEG [59]. On the psychological level numerous studies have demonstrated that hyperarousal and hyperarousability, as measured by questionnaires, is something very typical for patients with insomnia, which they also experience directly on a subjective level (e.g., racing thoughts whenever trying to sleep). The hyperarousal model of insomnia can be easily linked to neurobiological models of sleep and sleep regulation such as the flip-flop switch model of sleep regulation formulated by Saper et al. [60] (see also the first article in this volume and section “The flip-flop switch model and the role of the orexinergic system”).
Insomnia as a predictor of depression and suicidality
The renowned interest into the relationships between sleep and circadian rhythms and depression is mainly due to studies that have shown that insomnia/insomniac symptoms are independent predictors for depressive disorders [61–64] and suicidal ideation/ suicide and suicide attempts [65–68]. These epidemiologically based data have led to great interest in sleep continuity changes in the context of depression and other mental disorders. Thus, the quest for specific biological markers was given up in lieu of focussing on transdiagnostic overarching markers/mechanisms, which can be elucidated by subjective self-report, rendering research and clinical applicability easier and economic.
Sleep and insomnia: bi-directional relationships with depression
Given the fact, that on the one hand, almost all depressed patients display some kind of sleep alteration, and that on the other hand especially insomnia alone conveys an increased risk for depression and suicidality, the sleep-depression relationship needs to be conceptualized as bi-directional. Several observations converge with this view: When comparing etiological and pathophysiological explanations for insomnia and depression, a clear overlap is present—both conditions have been shown to be triggered by psychosocial stressors. Hyperarousal being regarded a psychological and a biological factor is present in both conditions. When considering treatment modalities, sedating antidepressants can be used for both conditions and CBT-I, the most effective insomnia treatment, has—to some extent—been derived from classical CBT for depression. On the other hand treating insomnia early has the potential to prevent or reduce depressive symptoms as promising preliminary data indicate. We suggest to acknowledge that both disorders can occur independently of each other, as evidenced by the fact that many patients with chronic insomnia never develop depression. Equally, insomnia might indicate a first step towards onset of psychopathology, sharing some underlying (epi-) genetic, personality and neurobiological changes typical for depression. Whether or not depression develops might depend on additional environmental triggers, such as psychosocial stress load, lifestyle, coping mechanisms and early preventative treatments. Given the fact that by and large only a minority of patients with insomnia is sufficiently treated (with CBT-I), there seems to be ample room to test the hypothesis whether stringent and early insomnia treatment might reduce the risk for becoming depressed in the long run.
Treatments of insomnia—a chance for prevention of depression?
As mentioned before, presently Cognitive-Behavioral Treatment for Insomnia (CBT-I) is acknowledged to be the first-line treatment for insomnia [54–56, 69]—thus opening the possibility for large-scale studies to test whether early and adequate treatment of insomnia may prevent psychiatric sequelae, i.e., depression or psychosis. First studies targeting this issue showed that online delivered CBT-I seems to be able to reduce insomnia and depression scores in subclinically depressed patients with insomnia [70]. In a similar vein, advances in the field of chronobiology have led to a renaissance of chronotherapeutic approaches for mental disorders [71–73]. These strategies are useful non-pharmacological preventive implementations into everyday lifestyle—regular sleep-wake rhythmicity, season-adapted daily food and physical activity, day structure, correct light exposure at the “right” time in indoor lighting—all strategies showing depression-preventive properties, which started to be empirically tested and confirmed but need replication.
Insomnia as a transdiagnostic symptom/ syndrome for psychopathology
A further boost for the field came from DSM-5 [50] with the establishment of the category of insomnia disorder, thus ascribing independent value to this symptom complex instead of considering it mainly a symptom of any kind of mental disorder. Still, in DSM-5, separation of condition by categories is current practise but clinical research inspired by RDoC (Research Domain oriented Criteria; www.nimh.nih.gov) is supporting a dimensional approach using constructs and domains to understand pathophysiology. RDoC has suggested a major domain named “arousal and regulatory systems” with the constructs “arousal”, “circadian rhythms” and “sleep-wakefulness” with a detailed listing of areas of interest for research from the level of molecules to circuits, behavior and paradigms. Two recently published meta-analysis on PSG derived sleep variables in insomnia and all different types of mental disorders [13, 74] support this concept and stress a transdiagnostic approach of sleep continuity disturbances/ insomnia towards mental illness. Instead of adhering to an approach, which sought to identify disease-relevant mechanisms through identifying biological markers for specific mental disorders, we postulate that (i) sleep and circadian rhythm disturbances occur independently of and predict/coincide with affective disorders, (ii) clinical psychopathological syndromes do not necessarily reflect homogenous pathophysiological origin, (iii) neuropsychiatric syndromes like depression and sleep/circadian disturbances are linked through common mechanistic origins [58, 75].
Neurobiology of sleep and circadian rhythms—relevance for depression
This chapter highlights recent developments in the field with emphasis on the so-called “flip-flop” switch model, the neuroplasticity hypothesis of sleep and depression, and neural biological timekeeping being pivotal in generating rhythmic behavior.
The flip-flop switch model and the role of the orexinergic system
Saper et al. [60, 76] have developed neurobiological models of sleep-wake and REM-NREM (non-rapid eye movement sleep) regulation (details see Fig. 4; see also the first article in this issue of NPPR) proposing bistable switch mechanisms between wake and sleep-promoting cell populations, as well as REM and NREM promoting clusters. Wakefulness is governed by a network of cell groups in the hypothalamus (including orexinergic neurons), basal forebrain, and brain stem, which activate the thalamus and the cerebral cortex. These wake-promoting centers include but extend beyond the cell groups in the reticular formation of the brain stem originally described as the ARAS (ascending reticular activating system) by Moruzzi and Magoun [77]. As the main sleep-promoting center, the VLPO (ventrolateral preoptic nucleus) is primarily active during sleep, gives output to all major wake-promoting centers in the hypothalamus and brain stem that participate in arousal and reduces their activity using the inhibitory neurotransmitters galanin and GABA (gamma-aminobutyric acid). The VLPO also receives afferent signals from each of the major monoaminergic systems and its neurons are inhibited by noradrenaline and serotonin. Saper and colleagues postulate that this mutual inhibitory circuit constitutes a “flip-flop switch” with sharp state transitions between wake and sleep. The state of this intricate network is governed by circadian and homeostatic processes and exerts sleep-wake regulation in animals and in humans. It may be speculated that a dysfunctional “key switch” plays a major role in the pathogenesis of primary insomnia. An imbalance between sleep-promoting areas in the Central Nervous System (CNS) (i.e., the VLPO, neurotransmitter: GABA) and arousal-inducing neurons (among others orexin neurons in the lateral hypothalamus) with a relative overactivity of the orexin system or a hypofunction of the VLPO might create an unphysiological hybrid state between sleep and wakefulness. This offers a possible neurobiological model for insomnia (see section “Circuit-based interrogation of a shared neurobiological pathogenesis of insomnia and depression using animal models”). We speculatively assume that an overactivation of the arousal system is present both in insomnia and in depression with hyperarousal explaining the close association between both conditions. Considering the role of the orexin system in regulating both the arousal and the affective system, it could be possible that enhanced production of orexin related to psychophysiological hyperarousal increases the risk of reacting with enhanced emotional activation to stressful situations.
Fig. 4.

This figure displays the relationships between normal and insomniac sleep, spectral analysis of sleep EEG and neuroanatomical structures as delineated from the “flip-flop” switch model. a Upper panel—healthy sleeper; lower panel—insomniac sleeper: It is shown that there is no grossly disturbed macrostructure of Non-REM-REM sleep cycling in insomniac patients, but an altered microstructure with many brief awakenings and arousals in insomnia especially in REM sleep (see arrows). b Here shown are spectral analytic data from healthy good sleepers (HGS) and people with insomnia, reflecting a significant increase in fast waves especially in the sigma and beta range. c Anatomical structures and pathways involved in the regulation of wake (left), Non-REM (middle), and REM sleep (right) according to “flip-flop” switch model (see text). Panels a and b of the figure are taken from ref. [53]—permission granted. Panel c is based on refs [60, 76, 193]—permission granted
The orexinergic system [78–80] also orchestrates other depression-related factors such as behavioral and neuroendocrine responses to stress, reward-seeking behaviors, energy homeostasis, learning and memory. The orexin system directly innervates and excites noradrenergic, dopaminergic, serotonergic, histaminergic and cholinergic neurons. It also has a major role in modulating the release of glutamate and other amino acid transmitters and in enhancing hippocampal neurogenesis. Contradicting our line of reasoning, it has to be mentioned that narcolepsy, a debilitating disorder of excessive hypersomnolence (coupled with the frequent occurrence of shortened REM latencies) has been shown to be related to orexin deficiency [81].
Sleep, depression, and neuroplasticity
The synaptic plasticity hypothesis of major depression posits that alterations of synaptic plasticity represent a final common pathway for the clinical manifestations of the disorder. The hypothesis is based on animal models of depression [40], postmortem studies in humans and on non-invasive indices of plasticity in humans [41]. Of particular interest, sleep has been identified as a potential modulator of synaptic plasticity [82]. Current models emphasize that sleep might promote the strengthening of newly induced information-bearing synaptic connections, while—through down selection of less relevant synapses—keeping overall network function stable [42, 83].
In particular, two links between sleep and depression are discussed. First, chronic sleep disruptions in the form of insomnia might disrupt synaptic plasticity and neural network function. Particularly, a dorsal executive network that includes the hippocampus and the prefrontal cortex appear to be particularly sensitivity to sleep loss, resulting in deficits such as attenuated hippocampus-dependent declarative memory formation in chronic insomnia. A ventral emotional network, which includes the amygdala, might be more resilient (for instance indexed by emotional fear conditioning) and might only decompensate with chronic severe sleep disruptions [84], relating to the clinical trajectory from cognitive to emotional complaints with chronic sleep disruptions. Second, it was recently proposed that therapeutic sleep deprivation—through a homeostatic, wake-related increase in overall synaptic strength—transiently shifts initially deficient net synaptic strength in patients with depression into a more favorable window of associative synaptic plasticity, related network function and behavior [44] (see also section “Biological timing and sleep deprivation in depression”). Proposed, but not sufficiently confirmed neural mechanisms include a BNDF, p11, Homer1a, and AMPA receptor cascade of rapid antidepressant treatment, potentially shared with the rapid antidepressant mechanism of ketamine [85]. While the level of evidence remains moderate and numerous questions open, such as specificity, these lines of research allow for deriving testable hypotheses, for instance on the effects of selective modulation of sleep slow waves through auditory or current stimulation, and potentially for the development of new, rapid acting antidepressive treatments.
