

E. coli infectious diarrhea is an important contributor to child mortality worldwide. However, diagnosing and thus treating E. coli infections remain challenging due to technical and other reasons associated with the limitations of the traditional culture-based techniques and the requirement to apply Koch’s postulates. In this study, we integrated traditional microbiology techniques with metagenomics and epidemiological data in order to identify cases of diarrhea where E. coli was most likely the causative disease agent and evaluate specific signatures in the disease-state gut microbiome that distinguish between diffuse adherent, enterotoxigenic, and enteropathogenic E. coli pathotypes. Therefore, our methodology and results should be highly relevant for diagnosing and treating diarrheal infections and have important applications in public health.
KEYWORDS: Escherichia coli, infectious diarrhea, gut microbiome, metagenomics, 16S rRNA, pathotypes, Ecuador, clinical metagenomics
ABSTRACT
Escherichia coli is a leading contributor to infectious diarrhea and child mortality worldwide, but it remains unknown how alterations in the gut microbiome vary for distinct E. coli pathotype infections and whether these signatures can be used for diagnostic purposes. Further, the majority of enteric diarrheal infections are not diagnosed with respect to their etiological agent(s) due to technical challenges. To address these issues, we devised a novel approach that combined traditional, isolate-based and molecular-biology techniques with metagenomics analysis of stool samples and epidemiological data. Application of this pipeline to children enrolled in a case-control study of diarrhea in Ecuador showed that, in about half of the cases where an E. coli pathotype was detected by culture and PCR, E. coli was likely not the causative agent based on the metagenome-derived low relative abundance, the level of clonality, and/or the virulence gene content. Our results also showed that diffuse adherent E. coli (DAEC), a pathotype that is generally underrepresented in previous studies of diarrhea and thus, thought not to be highly virulent, caused several small-scale diarrheal outbreaks across a rural to urban gradient in Ecuador. DAEC infections were uniquely accompanied by coelution of large amounts of human DNA and conferred significant shifts in the gut microbiome composition relative to controls or infections caused by other E. coli pathotypes. Our study shows that diarrheal infections can be efficiently diagnosed for their etiological agent and categorized based on their effects on the gut microbiome using metagenomic tools, which opens new possibilities for diagnostics and treatment.
IMPORTANCE E. coli infectious diarrhea is an important contributor to child mortality worldwide. However, diagnosing and thus treating E. coli infections remain challenging due to technical and other reasons associated with the limitations of the traditional culture-based techniques and the requirement to apply Koch’s postulates. In this study, we integrated traditional microbiology techniques with metagenomics and epidemiological data in order to identify cases of diarrhea where E. coli was most likely the causative disease agent and evaluate specific signatures in the disease-state gut microbiome that distinguish between diffuse adherent, enterotoxigenic, and enteropathogenic E. coli pathotypes. Therefore, our methodology and results should be highly relevant for diagnosing and treating diarrheal infections and have important applications in public health.
INTRODUCTION
Diarrheal diseases remain a major public health issue worldwide, especially in developing countries where poor sanitary conditions and limited access to clean water exacerbate the burden (1). Although most diarrheal cases self-resolve relatively quickly (2, 3) and hence the causative agent(s) is never identified, there are instances where acute diarrheal infections lead to death, making detailed investigations of the causative agents and their signatures necessary. Such cases include not only acute child diarrhea in the developing world but also foodborne outbreaks linked to contaminated food and other causes in developed countries (2–4).
Diagnostic testing for enteric pathogens has historically relied on culture-based techniques or culture-independent molecular assays such as PCR that are based on cultured material. Accordingly, reference (cultured) isolates are the foundation for public health surveillance (5–7). It is important to realize, however, that culture-based techniques are typically time-consuming and costly and require significant expertise, while they frequently miss moderately or distantly related relatives of the reference isolates, since many bacterial microorganisms are difficult to growth or are even nonculturable, although still viable. For example, some pathogens, including Escherichia coli, can quickly lose the ability to be cultured upon exposure to laboratory conditions (8). In addition, culture-based techniques may detect transient pathogens that are not related to the current diarrheal episode. Several of these limitations also apply to (culture-derived) culture-independent methods: in cases where PCR detection is applied for example, there is still a high risk of detecting traces of asymptomatic carriage or residual DNA from previous infections or missing divergent (target) sequence of close or distant relatives to the reference isolate (9, 10). Further, and perhaps more importantly, culture does not provide quantitative estimates of a pathogen's abundance, virulence potential, and diversity, and it does not allow the characterization of the gut microbiome response, which could be important for diagnosis.
