

Abstract
Background and Objectives: Bladder cancer (BC) is a complex tumor associated with high recurrence and mortality. To discover key molecular changes in BC, we analyzed next-generation sequencing data of BC and surrounding tissue samples from clinical specimens. Methods. Gene expression profiling datasets of bladder cancer were analyzed online. The Database for Annotation, Visualization, and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) was used to perform Gene Ontology (GO) functional and KEGG pathway enrichment analyses. Molecular Complex Detection (MCODE) in Cytoscape software (Cytoscape_v3.6.1) was applied to identify hub genes. Protein expression and survival data were downloaded from OncoLnc (http://www.oncolnc.org/). Gene expression data were obtained from the ONCOMINE website (https://www.oncomine.org/). Results. We identified 4211 differentially expressed genes (DEGs) by analysis of surrounding tissue vs. cancer tissue (SC analysis) and 410 DEGs by analysis of cancer tissue vs. recurrent tissue cluster (CR analysis). GO function analysis revealed enrichment of DEGs in genes related to the cytoplasm and nucleoplasm for both clusters, and KEGG pathway analysis showed enrichment of DEGs in the PI3K-Akt signaling pathway. We defined the 20 genes with the highest degree of connectivity as the hub genes. Cox regression revealed CCNB1, ESPL1, CENPM, BLM, and ASPM were related to overall survival. The expression levels of CCNB1, ESPL1, CENPM, BLM, and ASPM were 4.795-, 5.028-, 8.691-, 2.083-, and 3.725-fold higher in BC than the levels in normal tissues, respectively. Conclusions. The results suggested that the functions of CCNB1, ESPL1, CENPM, BLM, and ASPM may contribute to BC development and the functions of CCNB1, ESPL1, CENPM, and BLM may also contribute to BC recurrence.
1. Introduction
Bladder cancer (BC) is a common urogenital cancer, with an estimate of 80,470 new cases and 17,670 deaths in the United States in 2019 [1]. Bladder cancer patients are often diagnosed by cystoscopy for diagnostic testing prompted by haematuria. Approximately 80% of urinary bladder tumors are superficial papillary lesions but also can be multifocal and exhibit a tendency for recurrence: remaining tumors may invade the bladder wall and lead to distant metastases [2]. Treatment for BC includes transurethral resection of bladder tumor (TURBT), chemotherapy, or vaccine-based therapy directed to the bladder, cystectomy, radiotherapy, and chemotherapy [3]. However, BC is a complex disease associated with a high recurrence rate and high mortality, and its biology remains poorly understood [4].
There are several important risk factors for BC, such as cigarette smoking, occupational chemical exposure (especially to aromatic amines), water arsenic level, Schistosoma haematobium infection, and radiation therapy for pelvic malignancies [5]. Previous studies identified aspects of the molecular mechanism of BC development and recurrence. BC has been genetically associated with mutations of two genes, fibroblast growth factor receptor 3 (FGFR3, for low-grade, noninvasive papillary tumors), and tumor protein P53 (TP53, for high-grade, muscle-invasive tumors) [6]. Treatment with drugs targeting mutations in genes such as FGFR3, VEGF, signal transducer and activator of transcription 3, and CD24 has all shown preclinical activity [4]. Next-generation sequencing (NGS) has drastically increased the understanding of cancer processes including BC, and analyses of these data can provide insight into effective diagnostic and therapeutic BC treatments [7, 8].
There are significant BC molecular profiling data [9–12]. Researchers have explored screening of urine to detect DNA mutations as an alternative for urine cytology as a tool for the noninvasive detection and surveillance of BC [13]. Additionally, the analysis of frequently mutated genes in BC may suggest potential targets for personalized treatment and predict treatment response [8]. However, to date, it has been difficult to identify key genes related to BC from NGS data. To discover key molecules active in BC, we analyzed BC data from microarray experiments and NGS sequencing data of clinical specimens. Our results suggested CCNB1, ESPL1, CENPM, BLM, and ASPM may contribute to BC development and recurrence.
2. Materials and Methods
2.1. Online Data
The gene expression profiling datasets of bladder cancer were analyzed online (GEO; https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE13507). 58 normal tissues surrounding cancer, 165 primary bladder cancer, and 23 recurrent samples were measured in this array.
2.2. Identifying Differentially Expressed Genes
To analyze the microarray data, we compared the gene expression between 58 normal tissues surrounding cancer and 165 primary bladder cancer samples to identify genes involved with tumorigenesis, and gene expression comparison between 165 primary bladder cancer and 23 recurrent samples was also performed to screen genes that promote tumor recurrence. Differentially expressed genes were screened by adjusted p value or p value and fold change (FC). For comparison between surrounding tissue and cancer tissue, differentially expressed genes were restricted by adjusted p value <0.05 and |FC| > 4, and we defined these genes cluster SC (surrounding tissue vs. cancer tissue). For comparison between cancer tissue and recurrent tissue, differentially expressed genes were restricted by p value <0.05 and |FC| > 2, and we defined these genes cluster CR (cancer tissue vs. recurrent tissue).
2.3. Merging Data
We proposed two methods to process the clusters SC and CR: (1) tumorigenesis and recurrence were promoted by the same genes or proteins, the overlap between SC and CR were the key genes, and overlap genes were analyzed to perform Gene Ontology and KEGG pathway analysis and retrieve interacting genes; (2) tumorigenesis and recurrence were contributed by different genes, we would find key genes from clusters SC and CR individually, and SC and CR genes were individually analyzed to perform Gene Ontology and KEGG pathway analysis and retrieve interacting genes. For method 1, Venny 2.1.0 (http://bioinfogp.cnb.csic.es/tools/venny/index.html) was used to identify overlapping differentially expressed genes between SC and CR. The upregulated and downregulated genes were measured, respectively.
