

Summary
Anatomical homeostasis results from dynamic interactions between gene expression, physiology, and the external environment. Owing to its complexity, this cellular and organism-level phenotypic plasticity is still poorly understood. We establish planarian regeneration as a model for acquired tolerance to environments that alter endogenous physiology. Exposure to barium chloride (BaCl2) results in a rapid degeneration of anterior tissue in Dugesia japonica. Remarkably, continued exposure to fresh solution of BaCl2 results in regeneration of heads that are insensitive to BaCl2. RNA-seq revealed transcriptional changes in BaCl2-adapted heads that suggests a model of adaptation to excitotoxicity. Loss-of-function experiments confirmed several predictions: blockage of chloride and calcium channels allowed heads to survive initial BaCl2 exposure, inducing adaptation without prior exposure, whereas blockade of TRPM channels reversed adaptation. Such highly adaptive plasticity may represent an attractive target for biomedical strategies in a wide range of applications beyond its immediate relevance to excitotoxicity preconditioning.
Subject Areas: Marine Organism, Ion Activity, Cellular Physiology
Graphical Abstract
Highlights
-
•
Exposure to BaCl2 causes the heads of Dugesia japonica to degenerate
-
•
Prolonged exposure to BaCl2 results in regeneration of a BaCl2-insensitive head
-
•
Ion channel expression is altered in the head to compensate for excitotoxic stress
-
•
TRPMa is upregulated in BaCl2-treated animals; blocking TRPM prevents adaptation
Marine Organism; Ion Activity; Cellular Physiology
Introduction
A fundamental challenge faced by all living things is survival in an uncertain and changing environment. One of the fundamental goals of biology and medicine is to understand how physiology and morphology, whether at the cell or organism level, adjusts to stress—conditions that are not conducive to continued health and reproduction. Living tissues are confronted with continuous environmental challenge at all scales of size and organization, from exposure to agents that damage DNA and produce reactive oxygen species at the level of single cells to secreted factors and cell group activity that drive embryogenesis and repair/regeneration; to the physiological functions of immune, circulatory, and endocrine systems; and ultimately to the behavior of the entire animal. Thus, all components of an organism must continuously select from a very large space of possible transcriptional, protein-level, and physiological-level responses to adaptively maintain homeostasis along numerous parameters during its lifespan. Understanding this ubiquitous process is vital, with direct applications to reprotoxicology (the ability of embryos to regulate, or fail to regulate, their normal developmental sequence under diverse, potentially teratogenic influences), immunology, aging, and cancer. Harnessing this process is also a goal of regenerative medicine.
Major gaps remain in our understanding of the fundamental biology of computation in living media: how do cellular networks process real-time information toward near-optimal responses? On a population scale, recent work with bacterial persisters and yeast subjected to antibiotic and metabolic stress, respectively, reveals evolutionary aspects of epigenetic tolerance to stressors (Lopez Garcia de Lomana et al., 2017, Samani and Bell, 2016, Gonzalez and Bell, 2013, Kussell et al., 2005, Balaban et al., 2004, Tkachenko, 2018, Lambert and Kussell, 2014). However, experiments in Drosophila and other model systems reveal that, even within the lifetime of single individuals (i.e., not via selection), cells have the remarkable ability to execute appropriate responses to stressors (Soen et al., 2015, Elgart et al., 2015, Stern et al., 2012, Soen and Braun, 2000, Karin et al., 2016). This emerging body of work on cellular adaptation and plasticity (Stetina et al., 2015, Prymaczok et al., 2016) is complemented by similar robustness observed at the organ or even whole-organism scale (Pezzulo and Levin, 2015, Brunke and Hube, 2014, Sorek et al., 2013, Freddolino and Tavazoie, 2012).
One of the best examples of large-scale dynamic plasticity is regulative development, which is often able to adjust to drastic injury (Cooke, 1981, Tarkowski, 1961). One example can be seen in the processes that convert a tadpole face to that of a frog, which requires significant movements of the eyes and other organs to rearrange internal head structures into the correct species-specific outcome. It was recently shown that this is not a hardwired process in which the different components move in pre-determined paths (Vandenberg et al., 2012) and rely on a constant starting state to implement normal frog development. Tadpoles engineered to have scrambled anatomies (eyes, nostrils, and other structures in highly abnormal positions) still usually develop into normal frogs. Thus, the genome encodes a system with the ability to reach the required anatomical state from a range of diverse starting conditions, dynamically adapting to (evolutionarily) unexpected circumstances and readjusting the various tissues as needed to optimize subsequent survival.
Beyond development, metazoan survival throughout the lifespan requires a constant and active resistance to aging, wear and tear, and carcinogenic transformation (Rubin, 1985, Rubin, 2006, Rubin, 2007). Moreover, some species are capable of large-scale remodeling and pattern homeostasis throughout their lifespan. Planarians are a class of free-living flatworms that contains many highly regenerative members (Cebrià et al., 2018, Saló et al., 2009). The freshwater planarian Dugesia japonica is one such species, able to restore an entire body (including the brain) from a mere fragment, scaling the growth with exquisite precision and ceasing further growth and remodeling when an allometrically correct body plan is complete (Levin et al., 2019, Sahu et al., 2017, Saló et al., 2009). Planarians possess bilateral symmetry, stem cell populations (Hill and Petersen, 2015, Tanaka and Reddien, 2011), and a true centralized brain with numerous complex behavioral responses including learning (Sarnat and Netsky, 1985, Corning and Freed, 1968). They appear to have even conquered the ultimate stressor—aging (Sahu et al., 2017, Petralia et al., 2014). Much progress has been made on the biochemical (Reddien, 2018, Owlarn and Bartscherer, 2016) and physiological/bioelectrical (Lange and Steele, 1978, Chan et al., 2014, Durant et al., 2017, Sullivan et al., 2016, Beane et al., 2011, Beane et al., 2013) control of patterning during regeneration, making them an ideal model system in which one can investigate adaptive responses to novel stimuli that threaten homeostasis.
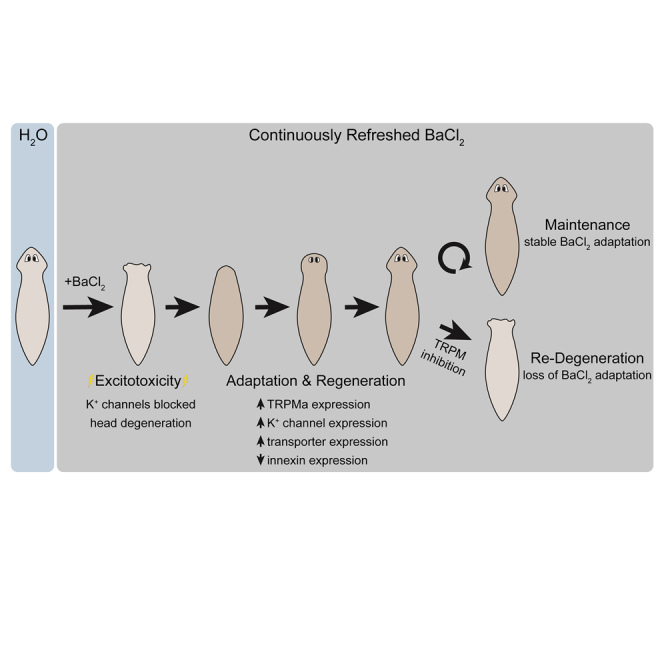
To establish a model for the study of adaptation at multiple levels of organization, we chose regeneration in D. japonica as a context to determine how cellular and body-wide plasticity must cooperate. Owing to the known dependence of planarian remodeling response on bioelectric events (Levin et al., 2019, Durant et al., 2017, Emmons-Bell et al., 2015, Beane et al., 2011, Beane et al., 2013, Oviedo et al., 2010), we challenged planaria with a novel stressor: the non-specific potassium channel blocker barium chloride (BaCl2). The inability to pass potassium ions led to a severe head degeneration. Remarkably, however, the planaria soon regenerated a BaCl2-tolerant head. We analyzed these BaCl2-adapted heads using molecular and physiologic approaches, which revealed an example of transcriptional and physiological plasticity with significant implications for basic biology and neural injury repair.
Results
When Exposed to BaCl2, Planaria Heads Rapidly Degenerate and Subsequently Regenerate with Newly Acquired BaCl2 Resistance
Given the ubiquitous importance of potassium channels in cell physiology, we first asked what would happen to planaria continuously exposed to broad K+ flux inhibitors. We cultured D. japonica flatworms in 1 mM barium chloride (BaCl2), a powerful, widely used, universal potassium channel blocker (Jiang and Mackinnon, 2000, Latorre et al., 1997), which can also affect other divalent cation channels and has been extensively used in invertebrates (Armstrong and Taylor, 1980, Eaton and Brodwick, 1980, Hanrahan et al., 1986). We observed rapid and complete degeneration of the entire head within 72 h (83% of observed worms showed degeneration, SD = 12% across all BaCl2 treatments, Figure 1Ab), consistent with the importance of potassium currents in maintenance and physiology of this complex organ. Remarkably, however, when left to regenerate in fresh BaCl2 solution, the worms regenerated normal heads that were insensitive to the toxic environmental conditions (Figures 1Ac and 1Ad). Although BaCl2 could potentially form insoluble ions with sulfate and carbonate ions present in the Poland Spring water, we ruled out adaptation via reduction of BaCl2 concentration in the media by making sure that media was frequently refreshed. Furthermore, degeneration of anterior tissues was never observed in control culture conditions (Figure 1Aa). Heads regenerated in BaCl2 retained normal size and morphology (Figure 1Ad), and the BaCl2-adapted worms exhibited motion that was not visually different from that of wild-type (WT) worms.
Figure 1.

Long-term Exposure to BaCl2 Results in Degeneration of Anterior Tissues and Subsequent Regeneration of Structures That Are Resistant to BaCl2
(A) Whole D. japonica worms after treatment in 1 mM BaCl2. (a) Normal worms before treatment. (b) Within 72 h of exposure to BaCl2, the anterior tissues of the planarian degrade and the head deprogresses. This occurs in 83% of worms, SD = 12%. Degeneration occurs through a contraction of tissue at the base of the wound, minimizing tissue loss. (c) After 15 days in BaCl2, D. japonica form a blastema and begin to regrow a head. (d) By 37 days of treatment, a new, BaCl2-insensitive head has formed and the worm is phenotypically normal. (a’–d’) 200x zoomed in images of the anterior portion of the worms shown in (a)–(d). Scale bar 0.5 mm. Results representative of three independent biological replicates, N > 50 for each replicate.
(B) (a) D. japonica worm after 35 days of BaCl2 treatment. This worm has degenerated and regenerated a head and is now insensitive to BaCl2. (b) D. japonica worms are then placed in water for 30 days with no obvious morphological effect. (c) However, upon 24 h of a second BaCl2 treatment, the head degenerates. (a’–c’) 200x zoomed in images of the anterior portion of the worms shown in (a)–(c). Scale bar 0.5 mm. Results representative of three independent biological replicates, N > 50 for each replicate.