Chronobiological timekeeping approaches
Compelling evidence suggests that mood disorders arise in part from alterations in the biological timekeeping system, triggered from within the body or from the body’s response to changes in the external environment. Each part of the human body has developed timekeeping mechanisms on every level, from molecular, cellular, neuronal, endocrine up to organ level, affecting the entire body. The timekeeping system is not hierarchical but has a multi-oscillatory structure [86]. Individuals with depression are likely to have internal oscillatory systems out of sync or dampened, but it is difficult to pinpoint the rhythms’ phase-relationships at every level. A classical psychopathological feature in this context is the diurnal mood variation [e.g., ref. [38]], observed in many patients with depression, which is indicated as a “morning low” in mood and a spontaneous alleviation of mood in the afternoon/ evening. Other well-known rhythmic features include seasonal modulation of onset of depression or rapid-cycling phenomena with strict periodicities [87]. Disrupting the body’s stable time patterns by shift work and jet lag can worsen or cause changes in mood. Various hypotheses have been suggested to explain these altered biological rhythms in affective disorders. One concerns disruption within the master pacemaker, the suprachiasmatic nuclei (SCN), causing mood disturbances [88], whereas another suggests that light directly from the retina and bypassing the SCN, targets brain regions to control mood [89]. Endogenous processes oscillate with an approximate cycle of 24 h, in adaption to the solar day-night cycle, and hence rhythms with this length are named ‘circadian’’ rhythms. However, many other functions have a more frequent activity pattern, called ‘ultradian’’ rhythms, for example the NREM-REM sleep cycles. At the behavioral level, human sleep-wake pattern displays circadian rhythmicity, while our food-intake displays an ultradian rhythmicity. Both are under the influence of the light–dark cycle whereby day light plays a crucial role in setting the activity of the SCN to the local environment. At the cellular level, circadian rhythms are generated by a molecular clockwork that consists of multiple transcriptional/translational feedback loops [86]. Nearly all tissues express circadian genes (e.g., Bmal1, Clock, Per, and Cry), which heterodimerize in specific combinations, and regulate the expression of many clock-controlled genes, as well as their own transcription in a feedback fashion. These oscillatory processes ensure cellular timekeeping within tissues, while the SCN generates self-sustaining circadian oscillations, which ensure the synchronization of bodily rhythms. Some studies from individuals with depression found abnormal signaling in SCN cells to correlate with disease duration [90, 91], while others reported altered nitric oxide signaling in the SCN [92], as well as in response to changes in the light–dark cycle [93] and nitric oxide has also been implicated in mood regulation [94]. Hence this substance may affect mood and circadian regulation independently and synergistically. While receiving and integrating information, the SCN maintains a functional circuit with the paraventricular nucleus (PVN), which is important for the control of pituitary hormones and melatonin secretion from the pineal gland [95]. Disturbances in the functioning of this circuit can potentially explain hormonal alterations associated with depression. Another structure with a direct connection to the SCN is the lateral habenula, which couples dopaminergic and serotonergic systems [96] and which is receptive to deep brain stimulation to treat depression [97–99]. The lateral habenula not only exhibits intrinsic neuronal oscillates but also responds to retinal light exposure directly [100] and by bringing it all together, it could be an important interface between retinal light, the pacemaker’s rhythms and multiple monoaminergic brain regions that control mood and motivational behaviors, stress and inflammatory systems, reward circuits, arousal and sleep. Melatonin secretion in the pineal is indirectly regulated by retinal light exposure involving the SCN and the PVN in a multi-synaptic pathway, and its secretion span feedbacks time-of-day information to MT1 and MT2 receptors, which are widespread in the brain, including the SCN, and in peripheral tissue [101]. Sleep deprivation (SD) with “lights on” suppresses melatonin synthesis in the pineal, dampens the neuronal firing rate in the SCN and attenuates light-induced phase-shifting. Moreover, SD changes circadian gene expression in brain regions involved in mood regulation, but not the SCN, while arousal due to SD when supposed to sleep, leads to an increased level of serotonin in the SCN [102].
Given these diverse relationships, it seems reasonable to test combinations of chronotherapeutic interventions on a larger level to derive an estimate of their clinical usefulness.
Synthesis—a model of the bi-directional relationships between sleep and depression
A comprehensive neuropsychobiological model linking the fields of insomnia and depression research is summarized in Fig. 5.
Fig. 5.

Comprehensive insomnia model (see text)—authors´ own conception
This neuropsychobiological model integrates different strands of research from the fields of genetics, neurobiology, personality research, psychophysiology and cognitive-behavioral research to provide a comprehensive understanding of the mechanisms involved in the etiology and pathophysiology of insomnia and its role for the development of psychopathology, especially depression. The basic structure of this model is taken from the 3P model of insomnia formulated by Spielman et al. [103]. The 3P´s stand for: Predisposing, Precipitating, and Perpetuating factors.
Predisposing factors stem from the fields of genetics, neurobiology and personality.
In short [104] genetic and epigenetic factors [105] have been proven to be involved in the etiology and pathophysiology of insomnia by family and twin studies. Two GWAS studies [106, 107] pointed to an involvement of MEIS1, which is also associated with the restless legs syndrome. Lane et al. [108] applied Mendelian randomization analysis and concluded that reliable evidence for a possible causal link between insomnia symptoms, coronary artery disease, depressive symptoms and well-being has been established.
Neurobiological mechanisms, like the flip-flop switch model (see section “The flip-flop switch model and the role of the orexinergic system”), the neuroplasticity hypothesis of sleep and depression (see section “Sleep, depression, and neuroplasticity”) and biological timekeeping mechanisms (see section “Chronobiological timekeeping approaches”) have to be taken into account when constructing a bridge between sleep, insomnia and depression on the cellular level.
With respect to personality variables as predisposing factors, sleep reactivity, the tendency to exhibit pronounced albeit transient sleep disturbance in response to stressful events, has been shown to be a major risk factor for chronic insomnia [109]. Moreover, several personality traits have been linked to insomnia, including neuroticism, perfectionism, sensitivity to anxiety symptoms and the tendency to internalize problems [110], some of which also seem to be depression-related.
Precipitating factors are significant life events, which facilitate the onset of acute episodes of insomnia. The most frequently reported triggers of acute episodes of insomnia are stressful life events that are related to threat of security to family, health, and work/school living, that lead to a negative emotional valence [111, 112]. Additionally, experimental work using stress-induction paradigms clearly showed that acute stress has a negative effect on sleep initiation and sleep maintenance. Needless to say that life events have also been linked with depression.
Perpetuating factors: With respect to hyperarousal, we extended the hyperarousal concept (see section “Insomnia and hyperarousal”) to a more specific hypothesis of REM sleep instability as a possible pathway to insomnia and depression [57]. The concept is based on PSG findings showing that primary insomnia is characterized by a decreased amount of REM sleep and increased EEG arousals during REM sleep [113]. REM sleep regulation is governed by a unique neuronal pattern, which requires a delicate balance between arousing and de-arousing brain activities to sustain this highly aroused sleep brain-state accompanied by muscle atonia. This activity is dominated by cholinergic input to higher brain areas and an inhibition of noradrenergic, serotonergic and orexinergic output. The hypothesis of “REM instability” suggests that a generally increased arousal level leads to a modest REM sleep reduction and fragmentation in primary insomnia. This in turn results in altered perception of mental processes taking place during the REM sleep state: instead of experiencing dreams, patients with insomnia, due to the nature of their pre-sleep concerns, emotions and cognitions, might experience their REM sleep mentation as more wake-like and centered on their main concern, i.e., insomnia [114]. With respect to the relationship with depression, we propose that a chronic fragmentation of REM sleep in insomnia might interfere with basal processes of emotion regulation. With persistence of the disorder and hence REM sleep reduction and fragmentation, at some point a REM sleep rebound (leading to shortened REM latency and increased REM density) and a more stable pattern of emotional reactivity characterized by enhanced responses to negative stimuli (both subjectively and objectively measured) may occur, which may facilitate the development of a depressive episode.
Behavioral perpetuating factors include prolonged time in bed, an irregular sleep-wake schedule and daytime napping [103]. Many patients with insomnia use excessive time in bed and daytime napping as compensatory strategies against their perceived sleep loss. However, these strategies lead to more-pronounced difficulties in sleep-onset and sleep-maintenance [103]. Classical conditioning has also been suggested as an important perpetuating factor in insomnia [115]. In particular, it has been suggested that the bed and the bedroom environment of patients with insomnia become conditioned to arousal and anxiety during an acute episode of insomnia leading to sleep initiation and maintenance problems even after the removal of the initial stressor. In addition to this, a large body of evidence shows that worry and rumination are involved in the maintenance of insomnia [116]. These unproductive thought processes are believed to be associated with a level of physiological arousal that is incompatible with sleep initiation and maintenance. Moreover, self-report studies on emotions in insomnia showed an increased experience of negative emotions in general and specifically at bedtime [117].
What is known about the relationships between sleep, insomnia and the regulation of emotion? Experimental data suggest that people with insomnia report more negative emotions both in general and close to sleep time as compared to good sleepers [118]. Very recent results from negative emotional stimulation show that this response is correlated with a reduced activity of the amygdala and with a moderate level of arousal when compared to good sleepers [119]. In summary, altered emotional responses, both subjective and physiological, have been shown for insomnia. The sensitivity of emotion regulation in insomnia may explain the close association between insomnia and depression. Considering possible biological mechanisms involved in the link between sleep alterations, insomnia, emotion regulation, and mood symptoms, orexin neurons have been found to be related to both sleep-wake regulation as orexin is inhibited during sleep by GABA and galaninergic neurons from the ventrolateral preoptic nucleus (VLPO) (see section “The flip-flop switch model and the role of the orexinergic system”) and affective processes. Consistently, efferent pathways from the amygdala to the lateral hypothalamic area (LHA) have been identified and orexin production has been shown to be activated by emotional events, especially related to emotional stress [120, 121].
Present management of sleep/ circadian disorders in depression
Subjectively reported sleep disturbances typically manifest in various forms, such as difficulties falling asleep and maintaining sleep, waking up too early and feeling worn, fatigued and sleepy during the day. Although these symptoms are described by DSM-5 as insomnia disorder, they are rarely investigated properly by general practitioners or mental health professionals—unfortunately at present it is far more probable that both insomnia and symptoms of circadian dysfunction are still more or less viewed as symptoms of an underlying mental disorder, which will, at best, remit when the “primary” (=underlying) disorder is properly treated or, at worst, treated with the wrong drugs. A main reason for this is that there is a serious lack in the knowledge about basic sleep and chronomedicine across fields in the medical professions, maybe because these disturbances are transdiagnostic. Insofar, there still is a strong need for further education of health professionals, in particular in psychiatry (hospital and community doctors, nurses, health advisors), in the domain of sleep and chronomedicine to enable them to recognize, properly diagnose and treat individuals with sleep problems [122]. Specific evidence-based treatment strategies incorporating chronomedicine (right drug, right dose, right time) for these disorders are available and might offer not only to improve the underlying sleep/circadian disorder but concomitantly ameliorate the outcome and course of mental disorders compared to standard psychiatric treatment.