2.4. Gene Ontology and KEGG Pathway Analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) was used to perform Gene Ontology (GO) functional and KEGG pathway enrichment analyses. p < 0.05 was considered as statistically significant.
2.5. Retrieving Interacting Genes
Search Tool for the Retrieval of Interacting Genes (STRING) is an online tool (https://string-db.org) designed to integrate information by consolidating known and predicted protein-protein association data. Molecular Complex Detection (MCODE) in Cytoscape software (Cytoscape_v3.6.1) was applied to screen hub genes. All identified differentially expressed genes described above were analyzed. The top 20 hub genes with connection degree >10 were selected.
2.6. Survival Analysis
The protein expression and raw survival data were downloaded from OncoLnc (http://www.oncolnc.org/). Overall survival and disease-free survival were analyzed by Gene Expression Profiling Interactive Analysis (GEPIA, online website: http://gepia.cancer-pku.cn/detail.php?gene).
2.7. Gene Expression Data
Gene expression data were obtained from ONCOMINE website (https://www.oncomine.org/). Cancer type was restricted by bladder cancer, and the expressions of CCNB1, ESPL1, CENPM, BLM, ASPM, JUN, and CDK6 were obtained.
2.8. Statistical Analysis
Clinical information was analyzed by SPSS 18.0 (IBM Corporation, Armonk, NY). A Cox regression model was conducted to perform univariate and multivariate analyses. The gene expressions were analyzed by GraphPad Prism 7.0. p < 0.05 is considered to reveal a statistically significant difference.
3. Results
Analysis was performed using data for 58 normal tissues surrounding cancer, 165 primary bladder cancer samples, and 23 recurrent cancer samples. We identified 4211 differentially expressed genes (DEGs) by analysis of surrounding tissue vs. cancer tissue (SC analysis) and 410 DEGs by analysis of cancer tissue vs. recurrent tissue cluster (CR analysis). There were 1657 and 258 upregulated DEGs in cluster SC and cluster CR, respectively, and 2514 and 152 individually downregulated DEGs in cluster SC and cluster CR. A comparison of these sets of genes revealed 148 overlap genes, including 91 upregulated and 57 downregulated DEGs (Figure 1). We next analyzed these genes by performing two kinds of functional analysis.
Figure 1.
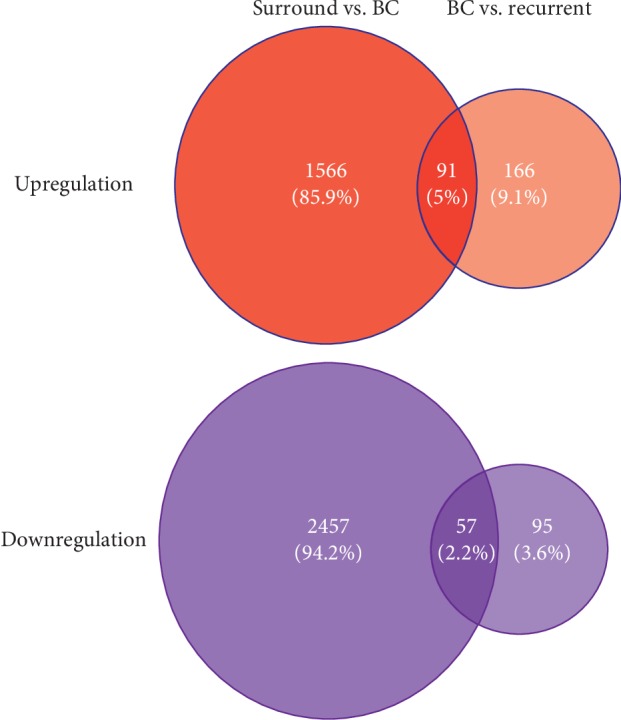
4211 differentially expressed genes (DEGs) were found by comparing 58 normal tissues surrounding cancer and 165 primary bladder cancer samples, and 410 DEGs were found by comparing 165 primary bladder cancer and 23 recurrent samples. There were 1657 and 258 upregulated DEGs and 2514 and 152 individually downregulated DEGs for each group. 91 and 57 overlap genes were found in up- and downregulation genes.
3.1. Gene Ontology and KEGG Pathway Analysis
In the first analysis, the 91 upregulated and 57 downregulated genes that were differentially expressed in both the comparison of cancer and surrounding tissues and the comparison of cancer and recurrent cancer tissues were analyzed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://david.ncifcrf.gov/). Gene Ontology (GO) functional and KEGG pathway enrichment analyses were performed. GO function analysis revealed enrichment of these DEGs in functions related to the cytoplasm and nucleoplasm. There is an enrichment of genes involved with protein binding and protein kinase binding, regulating cell division, DNA replication, and cyclin-dependent protein serine/threonine kinase activity. KEGG pathway analysis indicated that the identified DEGs are mainly enriched in the PI3K-Akt signaling pathway, microRNAs related to cancer, and the cell cycle. The 15 most enriched classes based on GO function analysis and the eight most enriched KEGG pathways are listed in Table 1.
Table 1.