Degeneration involved tissue progressively bubbling off from the very anterior tip of the worm to the plane of the photoreceptors (roughly 1 mm of tissue), with apparent muscular contraction around the degeneration site that minimized internal tissue loss (Figure 1Ab). The process was complete after 72 h in BaCl2 solution. Blastema formation was then initiated, and regenerative processes proceeded, although full regeneration took longer than normal (∼4 weeks, as opposed to ∼2 weeks, Figure 1Ac). After regeneration of the head, the worms were transferred into a fresh BaCl2 solution and showed no signs of degeneration. We conclude that BaCl2 exposure is acutely toxic to planarian anterior tissues but that the worms have the ability to produce new heads that are completely adapted to this novel, harsh condition.
Acquired Resistance to BaCl2 Is Lost after 30 Days in Water
We then investigated whether the acquired adaptation to BaCl2 was permanent or temporary. D. japonica worms were allowed to degenerate and regenerate heads in 1 mM BaCl2 over the course of approximately 35 days (Figure 1Ba). They were then transferred to Poland Spring water for 30 days, with the water refreshed every 7 days (Figure 1Bb). They were then subjected to a second round of 1 mM BaCl2 treatment. All worms displayed characteristic degeneration of anterior tissues after the second round of BaCl2 treatment, revealing that the insensitivity to BaCl2 is lost after 30 days in plain water (Figure 1Bc).
Transcriptionally Profiling the BaCl2-Adapted Head
What is different about the new, BaCl2-compatible heads when compared with the WT planarian head—what changes allow survival in BaCl2 after the initial adaptation? We hypothesized that the conferred resistance to BaCl2 treatment could be due to altered expression of ion channels or pumps that could compensate for, and buffer, the physiological perturbation. To test this hypothesis, we performed RNA sequencing (RNA-seq) and sub-network enrichment analysis (SNEA) on head samples from worms that were either treated with BaCl2 and allowed to regenerate insensitive heads or kept in water following the BaCl2 treatment. Table S1 provides all upregulated and downregulated transcripts identified. We report the total number of reads generated for each sample as well as a table of the alignment percentage of the reads mapped to the reference in Tables S2 and S3, respectively. GO and SNEA data are provided in Tables S4 and S5, respectively. A subnetwork enrichment was also conducted to enhance the overall dataset and to provide additional information on processes affected in the BaCl2-resistant heads (Table 1).
Table 1.
Categories of Transcripts Altered by BaCl2 Adaptation
| Process | Gene Set Seed | # Of Measured Neighbors | Median Change | p Value |
|---|---|---|---|---|
| Neural/Voltage | Cell membrane depolarization | 5 | −1.86 | 0.037 |
| Cell-cell signaling | 10 | −1.29 | 0.036 | |
| Anion transport | 17 | −1.17 | 0.049 | |
| Transmembrane potential | 85 | −1.09 | 0.032 | |
| Synapse structure | 8 | 1.05 | 0.019 | |
| Long-term synaptic depression | 50 | 1.16 | 0.010 | |
| Synaptogenesis | 83 | 1.17 | 0.045 | |
| Nervous system physiology | 26 | 1.24 | 0.047 | |
| Neuronal plasticity | 46 | 1.24 | 0.013 | |
| Transmission of nerve impulse | 61 | 1.28 | 0.012 | |
| Regulation of action potential | 35 | 1.42 | 0.005 | |
| Innervation | 31 | 1.88 | 0.003 | |
| Neuronal guidance | 5 | 2.17 | 0.033 | |
| Transmembrane signaling | 8 | 2.46 | 0.012 | |
| Na+ influx co-transport | 8 | 2.99 | 0.002 | |
| Neurotransmitter uptake | 5 | 3.73 | 0.019 | |
| Immune | T cell response | 71 | 1.04 | 0.041 |
| Monocyte recruitment | 19 | 1.42 | 0.027 | |
| Leukocyte recruitment | 25 | 1.52 | 0.016 | |
| Leukocyte migration | 29 | 1.58 | 0.043 | |
| Leukocyte accumulation | 12 | 1.88 | 0.035 | |
| Neutrophil adhesion | 18 | 1.88 | 0.042 | |
| Other | Melanocyte differentiation | 7 | −2.44 | 0.002 |
| Microtubule depolymerization | 9 | −2.26 | 0.011 | |
| Stem cell maintenance | 38 | −1.20 | 0.043 | |
| Stem cell proliferation | 50 | −1.15 | 0.005 | |
| Melanogenesis | 21 | 1.01 | 0.047 | |
| Memory | 127 | 1.05 | 0.036 | |
| Endothelial cell function | 54 | 1.06 | 0.034 | |
| Intracellular signaling cascade | 23 | 1.24 | 0.004 | |
| Endothelial cell development | 8 | 1.42 | 0.021 | |
| Positive chemotaxis | 12 | 2.63 | 0.029 |
Differentially expressed cell processes following degeneration and regeneration of a BaCl2-insensitive head, focusing on processes related to membrane potential homeostasis and regeneration. The number of items in the pathway, as well as the median fold change of the network and the p value, is reported for each enriched process. All subnetworks are presented in Table S5.
We detected a number of transcripts that were up- and downregulated after BaCl2 exposure (Table S1, q-value<0.05, change > log2 = 3), and genes that were only identified in one condition but not the other were listed as highly changed in the appropriate table. Significantly upregulated genes included metalloendopeptidase, palmitoyl transferase, Slc17a1, NPYR-14, neuroglian, aquaporin, Rab-protein 8, SMAD6/7, and Slc2a1 (Table 3). Downregulated genes included Propionyl-CoA synthetase, CyclinA-like protein, fibronectin, Mariner Mos1 transposase, L-lactate dehydrogenase, TBC1 domain family member 9B, SPSB1, Slc38a2, C3H-zinc finger-containing protein 1, ybox protein 4-like protein, plastin 1, and innexin (Table 4). Interestingly, some ion channels and pumps were differentially expressed (Tables S1, 3, and 4) (p < 0.05). For example, Transient receptor potential ion channel Ma Dj-TRPMa (Fragment) was upregulated, appearing in the BaCl2-adapted heads but below detection in the wild-type, as well as an uncharacterized voltage-gated potassium channel, Slc17a-1, and NPYR14.
Table 3.
Summary of Transcripts Upregulated in Heads Pre-Conditioned by BaCl2 Exposure
| Protein Name | TCONS | Gene Name | Log Fold Change | p Value | q Value |
|---|---|---|---|---|---|
| Metalloendopeptidase (EC 3.4.24.-) | TCONS_00013146 | NA | – | p < 0.001 | 0.004 |
| Voltage-gated potassium channel | TCONS_00008343 | NA | – | p < 0.001 | 0.020 |
| Palmitoyltransferase (EC 2.3.1.225) | TCONS_00010738 | CLF_106085 | – | p < 0.001 | 0.022 |
| GCR085 | TCONS_00007438 | gcr085 | – | 0.001 | 0.027 |
| Transient receptor potential ion channel Ma DjTRPMa (fragment) | TCONS_00000400 | NA | – | 0.001 | 0.030 |
| Slc17a-1 (Vesicular glutamate transporter) | TCONS_00006778 | vglut slc17a-1 | – | 0.001 | 0.030 |
| NPYR-14 (Neuropeptide Y receptor) | TCONS_00002461 | npyr-14 | – | 0.001 | 0.031 |
| Neuroglian | TCONS_00013039 | EGR_00,245 | – | 0.001 | 0.031 |
| Ubiquitin thioesterase (EC 3.4.19.12) | TCONS_00004623 | NA | – | 0.001 | 0.040 |
| Slc2a-1 (Glut1, glucose transporter) | TCONS_00003196 | slc2a-1 | 3.11838 | 0.001 | 0.030 |
| SMED-SMAD6/7-2 | TCONS_00000433 | NA | 2.31825 | 0.002 | 0.050 |
| Clone ZZD1463 mRNA sequence (Rab-protein 8) | TCONS_00005887 | Rab8 | 2.12996 | p < 0.001 | 0.004 |
| Aquaporin | TCONS_00007316 | aquaporin | 2.01898 | p < 0.001 | 0.004 |
Transcripts with greater than a 2-fold change in expression, significance of q < 0.05, with clear annotation are shown. A log fold change of “–” indicates transcripts that were only identified in BaCl2-treated worms and thus have no calculatable fold change. Bolded transcripts are associated with our excitotoxicity model. TCONS refers to the transcript ID in Table S1.
Table 4.