In the United States, a total of 38.4 million cases of foodborne illness per year cannot be attributed to specific causes, and the proportion caused by yet-to-be-described microbial agents is unknown (9, 11, 12). In developing country settings, even using highly sensitive methods such as qPCR to detect a broad array of pathogens, >10% of moderate-to-severe diarrhea cases cannot be traced to an etiologic agent, and the fraction of false-positive signal for cases with detected pathogens, due to transient pathogens for instance, remains essentially speculative (12). Culture-independent metagenomic approaches can be used to robustly assess diarrheal infections.
E. coli is a gut commensal of vertebrates, including humans (13); it can nevertheless cause a broad range of diseases, including intestinal and extraintestinal infections, through the acquisition of accessory genes. The relevance of this species in diarrheal disease has been extensively illustrated. For example, the Global Enteric Multicenter Study (GEMS) demonstrated that Shigella (a distinct lineage within the E. coli phylogenetic clade) and enterotoxigenic E. coli (ETEC) producing heat-stable toxin (ST), either alone or in combination with heat-labile toxin (LT), are among the most important pathogens associated with moderate-to-severe diarrhea in children younger than 5 years old in developing countries (11, 12). ETEC, enteropathogenic E. coli (EPEC), and Shigella are responsible for an estimated 18% of diarrhea deaths in children <5 years old globally (12). Despite the relevance of E. coli as a diarrheagenic pathogen, little is known about the effect(s) of pathogenic E. coli infections on the gut microbiome, such as how the structure and diversity of the community changes during active infections.
Although commensal E. coli is typically a relatively minor component of the colonic gut microbiome in humans, where it represents <0.1% of the total bacterial cells (estimated at ∼108 cells/g) (13, 14), during infection with an invading pathogenic E. coli strain or another enteric pathogen, the overall signal of E. coli in the gut microbiome can increase substantially (15, 16), allowing the detection and recovery of genomes based on culture-independent metagenomic techniques. In addition, quantifiable shifts in the proportion and diversity of the entire gut microbial community in response to the enteric infection disturbance have been observed (17–20). However, the number of samples analyzed in these previous studies were limited, and the methodological approaches employed were based almost exclusively on taxonomic marker genes such as 16S rRNA that have limited resolution. Furthermore, and perhaps more importantly, in all previous studies the etiological agent of infection was inferred by DNA sequencing and was not validated by an independent approach. Accordingly, it remains unclear whether different pathogens such as different E. coli pathotypes produce distinct alterations in the indigenous microbiome due to their characteristic mechanism of infection and virulence factors they excrete and employ during infection. Alterations of the gut microbiome composition upon infection have been shown to take place quickly, within 1 to 2 days or a few hours in some cases (16, 20). Thus, metagenomic sampling following the onset of diarrhea, even in a cross-sectional sampling strategy, could potentially provide insights into pathotype-specific signatures and quick results for diagnostics.