Gene ontology and KEGG pathway analysis of differentially expressed genes according to method 1.
| Category | Term | Count | % | p value | |
|---|---|---|---|---|---|
| GO analysis | GOTERM_CC_DIRECT | GO:0005654∼nucleoplasm | 42 | 28.1879194630872 | 1.22E − 05 |
| GOTERM_MF_DIRECT | GO:0005515∼protein binding | 92 | 61.744966442953 | 8.65E − 05 | |
| GOTERM_BP_DIRECT | GO:0051301∼cell division | 12 | 8.05369127516778 | 1.08E − 04 | |
| GOTERM_BP_DIRECT | GO:0006260∼DNA replication | 8 | 5.36912751677852 | 2.35E − 04 | |
| GOTERM_CC_DIRECT | GO:0005737∼cytoplasm | 60 | 40.2684563758389 | 2.68E − 04 | |
| GOTERM_MF_DIRECT | GO:0019901∼protein kinase binding | 11 | 7.38255033557047 | 7.94E − 04 | |
| GOTERM_BP_DIRECT | GO:0000082∼G1/S transition of mitotic cell cycle | 6 | 4.02684563758389 | 0.001302766851924 | |
| GOTERM_CC_DIRECT | GO:0000922∼spindle pole | 6 | 4.02684563758389 | 0.001440410268581 | |
| GOTERM_MF_DIRECT | GO:0004693∼cyclin-dependent protein serine/threonine kinase activity | 4 | 2.68456375838926 | 0.002393195271174 | |
| GOTERM_BP_DIRECT | GO:0001706∼endoderm formation | 3 | 2.01342281879194 | 0.003901253353245 | |
| GOTERM_CC_DIRECT | GO:0005813∼centrosome | 10 | 6.71140939597315 | 0.005184882974603 | |
| GOTERM_CC_DIRECT | GO:0005739∼mitochondrion | 20 | 13.4228187919463 | 0.005522684005422 | |
| GOTERM_BP_DIRECT | GO:0051591∼response to cAMP | 4 | 2.68456375838926 | 0.005751402857203 | |
| GOTERM_BP_DIRECT | GO:0098609∼cell-cell adhesion | 8 | 5.36912751677852 | 0.005854477268833 | |
| GOTERM_CC_DIRECT | GO:0005829∼cytosol | 38 | 25.5033557046979 | 0.007692882731082 | |
|
| |||||
| KEGG pathway | KEGG_PATHWAY | hsa04110: cell cycle | 7 | 4.6979865771812 | 0.00123536590764 |
| KEGG_PATHWAY | hsa05222: small cell lung cancer | 5 | 3.35570469798657 | 0.009218413749072 | |
| KEGG_PATHWAY | hsa04151: PI3K-Akt signaling pathway | 9 | 6.04026845637583 | 0.018153887794995 | |
| KEGG_PATHWAY | hsa05206: microRNAs in cancer | 8 | 5.36912751677852 | 0.020570720541055 | |
| KEGG_PATHWAY | hsa04115: p53 signaling pathway | 4 | 2.68456375838926 | 0.02717134771579 | |
| KEGG_PATHWAY | hsa04920: adipocytokine signaling pathway | 4 | 2.68456375838926 | 0.030418432882382 | |
| KEGG_PATHWAY | hsa04152: AMPK signaling pathway | 5 | 3.35570469798657 | 0.031444562545736 | |
| KEGG_PATHWAY | hsa04068: FoxO signaling pathway | 5 | 3.35570469798657 | 0.04111192229316 | |
BP: biological process; CC: cellular component; GO: gene ontology; MF: molecular function.
In the second analysis, we focused on the DEGs identified by the comparison of cancer and surrounding tissues or those identified by the comparison of cancer and recurrent cancer samples. Analysis of DEGs from the surrounding tissue vs. cancer tissue comparison should reflect key genes participating in tumorigenesis or bladder cancer development. GO function analysis of these genes found high enrichment of functions related to extracellular exosomes, extracellular space, and extracellular matrix. Protein binding, heparin binding, and integrin binding are the main functions of these genes, which participate in cell adhesion, extracellular matrix organization, and aging. KEGG pathway analysis indicated enrichment of these genes in HTLV-I infection, Staphylococcus aureus infection, and focal adhesion (Table 2). We next analyzed the DEGs identified by the comparison of cancer and recurrent cancer samples, which should include genes related to bladder cancer recurrence. GO function analysis revealed enrichment of these genes in functions related to the cytoplasm, cytosol, and nucleoplasm, and analysis of molecular function showed enrichment in protein binding. The most relevant enriched biological processes are angiogenesis and the G1/S transition of the mitotic cell cycle, and KEGG pathway analysis indicated enrichment of these genes in cancer pathways, the PI3K-Akt signaling pathway, and cell cycle (Table 2).
Table 2.