Summary of Transcripts Downregulated in Heads Pre-Conditioned by BaCl2 Exposure
| Protein Name | TCONS | Gene Name | Log Fold Change | p Value | q Value |
|---|---|---|---|---|---|
| Tubulin alpha chain (fragment) | TCONS_00000293 | TBA3 TR27993 | – | p < 0.001 | 0.004 |
| Marvel containing potential lipid | TCONS_00001520 | EgrG_000088200 | – | p < 0.001 | 0.004 |
| GCR041 | TCONS_00002574 | gcr041 | – | p < 0.001 | 0.004 |
| Propionyl-CoA synthetase | TCONS_00010061 | CLF_100045 | – | p < 0.001 | 0.004 |
| Propionyl-CoA synthetase | TCONS_00010062 | CLF_100045 | – | p < 0.001 | 0.004 |
| cAMP-dependent protein kinase regulatory subunit | TCONS_00014767 | PRKAR2A | – | p < 0.001 | 0.004 |
| CyclinA-like protein (fragment) | TCONS_00014792 | NA | – | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00000293 | NA | – | p < 0.001 | 0.004 |
| Uncharacterized protein C1orf177 homolog | TCONS_00015160 | CA177 TR105569 | – | p < 0.001 | 0.004 |
| Fibronectin type 3 and ankyrin repeat domains protein 1 (FANK1) | TCONS_00015183 | CLF_102920 | – | p < 0.001 | 0.004 |
| Mariner Mos1 transposase | TCONS_00015672 | HmN_000702600 | – | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00015804 | NA | – | p < 0.001 | 0.004 |
| L-lactate dehydrogenase (EC 1.1.1.27) | TCONS_00014875 | NA | – | p < 0.001 | 0.007 |
| TBC1 domain family member 9B | TCONS_00012203 | CLF_111893 | – | 0.001 | 0.031 |
| SPSB | TCONS_00002395 | Spsb | −8.9764 | p < 0.001 | 0.004 |
| SPSB | TCONS_00015862 | Spsb | −6.74196 | p < 0.001 | 0.004 |
| Slc38a-2 (Na+-coupled neutral amino acid transporter) | TCONS_00008866 | slc38a-2 | −5.10772 | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00000243 | MS3_11,213 | −5.00986 | p < 0.001 | 0.004 |
| Tubulin beta chain | TCONS_00015047 | TR83001 | −4.80567 | p < 0.001 | 0.043 |
| Tubulin beta chain | TCONS_00015847 | NA | −4.02875 | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00014885 | NA | −3.9266 | p < 0.001 | 0.004 |
| C3H-zinc finger-containing protein 1 | TCONS_00015349 | NA | −3.87679 | p < 0.001 | 0.004 |
| Phosphotransferase (EC 2.7.1.-) | TCONS_00015828 | slc25a-4 | −3.86765 | p < 0.001 | 0.004 |
| Y box protein 4-like protein | TCONS_00000137 | NA | −3.65495 | p < 0.001 | 0.004 |
| Protein FAM154A | TCONS_00015465 | EmuJ_000818000 | −3.60524 | p < 0.001 | 0.004 |
| Plastin-1 | TCONS_00015023 | CLF_110637 | −3.56204 | p < 0.001 | 0.016 |
| Innexin | TCONS_00005338 | inx | −3.54459 | p < 0.001 | 0.004 |
| Tubulin beta chain | TCONS_00000701 | NA | −3.41177 | p < 0.001 | 0.004 |
| Tubulin beta-2C chain | TCONS_00000158 | NA | −3.37944 | p < 0.001 | 0.004 |
| SJCHGC04177 protein | TCONS_00015105 | NA | −3.37057 | p < 0.001 | 0.004 |
| Tubulin alpha chain (fragment) | TCONS_00015786 | CLF_103359 | −3.25329 | p < 0.001 | 0.004 |
| Tubulin beta chain (fragment) | TCONS_00002700 | bt3 | −3.05291 | p < 0.001 | 0.004 |
| Expressed conserved protein | TCONS_00014926 | EmuJ_001095400 | −2.98249 | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00014888 | NA | −2.83912 | 0.001 | 0.034 |
| T-complex protein 1 subunit gamma (fragment) | TCONS_00015722 | T265_13,906 | −2.74806 | p < 0.001 | 0.013 |
| C3H-zinc finger-containing protein 1 | TCONS_00015007 | NA | −2.74023 | p < 0.001 | 0.007 |
| Plasminogen-1 (fragment) | TCONS_00014024 | NA | −2.66561 | p < 0.001 | 0.004 |
| SJCHGC05018 protein | TCONS_00000671 | NA | −2.65743 | p < 0.001 | 0.004 |
| Tubulin alpha chain (fragment) | TCONS_00000134 | atub2 | −2.34531 | p < 0.001 | 0.004 |
| Tubulin alpha chain | TCONS_00014770 | NA | −2.19894 | p < 0.001 | 0.024 |
| Trypsin-like serine protease | TCONS_00009159 | NA | −1.9831 | p < 0.001 | 0.004 |
Transcripts significantly downregulated by BaCl2 treatment with greater than a 2-fold change in expression, significance of q < 0.05, with clear identification are shown. A log fold change of “–” indicates transcripts that were only identified in untreated worms and thus have no calculatable fold change. Bolded transcripts are specifically predicted by our excitotoxicity model. TCONS refers to the transcript ID in Table S1.
More broadly, downregulated functional sets in BaCl2-resistant worms included transmembrane potential, anion transport, and cell membrane depolarization, whereas processes such as innervation, long-term synaptic depression, and Na+ influx co-transport were increased in mean expression (Figure 2A and Tables 1 and S6). Many processes related to the immune system were upregulated, including leukocyte accumulation, leukocyte recruitment, monocyte recruitment, and T cell response (Table 1). Enriched gene networks were involved in both cellular structure and differentiation, as well as cell membrane depolarization confirming transcriptional rewiring of the bioelectric machinery (Table 1). We conclude that BaCl2-adapted heads exhibit transcriptional changes reflective of several subsystems' adaptation to this novel bioelectrical condition and include proteins that could implement physiological homeostasis in response to BaCl2. Tables 1, S4, and S5 and Figure 2A all depict networks for cell processes that were altered in the BaCl2-adapted heads.
Figure 2.

Possible Mechanism of BaCl2-induced Head Degradation Via Excitotoxicity, and Subsequent Adaptation
(A) Pathway Studio v10.0 was used to perform pathway analysis of RNA-seq data for (a) anion transport and (b) transmission of nerve impulse, two pathways critical to deprogression and regeneration of the planaria head. Tables S6A and S6B contain details of all of the components listed here.
(B) qPCR validation of two transcripts identified as upregulated in the RNA-seq—(a) Dj-TRPMa and (b) Slc2a1. Points represent levels for individual worms normalized to GAPDH. Line indicates median. Unpaired t tests were performed to assess significance, **p < .01, *p < .05, n = 3 for each condition.
(C) Proposed regulatory networks detailing (a) normal/untreated state, (b) the main excitotoxicity-related feedback induced by BaCl2, and (c) proposed adaptations to BaCl2 treatment. Red lines with flat endpoints show an inhibitory/downregulation relationship, whereas blue lines with circular endpoints show an activating/upregulatory relationship. Under normal conditions, K+ channels hyperpolarize the cell to regulate membrane excitability and therefore maintain an open state of CaV channels and upregulate glutamate signaling (a). Ba2+ is proposed to induce excitotoxicity in neurons by blocking K+ channels, leading to significant Vmem depolarization, which activates voltage-gated Ca2+ channels (CaV) leading to sustained increases in cytosolic Ca2+, excess Ca2+-induced exocytosis of glutamate, and sustained activation of glutamate signaling, leading to further calcium entry to the cell (b, “excitotoxic positive feedback 1”). Ba2+-induced Vmem depolarization may also open innexin hemichannels, leading to further glutamate release (b). Extracellular glutamate may activate glutamate-gated chloride channels and increased cytosolic Ca2+ may activate Ca2+-gated chloride channels, leading to chloride secretion from cells and further Vmem depolarization (b, “excitotoxic positive feedback 2”). Sustained increases in cytosolic Ca2+ may initiate apoptosis via caspases, leading to neural degradation (b). Adaptation to Ba2+-induced excitotoxicity is proposed to occur via a variety of changes to gene expression, including upregulation of TRPMa and NPY receptors, which break positive feedback loops supporting excitotoxicity (c).
To ensure that the RNA-seq identified transcripts that were up- or downregulated in the BaCl2-adapted heads, we performed qPCR. qPCR for Dj-TRPMa using primers from (Inoue et al., 2014) showed a 2.4-fold increase in transcripts from BaCl2-adapted heads over WT (Figure 2Ba). qPCR for Slc2a1 showed a 1.3-fold increase in transcript abundance in BaCl2-adapted heads compared with WT (Figure 2Bb). Although a bit lower in magnitude than reported in the RNA-seq, both showed similar upregulation in BaCl2-adapted heads compared with WT. Thus, despite using a broad platyhelminthes transcriptome, we were able to identify transcripts that were indeed upregulated in BaCl2-adapted heads.
An Excitotoxicity-Based Model of BaCl2-Induced Neural Apoptosis and Adaptation
We next sought to understand the mechanism of BaCl2's effect and the striking ability of planaria to overcome it with a newly regenerated structure. One plausible explanation for the head degeneration effect is induction of BaCl2-induced drop in resting membrane potential and subsequent neural excitotoxicity in the neuron-rich head of the planarian (Walter et al., 2001, Wright, 2004). Our model is based on the premise that an excitotoxic cascade is initiated by neuronal depolarization induced by the broad potassium channel blocker BaCl2. In other systems, strong depolarization activates Vmem-gated Ca2+ channels, which causes neurotransmitter exocytosis (Mcmahon and Nicholls, 1993, Sihra et al., 1993). As glutamate signals extracellularly via Vmem-depolarizing ionotropic receptors, this further increases cytosolic Ca2+, leading to a positive feedback cycle (see “excitotoxic positive feedback” of Figure 2Cb). We also postulate that disruption of ion balance via BaCl2 should lead to the upregulation of channels and transporters that can help the tissues compensate by breaking positive feedback cycles.
The dynamics of our model under initial BaCl2 exposure and adapted state are shown in Figure 2C. The model makes several specific predictions, shown in Table 2. First, one of the immediate physiological effects of the BaCl2 should be a significant depolarization. Second, manipulation of several components (blockade of calcium channels and chloride channels, activation of dopamine signaling) should prevent the excitotoxicity storm and thus prevent head degeneration due to BaCl2 exposure. Third, blockade of TRPMa channels should counteract the adapted state and lead to head degradation even after adaptation to BaCl2. We thus tested each of these specific predictions in the BaCl2 degeneration and adaptation assay.
Table 2.
Summary of Targets Predicted from BaCl2-Induced Excitotoxicity Hypothesis and the Results of Experimental Interventions
| Intervention | Reasoning | Experiment |
|---|---|---|
| Dopamine signaling agonists (e.g., quinpirole, bromocriptine, cabergoline) | Agonists to dopamine signaling have shown mitigated excitotoxicity (Vaarmann et al., 2013, Cepeda et al., 1998, Odaka et al., 2014) | Dopamine agonist and serotonin 5-HT7-like receptor antagonist bromocriptine significantly delayed head degeneration in the majority of worms for the duration of the test (35 days) compared with control, which showed full degeneration within 24 h (Figures 4Ae and 4Af) |
| Cl- channel inhibitors | Previous studies have shown decreased neural death during excitotoxic cascades in the presence of chloride channel antagonists (Inoue et al., 2007, Chen et al., 1998, Rungta et al., 2015, Liang et al., 2007) | Chloride channel blockers NPPB and NFA significantly delayed head degeneration upon exposure to BaCl2. 12% of NPPB- and 17% of NFA-treated worms showed some degeneration by day 3 compared with 100% of controls (Figures 4Ag–4Aj) |
| Calcium channel antagonists | Excitotoxicity is mediated by high intracellular Ca2+ levels, a component of which arises from neuronal voltage-gated Ca2+ channels (Lai et al., 2014, Szydlowska and Tymianski, 2010). And calcium channel antagonists can prevent neuronal death (Prentice et al., 2015) | L-type calcium channel blocker Nicardipine delayed degeneration in 69% of worms (31% showed some degeneration) through 3 days of treatment compared with 100% degeneration in controls (Figures 4Ak and 4Al) |
| TRPM antagonists | TRP channels show complex involvement in excitotoxicity (Szydlowska and Tymianski, 2010, Zheng and Phelan, 2014, Aarts and Tymianski, 2005, Bengtson et al., 2004, Li et al., 2012). We found that a TRPM channel is significantly upregulated in Ba2+-adapted regenerated heads (Table 3A), suggesting TRPM inhibition may sabotage the mechanism by which planaria adapt to BaCl2 | TRPM antagonist AMTB leads to rapid (less than 24 h) head degeneration in BaCl2-adapted worms, whereas adapted worms maintained in BaCl2 did not degenerate (Figure 4B) |
Exposure to BaCl2 Results in Depolarization of Anterior Tissues
Although the molecular and cell-level activity of BaCl2 has been thoroughly characterized in in vitro preparations (Armstrong and Taylor, 1980, Hanrahan et al., 1986, Kurachi, 1986, Quayle et al., 1988), the effects of BaCl2 exposure on physiological function and/or regeneration in vivo are largely unknown. Our excitotoxicity model predicts that BaCl2 should initiate a significant depolarization of the head. To characterize the bioelectric state of the worm after exposure to the potassium channel blocker, we imaged D. japonica worms after 30 min in water (Figure 3Aa) and after 30 min in BaCl2 (Figure 3Ab) using the voltage reporter dye DiBAC4(3), which grows brighter with increasing depolarization. We have previously demonstrated that DiBAC4(3) is a reliable indicator of depolarization in D. japonica (Oviedo et al., 2008, Beane et al., 2011, Beane et al., 2013, Durant et al., 2017, Durant et al., 2019). Figure S1 shows that depolarization with 100 nM of the ionophore valinomycin in combination with 15 mM K+-gluconate can be visualized in D. japonica fragments within an hour of treatment. To compare relative depolarization while minimizing confounding factors, Control and BaCl2-treated worms were mounted and imaged together on the same PIC. After just 30 min in the BaCl2 solution, worms displayed a significant depolarization of anterior tissues (Figure 3Ab, quantified in 3B), as expected from a K+ channel blocker and predicted by our model. This depolarization was most appreciable in the most anterior 1/6 of the animal. Given the extensive depolarization after just 30 min in BaCl2, we conclude that the robust depolarizing effect of the drug is consistent with our model's prediction.
