

Extensive efforts are underway to comprehensively characterize changes in the human brain transcriptome in neurodevelopmental and neuropsychiatric disorders. Although bulk RNA-sequencing (RNA-seq) in postmortem human brain has identified many molecular associations with psychiatric disease, analysis of homogenate brain tissue can mask the heterogeneity of associations within and across specific cell types. Although computational approaches exist to deconvolute cell-type-specific effects from bulk data, these approaches largely control for cellular heterogeneity across samples [1] and do not pinpoint candidate cell types harboring transcriptional differences [2]. However, rapid progress towards characterizing the cell types that make up the brain has been achieved by major advances in single-cell (sc) and single-nuclei (sn) approaches, which are revealing molecular profiles of distinct cell populations. However, these methods still require tissue dissociation, which removes molecularly defined cell types from their spatial environment. This is problematic, because the ability to assess gene expression as a function of neuroanatomy and cytoarchitectural organization will be critical for interpreting molecular and genetic associations.
Methods such as laser-capture microdissection allow for transcriptome-wide profiling in a defined spatial area, but tissue is removed from the surrounding spatial context, making it difficult to analyze gradients of gene expression. The rapidly accelerating field of spatial transcriptomics utilizes techniques that examine the location of hundreds of gene targets in intact tissue slices. In situ sequencing and fluorescent in situ hybridization-based technologies have achieved high levels of multiplexing in single cells of mouse brain using padlock probes or barcoding strategies in combination with sequential rounds of probing, imaging, and stripping (reviewed in refs. [3, 4]). However, even for methodologies that can accommodate hundreds of transcripts simultaneously, molecular crowding within cells leads to fluorescence overlap posing significant microscopy and computational challenges [3, 5].
Although not yet sc in resolution, additional platforms such as Slide-seq [6] and SPATiAL transcriptomics [7] have emerged to provide positional context of genome-wide expression in intact tissue sections. SPATiAL generates transcriptome-wide RNA-seq data through capture of polyadenylated RNA on arrays containing positional molecular barcodes that are introduced during cDNA synthesis, which occurs on the surface of the tissue section. Barcoded cDNA is then cleaved, prepared into libraries, and sequenced on a standard Illumina platform. Each RNA is then mapped back to its spatial location within 1 of ~1000 different molecular positions in a 6 mm2 array. Although these approaches have not yet been widely applied to postmortem human brain, SPATiAL has been used to identify perturbations in transcriptional pathways for several normal and pathological human tissues, including the spinal cord [8].
Ultimately, understanding the molecular basis of psychiatric disease will require linking molecularly defined cell types to measures of function, including correlates of morphology, physiology, and connectivity. Analyzing gene expression within the existing tissue architecture in the human brain is the next step. These rapidly evolving spatial transcriptomics techniques open possibilities for combining spatial expression maps with sc/snRNA-seq data using spatial registration approaches to add anatomical dimensions to existing datasets and further refine cell-type classifications in the human brain. Importantly, these spatial transcriptomic approaches capture gene expression in the cytoplasm and neurites (Fig. 1), which has been largely missed by snRNA-seq approaches in human brain tissue. This is important, because synaptically localized mRNAs may be especially relevant for understanding genetic risk for psychiatric disorders as genome-wide association study signals are preferentially enriched for synaptic neuropil transcripts [9].
Fig. 1.
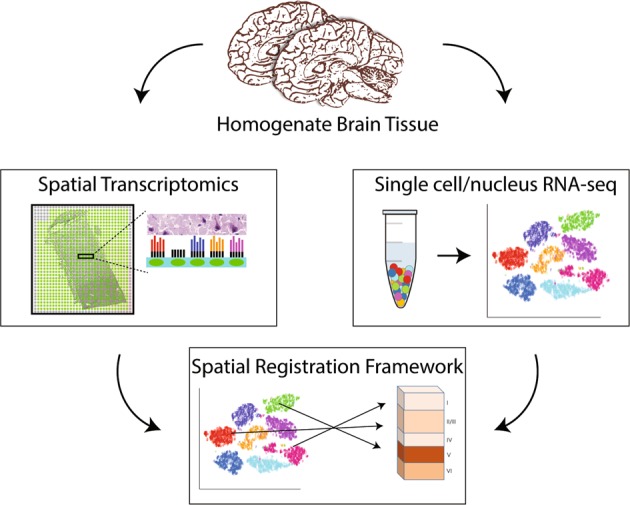
Spatial resolution of cell-type-specific gene expression in the human brain. Bulk homogenate tissue contains heterogeneous cell types with distinct spatial orientations. Spatial transcriptomic approaches, such as SPATiAL, use spotted arrays with positional molecular barcodes that tag cDNA synthesized in intact tissue sections with a spatial location allowing gene expression to be mapped back to a histological image (i.e., specific cortical layers). Parallel sc/sn RNA-seq approaches define gene expression in individual cells dissociated from homogenate tissue. Integrating spatial transcriptomic data with sc/snRNA-seq data opens possibilities for adding anatomical dimensions to existing datasets, to better understand cell-type-specific molecular profiles in the human brain during development and psychiatric disease
Funding and disclosure
Funding to support this research was provided by the Lieber Institute for Development.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng SC, Breeze CE, Beck S, Teschendorff AE. Identification of differentially methylated cell types in epigenome-wide association studies. Nat Methods. 2018;15:1059–66. doi: 10.1038/s41592-018-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lein E, Borm LE, Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017;358:64–69. doi: 10.1126/science.aan6827. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361:eaat5691. doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess DJ. Spatial transcriptomics coming of age. Nat Rev Genet. 2019;20:317. doi: 10.1038/s41576-019-0129-z. [DOI] [PubMed] [Google Scholar]
- 6.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 8.Maniatis S, Äijö T, Vickovic S, Braine C, Kang K, Mollbrink A, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93. doi: 10.1126/science.aav9776. [DOI] [PubMed] [Google Scholar]
- 9.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50:825–33. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]