Gene ontology and KEGG pathway analysis of differentially expressed genes in bladder cancer development and recurrence.
| Bladder cancer development | Bladder cancer recurrence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Category | Term | Count | % | p value | Category | Term | Count | % | p value | |
| GO analysis | GOTERM_CC_DIRECT | GO:0070062∼extracellular exosome | 331 | 25.5007704160246 | 6.61E − 26 | GOTERM_MF_DIRECT | GO:0005515∼protein binding | 233 | 57.2481572481572 | 5.48E − 07 |
| GOTERM_CC_DIRECT | GO:0031012∼+A1 | 69 | 5.31587057010785 | 2.26E − 19 | GOTERM_CC_DIRECT | GO:0005737∼cytoplasm | 152 | 37.3464373464373 | 1.25E − 06 | |
| GOTERM_CC_DIRECT | GO:0005615∼extracellular space | 164 | 12.6348228043143 | 1.98E − 13 | GOTERM_CC_DIRECT | GO:0005654∼nucleoplasm | 86 | 21.1302211302211 | 1.16E − 04 | |
| GOTERM_BP_DIRECT | GO:0030198∼extracellular matrix organization | 46 | 3.54391371340523 | 9.99E − 13 | GOTERM_CC_DIRECT | GO:0016020∼membrane | 70 | 17.1990171990172 | 2.74E − 04 | |
| GOTERM_CC_DIRECT | GO:0005578∼proteinaceous extracellular matrix | 54 | 4.16024653312788 | 1.67E − 12 | GOTERM_CC_DIRECT | GO:0005829∼cytosol | 96 | 23.5872235872235 | 4.19E − 04 | |
| GOTERM_CC_DIRECT | GO:0005925∼focal adhesion | 67 | 5.16178736517719 | 6.60E − 12 | GOTERM_CC_DIRECT | GO:0070062∼extracellular exosome | 83 | 20.3931203931203 | 6.83E − 04 | |
| GOTERM_CC_DIRECT | GO:0009986∼cell surface | 82 | 6.31741140215716 | 1.84E − 11 | GOTERM_MF_DIRECT | GO:0004693∼cyclin-dependent protein serine/threonine kinase activity | 6 | 1.47420147420147 | 7.00E − 04 | |
| GOTERM_BP_DIRECT | GO:0007155∼cell adhesion | 73 | 5.62403697996918 | 9.83E − 11 | GOTERM_MF_DIRECT | GO:0030332∼cyclin binding | 5 | 1.22850122850122 | 7.36E − 04 | |
| GOTERM_CC_DIRECT | GO:0005576∼extracellular region | 175 | 13.4822804314329 | 3.89E − 10 | GOTERM_BP_DIRECT | GO:0048146∼positive regulation of fibroblast proliferation | 7 | 1.71990171990172 | 8.99E − 04 | |
| GOTERM_MF_DIRECT | GO:0005515∼protein binding | 711 | 54.7765793528505 | 8.26E − 10 | GOTERM_BP_DIRECT | GO:0000082∼G1/S transition of mitotic cell cycle | 9 | 2.21130221130221 | 0.001364705455254 | |
| GOTERM_MF_DIRECT | GO:0008201∼heparin binding | 35 | 2.69645608628659 | 3.49E − 09 | GOTERM_BP_DIRECT | GO:0048839∼inner ear development | 6 | 1.47420147420147 | 0.00158628474829 | |
| GOTERM_MF_DIRECT | GO:0005178∼integrin binding | 27 | 2.08012326656394 | 8.57E − 09 | GOTERM_BP_DIRECT | GO:0031581∼hemidesmosome assembly | 4 | 0.982800982800982 | 0.001745599823436 | |
| GOTERM_BP_DIRECT | GO:0048146∼positive regulation of fibroblast proliferation | 19 | 1.46 E + 00 | 1.38E − 08 | GOTERM_BP_DIRECT | GO:0051591∼response to cAMP | 6 | 1.47420147420147 | 0.002664403703622 | |
| GOTERM_BP_DIRECT | GO:0007568∼aging | 33 | 2.54237288135593 | 1.37E − 07 | GOTERM_BP_DIRECT | GO:0001525∼angiogenesis | 13 | 3.19410319410319 | 0.002731528888927 | |
| GOTERM_BP_DIRECT | GO:0060333∼interferon-gamma-mediated signaling pathway | 20 | 1.54083204930662 | 2.89E − 07 | GOTERM_CC_DIRECT | GO:0005615∼extracellular space | 44 | 10.8108108108108 | 0.003130126302432 | |
|
| ||||||||||
| KEGG pathway | KEGG_PATHWAY | hsa05166: HTLV-I infection | 49 | 3.77503852080123 | 1.54E − 08 | KEGG_PATHWAY | hsa05200: pathways in cancer | 22 | 5.4054054054054 | 0.001345594815917 |
| KEGG_PATHWAY | hsa05150: Staphylococcus aureus infection | 20 | 1.54083204930662 | 1.67E − 08 | KEGG_PATHWAY | hsa04151: PI3K-Akt signaling pathway | 19 | 4.66830466830466 | 0.003848355864399 | |
| KEGG_PATHWAY | hsa04510: focal adhesion | 40 | 3.08166409861325 | 3.47E − 07 | KEGG_PATHWAY | hsa04923: regulation of lipolysis in adipocytes | 6 | 1.47420147420147 | 0.015090205294328 | |
| KEGG_PATHWAY | hsa05323: rheumatoid arthritis | 23 | 1.77195685670261 | 1.19E − 06 | KEGG_PATHWAY | hsa04110: cell cycle | 9 | 2.21130221130221 | 0.015736544810328 | |
| KEGG_PATHWAY | hsa05416: viral myocarditis | 18 | 1.38674884437596 | 1.41E − 06 | KEGG_PATHWAY | hsa05222: small cell lung cancer | 7 | 1.71990171990172 | 0.023482661953239 | |
| KEGG_PATHWAY | hsa04514: cell adhesion molecules (CAMs) | 30 | 2.31124807395993 | 2.37E − 06 | KEGG_PATHWAY | hsa00010: glycolysis/gluconeogenesis | 6 | 1.47420147420147 | 0.03037227765735 | |
| KEGG_PATHWAY | hsa05310: asthma | 12 | 0.924499229583975 | 1.17E − 05 | KEGG_PATHWAY | hsa03320: PPAR signaling pathway | 6 | 1.47420147420147 | 0.03037227765735 | |
| KEGG_PATHWAY | hsa04110: cell cycle | 26 | 2.00308166409861 | 1.49E − 05 | KEGG_PATHWAY | hsa04610: complement and coagulation cascades | 6 | 1.47420147420147 | 0.033914937801339 | |
| KEGG_PATHWAY | hsa04610: complement and coagulation cascades | 18 | 1.38674884437596 | 2.41E − 05 | KEGG_PATHWAY | hsa05205: proteoglycans in cancer | 11 | 2.7027027027027 | 0.036633631217231 | |
| KEGG_PATHWAY | hsa04612: antigen processing and presentation | 19 | 1.46379044684129 | 2.49E − 05 | KEGG_PATHWAY | hsa04921: oxytocin signaling pathway | 9 | 2.21130221130221 | 0.042657563729801 | |
BP: biological process; CC: cellular component; GO: gene ontology; MF: molecular function.