Figure 3.

Visualization of Relative Membrane Potential in WT and BaCl2-treated D. japonica Flatworms
(A) Voltage-sensitive dye was used to determine pattern of resting potentials in planaria. White arrowheads indicate the anterior of the worm. Images are pseudocolored to allow for ease of visualization of depolarization patterns, but worms were imaged in the same frame so as not to confound data after pseudocoloring, and all image analysis was done using raw un-colored images. (a) Untreated D. japonica flatworm imaged with DiBAC4(3) dye. (b) D. japonica flatworm imaged with DiBAC4(3) dye after 30 min in BaCl2. Scale bars, 0.5 mm.
(B) Quantification of average pixel intensities in untreated and BaCl2-treated worms. Bars represent mean ± SD. Welch's unpaired t test, ***p = 0.00002.
See also Figure S1.
Calcium and Chloride Blockers Prevent BaCl2-Induced Degeneration
The central component of our model is an excitatory storm and the attendant positive feedback loop involving ion channels and neurotransmitters (Figure 2C). Thus, it predicts that interrupting this loop (inducing neuroprotection) by targeting specific dopamine machinery (Vaarmann et al., 2013, Cepeda et al., 1998, Odaka et al., 2014), chloride channels (Hasbani et al., 1998, Rungta et al., 2015, Takeuchi et al., 2011, Chen et al., 1999), or L-type calcium channels (Szydlowska and Tymianski, 2010, Mark et al., 2001, Hasbani et al., 1998), should prevent degeneration.
To test these predictions, we exposed worms to a combination of BaCl2 and one of several blockers (previously characterized in planaria or other invertebrates), tracking the incidence of degeneration over 3 days and comparing with BaCl2-only controls. All drugs had no effect in the absence of BaCl2 (Figure S2). Since dopamine signaling is both a target of our model and one of the pathways identified by the RNA-seq as altered in the BaCl2-adapted worms (Table S5), we used bromocriptine mesylate (0.5 μM) to activate monoaminergic signaling. Bromocriptine has been shown in mammals to be a dopamine agonist (Liberante et al., 2016, Oda et al., 2008, Parmar et al., 1984, Schneider et al., 1984, Via et al., 2010) but in D. japonica appears to have broader functions, agonizing dopaminergic signaling (Chan et al., 2014) and antagonizing serotonergic signaling (Chan et al., 2016). Previous work in D. japonica has shown that dopamine and serotonin oppose one another in head regeneration (Chan et al., 2014), indicating that bromocriptine targets general monoaminergic pathways. We hypothesized that bromocriptine would modulate monoaminergic pathways, suppressing excitotoxic activity. We found that bromocriptine was effective at blocking degeneration with 74% of worms experiencing no degeneration until day 2 (Figures 4Ae and 4Af) and 50% of worms experiencing no degeneration up until day 22. Although this effect is noticeable, 38% of the worms did show head degeneration by day 22 and 9% of the worms died during this treatment. Most likely, this failure to protect the worms from degeneration is related to variability in individual worm response to the drug. NPPB (5 μM) and niflumic acid (1.25 μM) were used as calcium-activated chloride channel blockers. Both of these drugs have been shown to block Cl− channels both in mammalian cells and in C. elegans (Bush et al., 2009, Schriever et al., 1999, White and Aylwin, 1990, Wu and Hamill, 1992). NPPB delayed degeneration in most worms (82%) until day 6, at which point all worms died, suggesting only a partial rescue of the excitotoxic effect (Figures 4Ag and 4Ah). Niflumic acid, on the other hand, was able to prevent head degeneration or death up until day 2 in 92% of the treated planaria but did occasionally induce ectopic eyes (1.9%) (Figures 4Ai and 4Aj). Finally, nicardipine hydrochloride (2.5 μM) was used to block L-type calcium channels (Beane et al., 2011, Hockerman et al., 1997, Mendonca-Silva et al., 2006, Nogi et al., 2009). Nicardipine induced a delay in degeneration in 78% of worms through 2 days of treatment (Figures 4Ak and 4Al). Taken as a whole, exposure to these channel activity modifiers similarly prevented BaCl2-mediated head degeneration (Figure 4A, quantified in 4Am) and mostly also prevented BaCl2-induced death in the planaria, suggesting that these drugs directly acted on the mechanism by which BaCl2 is toxic. We conclude that, as predicted by our model, drugs predicted to break the positive feedback loop of excitotoxicity efficiently suppress the BaCl2-induced head deprogression.
Figure 4.

Targeting Ion Channels Allows Modulation of Degeneration and Adaptation
(A) A variety of drugs targeting ion channels were used to test our excitotoxicity hypothesis. (a and b) D. japonica worm before treatment (a) and after 2 days in water (b). (c and d) Planaria treated with 1 mM BaCl2 for 0 h (c) show no phenotype, but after 48 h, the head deprogresses (d). (e and f) Planaria in dopamine agonist bromocriptine (0.5 μM) and 1 mM BaCl2 solution for 0 (e) and 2 days (f). Bromocriptine is able to prevent head degeneration upon exposure to BaCl2 in 74% of worms (f). (g) exposure to BaCl2 and calcium-activated chloride channel blocker NPPB (5 μM) has no effect at the time of treatment, but within 2 days (h) NPPB has prevented head degeneration in 84% of worms. (i and j) Calcium-activated chloride channel blocker Niflumic acid (1.24 μM) exposure in combination with BaCl2 has no effect at 0 days (i) but prevents head degeneration in 92% of worms within 2 days (j). (k and l) L-type calcium channel blocker nicardipine hydrochloride (2.5 μM) treatment in combination with BaCl2 has no effect at the time of treatment (k) but prevents head degeneration in 78% of worms within 2 days (l). Scale bars, 0.5 mm. (m) Prevalence of head degeneration phenotype with each of the drug treatments listed in (e–l). The overwhelming majority of worms with head degeneration in the BaCl2-treated worms are replaced with the majority of worms not experiencing head degeneration when treated with ion channel modulators.
(B) Resensitization of worms to BaCl2. BaCl2-adapted D. japonica worms in (a) water, (b) water with 100 μM AMTB hydrochloride, or (c) water with BaCl2 do not induce head degeneration in BaCl2-adapted worms. (d) However, treatment with AMTB (100 μM) in addition to further treatment with BaCl2 induced head degeneration within 1.5 h. Scale bars, 0.5 mm. (e) Prevalence of head degeneration phenotypes in BaCl2-adapted, BaCl2-treated planaria with or without AMTB treatment, as shown in (c) and (d). Treatment with AMTB resulted in a near-complete change from normal heads to fully degenerated heads.
See also Figure S2.
Blocking Adaptation
Conversely, we next asked whether our model also suggested efficacious methods of counteracting the BaCl2-adapted state. A transcript identified at significant levels only in the BaCl2-adapted heads (Table 3) was TRPMa–a member of a family of channels that sense chemical, mechanical, and osmotic signals (opening with cell swelling) and convey signals to the genome via increased intracellular Ca2+. Thus, we next blocked TRPM via the well-characterized TRPM blocker AMTB (Lashinger et al., 2008, Yapa et al., 2018), which has previously been shown (and confirmed by RNAi) to be effective in inhibiting TRPMa in planaria (Inoue et al., 2014). Although exposure to 100 μM AMTB alone did not induce any head degeneration or death (Figure 4Bb), BaCl2-adapted planaria began degenerating their heads after just an hour and a half of exposure to AMTB (24 h time point shown in Figure 4Bd). By 24 h of exposure, 98% of heads had fully degenerated, whereas worms that were maintained in BaCl2 showed no degeneration (Figure 4Bc). This rate of degeneration is equivalent to the controls that had never experienced BaCl2 adaptation (Figures 1Aa, 4Aa, and 4Ab). Furthermore, in the presence of both BaCl2 and AMTB, 62% of the planaria died within 48 h, suggesting that the mechanism by which they adapted to the BaCl2 could not overcome both BaCl2 and AMTB. Thus, we conclude that, consistent with the known mechanisms of excitotoxicity, BaCl2 adaptation can be erased by TRPM blockade.
Discussion
Planarian Regeneration Can Compensate for Dramatic Physiological Perturbations
Habituation to extreme physiological stressors has been known since the classic experiments of Jollos, who showed that lineages of paramecia exposed to toxins or extreme heat gained resistance that persisted for hundreds of generations (Jollos, 1933, Jollos, 1934). However, very little information is available on the mechanism of such plasticity taking place in single organisms (i.e., not due to multi-generational selection) (Elgart et al., 2015). Here we established a model for studying the interplay of physiological and transcriptional plasticity on a short timescale. The planarian head degeneration model also has the advantage that it reveals a multi-scale phenomenon: the cellular stress must be coupled to organism-wide patterning networks, as the animal has to rebuild a complex new structure that will maintain its anatomical integrity while the cells within are forced to significantly alter their normal physiological function.
As a model system, we chose planaria because of their remarkable ability to regulate anatomy despite drastic injury (Saló et al., 2009, Owlarn and Bartscherer, 2016), targeting the function of K+ channels, as these are required for a wide range of processes at the level of cell behavior (Urrego et al., 2014, Pardo and Stuhmer, 2014) and body-wide patterning (Adams et al., 2016, Dahal et al., 2012, Simons et al., 2015, Masotti et al., 2015). Bioelectric processes underlie behavioral plasticity in the brain, suggesting the hypothesis that ionic mechanisms may be of considerable interest in understanding different kinds of adaptation (Pezzulo and Levin, 2015). Interestingly, recent work in Drosophila mutants revealed the importance of regulation of excitability to buffer against environmental changes (Kim et al., 2017).