Cognitive behavioral therapy for insomnia (CBT-I)
For non-pharmacological insomnia treatment the most extensive data basis exists now for CBT-I [69]. CBT-I has been primarily investigated in primary insomnia and studies are just gathering momentum to evaluate its efficacy and effectiveness in co-morbid insomnia. Most recent guidelines for insomnia treatment from the US and Europe [54–56] conclude that CBT-I is the first-line treatment, whereas recommendations for all investigated hypnotic drugs are not stronger than “weak”, and mostly for the short-term treatment (3–4 weeks; AASM, 2017) [123]. The AASM stresses that these rather low rankings for all existing hypnotics, especially benzodiazepines, benzodiazepine receptor agonists etc. reflect the fact that the Grade system (used to make the recommendations) downgrades the quality of evidence if the funding source is a pharmacological company. Furthermore, the AASM states that a weak recommendation should not be construed as an indication of ineffectiveness. The ultimate decision to prescribe sleep medication should be based on the clinician´s decision reflecting all individual circumstances, the patient´s shared consent and the availability of alternative treatment options in a given clinical setting. All of these guidelines [54–56, 123] were based on thorough analyses of the relevant literature. Thus, either published meta-analyses (based on randomized clinical trials/ RCT)) or single RCTs were considered to evaluate the evidence. Insofar, the above-mentioned guidelines can be considered as based on the highest levels of presently available evidence. These new guidelines also indicate that CBT-I has significant long-term effects beyond the acute treatment phase, in that respect being superior to pharmacological treatment for primary insomnia. CBT-I encompasses relaxation techniques, sleep hygiene, stimulus control, sleep restriction and cognitive techniques to reduce nocturnal ruminations [124]. In psychological models of insomnia like for example the 3P Model [103], the model of Morin [52] or the cognitive models of Harvey and Espie [125, 126] components of CBT-I can be easily matched to the psychological components of the pathophysiology of insomnia. It is also of interest that the 2-process model of sleep-wake regulation and the concept of sleep propensity/sleep pressure [25] have been at the core of informing behavioral strategies for insomnia, i.e., sleep restriction: this strategy is derived from the clinical observation that patients with chronic insomnia prolong time in bed to achieve more sleep—a behavior, which unfortunately perpetuates insomnia instead of relieving it. By shortening time in bed (as suggested by sleep restriction), in contrast, the homeostatic sleep drive is stimulated and subsequent sleep quality improved [127, 128]. Considering sleep restriction as a kind of partial sleep deprivation, it is interesting to note that total sleep deprivation on its own is a rapid, albeit transient, treatment modality in depression (see section “Biological timing and sleep deprivation in depression”).
Hypnotics, chronobiotics, antidepressants
Regarding prescription drugs such as benzodiazepines (BZ)/benzodiazepine receptor agonists (BZRA), melatoninergic agents, suvorexant, sedating antidepressants and atypical antipsychotics are used in the wider field of insomnia and for sleep problems within the context of depressive disorders. Evidence from clinical studies is strong for effective short-term administration (3-4 weeks) of BZ/BZRA for insomnia [55, 56, 123]. Doubtlessly, hypnotic agents like BZ, BZRA, or suvorexant will produce stable positive effects on patient´s sleep over a window of 3–4 weeks; however, given the fact that practically all performed studies were sponsored by the pharmaceutical industry, overall recommendations for example by the AASM [123] could not be beyond “weak”. Many patients afflicted with insomnia suffer chronically and there is doubt for effective long-term treatment beyond four weeks with these drugs. A major concern with long-term BZ/BZRA treatment is the issue of adverse events (e.g., traffic safety, mnestic effects, paradoxical behavior during the night, intoxication, falls in the elderly etc [129–131]) and the development of tolerance/rebound and abuse/dependency [132–134]. These and related issues concerning the risks of BZ/ BZRA for increased mortality [135, 136] are discussed controversially, because other studies [137, 138] highlighted the likely role of confounding factors when relating risk of cancer or mortality to BZ/BZRA intake. Nevertheless, especially the issue of potential risks concerning abuse or dependency probably are responsible for the decision that treatment of insomnia with these agents beyond 3–4 weeks is not endorsed in Europe by drug regulating authorities. A different stance has been taken by the FDA (US American Food and drug administration) in recent years granting long-term use for all newly admitted hypnotics since 2004 (e.g., eszopiclone).
Melatoninergic agents like ramelteon [139] have been admitted to the US market. In Europe, a prolonged release formulation of melatonin Circadin, can be prescribed for long-term (3 months) treatment of insomnia in the over 55-years-old [EMEA: http://www.emea.europa.eu/humandocs/PDFs/EPAR/circadin/H-695-de1.pdf]. The benefit of melatonin treatments with respect to circadian rhythm disturbances, like jet lag and phase delay [140] has been proven. However, the evidence for melatonin being beneficial in the treatment of insomnia is not very convincing [141–145].
Sedating antidepressants (SED; e.g., amitriptyline, doxepin, trimipramine, trazodone, mirtazapine, agomelatine or atypical antipsychotics (AP; quetiapine, olanzapine) are frequently used for symptoms of sleep disturbances in patients with mental disorders, i.e., major depression or bipolar disorder or schizophrenia [146]. They are not indicated for the treatment of insomnia without mental comorbidity. However, recent pharmaco-epidemiological studies revealed that in the US (for example trazodone) or in Europe (for example trimipramine, mirtazapine) sedating antidepressants are increasingly used in that field [147, 148]. The efficacy and effectiveness of SED and AP to treat major mental disorders is undisputed—however, there still is a lack of data from large controlled trials, apart from a few pilot studies [149–151] and a recent meta-analysis [152], demonstrating clearly that these compounds are superior to placebo when addressing insomnia alone and sleep disturbances in mental disorders. Interestingly, doxepin, one of the “oldest” available SEDs, has been shown to be effective at very low doses in primary insomnia [153] and thus gained approval by US Food and Drug Administration. With respect to AP even less evidence is available—studies are available investigating the effects of AP on sleep in schizophrenia [154] and in insomnia [155, 156]. At present, it is safe to conclude that most AP are clinically useful to combat sleep problems in a subset of patients with schizophrenia, but not for insomnia without mental comorbidity.
Behavioral chronotherapy and chronopharmacology
Behavioral chronotherapeutic approaches encompass bright light therapy, dark therapy, dawn simulation, sleep-wake manipulations (sleep deprivation, sleep phase advance/delay), often in combination with precisely personalized timed low-dose fast-release melatonin agents, as well as social rhythm therapy [72, 73]. Application of bright light, usually for periods of 30–60 min at an intensity of 5000–10.000 lux in the morning (dependent on indication) or the use of a dawn simulator with a slow increase in light over 30 min around wake-time, is used therapeutically in circadian sleep-wake rhythm disorders, such as phase advance/delay, seasonal affective disorders (SAD), major depression and geriatric disorders (e.g., dementia). Dark therapy is successfully used to prevent manic phases in patients with bipolar disorder [157]. Sleep-wake manipulations include total and partial sleep deprivation (SD) and phase shifts of the sleep phase (sleep phase advance therapy). This type of manipulation is dominantly used in patients with major depression and especially those suffering from melancholia displaying clinical features like diurnal mood variations. Timed light exposure and type of light should be considered as important during sleep deprivation since light affects the circadian timing system. Wu and Bunney [34] published a systematic review on SD demonstrating that total SD produces a clinically significant, albeit short-lived, remission in mood in as many as 60–70% of patients with severe depression. Social rhythm therapy (SRT) aims at affective disorders, especially bipolar depression. In combination with specific psychotherapy (interpersonal therapy) and the administration of mood stabilizers (e.g., lithium), SRT aims at reducing the impact of stressful life events on the clinical course of the disorder by addressing issues of lifestyle regularity [158, 159]. With respect to circadian disturbances, studies have assessed melatonin’s efficacy in the visually impaired (diminished light input to the pacemaker), jet lag syndrome, shift work, phase advance/ delay, depression and neurodevelopmental disorders like autism with promising results [140, 160–163].
Another very relevant aspect of chronomedicine is Chronopharmacology, the timing of drug intake. Antidepressants in particular involve a number of neurotransmitters that are acting directly on the SCN or in circuits, which are regulated by the timekeeping system, for example the SCN has serotonin receptors and lithium is known to prolong the circadian period.
Unfortunately, up to now no data are available how frequent chronotherapeutic approaches are used for the treatment of circadian and other disorders—this is in contrast to prescribed medications and psychotherapeutic treatments, where usually sales figures or statistics derived from national health care systems can give at least a rough indication how frequently a certain intervention is applied. This is in part due to the fact that for example an intervention like sleep deprivation or light therapy in a depressed patient applied in a hospital will produce no tangible costs to figure in a statistic—thus apart from enhancing awareness for circadian/chronobiological disorders it will also be a challenge to collect data on the specific treatment modalities in clinical care to get an impression of their effectiveness in routine clinical care.
Non-invasive brain stimulation
Recent work suggests that arousal and sleep can be modulated by targeting a cortico-thalamic top-down pathway of sleep-wake regulation through different types of non-invasive brain stimulation (NIBS), including transcranial magnetic stimulation (TMS), transcranial current stimulation (tCS) or sensory stimulation, such as auditory stimulation [164]. Of note, patients in the diagnostic category of major depression might suffer from different sleep complaints along the domain of hyper- (insomnia) and hypoarousal (prolonged sleep duration). This domain may be suitable for structuring future NIBS research and treatment development, because it can be characterized on different levels, including the genetic, molecular, neurocircuitry and behavioral level. Of particular interest, it might be possible to identify more fine-graded alterations in individual patients, such as alterations of sleep slow waves, sleep spindles or local aspects of sleep that might be targeted with specific NIBS techniques. To date, several NIBS approaches have been used to modify sleep: Transcranial magnetic stimulation (TMS) uses a magnetic field, which modulates cortical activity. For instance, specific EEG patterns such as sleep slow waves and spindles can be triggered using this technique [45, 46]. However, the TMS setting during sleep is demanding and limited to specific research questions. Transcranial current stimulation (tCS) can induce local shifts in cortical excitability [165] and has the potential to affect distinct aspects of sleep. For instance, anodal tDCS during slow wave sleep improved declarative memory [166]. Moreover, anodal tDCS can reduce total sleep time by ~25 min in healthy humans [167], but does not modify sleep in patients with insomnia, indicative of brain-state-dependent effects of the stimulation protocol [168]. While tDSC is easily applicable, the clinical relevance of the observed effects remains unclear. Auditory stimulation applied in a brain-state-informed protocol (auditory closed-loop stimulation) can enhance sleep slow waves and sleep-related memory consolidation [45] or selectively suppress slow waves and modulate neurophysiological measures of neuroplasticity [46]. These approaches bear the potential to target distinct aspects of sleep, to increase our mechanistic understanding and, potentially, to develop new sleep-based treatments for depression. Yet currently, no clinically relevant sleep-based interventions in this area are known.