3.2. Hub Gene Analysis
We used STRING for investigating and integrating interaction between proteins. Data were exported for further analysis by Cytoscape. We defined the top 20 genes with the highest degree of connectivity as the hub genes. For method 1, 20 hub genes are shown in Figure 2(a). Also, hub genes in clusters SC and CR are shown in Figures 2(b) and 2(c).
Figure 2.

(a) According to method 1, 20 hub genes were discovered from overlapping differentially expressed genes between SC (surrounding tissue vs. cancer tissue) and CR (cancer tissue vs. recurrent tissue). (b) 20 hub genes were discovered from SC. (c) 20 hub genes were discovered from CR.
3.3. Clinical Analysis
Kaplan–Meier analysis was performed for the identified hub genes using the DAVID website. We defined the 20 genes with the highest degree of connectivity as hub genes and determined hub genes for the SC comparison and for the CR comparison. For the 20 hub genes identified in the SC analysis, JUN and CDK6 were associated with the overall survival of bladder cancer patients (Figures 3(j) and 3(o)). High JUN expression increased the risk of death by 40% relative to low JUN expression (p=0.041), and high CDK6 expression increased the risk of death by 50% compared to low CDK6 expression (p=0.013). Overall survival analysis of other hub genes did not exhibit statistical significance for high and low expressions (Figures 3(a)–3(i), 3(k)–3(n), and 3(p)–3(t)).
Figure 3.

Overall survival analysis for 20 hub genes from method 1: (a) CCNB1, (b) CCNB2, (c) ESPL1, (d) CDC45, (e) FANCI, (f) MCM10, (g) CDT1, (h) PRC1, (i) MKI67, (j) JUN, (k) ASP1B, (l) CENPM, (m) SGOL1, (n) CDC25A, (o) CDK6, (p) CDCA3, (q) TACC3, (r) TROAP, (s) CDK4, and (t) BLM. For the 20 hub genes, only (j) JUN (HR = 1.4, p=0.041) and (o) CDK6 (HR = 1.5, p=0.013) showed statistical significance that higher expression patients indicated poor overall survival.
We also determined 20 hub genes for the CR analysis. None of these hub genes were associated with overall survival (Supplement ). We next analyzed the hub genes and their association with disease-free survival (DFS) instead of overall survival. In this analysis, we found an association of CDK6 with DFS of bladder cancer patients (Supplement ).
We then downloaded the raw data from OncoLnc for further analysis. Cox regression revealed that CCNB1, ESPL1, CENPM, BLM, and ASPM are related to overall survival (Supplement ). Of these, CCNB1, ESPL1, CENPM, and BLM were identified as hub genes from cluster CR, and ASPM was identified as a hub gene from cluster SC (Supplement ).
3.4. Gene Expression in BC
The expressions of CCNB1, ESPL1, CENPM, BLM, ASPM, and two other genes (JUN and CDK6) associated with bladder cancer patient overall survival are shown in Figure 4 and Supplement . The expression levels of CCNB1, ESPL1, CENPM, BLM, and ASPM were 4.795-, 5.028-, 8.691-, 2.083-, and 3.725-fold higher in BC than in normal tissues (p = 3.86E−13, 5.92E−20, 5.91E−26, 5.19E−14, and 2.56E−13). The expressions of JUN and CDK6 were not significantly different between BC and normal tissues (p = 0.639 and 0.466).
Figure 4.

(a) CCNB1, (b) ESPL1, (c) CENPM, (d) BLM, and (e) ASPM were 4.795-, 5.028-, 8.691-, 2.083-, and 3.725-fold higher in BC than in normal tissues (p = 3.86E−13, 5.92E−20, 5.91E−26, 5.19E−14, and 2.56E−13, respectively).