Upon exposure to BaCl2, a potent non-specific K+ channel blocker, planaria experience a striking degradation of the entire head. Remarkably, they are then able to produce a new head that is insensitive to the BaCl2 (Figure 1A). We characterized this example of large-scale adaptation to a drastic physiological challenge by regenerative processes.
Extended Time in Water Results in a Loss of BaCl2 Resistance
The acquired resistance to BaCl2 is not permanent. When BaCl2-insensitive worms are placed in water for 30 days, they lose their resistance to BaCl2, and upon a second exposure to BaCl2 will undergo degeneration and regeneration of anterior tissues again (Figure 1B). Interestingly, 30 days is approximately the time required for cellular turnover in planaria (Pellettieri and Sánchez Alvarado, 2007); it is not known yet whether the BaCl2 adaptation and de-adaptation is in some way tied to the temporal profile of neoblast activity and somatic cell turnover. Taken together, this temporal profile of adjustment to novel stimuli, and its return to normal, highlights the ability of living systems to adaptively, flexibly integrate information from the internal and external environments of the organism via the interplay between physiological information and genetic programs. The fact that BaCl2 adaptation spontaneously reverses suggests that the adapted state is not a stable attractor in the transcriptional landscape (Huang et al., 2005, Sullivan et al., 2016); instead, it is compatible with a dynamic monitoring system that adjusts to novel stressors but can also detect their cessation. Future development of transgenic strains of planaria expressing novel physiological fluorescent reporters will be invaluable in tracing the exact temporal profiles of signaling cascades during this process in vivo.
BaCl2 Adaptation Involves a Unique Transcriptional Signature
To investigate the mechanism of head degeneration and subsequent BaCl2 tolerance, we analyzed transcriptomes comparing WT heads with BaCl2-adapted heads. Importantly, we analyzed completely regenerated heads, so that the profiles would reflect not mechanisms involved in head regeneration per se, but differences in mature normal and BaCl2-insensitive heads. RNA-seq analysis revealed a number of transcriptional differences that distinguish WT (BaCl2-sensitive) heads from BaCl2-adapted heads (Tables 3 and S1). Unfortunately, no gain-of-function technology exists in planaria that can be used to misexpress transcripts; however, we exploited the druggable nature of some of the key targets to individually modulate both the degeneration and adaptation phases (Figure 4). It must be noted that important changes can occur at the level of physiology, not transcription, and it is likely that additional mechanisms of plasticity that are invisible to RNA-seq remain to be characterized in future work.
Transcriptional Rewiring of Bioelectric Networks
One of the most salient aspects of the RNA-seq dataset was the identification of changes in genes involved in regulating bioelectric state (Table 1). Overall, 1.98% of the transcripts identified by RNA-seq in planaria were affected by BaCl2 exposure (q < 0.05, >2-fold change). A number of channels and pumps are altered in the transition from BaCl2 sensitivity to adaptation, including calcium-sensitive transporters, solute transporters, voltage-gated channels, and others (Tables S1, 1, and 3). Interestingly, many of these channels are transporters, suggesting that the physiological buffering of a depolarizing treatment is dependent on employing alternative means of transporting ions into and out of anterior cells and tissues. Newly developed platforms for bioelectric simulation will enable construction and analysis of quantitative physiological models linking post-translational ion flow dynamics (channel opening/closing) to transcriptional regulation and may explain how the observed changes in the electrogenic mRNA profile can compensate for BaCl2 exposure (Pietak and Levin, 2016, Cervera et al., 2015, Cervera et al., 2016a, Cervera et al., 2016b). Future functional analysis will also investigate physiological repercussions of detected transcriptional changes.
An Excitotoxicity Model of Degeneration and Adaptation
To understand this striking phenomenon, we formulated and tested a model based on excitotoxicity (Mark et al., 2001, Szydlowska and Tymianski, 2010). We propose that depolarized Vmem resulting from Ba2+ block of K+ channels (Walter et al., 2001), as well as the Ba2+ ion's ability to substitute for Ca2+ in a variety of processes (Condrescu et al., 1997, Dingledine et al., 1992, Zhou et al., 2012, Ni et al., 2014), induces glutamate-mediated excitotoxicity (Figure 2C). It was hypothesized that Ba2+ induces a strong depolarization of Vmem by blocking K+ channels (Wright, 2004, Walter et al., 2001), which in turn activates voltage-gated Ca2+ channels, leading to increases in both intracellular Ca2+ and potentially Ba2+ (Mark et al., 2001, Szydlowska and Tymianski, 2010). Increased Ca2+ and Ba2+ then induce glutamate exocytosis, which may signal at ionotropic glutamate receptors to further depolarize Vmem and increase cytosolic Ca2+ and Ba2+ in positive feedback (Mcmahon and Nicholls, 1993, Sihra et al., 1993, Mark et al., 2001, Lau and Tymianski, 2010), leading to an excitotoxic cascade and neural death via Ca2+ activation of apoptosis (Zhang and Bhavnani, 2005, Mcmahon and Nicholls, 1993). One of the key predictions of our model is that a strong anterior depolarization should be present early in the process, which was indeed observed (Figure 3).
Remarkably, the organism is able to alter its transcriptional profile to regenerate new heads that are resistant to BaCl2. Neuroprotective changes in BaCl2-adapted heads include increased expression of neuropeptide Y receptors and dramatically decreased expression of innexins (invertebrate gap junctions), both of which have been previously shown to inhibit glutamate-induced excitotoxicity. Increased NPY signaling has been found to modulate excitotoxicity (Greber et al., 1994, Grundemar et al., 1991a, Grundemar et al., 1991b, Whittaker et al., 1999, Silva et al., 2005) and may act through inhibition of Ca2+ channels (Grundemar et al., 1991a, Mccullough et al., 1998). Past work in numerous systems has shown a strong neuroprotective effect of gap junction blockade in excitotoxic cascades, where gap junction inhibition has shown significant reduction in neural death (Wang et al., 2010, Wang et al., 2012, Galinsky et al., 2017, Takeuchi et al., 2011, Thompson, 2015, Belousov et al., 2017). Other changes include increased expression of a TRPM channel, increased aquaporin channels, and increased levels of a voltage-gated K+ channel, which appear to convey neuroprotective adaptations to the regenerated heads. Decreases in glutamine transporter Slc38a2 (which increases neural glutamate via the glutamine-glutamate cycle [Bak et al., 2006]) were also found. These findings reveal the transcription-level acquisition of tolerance to a significant toxic challenge by planarian regeneration. Additional adaptive changes, at the protein or physiological levels, may also have occurred and will be studied in subsequent work.
Ion Channels' Roles in Degeneration and Adaptation
Our model implicated specific classes of targets in the degeneration and adaptation process. Indeed, we found that blockade of calcium and chloride channels were able to counteract the toxic effects of BaCl2 exposure (Figures 4Ag–4Al). Likewise, monoaminergic activation was also able to prevent degeneration (Figures 4Ae and 4Af). These results demonstrate that an adaptive stress response (BaCl2 insensitivity) can be artificially induced without prior exposure to the actual stressor. Importantly, our model also enabled identification of a simple intervention to reverse the adapted state—TRPMa channel blockers rapidly induced BaCl2-sensitivity to BaCl2-adapted heads (Figure 4B), revealing the induction of TRPMa after BaCl2 exposure as a key functional step in the observed regenerative plasticity. Although any pharmacological reagent may have additional targets, the use of several diverse channel drugs to cleanly abrogate a very specific process, and improve tissue health and integrity (i.e., not simply cause toxicity), supports the predictive value of our model. Any future models of this process can likewise be evaluated by their ability to identify reagents that enable modulation of the plasticity process. The use of small molecule drugs to target these kinds of processes in vivo is an important complement to genetic manipulation studies, as it may allow therapeutic applications that do not require gene therapy. Targeting both phases of the adaptive response (the initial toxicity and the resulting adapted state), as shown in our data, represents an important strategy in applications.
Conclusion
Planaria are champions of plasticity and robustness, not only repairing their anatomy after amputation in normal conditions, but also apparently able to adjust their physiology to survive and regenerate under significant physiological perturbation. This occurs within an individual's lifetime, not via population selection, revealing the ability of tissues to activate appropriate responses to relieve physiological stressors. Using this and similar tractable models will allow a better understanding of the dynamics of biological circuits, not only hardwired activity emergent from genetically encoded features, but also flexible adaptation to unpredictable stimuli. The study of the mechanisms and algorithms that enable this property blurs the line between developmental genetics, adult physiology, and the kind of plasticity studied in neuroscience. Interestingly, our NGS analysis implicated targets belonging to the Memory/Neuroplasticity categories (Table 1), suggesting the hypothesis, which we will test in future work, that regenerative and physiological plasticity are related to (and perhaps early forms of) cognitive plasticity in higher animals (Baluška and Levin, 2016).
We chose BaCl2 for two reasons. First, because as a very broad K+ channel blocker, it would challenge tissues with a stress that could not easily be overcome through simple redundancy among potassium channel family members. Second, because to our knowledge, exposure to significant quantities of BaCl2 is not something planaria encounter in the wild. Thus, it is plausible that plasticity to BaCl2 toxicity is not a genetic response that has been specifically selected for; rather than a hardwired response to a common feature of the planarian environment over evolutionary time, it is likely that the remarkable adaptation we observe is an example of a more general and still poorly understood feature of biology: robustness in the face of novel stresses. The algorithms and mechanisms by which living systems match transcriptional and physiological responses to environmental challenges represent a key intellectual challenge for the coming decade, with significant implications both for biomedicine and for the design of highly resilient systems in engineering.
Understanding cellular adaptation to stress may have implications in addition to shedding light on evolvability and regulative morphogenesis. Although other species may not exhibit the convenient head degeneration phenotype, ionic disbalance and stress response is a universal biological and biomedical phenomenon. Moreover, preconditioning (exposure to preinjury stressors to achieve induction of tolerance) (Yokobori et al., 2013) is an important topic in the biomedicine of brain injury and epilepsy (Blondeau et al., 2000, Fern et al., 2014), where ion channels are already beginning to be viewed as therapeutic targets for neuroprotection (Skaper, 2011). Future work on the modulation of regeneration via developmental pharmacology will exploit novel biomaterials, computational models, and ion channel drugs to transition these advances toward biomedical applications (Herrera-Rincon et al., 2018, Churchill et al., 2018, Pai et al., 2018).