Key directions for future research and clinical practice
Establish CBT-I as preventive strategy for depression
Unfortunately, apart from research studies, CBT-I up to now has not been established in routine clinical practice or care in the US or European countries. In order to overcome this problem a stepped-care approach has been suggested by Espie [169]. According to this model, in a first step self-help approaches in bookform or internet-based programs can be used by afflicted individuals with mild to moderate symptoms [170]. In a next step, community-based approaches involving group courses given by trained nurses would reach larger numbers of patients face-to-face [171]. Contact with a qualified clinical psychologist/ psychiatrist or sleep specialist would be reserved for the most severe cases to administer state of the art CBT-I or abbreviated forms of this treatment [172, 173]. Very recently, a large- scale study with digital cognitive-behavioral therapy for insomnia found sustained effects of improved psychological well-being and sleep-related quality of life resulting from an 8-week sleep-hygiene education program [174]. This provides first evidence that psychoeducation via web and/or mobile channels in addition to usual treatment is an easy-to-use and effective element in a stepped-care approach for insomnia.
Furthermore, the central question to address will be the efficacy and effectiveness of CBT-I for insomnia co-morbid with mental disorders—there is promising evidence now even in the form of meta-analysis [175, 176]. Thus, larger-scale longitudinal studies are needed to prove that CBT-I adjuvant to standard psycho-and pharmacotherapy in different populations of psychiatric patients not only positively influences insomniac symptoms but beyond improves the general outcome of the disorder. In the same vein, the often-quoted assumption that insomnia treatment may prevent psychiatric sequelae in chronic insomniacs [62] needs to be proven—data from the “GoodNight” study [177] point to a first empirical confirmation of this assumption [70].
Integrate chronobiological interventions into the armamentarium of antidepressant treatments
It represents a timely and urgently needed step to translate the impressive basic science knowledge on circadian rhythmicity to the clinic. This translation needs to address the following points: (i) provide guidance for a clinically useful estimation of the individual circadian phase (e.g., through the wake-up time or mid sleep phase), (ii) provide recommendations for the application of melatonin and light with regard to the individual phase response curve, (iii) provide recommendations on the duration, monitoring and optimization of the treatment (e.g., phase-shift during therapy), (iv) test for effects on circadian rhythms, sleep, mental disorders and broader health outcomes. Although the points outlined above appear trivial referred to the deciphering of the molecular mechanisms of circadian rhythms, there is to date, to our knowledge, no practical recommendation for implementing circadian interventions into broader clinical practice,
Novel molecules for depression and sleep disturbances
As can be deduced from the section on the regulation of sleep and its neuropharmacology novel pharmaceutics may target noradrenergic, serotonergic, histaminergic, adenosinergic, melatoninergic, or orexinergic neurotransmission/ pathways to go beyond an influence on gaba-ergic neurotransmission. An already available new line of treatment consists in the application of orexin receptor agonists like suvorexant, which has just been recently admitted to the market in the US. This type of medication is based on orexin receptor antagonism and has been proven to produce clinically relevant effects on sleep in insomnia [178, 179].
Potential for non-invasive brain stimulation approaches in the treatment of insomnia and depression
In addition to psycho-/pharmacotherapy and chronotherapeutic approaches, non-invasive brain stimulation techniques, including thermo-stimulation and transcranial direct current stimulation (tDCS), seem to have the potential to improve sleep. These techniques are able to induce local activity changes in specific cortical areas, which might modulate arousal and sleep via cortico-thalamo-cortical feedback loops. First proof-of-concept studies exist for the short-term induction of EEG slow waves by tDCS in healthy subjects [166] and a dose-dependent improvement of sleep latency and efficiency by frontal cerebral thermo-stimulation [180].
Another evolving strategy transforms recordings of brain activity patterns, such as EEG or real-time fMRI signals, into insomnia treatments by neurofeedback. These techniques aim at characterizing insomnia-related brain activity in order to allow patients to learn to control/ suppress specific patterns of brain activity with biofeedback [181, 182]. Further work is necessary to evaluate whether neurofeedback can be helpful to treat insomnia and depression.
Circuit-based interrogation of a shared neurobiological pathogenesis of insomnia and depression using animal models
Rodent models of depression are widely used in drug discovery studies and a variety of tests has been developed to assess depressive behavior in rodents [183, 184]. Surprisingly, sleep is rarely considered as outcome parameter in drug studies using animal models of depression. However, a cage exchange paradigm has been developed to investigate neural circuitry of stress-induced insomnia in rats inducing sleep disruption through mild stress and novelty of a depression [185]. This paradigm reproduces several of the polysomnographic features, which often accompany acute insomnia, namely sleep fragmentation, increased sleep onset latency, decreased NREM and REM sleep and an increase in high-frequency EEG activity during NREM sleep Interestingly, the regional assessment of neural activity using FOS expression, indicates a simultaneous activation of the sleep-promoting VLPO and arousal systems, leaving the sleep-wake regulatory switch in the brain in a hybrid state [185]. While these findings were acquired before the broad application of optogenetics to the neurobiological sleep research, the model provides a valuable basis for further delineation of neurophysiological mechanisms of insomnia in a rodent model
An intriguing opportunity to further disentangle the presumed hybrid state of sleep regulatory systems in insomnia arises from recent findings that many important aspects of sleep are regulated on a microscale [186, 187]. Sleep homeostasis, the neuronal recovery process during NREM sleep, which is directly reflected by slow wave activity (0.5–4.0 Hz) during NREM, can strongly vary between individual regions depending on the previous use of cortical areas during wakefulness [188–190]. A hybrid pattern of signals characteristic for wakefulness and sleep can appear in the EEG of a wake and behaving animal after extensive training or prolonged wakefulness. Hybrid states of sleep microarchitecture have now also been reported in humans using intracerebral recordings acquired from patients undergoing diagnostic assessment for epilepsy [191, 192]. Taken together, evidence from intracerebral recordings indicates a complex microarchitecture of vigilance states, which is not always observable on EEG recordings. Hybrid states on the microscale of either local cortical activation during sleep [191] or local cortical OFF states during wakefulness [190, 192] can have remarkable behavioral consequences. These findings from basic research provide a putative explanation for the above-mentioned notion that subjective complaints of disrupted or non-restorative sleep are necessarily accompanied by polysomnographic findings (see section “What is insomnia?”). Further highly relevant basic research strategies encompass the modulation of neuroplasticity by sleep and the role of sleep as a key modulator of the neuro-immune axis.
Funding and disclosure
None of the authors declares any competing financial interest in relation to the work described. Concerning the pharmaceutical industry, DR received honoraria from HEEL pharmaceuticals in 2017/2018 for one congress presentation each. DR is a member of the executive board of Freiburg Institute for Behavioral Therapy and thus receives honoraria for regular meetings, examinations, and presentations in this context. DR frequently lectures at hospitals, universities, and other institutions of education about sleep, insomnia, and mental health—frequently, he receives a honorarium from these institutions for his work. DR receives royalties from several publishing houses for his books on sleep and insomnia. DR receives payment from the European Sleep Research Society for his duty as Editor in Chief of the Journal of Sleep Research. The other authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Burton Robert. The Anatomy of Melancholy. In: Faulkner Thomas C., Kiessling Nicolas K., Blair Rhonda L., editors. The Anatomy of Melancholy, Vol. 1: Text. 1621. pp. lxi–lxi. [Google Scholar]
- 2.Strobl P. Die Macht des Schlafes in der griechisch-römischen Welt. Verlag Dr. Kovac: Hamburg; 2002.
- 3.Kraepelin E. Psychiatrie. JA Barth Publishing House (8th. edition); 1909.