4. Discussion
In this analysis, we defined differentially expressed genes for the SC comparison of surrounding tissue vs. cancer tissue and for the CR comparison of cancer tissue vs. recurrent tissue and considered the identified DEGs contributing to BC development and contributing to BC recurrence, respectively. Genes found in both SC and CR analyses affect both BC development and recurrence, and key genes identified in either SC analysis or CR analysis but not in both analyses are genes that affect either BC development or recurrence, respectively. GO function analysis discovered DEGs are mainly enriched in cytoplasm and nucleoplasm for both clusters, and KEGG pathway analysis indicated high enrichment of DEGs in the PI3K-Akt signaling pathway. We found that CCNB1, ESPL1, CENPM, BLM, and ASPM may be associated with BC development, and CCNB1, ESPL1, CENPM, and BLM may be associated with BC recurrence. It was interesting that our analysis revealed four genes, CCNB1, ESPL1, CENPM, and BLM, which are associated with both BC development and recurrence. Although JUN and CDK6 were not associated with BC development or recurrence, they may be prognostic factors for overall survival (Figure 3(j), 3(o)). The p value was unadjusted for tumor recurrence, and without a correction for multiple tests, the results are meaningful but not conclusive for recurrent tumors.
Among the identified genes, we found CCNB1 was 4.8-fold more highly expressed in BC compared to the level in normal tissues (p = 3.86E−13). CCNB1 is an important cell cycle protein and is a key regulator of the G2/M checkpoint. High levels of CCNB1 usually lead to cell immortalization, resulting in aneuploidy, which contributes to chromosomal instability and is related to the aggressive nature of certain cancers [14]. The involvement of CCNB1 with BC was demonstrated previously [15–19]. Three bioinformatics analyses indicated that CCNB1 was a key gene in BC, consistent with our findings [17–19]; however, other hub genes reported previously such as KIF4A, TPX2, BUB1B, CDK1, ISG15, KIF15, RAD54L, and TRIP13 were not identified in our analysis. CCNB1 has been positively correlated with cell proliferation, invasion, and migration [20]. Gene expression profiling in 102 patients with non-muscle-invasive BC identified an association of CCNB1 with disease recurrence [16], and other analyses showed a positive correlation of CCNB1 with pathological stage and metastasis [20]. Cytological experiments may be required to confirm the function of CCNB1 in BC cells.
Our analysis discovered ESPL1 was expressed at a level 5.0-fold higher in BC than the level in normal tissues (p = 5.92E−20). ESPL1, also known as extra spindle poles-like 1 protein or separin, plays a central role in chromosome segregation by cleaving the cohesin complex at the onset of anaphase, and altered ESPL1 activity is correlated with aneuploidy and cancer [21]. Genomic analysis of transitional cell carcinoma (TCC) by both whole-genome and whole-exome sequencing of 99 individuals with TCC found frequent alterations in ESPL1 [22]. ESPL1 expression was negatively correlated with gastric adenocarcinoma pathologic stage progression, and the high expression of ESPL1 was significantly correlated with favorable outcomes [23]. In contrast, ESPL1 functions as an oncogene rather than as an antioncogene in breast cancer [24]. Further work is required to resolve the conflicting roles of ESPL1 in cancer and determine its function in BC.
CENPM was also identified as a key gene associated with BC. CENPM showed an 8.7-fold higher expression in BC compared to the levels in normal tissues (p = 5.91E−26). A study comparing the effects of garlic extracts and cisplatin for the treatment of BC identified 515 common anticancer genes, including CENPM. BC patients with low expression of CENPM showed significantly better progression-free survival than those with high expression of CENPM [25]. CENPM encodes centromere protein M, which is a component of the CENPA-NAC (nucleosome-associated) complex. The complex plays a central role in the assembly of kinetochore proteins, mitotic progression, and chromosome segregation [26]. Thus, we speculated that CENPM may be an important gene in BC development and recurrence.
BLM participates in DNA replication and repair and plays an important role in the maintenance of genome stability [27, 28]. Mutations altering BLM function are associated with highly elevated cancer susceptibility [29]. Its roles in BC are unknown, and our research suggests BLM function may be related to BC development and recurrence. The expression of BLM was 2.1-fold higher in BC than the level in normal tissues (p = 5.19E−14), but further research will be required to uncover the underlying mechanisms.
ASPM is the only gene that we found involved that was associated with BC development but not recurrence (Supplement ). ASPM exhibited a 3.7-fold higher expression level in BC than the level in normal tissues (p = 2.56E−13). Abnormal spindle-like microcephaly-associated protein is encoded by ASPM and is involved in mitotic spindle regulation and the coordination of mitotic processes [30]. Recently, another study reported significant overexpression of ASPM in bladder cancer that was associated with invasive pathological characteristics [31]. These results support our findings linking ASPM function to BC.
There are some limitations of this analysis that are worth noting. First, this research was based on data from a single gene array, so the inclusion of other expression data would strengthen the conclusions. Second, altered expression levels of these genes in BC have not been verified by biological methods, so additional experiments to knock down or overexpress these genes should be conducted. Finally, a major drawback of this study is insufficient evidence to suggest changes at the protein level, since the analysis was based only on mRNA expression data and protein interactions were predicted by STRING.
In conclusion, our study suggested CCNB1, ESPL1, CENPM, BLM, and ASPM may be associated with BC development, and CCNB1, ESPL1, CENPM, and BLM may be associated with BC recurrence. The functions of most of these candidate genes have not been the focus of previous studies of BC, and their functions in this cancer should be verified by in vivo and in vitro experiments.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (grant number 81860454) and Jiangxi Provincial Science and Technology Program (grant number 20181BAB205053).
Data Availability
No data were used to support this study.
Disclosure
Qingke Chen, Jieping Hu, and Jun Deng are co-first authors.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
G.J and C.Q.K performed the research, H.J.P and D.J analyzed the data, G.J and F.B designed the study, and H.J.P and G.J wrote the paper.