During the interaction with the intestinal epithelial cells, in the phase of attachment and colonization, distinctive machineries of pathogenicity are known to be used by each of the six known E. coli pathotypes, depending on whether the pathogen invades the cell, produces biofilms, or secretes toxins (21). For example, ETEC is thought to adhere to the small bowel mucosa and deliver secretory enterotoxins (22). Enterohemorrhagic E. coli (EHEC) adheres to the colonic mucosa and transduces a signal, resulting in secretory diarrhea. Concurrently, the organism releases Shiga toxin, resulting in local and systemic effects (21). Enteroaggregative E. coli (EAEC) adheres to intestinal epithelial cells and produces a thick mucous gel (biofilm) and causes intestinal secretion and damage (23). Diffusely adherent E. coli (DAEC) has been shown to elicit elongation of microvilli in vitro, although this effect has not been demonstrated in vivo, and has been considered not as virulent as other pathotypes (24). EPEC elicits the attaching and effacing lesion in the small bowel, resulting in intestinal secretion (25) and enteroinvasive E. coli (EIEC) invades the colonic mucosa, giving rise to inflammatory enteritis (26). Among the six pathotypes, EPEC and ETEC have received special attention in developing countries because they are believed to be major pathogens causing diarrhea in children 5 years old and younger (1, 14). It remains to be elucidated whether these biological differences in infectious mechanisms disturb the gut microbial community in different, distinguishable ways.
We devised a novel bioinformatic approach that combined traditional, isolate-based, and PCR techniques with metagenomic and epidemiological data to identify diarrheal cases where E. coli was most likely the causative agent and evaluate whether pathotype-specific signatures in the disease-state gut microbiome exist that distinguish among different E. coli infections. For this purpose, we took advantage of a large epidemiological, case-control study of diarrhea that was carried out over a period of 18 months in Northern coastal Ecuador (named EcoZUR for “E. coli en Zonas Urbanas y Rurales”). The study was uniquely suited to address these questions as it included data on diarrheal disease outcome, a large collection of pathogenic E. coli isolates from diarrheal and nondiarrheal (control) samples, and other sociodemographic and clinical data from study subjects. Most genetic studies of pathotypes have been carried out using collections of isolates unrelated in time and space. Our study circumvents these previous limitations and allowed us to also observe E. coli strain relatedness.
We previously reported on risk factors for diarrhea and pathotype distribution (27) in the EcoZUR study. Here, we report on a subset of the EcoZUR samples that comprised cases of infected children with three major E. coli pathotypes (DAEC, ETEC, and EPEC) and their age-matched controls (no diarrhea). Our study addressed three main objectives: (i) to describe and compare the overall gut microbiome diversity between cases of diarrhea and controls using both 16S rRNA marker genes and whole shotgun metagenomic data; (ii) to identify cases of diarrhea where E. coli was presumably the etiological agent based on a combination of metagenomics, isolate genome sequencing, and epidemiological data; and (iii) to determine whether pathogen-specific signatures in the disease gut microbiome exist that distinguish between DAEC, EPEC, and ETEC infections.
RESULTS
Study design and demographics of the study population.
We analyzed samples collected in the EcoZUR study, a case-control study of diarrhea carried out in four sites across a rural-urban gradient in northern Ecuador between April 2014 and September 2015. Participants were recruited from Ecuadorian Ministry of Health hospitals and/or clinics in Quito, Ecuador’s capital, with approximately 1.6 million inhabitants; Esmeraldas, a coastal city in the northwest of Ecuador with ∼160,000 inhabitants; Borbón, a town in Esmeraldas Province with ∼7,000 inhabitants; and rural villages located in the Borbón region with <800 inhabitants each. Cases comprised individuals who presented with diarrhea, defined as three or more loose stools in a 24-h period, and controls comprised age- and site-matched individuals for any other complaint, with no record of diarrhea or vomiting for the previous 7 days. Both cases and controls were excluded if they reported antibiotic usage in the prior week or if they had not lived in the study location for at least 6 months. Data on demographics, medical history, water, sanitation, hygiene (WASH) practices, animal contacts, and recent travel history were collected from all participants.
Cases and controls were similar on key demographic variables (e.g., age and race), indicating that the case-control matching was robust (27). Sociodemographic and WASH conditions varied as expected across the rural-urban gradient, with urban sites reporting higher levels of improved sanitation and improved drinking water and rural sites reporting a higher proportion of individuals receiving government assistance, and a lower proportion of individuals with fixed employment and higher education levels. Additional information and previous results from the EcoZUR study can be found elsewhere (Smith et al. [27]). Briefly, we found that travel to urban destinations was associated with higher risk of diarrhea and diarrheagenic E. coli infections.