- 4.Gustavsson A, Svensson M, Jacobi F, Allgulander C, Alonso J, Beghi E, et al. Cost of disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21:718–79. doi: 10.1016/j.euroneuro.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS. Grand challenges in global mental health. Nature. 2011;475:27–30. doi: 10.1038/475027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aserinsky E, Kleitman N. Regularly occuring periods of eye motility concomitant phenomena during sleep. Science. 1963;118:273–4. doi: 10.1126/science.118.3062.273. [DOI] [PubMed] [Google Scholar]
- 7.Kupfer DJ, Foster FG. Interval between onset of sleep and rapid eye movement sleep as an indicator of depression. Lancet. 1972;2:648–9. doi: 10.1016/s0140-6736(72)92090-9. [DOI] [PubMed] [Google Scholar]
- 8.Kupfer DJ. REM latency: a psychobiological marker for primary depressive disease. Biol Psychiat. 1976;11:159–74. [PubMed] [Google Scholar]
- 9.Strawbridge R, Young AH, Cleare AJ. Biomarkers for depression: recent insights, current challenges and future prospects. Neuropsychiatr Dis Treat. 2017;13:1245–62. doi: 10.2147/NDT.S114542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riemann D, Berger M, Voderholzer U. Sleep in depression: results from psychobiological studies. Biol Psychol. 2001;57:67–103. doi: 10.1016/s0301-0511(01)00090-4. [DOI] [PubMed] [Google Scholar]
- 11.Thase ME, Kupfer DJ, Spiker DG. Electroencephalographic sleep in secondary depression: a revisit. Biol Psychiat. 1984;19:805–14. [PubMed] [Google Scholar]
- 12.Benca RM, Obermeyer WH, Thisted RA, et al. Sleep and psychiatric disorders: a meta-analysis. Arch Gen Psychiat. 1992;49:651–68. doi: 10.1001/archpsyc.1992.01820080059010. [DOI] [PubMed] [Google Scholar]
- 13.Baglioni C, Nanovska S, Regen W, Spiegelhalder K, Feige B, Nissen C, et al. Sleep and mental disorders: a meta-analysis of the last 20 years of polysomnographic research. Psychol Bull. 2016;142:969–90. doi: 10.1037/bul0000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riemann D, Nissen C. Sleep and psychotropic drugs. In: Morin CM, Espie CA, editors. The Oxford Handbook of sleep and sleep disorders. Oxford: Oxford University Press; 2012. [Google Scholar]
- 15.Allocca G, Ma S, Martelli D, Cerri M, Del Vecchio F, Bastianini S, et al. Validation of ‘Somnivore’, a machine learning algorithm for automated scoring and analysis of polysomnography data. Front Neurosci. 2019. (In press). [DOI] [PMC free article] [PubMed]
- 16.Sitaram N, Nurnberger JI, Gershon ES, Gillin JC. Cholinergic regulation of mood and REM sleep: potential model and marker of vulnerability to affective disorder. Am J Psychiatry. 1982;139:571–6. doi: 10.1176/ajp.139.5.571. [DOI] [PubMed] [Google Scholar]
- 17.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–8. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 18.McCarley RW. REM sleep and depression: common neurobiological control mechanisms. Am J Psychiat. 1982;139:565–70. doi: 10.1176/ajp.139.5.565. [DOI] [PubMed] [Google Scholar]
- 19.Riemann D, Hohagen F, Bahro M, Lis S, Stadtmüller G, Gann H, et al. Cholinergic neurotransmission, REM sleep and depression. J Psychosom Res. 1994;38:15–25. doi: 10.1016/0022-3999(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 20.Berger M, Riemann D, Höchli D, Spiegel R. The cholinergic rapid eye movement induction test with RS-86: state or trait marker of depression? Arch Gen Psychiatry. 1989;46:421–8. doi: 10.1001/archpsyc.1989.01810050035006. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber W, Lauer CJ, Krumrey K, Holsoboer F, Krieg JC, Cholinergic REM. induction test in subjects at high risk for psychiatric disorder. Biol Psychiatry. 1992;32:79–90. doi: 10.1016/0006-3223(92)90144-o. [DOI] [PubMed] [Google Scholar]
- 22.Lauer CJ, Modell S, Schreiber W, Krieg JC, Holsboer F. Prediction of the development of a first major depressive disordere with a rapid eye movement sleep inducing. Test using the cholinergic agonist RS86. J Clin Psychopharmacol. 2004;24:356–7. doi: 10.1097/01.jcp.0000125744.22031.3a. [DOI] [PubMed] [Google Scholar]
- 23.Wehr TA, Wirz-Justice A. Circadian rhythm mechanisms in affective ilnesss and in antidepressant drug action. Pharmacopsychiat. 1982;15:31–9. doi: 10.1055/s-2007-1019506. [DOI] [PubMed] [Google Scholar]
- 24.Pflug B, Toelle R. Therapie endogener Depressionen durch Schlafentzug. Nervenarzt. 1971;42:117–24. [PubMed] [Google Scholar]
- 25.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 26.Borbely AA, Wirz-Justice A. Sleep deprivation and depression. A hypothesis derived from a model of sleep regulation. Hum Neurobiol. 1982;1:205–10. [PubMed] [Google Scholar]
- 27.Daan S, Beersma DGM, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–83. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 28.Wehr TA, Wirz-Justice A, Goodwin FK. Phase advance of the circadian sleep-wake cycle as an antidepressant. Science. 1979;206:710–3. doi: 10.1126/science.227056. [DOI] [PubMed] [Google Scholar]
- 29.Borbély AA, Daan S, Wirz-Justice A, Deboer T. The two-process model of sleep regulation: a reappraisal. J Sleep Res. 2016;25:131–43. doi: 10.1111/jsr.12371. [DOI] [PubMed] [Google Scholar]
- 30.Germain A, Kupfer DJ. Circadian rhythm disturbances in depression. Hum Psychopharmacol. 2008;23:571–85. doi: 10.1002/hup.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewy AJ, Sack RL, Miller SL, Hoban TM. Antidepressant and circadian phase-shifting effects of light. Science. 1987;235:352–4. doi: 10.1126/science.3798117. [DOI] [PubMed] [Google Scholar]
- 32.Ehlers CL, Frank E, Kupfer DJ. Social zeitgebers and biological rhythms. A unified approach to undesrstanding the etiology of depression. Arch Gen Psychiatry. 1988;45:948–52. doi: 10.1001/archpsyc.1988.01800340076012. [DOI] [PubMed] [Google Scholar]
- 33.Bunney BG, Bunney WE. Mechanisms of rapid antidepressant effects of sleep deprivation therapy: clock genes and circadian rhythms. Biol Psychiatry. 2013;73:1164–71. doi: 10.1016/j.biopsych.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Wu JC, Bunney WE. The biological basis of an antidepressant response to sleep deprivation and relapse: review and hypothesis. Am J Psychiat. 1990;147:14–21. doi: 10.1176/ajp.147.1.14. [DOI] [PubMed] [Google Scholar]
- 35.Dallaspezia S, Benedetti F. Chronobiology of bipolar disorder: therapeutic implication. Curr Psychiatry Rep. 2015;17:606. doi: 10.1007/s11920-015-0606-9. [DOI] [PubMed] [Google Scholar]
- 36.Riemann D, Wiegand M, Lauer C, Berger M. Naps after total sleep deprivation in depressed patients: Are they depressiogenic? Psychiatry Res. 1993;49:109–20. doi: 10.1016/0165-1781(93)90099-3. [DOI] [PubMed] [Google Scholar]
- 37.Landsness EC, Goldstein MR, Peterson MJ, Tononi J, Benca RM. Antidepressants effects of selective slow wave deprivation in major depression: a high-density EEG investigation. J Psychiat Res. 2011;45:1019–26. doi: 10.1016/j.jpsychires.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riemann D, Wiegand M, Berger M. Are there predictors for sleep deprivation response in depressed patients? Biol Psychiatry. 1991;29:707–10. doi: 10.1016/0006-3223(91)90145-c. [DOI] [PubMed] [Google Scholar]
- 39.Svetska J. Sleep deprivation therapy. Neuro Endocrin Lett. 2008;29:65–92. [PubMed] [Google Scholar]
- 40.Castren E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry. 2013;70:983–9. doi: 10.1001/jamapsychiatry.2013.1. [DOI] [PubMed] [Google Scholar]
- 41.Kuhn MA, Mainberger F, Feige B, Maier J, Mall V, Jung NH, et al. State-dependent partial occlusion of cortical LTP-like plasticity in major depression. Neuropsychopharmacology. 2016;41:1521–9. doi: 10.1038/npp.2015.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, et al. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat. Commun. 2016;7:12455. doi: 10.1038/ncomms12455.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf E, Kuhn M, Normann C, Biber K, van Calker D, Riemann D, et al. Sleep and plasticity: potential mechanisms of therapeutic sleep deprivation in major depression. Sleep Med Rev. 2016;30:53–62. doi: 10.1016/j.smrv.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Ngo HV, Martinetz T, Born J, Mölle M. Auditory closed loop stimulation of the sleep slow oscilation enhances memory. Neuron. 2013;78:545–53. doi: 10.1016/j.neuron.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 46.Fattinger S, de Beukelaar TT, Ruddy KL, Volk C, Heyse NC, Herbst JA, et al. Deep sleep maintains learning efficiency of the human brain. Nat Commun. 2017;8:15405. doi: 10.1038/ncomms15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nofzinger EA, Buysse DJ, Germain A, Carter C, Luna B, Price JC, et al. Increased activation of anterior paralimbic and executive cortex from waking to rapid eye movement sleep in depression. Arch Gen Psychiatry. 2004;61:695–702. doi: 10.1001/archpsyc.61.7.695. [DOI] [PubMed] [Google Scholar]
- 48.Nofzinger EA, Buysse DJ, Germain A, et al. Alterations in regional cerebral glucose metabolism across waking and non-rapid eye movement sleep in depression. Arch Gen Psychiatry. 2005;62:387–96. doi: 10.1001/archpsyc.62.4.387. [DOI] [PubMed] [Google Scholar]
- 49.Nissen Christoph, Nofzinger Eric. Sleep and Psychosomatic Medicine. 2007. Sleep and depression; pp. 51–65. [Google Scholar]
- 50.American Psychiatric Association (APA). Diagnostic and statistical manual of mental disorders. 5th ed.: DSM-5. APA Publishing: Washington DC; 2013.