Supplementary Materials
Supplement figure 1: overall survival analysis for 20 hub genes from cluster SC (surrounding tissue vs. cancer tissue) (A: CDK1; B. CCNB1; C: CDC20; D: CCNA2; E: AURKB; F: EZH2; G: KIF2C; H: AURKA; I: TOP2A; J: BUB1B; K: BUB1; L: CCNB2; M: CDCA8; N: KIF20A; O: TPX2; P: CDC45; Q: FOXM1; R: KIF4A; S: ASPM; T: NDC80). None of the hub genes showed statistical significance in comparing overall survival for high and low expression. Supplement figure 2: disease-free survival analysis for 20 hub genes from cluster CR (cancer tissue vs. recurrent tissue) (A: CCNB1; B: JUN; C: CCNB2; D: ESPL1; E: RHOA; F: CDC45; G: MKI67; H: CDK4; I: AR; J: CDT1; K: FANCI; L: PRC1; M: MCM10; N: CDC25A; O: ASF1B; P: CDK6; Q: BLM; R: CENPM; S: CDKN1A; T: CCR7). Only (P) CDK6 (HR = 1.4, p = 0.015) showed statistical significance that higher expression patients indicated poor disease-free survival. Supplement figure 3: the expressions of (A) JUN and (B) CDK6 were not significantly different between BC and normal tissues (p = 0.639 and 0.466). Supplement Table 1: Cox regression for hub genes in bladder cancer and clinical info.
References
- 1.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2019. CA: A Cancer Journal for Clinicians. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Czerniak B., Dinney C., McConkey D. Origins of bladder cancer. Annual Review of Pathology: Mechanisms of Disease. 2016;11(1):149–174. doi: 10.1146/annurev-pathol-012513-104703. [DOI] [PubMed] [Google Scholar]
- 3.Bladder Cancer: Diagnosis and Management of Bladder Cancer. NICE (2015) Bladder cancer: diagnosis and management of bladder cancer. BJU International. 2017;120(6):755–765. doi: 10.1111/bju.14045. [DOI] [PubMed] [Google Scholar]
- 4.Kamat A. M., Hahn N. M., Efstathiou J. A., et al. Bladder cancer. The Lancet. 2016;388(10061):2796–2810. doi: 10.1016/s0140-6736(16)30512-8. [DOI] [PubMed] [Google Scholar]
- 5.Cumberbatch M. G. K., Noon A. P., On behalf of the EAU Young Academic Urologists—Urothelial Cancer Working Party Epidemiology, aetiology and screening of bladder cancer. Translational Andrology and Urology. 2019;8(1):5–11. doi: 10.21037/tau.2018.09.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulia C., Baldassarra S., Signore F., et al. Role of non-coding RNAs in the etiology of bladder cancer. Genes. 2017;8(11) doi: 10.3390/genes8110339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandrasekar T., Erlich A., Zlotta A. R. Molecular characterization of bladder cancer. Current Urology Reports. 2018;19(12):p. 107. doi: 10.1007/s11934-018-0853-5. [DOI] [PubMed] [Google Scholar]
- 8.Pietzak E. J., Bagrodia A., Cha E. K., et al. Next-generation sequencing of nonmuscle invasive bladder cancer reveals potential biomarkers and rational therapeutic targets. European Urology. 2017;72(6):952–959. doi: 10.1016/j.eururo.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y., Noon A. P., Aguiar Cabeza E., et al. Next-generation RNA sequencing of archival formalin-fixed paraffin-embedded urothelial bladder cancer. European Urology. 2014;66(6):982–986. doi: 10.1016/j.eururo.2014.07.045. [DOI] [PubMed] [Google Scholar]
- 10.Alva A., Friedlander T., Clark M., et al. Circulating tumor cells as potential biomarkers in bladder cancer. Journal of Urology. 2015;194(3):790–798. doi: 10.1016/j.juro.2015.02.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim P. H., Cha E. K., Sfakianos J. P., et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. European Urology. 2015;67(2):198–201. doi: 10.1016/j.eururo.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fantini D., Meeks J. J. Genomic classification and risk stratification of bladder cancer. World Journal of Urology. 2018;37(9):1751–1757. doi: 10.1007/s00345-018-2558-2. [DOI] [PubMed] [Google Scholar]
- 13.Stasik S., Salomo K., Heberling U., et al. Evaluation of TERT promoter mutations in urinary cell-free DNA and sediment DNA for detection of bladder cancer. Clinical Biochemistry. 2019;64:60–63. doi: 10.1016/j.clinbiochem.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Chai N., Xie H.-H., Yin J.-P., et al. FOXM1 promotes proliferation in human hepatocellular carcinoma cells by transcriptional activation of CCNB1. Biochemical and Biophysical Research Communications. 2018;500(4):924–929. doi: 10.1016/j.bbrc.2018.04.201. [DOI] [PubMed] [Google Scholar]
- 15.Liu A., Zeng S., Lu X., et al. Overexpression of G2 and S phase-expressed-1 contributes to cell proliferation, migration, and invasion via regulating p53/FoxM1/CCNB1 pathway and predicts poor prognosis in bladder cancer. International Journal of Biological Macromolecules. 2019;123:322–334. doi: 10.1016/j.ijbiomac.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 16.Kim S.-K., Roh Y.-G., Park K., et al. Expression signature defined by FOXM1-CCNB1 activation predicts disease recurrence in non-muscle-invasive bladder cancer. Clinical Cancer Research. 2014;20(12):3233–3243. doi: 10.1158/1078-0432.ccr-13-2761. [DOI] [PubMed] [Google Scholar]