Limitations of the Study
One of the limitations of the study is that assays are not yet available in planaria to directly observe the physiological effects of all of the reagents used. Most of the pharmacological tools we applied have been used in planaria, and all have been utilized in other invertebrates, but NPPB and Niflumic acid have not heretofore been characterized in planaria. Likewise, there is currently no working technology for introducing foreign genes into planaria, preventing the use of molecular-genetic (dominant negative and fluorescent reporter) tools to directly observe the dynamics of the dopamine pathway under our various conditions. Furthermore, it is possible that some of the genes listed in Tables 3 and 4 as expressed in one condition but not detected in the other may reflect technical or statistical failures to detect transcript. Finally, it will be important in future work to conduct a large-scale RNAi effort targeting all of the genes identified by our RNA-seq to determine whether additional mechanisms of tissue adaptation to stress are also critical beyond those implicated in the specific model we propose.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the members of the Levin lab and Nicolas Roleau for useful discussions and Hans Gonzembach and the diligent undergraduates including Hannah Stowe, Quynh Anh Phan, Si Kun Wang, Sara E. Mitchell, Tien Hoang, John Fernandez, and Carolyn H. Nguyen who have helped us tend to our worm colony. We also thank members of the Agata lab for providing us with the Dj-TRPMa primer sequences. We gratefully acknowledge support by an Allen Discovery Center award from the Paul G. Allen Frontiers Group (No. 12171), the Templeton World Charity Foundation (No. TWCF0089/AB55), and the Barton Family Foundation. Research was sponsored by the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement Number HR0011-18-2-0022. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. Approved for public release; distribution is unlimited.
Author Contributions
M.L. and M.E.-B. conceived and planned the study. M.E.-B., F.D., A.T., D.D., and J.M. performed planarian experiments and analyzed data. K.M., A.T., and D.D. performed molecular biology (QPCR validation). A.P. built and analyzed the excitotoxicity model. C.J.M. and A.K. analyzed RNA-seq data. M.E.-B, A.K., A.P., and M.L. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: December 20, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.11.014.
Supplemental Information
Provided are the genes that are involved in the corresponding network, median fold change of the network, and the p value.
References
- Aarts M.M., Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- Adams D.S., Uzel S.G., Akagi J., Wlodkowic D., Andreeva V., Yelick P.C., Devitt-Lee A., Pare J.F., Levin M. Bioelectric signalling via potassium channels: a mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J. Physiol. 2016;594:3245–3270. doi: 10.1113/JP271930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M., Taylor S.R. Interaction of barium ions with potassium channels in squid giant axons. Biophys. J. 1980;30:473–488. doi: 10.1016/S0006-3495(80)85108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L.K., Schousboe A., Waagepetersen H.S. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- Baluška F., Levin M. On having No head: cognition throughout biological systems. Front. Psychol. 2016;7:902. doi: 10.3389/fpsyg.2016.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beane W.S., Morokuma J., Adams D.S., Levin M. A Chemical genetics approach reveals H,K-ATPase-mediated membrane voltage is required for planarian head regeneration. Chem. Biol. 2011;18:77–89. doi: 10.1016/j.chembiol.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beane W.S., Morokuma J., Lemire J.M., Levin M. Bioelectric signaling regulates head and organ size during planarian regeneration. Development. 2013;140:313–322. doi: 10.1242/dev.086900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belousov A.B., Fontes J.D., Freitas-Andrade M., Naus C.C. Gap junctions and hemichannels: communicating cell death in neurodevelopment and disease. BMC Cell Biol. 2017;18:4. doi: 10.1186/s12860-016-0120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson C.P., Tozzi A., Bernardi G., Mercuri N.B. Transient receptor potential-like channels mediate metabotropic glutamate receptor EPSCs in rat dopamine neurones. J. Physiol. 2004;555:323–330. doi: 10.1113/jphysiol.2003.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau N., Plamondon H., Richelme C., Heurteaux C., Lazdunski M. K(ATP) channel openers, adenosine agonists and epileptic preconditioning are stress signals inducing hippocampal neuroprotection. Neuroscience. 2000;100:465–474. doi: 10.1016/s0306-4522(00)00304-3. [DOI] [PubMed] [Google Scholar]
- Brunke S., Hube B. Adaptive prediction as a strategy in microbial infections. PLoS Pathog. 2014;10:e1004356. doi: 10.1371/journal.ppat.1004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush E., Foreman R., Walker R.J., Holden-Dye L. The actions of chloride channel blockers, barbiturates and a benzodiazepine on Caenorhabditis elegans glutamate- and ivermectin-gated chloride channel subunits expressed in Xenopus oocytes. Invert. Neurosci. 2009;9:175–184. doi: 10.1007/s10158-010-0096-8. [DOI] [PubMed] [Google Scholar]
- Cebrià F., Adell T., Saló E. Rebuilding a planarian: from early signaling to final shape. Int. J. Dev. Biol. 2018;62:537–550. doi: 10.1387/ijdb.180042es. [DOI] [PubMed] [Google Scholar]
- Cepeda C., Colwell C.S., Itri J.N., Gruen E., Levine M.S. Dopaminergic modulation of early signs of excitotoxicity in visualized rat neostriatal neurons. Eur. J. Neurosci. 1998;10:3491–3497. doi: 10.1046/j.1460-9568.1998.00357.x. [DOI] [PubMed] [Google Scholar]
- Cervera J., Manzanares J.A., Mafe S. Electrical coupling in ensembles of nonexcitable cells: modeling the spatial map of single cell potentials. J. Phys. Chem. B. 2015;119:2968–2978. doi: 10.1021/jp512900x. [DOI] [PubMed] [Google Scholar]
- Cervera J., Alcaraz A., Mafe S. Bioelectrical signals and ion channels in the modeling of multicellular patterns and cancer biophysics. Sci. Rep. 2016;6:20403. doi: 10.1038/srep20403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera J., Meseguer S., Mafe S. The interplay between genetic and bioelectrical signaling permits a spatial regionalisation of membrane potentials in model multicellular ensembles. Sci. Rep. 2016;6:35201. doi: 10.1038/srep35201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.D., Agbedanu P.N., Zamanian M., Gruba S.M., Haynes C.L., Day T.A., Marchant J.S. 'Death and axes': unexpected Ca(2+) entry phenologs predict new anti-schistosomal agents. PLoS Pathog. 2014;10:e1003942. doi: 10.1371/journal.ppat.1003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.D., Grab T., Marchant J.S. Kinetic profiling an abundantly expressed planarian serotonergic GPCR identifies bromocriptine as a perdurant antagonist. Int. J. Parasitol. Drugs Drug Resist. 2016;6:356–363. doi: 10.1016/j.ijpddr.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Olney J.W., Lukasiewicz P.D., Almli T., Romano C. Ca2+-independent excitotoxic neurodegeneration in isolated retina, an intact neural net: a role for Cl- and inhibitory transmitters. Mol. Pharmacol. 1998;53:564–572. doi: 10.1124/mol.53.3.564. [DOI] [PubMed] [Google Scholar]
- Chen Q., Moulder K., Tenkova T., Hardy K., Olney J.W., Romano C. Excitotoxic cell death dependent on inhibitory receptor activation. Exp. Neurol. 1999;160:215–225. doi: 10.1006/exnr.1999.7179. [DOI] [PubMed] [Google Scholar]
- Churchill C.D.M., Winter P., Tuszynski J.A., Levin M. EDEn – electroceutical design environment: an ion channel database with small molecule modulators and tissue expression information. iScience. 2018;11:42–56. doi: 10.1016/j.isci.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condrescu M., Chernaya G., Kalaria V., Reeves J.P. Barium influx mediated by the cardiac sodium-calcium exchanger in transfected Chinese hamster ovary cells. J. Gen. Physiol. 1997;109:41–51. doi: 10.1085/jgp.109.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke J. Scale of body pattern adjusts to available cell number in amphibian embryos. Nature. 1981;290:775–778. doi: 10.1038/290775a0. [DOI] [PubMed] [Google Scholar]
- Corning W.C., Freed S. Planarian behaviour and biochemistry. Nature. 1968;219:1227–1229. doi: 10.1038/2191227a0. [DOI] [PubMed] [Google Scholar]
- Dahal G.R., Rawson J., Gassaway B., Kwok B., Tong Y., Ptacek L.J., Bates E. An inwardly rectifying K+ channel is required for patterning. Development. 2012;139:3653–3664. doi: 10.1242/dev.078592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R., Hume R.I., Heinemann S.F. Structural determinants of barium permeation and rectification in non-NMDA glutamate receptor channels. J. Neurosci. 1992;12:4080–4087. doi: 10.1523/JNEUROSCI.12-10-04080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant F., Bischof J., Fields C., Morokuma J., Lapalme J., Hoi A., Levin M. The role of early bioelectric signals in the regeneration of planarian anterior/posterior polarity. Biophys. J. 2019;116:948–961. doi: 10.1016/j.bpj.2019.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant F., Morokuma J., Fields C., Williams K., Adams D.S., Levin M. Long-term, stochastic editing of regenerative anatomy via targeting endogenous bioelectric gradients. Biophys. J. 2017;112:2231–2243. doi: 10.1016/j.bpj.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton D.C., Brodwick M.S. Effects of barium on the potassium conductance of squid axon. J. Gen. Physiol. 1980;75:727–750. doi: 10.1085/jgp.75.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgart M., Snir O., Soen Y. Stress-mediated tuning of developmental robustness and plasticity in flies. Biochim. Biophys. Acta. 2015;1849:462–466. doi: 10.1016/j.bbagrm.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Emmons-Bell M., Durant F., Hammelman J., Bessonov N., Volpert V., Morokuma J., Pinet K., Adams D.S., Pietak A., Lobo D., Levin M. Gap junctional blockade stochastically induces different species-specific head anatomies in genetically wild-type Girardia dorotocephala flatworms. Int. J. Mol. Sci. 2015;16:27865–27896. doi: 10.3390/ijms161126065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R.F., Matute C., Stys P.K. White matter injury: ischemic and nonischemic. Glia. 2014;62:1780–1789. doi: 10.1002/glia.22722. [DOI] [PubMed] [Google Scholar]
- Freddolino P.L., Tavazoie S. Beyond homeostasis: a predictive-dynamic framework for understanding cellular behavior. Annu. Rev. Cell Dev. Biol. 2012;28:363–384. doi: 10.1146/annurev-cellbio-092910-154129. [DOI] [PubMed] [Google Scholar]