- 51.Riemann D, Spiegelhalder K, Feige B, Voderholzer U, Berger M, Perlis M, et al. The hyperarousal model of insomnia: A review of the concept and its evidence. Sleep Med Rev. 2010;14:19–31. doi: 10.1016/j.smrv.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Morin C, Drake C, Harvey A, Krystal AD, Manber R, Riemann D et al. Insomnia disorder. Nature Reviews Disease Primers. 2015 epub first, 10.1038/nrdp.2015.26. [DOI] [PubMed]
- 53.Ohayon MM. Epidemiology of insomnia: What we know and what we still need to learn. Sleep Med Rev. 2002;6:97–111. doi: 10.1053/smrv.2002.0186. [DOI] [PubMed] [Google Scholar]
- 54.Qaseem A, Kansagara D, Forciea MA, Cooke M, Denberg TD. Management of chronic insomnia disorder in adults: A clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;165:889–97. doi: 10.7326/M15-2175. [DOI] [PubMed] [Google Scholar]
- 55.Riemann D, Baglioni C, Bassetti C, Bjorvatn B, Dolenc Groselj L, Hertenstein E, et al. European guideline for the diagnosis and treatment of insomnia. J Sleep Res. 2017;26:675–700. doi: 10.1111/jsr.12594. [DOI] [PubMed] [Google Scholar]
- 56.Riemann D., Baum E., Cohrs S., Crönlein T., Hajak G., Hertenstein E., Klose P., Langhorst J., Mayer G., Nissen C., Pollmächer T., Rabstein S., Schlarb A., Sitter H., Weeß H.-G., Wetter T., Spiegelhalder K. S3-Leitlinie Nicht erholsamer Schlaf/Schlafstörungen. Somnologie. 2017;21(1):2–44. [Google Scholar]
- 57.Riemann D, Spiegelhalder K, Nissen C, Hirscher V, Baglioni C, Feige B. REM sleep instability—a new pathway for insomnia? Pharmacopsychiatry. 2012;45:167–76. doi: 10.1055/s-0031-1299721. [DOI] [PubMed] [Google Scholar]
- 58.Riemann D, Nissen C, Palagini L, Otte A, Perlis M, Spiegelhalder K. The neurobiology, investigation and treatment of chronic insomnia. Lancet Neurol. 2015;14:547–58. doi: 10.1016/S1474-4422(15)00021-6. [DOI] [PubMed] [Google Scholar]
- 59.Perlis M, Giles D, Mendelson W, Bootzin R, Wyatt J. Psychophysiological insomnia: The behavioural model and a neurocognitive perspective. J Sleep Res. 1997;6:179–88. doi: 10.1046/j.1365-2869.1997.00045.x. [DOI] [PubMed] [Google Scholar]
- 60.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 61.Riemann D, Voderholzer U. Primary insomnia: a risk factor to develop depression? J Affect Disord. 2003;76:255–9. doi: 10.1016/s0165-0327(02)00072-1. [DOI] [PubMed] [Google Scholar]
- 62.Riemann D. Does effective management of sleep disorders reduce depressive symptoms and the risk of depression? Drugs. 2009;69:43–64. doi: 10.2165/11531130-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 63.Baglioni C, Battagliese G, Feige B, Nissen C, Voderholzer U, Lombardo C, et al. Insomnia as a predictor of depression: A meta-analytic evaluation of longitudinal epidemiological studies. J Affect Disord. 2011;135:10–9. doi: 10.1016/j.jad.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 64.Li L, Wu C, Gan Y, Qu X, Lu Z. Insomnia and the risk of depression: a meta-analysis of prospective cohort studies. BMC Psychiat. 2016;16:375. doi: 10.1186/s12888-016-1075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bjørngaard JH, Bjerkeset O, Romundstad P, Gunnell D. Sleeping problems and suicide in 75,000 Norwegian adults: A 20 year follow-up of the HUNT I Study. Sleep. 2011;34:1155–9. doi: 10.5665/SLEEP.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pigeon WR, Pinquart M, Conner K. Meta-analysis of sleep disturbance and suicidal thoughts and behaviors. J Clin Psychiat. 2012;73:e1160–7. doi: 10.4088/JCP.11r07586. [DOI] [PubMed] [Google Scholar]
- 67.Malik S, Kanwar A, Sim LA, Prokop LJ, Wang Z, Benkhadra K, et al. The association between sleep disturbances and suicidal behaviors in patients with psychiatric diagnoses: a systematic review and meta-analysis. Syst Rev. 2014;3:18. doi: 10.1186/2046-4053-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernert RA, Turvey CL, Cornwell Y, Joiner TE. Association of poor subjective sleep quality with risk for death by suicide during a 10-year period. JAMA Psychiatry. 2014;71:1129–37. doi: 10.1001/jamapsychiatry.2014.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13:205–14. doi: 10.1016/j.smrv.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Christensen H, Batterham PJ, Gosling JA, Ritterband LM, Griffiths KM, Thorndike FP, et al. Indicated prevention of major depressive disorder: A randomised controlled effectiveness trial of an online insomnia programme (SHUTi): The GoodNight study. Lancet Psychiatry. 2016;3:333–41. doi: 10.1016/S2215-0366(15)00536-2. [DOI] [PubMed] [Google Scholar]
- 71.Wirz-Justice A, Terman M, Oren DA, Goodwin FK, Kripke DF, Whybrow PC, et al. Brightening depression. Science. 2004;303:467–9. doi: 10.1126/science.303.5657.467c. [DOI] [PubMed] [Google Scholar]
- 72.Wirz-Justice A, Benedetti F, Terman M. Chronotherapeutics for affective disorders. 2nd ed. Basel: Karger; 2013. [Google Scholar]
- 73.Benedetti F, Barbini B, Colombo C, Smeraldi E. Chronotherapeutics in a psychiatric ward. Sleep Med Rev. 2007;11:509–22. doi: 10.1016/j.smrv.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 74.Baglioni C, Regen W, Tehgen A, Spiegelhalder K, Feige B, Nissen C, et al. Sleep changes in the disorder of insomnia: A meta-analysis of polysomnographic studies. Sleep Med Rev. 2014;18:195–213. doi: 10.1016/j.smrv.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 75.Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 2010;11:589–99. doi: 10.1038/nrn2868. [DOI] [PubMed] [Google Scholar]
- 76.Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–94. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- 77.Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electro Clin Neurophysiol. 1949;1:455–73. [PubMed] [Google Scholar]
- 78.Li J, Hu Z, de Lecea L. The hypocretins/orexins: integrators of multiple physiological functions. Br J Pharmacol. 2014;171:332–50. doi: 10.1111/bph.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chieffi S, Carotenuto M, Monda V, Valenzano A, Villano I, Precenzano F, et al. Orexin System: The key for a healthy life. Front Physiol. 2017;8:357. doi: 10.3389/fphys.2017.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yeoh JW, Campbell EJ, James MH, Graham BA, Dayas CV. Orexin antagonists for neuropsychiatric disease: progress and potential pitfalls. Front Neurosci. 2014;8:36. doi: 10.3389/fnins.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liblau RS, Vassali A, Seifinejad A, Tafti M. Hypocretin (orexin) biology and the pathophysiology of narcolepsy with cataplexy. Lancet Neurol. 2015;14:318–28. doi: 10.1016/S1474-4422(14)70218-2. [DOI] [PubMed] [Google Scholar]
- 82.Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 83.Maier JG, Kuhn M, Mainberger F, Guo S, Nachtsheim K, Bucsenec U, et al. Sleep orchestrates local plasticity and global stability of neural assemblies in the human cortex. Sleep. 2018;41:A43. doi: 10.1093/sleep/zsy263. [DOI] [PubMed] [Google Scholar]
- 84.Kuhn M, Hertenstein E, Feige B, Landmann N, Spiegelhalder K, Baglioni C, et al. Declarative virtual water maze learning and emotional fear conditioning in primary insomnia. J Sleep Res. 2018;27:e12693. doi: 10.1111/jsr.12693. [DOI] [PubMed] [Google Scholar]
- 85.Van Calker D, Serchov T, Normann C, Biber K. recent insights into antidepressant therapy: distinct pathways and potential common mechanisms in the treatment of depressive syndromes. Neurosci Biobehav Rev. 2018;88:63–72. doi: 10.1016/j.neubiorev.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 86.Dunlap JC, Loros JJ, DeCoursey P. Chronobiology—biological timekeeping. Sunderland: Sinauer Association; 2004.
- 87.Akiskal Hagop S., Bourgeois Marc L., Angst Jules, Post Robert, Möller Hans-Jürgen, Hirschfeld Robert. Re-evaluating the prevalence of and diagnostic composition within the broad clinical spectrum of bipolar disorders. Journal of Affective Disorders. 2000;59:S5–S30. doi: 10.1016/s0165-0327(00)00203-2. [DOI] [PubMed] [Google Scholar]
- 88.Fernandes DC, Fogerson PM, Ospri LL, Thomsen MB, Layne RM, Severin D, et al. Light affects mood and learning through distinct retina-brain pathways. Cell. 2018;175:71–84. doi: 10.1016/j.cell.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang L, Peng Y, Yang Y, Huang X, Fu Y, Tao Q, et al. A visual circuit related to habenula underlies the antidepressive effects of light therapy. Neuron. 2019;102:128–42. doi: 10.1016/j.neuron.2019.01.037. [DOI] [PubMed] [Google Scholar]
- 90.Zhou JN, Riemersma RF, Unmehopa UA, Hoogendijk WJ, van Heerikhuize JJ, Hofman MA, et al. Alterations in arginine vasopressin neurons in the suprachiasmatic nucleus in depression. Arch Gen Psychiatry. 2001;58:655–62. doi: 10.1001/archpsyc.58.7.655. [DOI] [PubMed] [Google Scholar]
- 91.Wu YH, Ursinus J, Zhou JN, Scheer FA, Ai-Min B, Jockers R, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord. 2013;148:357–67. doi: 10.1016/j.jad.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 92.Bernstein HG, Heinemann A, Krell D, Mawrin C, Bielau H, Danos P, et al. Further immunohistochemical evidence for impaired NO signaling in the hypothalamus of depressed patients. Ann N Y Acad Sci. 2002;973:91–3. doi: 10.1111/j.1749-6632.2002.tb04613.x. [DOI] [PubMed] [Google Scholar]
- 93.Plano SA, Golombek DA, Chiesa JJ. Circadian entrainment to light-dark cycles involves extracellular nitric oxide communication within the suprachiasmatic nuclei. Eur J Neurosci. 2010;31:876–82. doi: 10.1111/j.1460-9568.2010.07120.x. [DOI] [PubMed] [Google Scholar]
- 94.Dhir A, Kulkarni K. Nitric oxide and major depression. Nitric Oxide. 2011;30:125–31. doi: 10.1016/j.niox.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 95.Kalsbeek A, Palm IF, La Fleur SE, Scheer FA, Perreau-Lenz S, Ruiter M, et al. SCN outputs and the hypothalamic balance of life. J Biol Rhythms. 2006;21:458–69. doi: 10.1177/0748730406293854. [DOI] [PubMed] [Google Scholar]
- 96.Mendoza J. Circadian neurons in the lateral habenula: clocking motivated behaviors. Pharm Biochem Behav. 2017;162:55–61. doi: 10.1016/j.pbb.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 97.Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, Unterberg AW, et al. Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol Psychiatry. 2010;67:e9–e11. doi: 10.1016/j.biopsych.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 98.Clemm von Hohenberg C, Weber-Fahr W, Lebhardt P, Ravi N, Braun U, et al. Lateral habenula perturbation reduces default-mode network connectivity in a rat model of depression. Transl Psychiatry. 2018;8:68. doi: 10.1038/s41398-018-0121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gosnell SN, Curtis KN, Velasquez K, Fowler JC, Madan A, Goodman W, et al. Habnular connectivity may predict treatment response in depressed psychiatric inpatients. J Aff Dis. 2019;242:211–9. doi: 10.1016/j.jad.2018.08.026. [DOI] [PubMed] [Google Scholar]