- 17.Yan X., Liu X. P., Guo Z. X., Liu T. Z., Li S. Identification of hub genes associated with progression and prognosis in patients with bladder cancer. Frontiers in Genetics. 2019;10:p. 408. doi: 10.3389/fgene.2019.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ai X., Jia Z.-M., Wang J., et al. Bioinformatics analysis of the target gene of fibroblast growth factor receptor 3 in bladder cancer and associated molecular mechanisms. Oncology Letters. 2015;10(1):543–549. doi: 10.3892/ol.2015.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan X., Guo Z. X., Liu X. P., et al. Four novel biomarkers for bladder cancer identified by weighted gene coexpression network analysis. Journal of Cellular Physiology. 2019;234(10):19073–19087. doi: 10.1002/jcp.28546. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z., Zhang G., Gao Z., et al. Comprehensive analysis of differentially expressed genes associated with PLK1 in bladder cancer. BMC Cancer. 2017;17(1):p. 861. doi: 10.1186/s12885-017-3884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar R. Separase: function beyond cohesion cleavage and an emerging oncogene. Journal of Cellular Biochemistry. 2017;118(6):1283–1299. doi: 10.1002/jcb.25835. [DOI] [PubMed] [Google Scholar]
- 22.Guo G., Sun X., Chen C., et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nature Genetics. 2013;45(12):1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D., Zhu H., Guo M., et al. Expression and prognostic value of cell-cycle-associated genes in gastric adenocarcinoma. BMC Gastroenterology. 2018;18(1):p. 81. doi: 10.1186/s12876-018-0811-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finetti P., Guille A., Adelaide J., Birnbaum D., Chaffanet M., Bertucci F. ESPL1 is a candidate oncogene of luminal B breast cancers. Breast Cancer Research and Treatment. 2014;147(1):51–59. doi: 10.1007/s10549-014-3070-z. [DOI] [PubMed] [Google Scholar]
- 25.Kim W. T., Seo S.-P., Byun Y. J., et al. The anticancer effects of garlic extracts on bladder cancer compared to cisplatin: a common mechanism of action via centromere protein M. The American Journal of Chinese Medicine. 2018;46(3):689–705. doi: 10.1142/s0192415x18500362. [DOI] [PubMed] [Google Scholar]
- 26.Izuta H., Ikeno M., Suzuki N., et al. Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP-A chromatin of human cells. Genes to Cells. 2006;11(6):673–684. doi: 10.1111/j.1365-2443.2006.00969.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang H., Li S., Zhang H., Wang Y., Hao S., Wu X. BLM prevents instability of structure-forming DNA sequences at common fragile sites. PLoS Genetics. 2018;14(11) doi: 10.1371/journal.pgen.1007816.e1007816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wan L., Han J., Liu T., et al. Scaffolding protein SPIDR/KIAA0146 connects the Bloom syndrome helicase with homologous recombination repair. Proceedings of the National Academy of Sciences. 2013;110(26):10646–10651. doi: 10.1073/pnas.1220921110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broberg K., Huynh E., Schlawicke Engstrom K., et al. Association between polymorphisms in RMI1, TOP3A, and BLM and risk of cancer, a case-control study. BMC Cancer. 2009;9(1):p. 140. doi: 10.1186/1471-2407-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang K., Rezabkova L., Hua S., et al. Microtubule minus-end regulation at spindle poles by an ASPM-katanin complex. Nature Cell Biology. 2017;19(5):480–492. doi: 10.1038/ncb3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Z., Zhang Q., Luh F., Jin B., Liu X. Overexpression of the ASPM gene is associated with aggressiveness and poor outcome in bladder cancer. Oncology Letters. 2019;17(2):1865–1876. doi: 10.3892/ol.2018.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement figure 1: overall survival analysis for 20 hub genes from cluster SC (surrounding tissue vs. cancer tissue) (A: CDK1; B. CCNB1; C: CDC20; D: CCNA2; E: AURKB; F: EZH2; G: KIF2C; H: AURKA; I: TOP2A; J: BUB1B; K: BUB1; L: CCNB2; M: CDCA8; N: KIF20A; O: TPX2; P: CDC45; Q: FOXM1; R: KIF4A; S: ASPM; T: NDC80). None of the hub genes showed statistical significance in comparing overall survival for high and low expression. Supplement figure 2: disease-free survival analysis for 20 hub genes from cluster CR (cancer tissue vs. recurrent tissue) (A: CCNB1; B: JUN; C: CCNB2; D: ESPL1; E: RHOA; F: CDC45; G: MKI67; H: CDK4; I: AR; J: CDT1; K: FANCI; L: PRC1; M: MCM10; N: CDC25A; O: ASF1B; P: CDK6; Q: BLM; R: CENPM; S: CDKN1A; T: CCR7). Only (P) CDK6 (HR = 1.4, p = 0.015) showed statistical significance that higher expression patients indicated poor disease-free survival. Supplement figure 3: the expressions of (A) JUN and (B) CDK6 were not significantly different between BC and normal tissues (p = 0.639 and 0.466). Supplement Table 1: Cox regression for hub genes in bladder cancer and clinical info.
Data Availability Statement
No data were used to support this study.