- Galinsky R., Davidson J.O., Lear C.A., Bennet L., Green C.R., Gunn A.J. Connexin hemichannel blockade improves survival of striatal GABA-ergic neurons after global cerebral ischaemia in term-equivalent fetal sheep. Sci. Rep. 2017;7:6304. doi: 10.1038/s41598-017-06683-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A., Bell G. Evolutionary rescue and adaptation to abrupt environmental change depends upon the history of stress. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;368:20120079. doi: 10.1098/rstb.2012.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber S., Schwarzer C., Sperk G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br. J. Pharmacol. 1994;113:737–740. doi: 10.1111/j.1476-5381.1994.tb17055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundemar L., Wahlestedt C., Reis D.J. Long-lasting inhibition of the cardiovascular responses to glutamate and the baroreceptor reflex elicited by neuropeptide Y injected into the nucleus tractus solitarius of the rat. Neurosci. Lett. 1991;122:135–139. doi: 10.1016/0304-3940(91)90211-b. [DOI] [PubMed] [Google Scholar]
- Grundemar L., Wahlestedt C., Reis D.J. Neuropeptide Y acts at an atypical receptor to evoke cardiovascular depression and to inhibit glutamate responsiveness in the brainstem. J. Pharmacol. Exp. Ther. 1991;258:633–638. [PubMed] [Google Scholar]
- Hanrahan J.W., Wills N.K., Phillips J.E., Lewis S.A. Basolateral K-channels in an insect epithelium - channel density, conductance, and block by barium. J. Gen. Physiol. 1986;87:443–466. doi: 10.1085/jgp.87.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbani M.J., Hyrc K.L., Faddis B.T., Romano C., Goldberg M.P. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp. Neurol. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- Herrera-Rincon C., Golding A.S., Moran K.M., Harrison C., Martyniuk C.J., Guay J.A., Zaltsman J., Carabello H., Kaplan D.L., Levin M. Brief local application of progesterone via a wearable bioreactor induces long-term regenerative response in adult Xenopus hindlimb. Cell Rep. 2018;25:1593–1609.e7. doi: 10.1016/j.celrep.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E.M., Petersen C.P. Wnt/Notum spatial feedback inhibition controls neoblast differentiation to regulate reversible growth of the planarian brain. Development. 2015;142:4217–4229. doi: 10.1242/dev.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockerman G.H., Peterson B.Z., Johnson B.D., Catterall W.A. Molecular determinants of drug binding and action on L-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 1997;37:361–396. doi: 10.1146/annurev.pharmtox.37.1.361. [DOI] [PubMed] [Google Scholar]
- Huang S., Eichler G., Bar-Yam Y., Ingber D.E. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. Rev. Lett. 2005;94:128701–212802. doi: 10.1103/PhysRevLett.94.128701. [DOI] [PubMed] [Google Scholar]
- Inoue H., Ohtaki H., Nakamachi T., Shioda S., Okada Y. Anion channel blockers attenuate delayed neuronal cell death induced by transient forebrain ischemia. J. Neurosci. Res. 2007;85:1427–1435. doi: 10.1002/jnr.21279. [DOI] [PubMed] [Google Scholar]
- Inoue T., Yamashita T., Agata K. Thermosensory signaling by TRPM is processed by brain serotonergic neurons to produce planarian thermotaxis. J. Neurosci. 2014;34:15701–15714. doi: 10.1523/JNEUROSCI.5379-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Mackinnon R. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 2000;115:269–272. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jollos V. Further experimental tests on the problem of type deformation. Naturwissenschaften. 1933;21:455–456. [Google Scholar]
- Jollos V. Dauermodifikationen und mutationen bei protozoen. Arch. Protistenk. 1934;83:197–219. [Google Scholar]
- Karin O., Swisa A., Glaser B., Dor Y., Alon U. Dynamical compensation in physiological circuits. Mol. Syst. Biol. 2016;12:886. doi: 10.15252/msb.20167216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.Z., Vienne J., Rosbash M., Griffith L.C. Non-reciprocal homeostatic compensation in Drosophila potassium channel mutants. J. Neurophysiol. 2017;117:2125–2136. doi: 10.1152/jn.00002.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi Y. Blocking effects of cesium and barium on the inward-rectifier K channel in the ventricular cell-membrane of the Guinea-pig. Jpn. Circ. J. 1986;50:512. [Google Scholar]
- Kussell E., Kishony R., Balaban N.Q., Leibler S. Bacterial persistence: a model of survival in changing environments. Genetics. 2005;169:1807–1814. doi: 10.1534/genetics.104.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai T.W., Zhang S., Wang Y.T. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Lambert G., Kussell E. Memory and fitness optimization of bacteria under fluctuating environments. PLoS Genet. 2014;10:e1004556. doi: 10.1371/journal.pgen.1004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C.S., Steele V.E. The mechanism of anterior-posterior polarity control in planarians. Differentiation. 1978;11:1–12. doi: 10.1111/j.1432-0436.1978.tb00965.x. [DOI] [PubMed] [Google Scholar]
- Lashinger E.S., Steiginga M.S., Hieble J.P., Leon L.A., Gardner S.D., Nagilla R., Davenport E.A., Hoffman B.E., Laping N.J., Su X. AMTB, a TRPM8 channel blocker: evidence in rats for activity in overactive bladder and painful bladder syndrome. Am. J. Physiol. Renal Physiol., 2008;295:F803–F810. doi: 10.1152/ajprenal.90269.2008. [DOI] [PubMed] [Google Scholar]
- Latorre R., Hurst R., Diaz F., Toro L., Stefani E. Barium as a probe of the molecular architecture of the pore of K+ channels. In: Latorre R., Saez J., editors. From Ion Channels to Cell-To-Cell Conversations. Plenum Press; 1997. pp. 129–146. [Google Scholar]
- Lau A., Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Levin M., Pietak A.M., Bischof J. Planarian regeneration as a model of anatomical homeostasis: recent progress in biophysical and computational approaches. Semin. Cell Dev. Biol. 2019;87:125–144. doi: 10.1016/j.semcdb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Huang J., Du W., Jia C., Yao H., Wang Y. TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. J. Neurochem. 2012;123:1010–1018. doi: 10.1111/jnc.12045. [DOI] [PubMed] [Google Scholar]
- Liang D., Bhatta S., Gerzanich V., Simard J.M. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg. Focus. 2007;22:E2. doi: 10.3171/foc.2007.22.5.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberante F.G., Pouryahya T., Mcmullin M.F., Zhang S.D., Mills K.I. Identification and validation of the dopamine agonist bromocriptine as a novel therapy for high-risk myelodysplastic syndromes and secondary acute myeloid leukemia. Oncotarget. 2016;7:6609–6619. doi: 10.18632/oncotarget.6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Garcia de Lomana A., Kaur A., Turkarslan S., Beer K.D., Mast F.D., Smith J.J., Aitchison J.D., Baliga N.S. Adaptive prediction emerges over short evolutionary time scales. Genome Biol. Evol. 2017;9:1616–1623. doi: 10.1093/gbe/evx116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark L.P., Prost R.W., Ulmer J.L., Smith M.M., Daniels D.L., Strottmann J.M., Brown W.D., Hacein-Bey L. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 2001;22:1813–1824. [PMC free article] [PubMed] [Google Scholar]
- Masotti A., Uva P., Davis-Keppen L., Basel-Vanagaite L., Cohen L., Pisaneschi E., Celluzzi A., Bencivenga P., Fang M., Tian M. Keppen-lubinsky syndrome is caused by mutations in the inwardly rectifying K(+) channel encoded by KCNJ6. Am. J. Hum. Genet. 2015;96:295–300. doi: 10.1016/j.ajhg.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccullough L.A., Egan T.M., Westfall T.C. Neuropeptide Y inhibition of calcium channels in PC-12 pheochromocytoma cells. Am. J. Physiol. 1998;274:C1290–C1297. doi: 10.1152/ajpcell.1998.274.5.C1290. [DOI] [PubMed] [Google Scholar]
- Mcmahon H.T., Nicholls D.G. Barium-evoked glutamate release from Guinea-pig cerebrocortical synaptosomes. J. Neurochem. 1993;61:110–115. doi: 10.1111/j.1471-4159.1993.tb03543.x. [DOI] [PubMed] [Google Scholar]
- Mendonca-Silva D.L., Novozhilova E., Cobbett P.J., Silva C.L., Noel F., Totten M.I., Maule A.G., Day T.A. Role of calcium influx through voltage-operated calcium channels and of calcium mobilization in the physiology of Schistosoma mansoni muscle contractions. Parasitology. 2006;133:67–74. doi: 10.1017/S0031182006000023. [DOI] [PubMed] [Google Scholar]
- Ni Y.L., Kuan A.S., Chen T.Y. Activation and inhibition of TMEM16A calcium-activated chloride channels. PLoS One. 2014;9:e86734. doi: 10.1371/journal.pone.0086734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi T., Zhang D., Chan J.D., Marchant J.S. A novel biological activity of praziquantel requiring voltage-operated Ca2+ channel beta subunits: subversion of flatworm regenerative polarity. PLoS Negl. Trop. Dis. 2009;3:e464. doi: 10.1371/journal.pntd.0000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T., Kume T., Izumi Y., Takada-Takatori Y., Niidome T., Akaike A. Bromocriptine, a dopamine D(2) receptor agonist with the structure of the amino acid ergot alkaloids, induces neurite outgrowth in PC12 cells. Eur. J. Pharmacol. 2008;598:27–31. doi: 10.1016/j.ejphar.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Odaka H., Numakawa T., Adachi N., Ooshima Y., Nakajima S., Katanuma Y., Inoue T., Kunugi H. Cabergoline, dopamine D2 receptor agonist, prevents neuronal cell death under oxidative stress via reducing excitotoxicity. PLoS One. 2014;9:e99271. doi: 10.1371/journal.pone.0099271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviedo N.J., Morokuma J., Walentek P., Kema I.P., Gu M.B., Ahn J.M., Hwang J.S., Gojobori T., Levin M. Long-range neural and gap junction protein-mediated cues control polarity during planarian regeneration. Dev. Biol. 2010;339:188–199. doi: 10.1016/j.ydbio.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviedo N.J., Nicolas C.L., Adams D.S., Levin M. Live imaging of planarian membrane potential using DiBAC4(3) Cold Spring Harb. Protoc. 2008;2008 doi: 10.1101/pdb.prot5055. pdb.prot5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owlarn S., Bartscherer K. Go ahead, grow a head! A planarian's guide to anterior regeneration. Regeneration (Oxf.) 2016;3:139–155. doi: 10.1002/reg2.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai V.P., Pietak A., Willocq V., Ye B., Shi N.Q., Levin M. HCN2 Rescues brain defects by enforcing endogenous voltage pre-patterns. Nat. Commun. 2018;9:998. doi: 10.1038/s41467-018-03334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo L.A., Stuhmer W. The roles of K(+) channels in cancer. Nat. Rev. Cancer. 2014;14:39–48. doi: 10.1038/nrc3635. [DOI] [PubMed] [Google Scholar]
- Parmar N.S., Tariq M., Ageel A.M. Effect of bromocriptine, a dopamine receptor agonist, on the experimentally induced gastric ulcers in albino rats. Life Sci. 1984;35:2035–2039. doi: 10.1016/0024-3205(84)90560-5. [DOI] [PubMed] [Google Scholar]
- Pellettieri J., Sánchez Alvarado A. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu. Rev. Genet. 2007;41:83–105. doi: 10.1146/annurev.genet.41.110306.130244. [DOI] [PubMed] [Google Scholar]