- 100.Langel J, Ikeno T, Yan L, Nunez AA, Smale L. Distributions of GABAergic and glutamatergic neurons in the brains of a diurnal and nocturnal rodent. Brain Res. 2018;1700:152–9. doi: 10.1016/j.brainres.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 101.Lacoste B, Angeloni D, Dominguez-Lopez S, Calderoni S, Mauro A, Fraschini F, et al. Anatomical and cellular localization of melatonin MT1 and MT2 receptors in the adult rat brain. J Pineal Res. 2015;58:397–417. doi: 10.1111/jpi.12224. [DOI] [PubMed] [Google Scholar]
- 102.Ciarleglio CM, Resuehr HE, McMahon DG. Interactions of the serotonin and circadian systems: nature and nurture in rhythms and blues. Neuroscience. 2011;197:8–16. doi: 10.1016/j.neuroscience.2011.09.036. [DOI] [PubMed] [Google Scholar]
- 103.Spielman AJ, Caruso LS, Glovinsky PB. Behavioral perspective on insomnia. Psychiatr Clin North Am. 1987;10:541–53. [PubMed] [Google Scholar]
- 104.Palagini L, Biber K, Riemann D. The genetics of insomnia—evidence for epigenetic mechanisms. Sleep Med Rev. 2014;18:225–35. doi: 10.1016/j.smrv.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 105.Lahtinen A, Puttonen S, Vanttola P, Vitasalo K, Sulkava S, Perjakova N, et al. A distinctive DNA methylation pattern in insufficient sleep. Sci Rep. 2019;9:1193. doi: 10.1038/s41598-018-38009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hammerschlag Anke R, Stringer Sven, de Leeuw Christiaan A, Sniekers Suzanne, Taskesen Erdogan, Watanabe Kyoko, Blanken Tessa F, Dekker Kim, te Lindert Bart H W, Wassing Rick, Jonsdottir Ingileif, Thorleifsson Gudmar, Stefansson Hreinn, Gislason Thorarinn, Berger Klaus, Schormair Barbara, Wellmann Juergen, Winkelmann Juliane, Stefansson Kari, Oexle Konrad, Van Someren Eus J W, Posthuma Danielle. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nature Genetics. 2017;49(11):1584–1592. doi: 10.1038/ng.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lane JM, Liang J, Vlasac I, Anderson SG, Bechtold DA, Bowden J, et al. Genom-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat Genet. 2017;49:274–81. doi: 10.1038/ng.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lane JM, Jones S, Dashti HS, Wood AR, Aragam K, Van Hees V, et al. Biological and clinical insights from genetics of insomnia symptoms. Nat Genet. 2019;51:387–93. doi: 10.1038/s41588-019-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drake CL, Pillai V, Roth T. Stress and sleep reactivity: a prospective investigation of the stress-diathesis model of insomnia. Sleep. 2014;37:1295–304. doi: 10.5665/sleep.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van de Laar M, Verbeek I, Pevernagie D, Aldenkamp A, Overeem S. The role of personality traits in insomnia. Sleep Med Rev. 2010;14:61–8. doi: 10.1016/j.smrv.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 111.Bastien CH, Vallières A, Morin CM. Precipitating factors of insomnia. Behav Sleep Med. 2004;2:50–62. doi: 10.1207/s15402010bsm0201_5. [DOI] [PubMed] [Google Scholar]
- 112.Healey E, Kales A, Monroe LJ, Bixler EO, Chamberlin K, Soldatos CB. Onset of insomnia: Role of life-stress events. Psychosom Med. 1981;43:439–51. doi: 10.1097/00006842-198110000-00007. [DOI] [PubMed] [Google Scholar]
- 113.Feige B, Al-Shajlawi A, Nissen C, Voderholzer U, Hornyak M, Spiegelhalder K, et al. Does REM sleep contribute to subjective wake time in primary insomnia? A comparison of polysomnographic and subjective sleep in 100 patients. J Sleep Res. 2008;17:180–90. doi: 10.1111/j.1365-2869.2008.00651.x. [DOI] [PubMed] [Google Scholar]
- 114.Feige B, Nanovska S, Baglioni C, Bier B, Cabrera L, Diemers L, et al. Insomnia—perchance a dream? Results from a NonREM/ REM sleep awakening study in good sleepers and patients with insomnia. Sleep 2018;41:10.1093/sleep/zsy032. [DOI] [PubMed]
- 115.Bootzin Richard R., Epstein Dana, Wood James M. Case Studies in Insomnia. Boston, MA: Springer US; 1991. Stimulus Control Instructions; pp. 19–28. [Google Scholar]
- 116.Carney CE, Harris AL, Moss TG, Edinger JD. Distinguishing rumination from worry in clinical insomnia. Behav Res Ther. 2010;48:540–6. doi: 10.1016/j.brat.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baglioni C, Spiegelhalder K, Regen W, Feige B, Nissen C, Lombardo C, et al. Insomnia disorder is associated with increased amygdala reactivity to insomnia-related stimuli. Sleep. 2014;37:1907–17. doi: 10.5665/sleep.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baglioni C, Spiegelhalder K, Lombardo C, Riemann D. Sleep and emotions: a focus on insomnia. Sleep Med Rev. 2010;14:227–38. doi: 10.1016/j.smrv.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 119.Baglioni C, Lombardo C, Bux E, Hansen S, Salveta C, Biello S, et al. Psychophysiological reactivity to sleep-related emotional stimuli in primary insomnia. Behav Res Ther. 2010;48:467–75. doi: 10.1016/j.brat.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 120.LeDoux J. The amygdala. Curr Biol. 2007;17:868–74. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 121.Sakurai T, Mieda M. Connectomics of orexin-producing neurons: Interface of systems of emotion, energy homeostasis and arousal. Trends Pharm Sci. 2011;32:451–62. doi: 10.1016/j.tips.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 122.Ballesta A, Innominato PF, Dallmann R, Rand DA, Lévi FA. Systems chronotherapeutics. Pharm Rev. 2017;69:161–99. doi: 10.1124/pr.116.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sateia MJ, Buyyse DJ, Krystal AD, Neubauer DN, Heald JL. Clinical practice guidelines for the pharmacologic treatment of chronic insomnia in adults: an American academy of sleep medicine clinical practice guideline. J Clin Sleep Med. 2017;13:307–49. doi: 10.5664/jcsm.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Perlis ML, Jungquist C, Smith MT, Posner D. Cognitive behavioural treatment of insomnia - A session-by-session guide. Springer: New York; 2005.
- 125.Harvey AG. A cognitive model of insomnia. Behav Res Ther. 2002;40:869–93. doi: 10.1016/s0005-7967(01)00061-4. [DOI] [PubMed] [Google Scholar]
- 126.Espie CA, Broomfield NM, MacMahon KM, Macphee LM, Taylor LM. The attention-inattention-effort pathway in the development of psychophysiologic insomnia: an invited theoretical review. Sleep Med Rev. 2006;10:215–45. doi: 10.1016/j.smrv.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 127.Miller CB, Espie CA, Epstein DR, Friedmann L, Morin CM, Pigeon WR, et al. The evidence base of sleep restriction therapy for treating insomnia disorder. Sleep Med Rev. 2014;18:415–24. doi: 10.1016/j.smrv.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 128.Maurer LF, Espie CA, Kyle SD. How does sleep restriction therapy for insomnia work? A systematic review of mechanistic evidence and the introduction of the Triple-R model. Sleep Med Rev. 2018;42:127–38. doi: 10.1016/j.smrv.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 129.Mendelson W. The treatment of chronic insomnia: drug indications, chronic use and abuse liability. Summary of a 2001 New Clinical Drug Evaluation Unit Meeting Symposium. Sleep Medicine Reviews. 2004;8(1):7–17. doi: 10.1016/S1087-0792(03)00042-X. [DOI] [PubMed] [Google Scholar]
- 130.Barker MJ, Greenwood KM, Jackson M, Crowe SF. Persistence of cognitive effects after withdrawal from longterm benzodiazepine use: ameta-analysis. Arch Clin Neuropsychol. 2004;19:437–54. doi: 10.1016/S0887-6177(03)00096-9. [DOI] [PubMed] [Google Scholar]
- 131.Dassanayake T, Micie P, Carter G, Jones A. Effects of benzodiazepines, antidepressants, and opoids on driving. A systematic review and meta-analysis of epidemiological and experimental evidence. Drug Saf. 2011;34:125–56. doi: 10.2165/11539050-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 132.Hajak G, Müller WE, Wittchen HU, Pittrow D, Kirch W. Abuse and dependence potential for the non-benzodiazepine hypnotics Zolpidem and Zopiclone - A review of case reports and epidemiological data. Addiction. 2003;98:1371–78. doi: 10.1046/j.1360-0443.2003.00491.x. [DOI] [PubMed] [Google Scholar]
- 133.Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18:249–55. doi: 10.1097/01.yco.0000165594.60434.84. [DOI] [PubMed] [Google Scholar]
- 134.MacFarlane J, Morin CM, Montplaisir J. Hypnotics in insomnia: the experience of zolpidem. Clin Ther. 2014;36:1676–701. doi: 10.1016/j.clinthera.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 135.Kripke DF, Garfinkel I, Wingard DL. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002;59:131–6. doi: 10.1001/archpsyc.59.2.131. [DOI] [PubMed] [Google Scholar]
- 136.Kripke DF. Do hypnotics cause death and cancer? The burden of proof. Sleep Med. 2009;10:275–6. doi: 10.1016/j.sleep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 137.Neutel CL, Johansen HL. Association between hypnotic use and increased mortality: causation or confounding? Eur J Pharm. 2015;71:637–42. doi: 10.1007/s00228-015-1841-z. [DOI] [PubMed] [Google Scholar]
- 138.Pottegard A, Friis S, Andersen M, Hallas J. Use of benzodiazepines or benzodiazepine related drugs and the risk of cancer: a population-based case-control study. Br J Clin Pharm. 2013;75:1356–64. doi: 10.1111/bcp.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McGechan A, Wellington K. Ramelteon. CNS Drugs. 2005;19:1057–65. doi: 10.2165/00023210-200519120-00007. [DOI] [PubMed] [Google Scholar]
- 140.Herxheimer A, Petrie KJ. Melatonin for the prevention and treatment of jet lag. Issue 4. Oxford: The Cochrane Library; 2002 [DOI] [PubMed]
- 141.Buscemi N, Vandermeer B, Hooton N, Pandiaya R, Tjosvold L, Hartling L, et al. The efficacy and safety of exogenous melatonin for primary sleep disorders. J Gen Int Med. 2005;20:1151–8. doi: 10.1111/j.1525-1497.2005.0243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Buscemi N, Vandermeer B, Hooton N, Pandiaya R, Tjosvold L, Hartling L, et al. Efficacy and safety of exogenous melatonin for secondary sleep disorders accompanying sleep restriction: meta-analysis. Brit Med J. 2006;332:1–9. doi: 10.1136/bmj.38731.532766.F6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sateia MJ, Kirby-Long P, taylor JL. Efficacy and clinical safety of ramelteon: an evidence-based review. Sleep Med Rev. 2008;12:319–32. doi: 10.1016/j.smrv.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 144.Frase Lukas, Nissen Christoph, Riemann Dieter, Spiegelhalder Kai. Making sleep easier: pharmacological interventions for insomnia. Expert Opinion on Pharmacotherapy. 2018;19(13):1465–1473. doi: 10.1080/14656566.2018.1511705. [DOI] [PubMed] [Google Scholar]