- Petralia R.S., Mattson M.P., Yao P.J. Aging and longevity in the simplest animals and the quest for immortality. Ageing Res. Rev. 2014;16:66–82. doi: 10.1016/j.arr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzulo G., Levin M. Re-membering the body: applications of computational neuroscience to the top-down control of regeneration of limbs and other complex organs. Integr. Biol. (Camb.) 2015;7:1487–1517. doi: 10.1039/c5ib00221d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietak A., Levin M. Exploring instructive physiological signaling with the bioelectric tissue simulation engine. Front. Bioeng. Biotechnol. 2016;4:55. doi: 10.3389/fbioe.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice H., Modi J.P., Wu J.Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell Longev. 2015;2015:964518. doi: 10.1155/2015/964518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prymaczok N.C., Pasqualino V.M., Viau V.E., Rodriguez E.M., Medesani D.A. Involvement of the crustacean hyperglycemic hormone (CHH) in the physiological compensation of the freshwater crayfish Cherax quadricarinatus to low temperature and high salinity stress. J. Comp. Physiol. B. 2016;186:181–191. doi: 10.1007/s00360-015-0954-0. [DOI] [PubMed] [Google Scholar]
- Quayle J.M., Standen N.B., Stanfield P.R. The voltage-dependent block of ATP-sensitive potassium channels of frog skeletal muscle by caesium and barium ions. J. Physiol. 1988;405:677–697. doi: 10.1113/jphysiol.1988.sp017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien P.W. The cellular and molecular basis for planarian regeneration. Cell. 2018;175:327–345. doi: 10.1016/j.cell.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin H. Cancer as a dynamic developmental disorder. Cancer Res. 1985;45:2935–2942. [PubMed] [Google Scholar]
- Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? BioEssays. 2006;28:515–524. doi: 10.1002/bies.20403. [DOI] [PubMed] [Google Scholar]
- Rubin H. Ordered heterogeneity and its decline in cancer and aging. Adv. Cancer Res. 2007;98:117–147. doi: 10.1016/S0065-230X(06)98004-X. [DOI] [PubMed] [Google Scholar]
- Rungta R.L., Choi H.B., Tyson J.R., Malik A., Dissing-Olesen L., Lin P.J.C., Cain S.M., Cullis P.R., Snutch T.P., Macvicar B.A. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–621. doi: 10.1016/j.cell.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Sahu S., Dattani A., Aboobaker A.A. Secrets from immortal worms: what can we learn about biological ageing from the planarian model system? Semin. Cell Dev. Biol. 2017;70:108–121. doi: 10.1016/j.semcdb.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Saló E., Abril J.F., Adell T., Cebrià F., Eckelt K., Fernandez-Taboada E., Handberg-Thorsager M., Iglesias M., Molina M.D., Rodríguez-Esteban G. Planarian regeneration: achievements and future directions after 20 years of research. Int. J. Dev. Biol. 2009;53:1317–1327. doi: 10.1387/ijdb.072414es. [DOI] [PubMed] [Google Scholar]
- Samani P., Bell G. The ghosts of selection past reduces the probability of plastic rescue but increases the likelihood of evolutionary rescue to novel stressors in experimental populations of wild yeast. Ecol. Lett. 2016;19:289–298. doi: 10.1111/ele.12566. [DOI] [PubMed] [Google Scholar]
- Sarnat H.B., Netsky M.G. The brain of the planarian as the ancestor of the human brain. Can. J. Neurol. Sci. 1985;12:296–302. doi: 10.1017/s031716710003537x. [DOI] [PubMed] [Google Scholar]
- Schneider M.B., Murrin L.C., Pfeiffer R.F., Deupree J.D. Dopamine receptors: effects of chronic L-dopa and bromocriptine treatment in an animal model of Parkinson's disease. Clin. Neuropharmacol. 1984;7:247–257. [PubMed] [Google Scholar]
- Schriever A.M., Friedrich T., Pusch M., Jentsch T.J. CLC chloride channels in Caenorhabditis elegans. J. Biol. Chem. 1999;274:34238–34244. doi: 10.1074/jbc.274.48.34238. [DOI] [PubMed] [Google Scholar]
- Sihra T.S., Piomelli D., Nichols R.A. Barium evokes glutamate release from rat brain synaptosomes by membrane depolarization: involvement of K+, Na+, and Ca2+ channels. J. Neurochem. 1993;61:1220–1230. doi: 10.1111/j.1471-4159.1993.tb13612.x. [DOI] [PubMed] [Google Scholar]
- Silva A.P., Xapelli S., Pinheiro P.S., Ferreira R., Lourenço J., Cristóvão A., Grouzmann E., Cavadas C., Oliveira C.R., Malva J.O. Up-regulation of neuropeptide Y levels and modulation of glutamate release through neuropeptide Y receptors in the hippocampus of kainate-induced epileptic rats. J. Neurochem. 2005;93:163–170. doi: 10.1111/j.1471-4159.2004.03005.x. [DOI] [PubMed] [Google Scholar]
- Simons C., Rash L.D., Crawford J., Ma L., Cristofori-Armstrong B., Miller D., Ru K., Baillie G.J., Alanay Y., Jacquinet A. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 2015;47:73–77. doi: 10.1038/ng.3153. [DOI] [PubMed] [Google Scholar]
- Skaper S.D. Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol. Disord. Drug Targets. 2011;10:44–56. doi: 10.2174/187152711794488638. [DOI] [PubMed] [Google Scholar]
- Soen Y., Braun E. Scale-invariant fluctuations at different levels of organization in developing heart cell networks. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2000;61:R2216–R2219. doi: 10.1103/physreve.61.r2216. [DOI] [PubMed] [Google Scholar]
- Soen Y., Knafo M., Elgart M. A principle of organization which facilitates broad Lamarckian-like adaptations by improvisation. Biol. Direct. 2015;10:68. doi: 10.1186/s13062-015-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek M., Balaban N.Q., Loewenstein Y. Stochasticity, bistability and the wisdom of crowds: a model for associative learning in genetic regulatory networks. PLoS Comput. Biol. 2013;9:e1003179. doi: 10.1371/journal.pcbi.1003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S., Fridmann-Sirkis Y., Braun E., Soen Y. Epigenetically heritable alteration of fly development in response to toxic challenge. Cell Rep. 2012;1:528–542. doi: 10.1016/j.celrep.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Stetina T., Kostal V., Korbelova J. The role of inducible Hsp70, and other heat shock proteins, in adaptive complex of cold tolerance of the fruit fly (Drosophila melanogaster) PLoS One. 2015;10:e0128976. doi: 10.1371/journal.pone.0128976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K.G., Emmons-Bell M., Levin M. Physiological inputs regulate species-specific anatomy during embryogenesis and regeneration. Commun. Integr. Biol. 2016;9:e1192733. doi: 10.1080/19420889.2016.1192733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowska K., Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Takeuchi H., Mizoguchi H., Doi Y., Jin S., Noda M., Liang J., Li H., Zhou Y., Mori R., Yasuoka S. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PLoS One. 2011;6:e21108. doi: 10.1371/journal.pone.0021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka E.M., Reddien P.W. The cellular basis for animal regeneration. Dev. Cell. 2011;21:172–185. doi: 10.1016/j.devcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski A.K. Mouse chimaeras developed from fused eggs. Nature. 1961;190:857–860. doi: 10.1038/190857a0. [DOI] [PubMed] [Google Scholar]
- Thompson R.J. Pannexin channels and ischaemia. J. Physiol. 2015;593:3463–3470. doi: 10.1113/jphysiol.2014.282426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkachenko A.G. Stress responses of bacterial cells as mechanism of development of antibiotic tolerance (review) Appl. Biochem. Microbiol. 2018;54:108–127. [Google Scholar]
- Urrego D., Tomczak A.P., Zahed F., Stuhmer W., Pardo L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014;369:20130094. doi: 10.1098/rstb.2013.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A., Kovac S., Holmstrom K.M., Gandhi S., Abramov A.Y. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 2013;4:e455. doi: 10.1038/cddis.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg L.N., Adams D.S., Levin M. Normalized shape and location of perturbed craniofacial structures in the Xenopus tadpole reveal an innate ability to achieve correct morphology. Dev. Dyn. 2012;241:863–878. doi: 10.1002/dvdy.23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via M.A., Chandra H., Araki T., Potenza M.V., Skamagas M. Bromocriptine approved as the first medication to target dopamine activity to improve glycemic control in patients with type 2 diabetes. Diabetes Metab. Syndr. Obes. 2010;3:43–48. doi: 10.2147/dmsott.s9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S.J., Shirley D.G., Folkerd E.J., Unwin R.J. Effects of the Potassium channel blocker barium on sodium and potassium transport in the rat loop of Henle in vivo. Exp. Physiol. 2001;86:469–474. doi: 10.1113/eph8602210. [DOI] [PubMed] [Google Scholar]
- Wang Y., Denisova J.V., Kang K.S., Fontes J.D., Zhu B.T., Belousov A.B. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J. Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Song J.H., Denisova J.V., Park W.M., Fontes J.D., Belousov A.B. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J. Neurosci. 2012;32:713–725. doi: 10.1523/JNEUROSCI.3872-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M.M., Aylwin M. Niflumic and flufenamic acids are potent reversible blockers of Ca2(+)-activated Cl- channels in Xenopus oocytes. Mol. Pharmacol. 1990;37:720–724. [PubMed] [Google Scholar]
- Whittaker E., Vereker E., Lynch M.A. Neuropeptide Y inhibits glutamate release and long-term potentiation in rat dentate gyrus. Brain Res. 1999;827:229–233. doi: 10.1016/s0006-8993(99)01302-5. [DOI] [PubMed] [Google Scholar]
- Wright S.H. Generation of resting membrane potential. Adv. Physiol. Educ. 2004;28:139–142. doi: 10.1152/advan.00029.2004. [DOI] [PubMed] [Google Scholar]
- Wu G., Hamill O.P. NPPB block of Ca(++)-activated Cl- currents in Xenopus oocytes. Pflugers Arch. 1992;420:227–229. doi: 10.1007/BF00374996. [DOI] [PubMed] [Google Scholar]
- Yapa K., Deuis J., Peters A.A., Kenny P.A., Roberts-Thomson S.J., Vetter I., Monteith G.R. Assessment of the TRPM8 inhibitor AMTB in breast cancer cells and its identification as an inhibitor of voltage gated sodium channels. Life Sci. 2018;198:128–135. doi: 10.1016/j.lfs.2018.02.030. [DOI] [PubMed] [Google Scholar]
- Yokobori S., Mazzeo A.T., Hosein K., Gajavelli S., Dietrich W.D., Bullock M.R. Preconditioning for traumatic brain injury. Transl Stroke Res. 2013;4:25–39. doi: 10.1007/s12975-012-0226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Bhavnani B.R. Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci. 2005;6:13. doi: 10.1186/1471-2202-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F., Phelan K.D. The role of canonical transient receptor potential channels in seizure and excitotoxicity. Cells. 2014;3:288–303. doi: 10.3390/cells3020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zeng X.H., Lingle C.J. Barium ions selectively activate BK channels via the Ca2+-bowl site. Proc. Natl. Acad. Sci. U S A. 2012;109:11413–11418. doi: 10.1073/pnas.1204444109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Provided are the genes that are involved in the corresponding network, median fold change of the network, and the p value.