- 145.Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med Rev. 2014;15:385–92. doi: 10.1016/j.sleep.2013.11.788. [DOI] [PubMed] [Google Scholar]
- 146.Davis KL, Charney D, Coyle JT, Nemeroff C. Neuropsychopharmacology. Philadelphia: Lippincott, Williams & Wilkins; 2002. [Google Scholar]
- 147.Walsh JK, Schweitzer PK. Ten-year trends in the pharmacological treatment of insomnia. Sleep. 1999;22:371–5. [PubMed] [Google Scholar]
- 148.Walsh JK. Drugs used to treat insomnia in 2002: regulatory-based rather than evidence-based medicine. Sleep. 2004;27:1441. doi: 10.1093/sleep/27.8.1441. [DOI] [PubMed] [Google Scholar]
- 149.Walsh JK, Erman M, Erwin M, et al. Subjective hypnotic efficacy of trazodone and zolpidem in DMS-III-R primary insomnia. Hum Psychopharmacol. 1998;13:191–8. [Google Scholar]
- 150.Hajak G, Rodenbeck A, Voderholzer U, Riemann D, Cohrs S, Hohagen F, et al. Doxepin in the treatment of primary insomnia: A placebo-controlled, double-blind, polysomnographic study. J Clin Psychiat. 2001;62:453–63. doi: 10.4088/jcp.v62n0609. [DOI] [PubMed] [Google Scholar]
- 151.Riemann D, Voderholzer U, Cohrs S, Rodenbeck A, Hajak G, Rüther E, et al. Trimipramine in primary insomnia: results of a polysomnographic double-blind controlled study. Pharmacopsychiatry. 2002;35:165–4. doi: 10.1055/s-2002-34119. [DOI] [PubMed] [Google Scholar]
- 152.Winkler A, Auer C, Doering BK, Rief W. Drug treatment of primary insomnia: a meta-analysis of polysomnographic radomized controlled trials. CNS Drugs. 2014;28:799–816. doi: 10.1007/s40263-014-0198-7. [DOI] [PubMed] [Google Scholar]
- 153.Roth T, Rogowski R, Hull S, Schwartz H, Koshorek G, Corsev B, et al. Efficacy and safety of doxepin 1 mg, 3 mg, and 6 mg in adults with primary insomnia. Sleep. 2007;30:1555–61. doi: 10.1093/sleep/30.11.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cohrs S. Sleep disturbances in patients with schizophrenia. CNS Drugs. 2008;22:939–62. doi: 10.2165/00023210-200822110-00004. [DOI] [PubMed] [Google Scholar]
- 155.McCall C, Vaughn McCall W. What is the role of sedating antidepressants, antipsychotics and antoconvulsants in the management of insomnia? Curr Psychiatry Rep. 2012;14:494–502. doi: 10.1007/s11920-012-0302-y. [DOI] [PubMed] [Google Scholar]
- 156.Anderson SL, Vande Griend JV. Quetiapine for insomnia: a review of the literature. Am J Health-Syst. 2014;71:394–402. doi: 10.2146/ajhp130221. [DOI] [PubMed] [Google Scholar]
- 157.Barbini B, Benedetti F, Colombo C, Dotoli D, Bernasconi A, Cigala-Fulgosi M, et al. Dark therapy for mania: a pilot study. Bipolar Disord. 2005;7:98–101. doi: 10.1111/j.1399-5618.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- 158.Frank E, Swartz HA, Kupfer DJ. Interpersonal and social rhythm therapy: managing the chaos of bipolar diorder. Biol Psychiat. 2000;48:593–604. doi: 10.1016/s0006-3223(00)00969-0. [DOI] [PubMed] [Google Scholar]
- 159.Frank E, Kupfer DJ, Thase M, Mallinger AG, Swartz HA, Fagioloni AM, et al. Two-year outcomes for interpersonal and social rhythm therapy in individuals with bipolar I disorder. Arch Gen Psychiat. 2005;62:996–1004. doi: 10.1001/archpsyc.62.9.996. [DOI] [PubMed] [Google Scholar]
- 160.Van Geilswijk IM, Korzilius HP, Smits MG. The use of exogenous melatonin in delayed sleep phase disorder: a meta-analysis. Sleep. 2010;33:1605–14. doi: 10.1093/sleep/33.12.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liira J, Verbeek JH, Costa G, Driscoll TR, Sallinen M, Isotalo LK, et al. Pharmacological interventions for sleepiness and sleep disturbances caused by shift work. Cochr Database Syst Rev. 2014; CD009776. [DOI] [PMC free article] [PubMed]
- 162.Spadoni G, Bedini A, Rivara S, Mor S. Melatonin receptor agonists: new options for insomnia and depression treatment. CNS Neurosci Ther. 2011;17:733–41. doi: 10.1111/j.1755-5949.2010.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Phillips L, Appleton RE. Systematic review of melatonin treatment in children with neurodevelopmental disabilities and sleep impairment. Dev Med Child Neurol. 2004;45:771–5. doi: 10.1017/s001216220400132x. [DOI] [PubMed] [Google Scholar]
- 164.Krone L, Frase L, Piosczyk H, Kuhn M, Selhausen P, Zittel S, et al. Top-down control of arousal and sleep and potential clinical implications. Sleep Med Rev. 2017;31:17–24. doi: 10.1016/j.smrv.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 165.Nitsche MA, Paulus W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol. 2000;527:633–9. doi: 10.1111/j.1469-7793.2000.t01-1-00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marshall L, Helgadóttir H, Mölle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–3. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- 167.Frase L, Piosczyk H, Zittel S, Jahn F, Selhausen P, Krone L, et al. The modulation of arousal and sleep continuity by transcranial direct current stimulation (tDCS) Neuropsychopharmacology. 2016;41:2577–86. doi: 10.1038/npp.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Frase L, Selhausen P, Krone L, Zittel S, Jahn F, Feige B, et al. Transcranial direct current stimulation (tDCS) in insomnia disorder. Brain Stimulation. 2019 (in press).
- 169.Espie CA. “Stepped care”: a health technology solution for delivering cognitive behavioral therapy as a first line insomnia treatment. Sleep. 2009;32:1549–58. doi: 10.1093/sleep/32.12.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Espie CA, Kyle S, Williams C, Ong J, Douglas NJ, Hames P, et al. A randomized, placebo-controlled trial of online cognitive-behaviroal therapy for chronic insomnia disorder delivered via an automated media-rich web application. Sleep. 2012;35:769–81. doi: 10.5665/sleep.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Espie CA, MacMahon KMA, Kelly HL, Broomfield NM, Douglas NJ, Engleman HM, et al. Randomized clinical effectiveness trial of nurse-administered small-group cognitive behavior therapy for persistent insomnia in general practice. Sleep. 2007;30:574–84. doi: 10.1093/sleep/30.5.574. [DOI] [PubMed] [Google Scholar]
- 172.Buysse DJ, Germain A, Moul DE, Franzen PL, Brar LK, Fletcher ME, et al. Efficacy of brief behavioral treatment for chronic insomnia in older adults. Arch Intern Med. 2011;171:887–95. doi: 10.1001/archinternmed.2010.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ellis J, Cushing T, Germain A. Treating acute insomnia: a randomized controlled trioal of a “single-shot” of cognitive behavioral therapy for insomnia. Sleep. 2015;38:971–8. doi: 10.5665/sleep.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Espie CA, Emsley R, Kyle SD, Gordon C, Drake CL, Siriwardena AN, et al. Effect of digital cognitive behavioral therapy for insomnia on health, psychological well-being, and sleep-related quality of life: a randomized clinical trial. JAMA Psychiatry. 2019;19:21–30. doi: 10.1001/jamapsychiatry.2018.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Geiger-Brown JM, Rogers VE, Liu W, Ludeman EM, Downton KD, Diaz-Abad M. Cognitive behavioral therapy in persons with comorbid insomnia: a meta-analysis. Sleep Med Rev. 2015;23:54–67. doi: 10.1016/j.smrv.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 176.Wu JQ, Appleman ER, Salazar RD, Ong J. Cognitive behavioral therapy for insomnia comorbid with psychiatric and medical conditions: a meta-analysis. JAMA Intern Med. 2015;175:1461–72. doi: 10.1001/jamainternmed.2015.3006. [DOI] [PubMed] [Google Scholar]
- 177.Gosling JA, Glozier N, Griffiths K, Ritterband L, Thorndike F, Mackinnon A, et al. The GoodNight study—online CBT for insomnia for the indicated prevention of depression: study protocol for a randomised controlled trial. Trials. 2014;15:56. doi: 10.1186/1745-6215-15-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Michelson D, Snyder E, Paradis E, Chengau-Lin M, Snavely DB, Hutzelman J, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:461–71. doi: 10.1016/S1474-4422(14)70053-5. [DOI] [PubMed] [Google Scholar]
- 179.Riemann D, Spiegelhalder K. Orexin receptor antagonists: a new treatment for insomnia? Lancet Neurol. 2014;13:441–3. doi: 10.1016/S1474-4422(13)70311-9. [DOI] [PubMed] [Google Scholar]
- 180.Nofzinger E, Buysse DJ. Frontal cerebral thermal transfer as a treatment for insomnia: a dose-ranging study. Sleep. 2011;34:A0534. [Google Scholar]
- 181.Sulzer J, Haller S, Scharnowski F, Weiskopf N, Birbaumer N, Biefasi ML, et al. Real-time fMRI neurofeedback: progress and challenges. Neuroimage. 2013;76:386–99. doi: 10.1016/j.neuroimage.2013.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schabus M, Heib DP, Lechinger J, Griessenberger H, Klimesch W, Pawlizki A, et al. Enhancing sleep quality and memory in insomnia using instrumental sensorimotor rhythm conditioning. Biol Psychol. 2014;95:126–34. doi: 10.1016/j.biopsycho.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 183.O’Leary OF, Cryan JF. Towards translational rodent models of depression. Cell Tissue Res. 2013;354:141–53. doi: 10.1007/s00441-013-1587-9. [DOI] [PubMed] [Google Scholar]
- 184.Ramaker MJ, Dulawa SC. Identifying fast-onset antidepressants using rodent models. Mol Psychiatry. 2017;22:656. doi: 10.1038/mp.2017.36. [DOI] [PubMed] [Google Scholar]
- 185.Cano G, Mochizuki T, Saper CB. Neural circuitry of stress-induced insomnia in rats. J Neurosci. 2008;28:10167–84. doi: 10.1523/JNEUROSCI.1809-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Krueger JM, Rector DM, Roy S, Van Dongen HPA, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–9. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Krueger JM, Tononi G. Local use-dependent sleep; synthesis of the new paradigm. Curr Top Med Chem. 2011;11:2490–2. doi: 10.2174/156802611797470330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kattler H, Dijk DJ, Borbély AA. Effect of unilateral somatosensory stimulation prior to sleep on the sleep EEG in humans. J Sleep Res. 1994;3:159–64. doi: 10.1111/j.1365-2869.1994.tb00123.x. [DOI] [PubMed] [Google Scholar]
- 189.Huber R, Felice Ghilardi M, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- 190.Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–7. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nobili L, Ferrara M, Moroni F, De Gennaro L, Russo GLo, Campus C, et al. Dissociated wake-like and sleep-like electro-cortical activity during sleep. Neuroimage. 2011;58:612–9. doi: 10.1016/j.neuroimage.2011.06.032. [DOI] [PubMed] [Google Scholar]
- 192.Nir Y, Andrillon T, Marmelshtein A, Suthana N, Cirelli C, Tononi G, et al. Selective neuronal lapses precede human cognitive lapses following sleep deprivation. Nat Med. 2017;23:1474–80. doi: 10.1038/nm.4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]