Fresh stool samples were incubated in E. coli-specific media (MacConkey agar media) and tested for different pathotypes based on the presence of specific virulence genes determined by PCR (Table 1; see additional details in Materials and Methods). All diarrhea samples that resulted in a PCR-positive signal for the presence of any marker gene characterizing DAEC, ETEC, or EPEC pathotypes and obtained from young children between 1 and 6 years old were selected for further analysis. These samples were matched with randomly selected control samples within the same age and location categories, without any diarrheagenic E. coli infection detected through PCR. This strategy resulted in a total of 80 samples that were taxonomically screened by amplicon sequencing of the 16S rRNA gene (38 diarrhea and 42 control samples). All diarrhea samples and a randomly selected subset of 23 control samples were subjected to whole shotgun metagenomic sequencing (see Fig. S1 and Table S1 in the supplemental material). The diarrheal samples included 16 samples that were PCR positive for afaB , the virulence marker of DAEC; 10 samples that were positive for bfpA, the marker gene for typical EPEC; and 12 samples that were positive for eltA and/or sta, marker genes for ETEC. All E. coli isolates recovered from the selected diarrheal samples were also sequenced for genomic characterization and comparison (see Table S3 in the supplemental material).
TABLE 1.
Primers used for PCR to detect diarrheagenic E. coli virulence genes
| E. coli pathotype | Gene | Primer sequence (5′–3′) | Size (bp) | Reference |
|---|---|---|---|---|
| Enteroaggregative E. coli (EAEC) | aggR | GTATACACAAAAGAAGGAAGC | 254 | 71 |
| ACAGAATCGTCAGCATCAGC | ||||
| Enterotoxigenic E. coli (ETEC) | lt | GCGACAAATTATACCGTGCT | 708 | 72 |
| CCGAATTCTGTTATATATGT | ||||
| sta | CTGTATTGTCTTTTTCACCT | 182 | 72 | |
| GCACCCGGTACAAGCAGGAT | ||||
| Typical enteropathogenic E. coli (EPECt) | bfp | CAATGGTGCTTGCGCTTGCT | 324 | 72 |
| GCCGCTTTATCCAACCTGGT | ||||
| Atypical enteropathogenic E. coli (EPECa) | eaeA | GACCCGGCACAAGCATAAGC | 384 | 73 |
| CCACCTGCAGCAACAAGAGG | ||||
| Typical enteroinvasive E. coli (EIEC) | ipaH | GCTGGAAAAACTCAGTGCCT | 424 | 72 |
| CCAGTCCGTAAATTCATTCT | ||||
| Diffusely adherent E. coli (DAEC) | afaB | GCTGGGCAGCAAACTGATAACTCTC | 750 | 74 |
| CATCAAGCTCTTTGTTCGTCCGCCG | ||||
| Shiga-toxin-producing E. coli (STEC) | stx1 | ATAAATCGCCATTCGTTGACTAC | 180 | 73 |
| AGAACGCCCACTGAGATCATC | ||||
| stx2 | GGCACTGTCTGAAACTGCTCC | 255 | 73 | |
| TCGCCAGTTATCTGACATTCTG |
High frequency of coeluting human DNA in diarrhea samples.
Analysis of the composition of metagenomic reads showed that specimens collected in this study differed in the proportional amount of microbial and human DNA sequenced. The average percentage of human reads detected in the diarrhea group was 17.8%, while in the control group it was only 0.07%. Samples with a large fraction of detected human contamination belonged mostly to DAEC-positive (as determined by culture and PCR test for pathotype-specific genes) and EPEC-positive groups (Fig. 1A). Our analysis also revealed no correlation between the fraction of human reads detected in our libraries and the severity of the disease, measured as the number of days with diarrhea previous to the sampling day or the detection of blood in the specimen. However, we did observe a significant increase in the fraction of human reads with detection of mucus in stool specimens versus those with no mucus detected (Welch’s two-sample test, P = 0.004).
FIG 1.

Abundance of human reads and estimated coverage of the metagenomic data sets obtained in this study. (A) Assignment of recovered metagenomic raw reads to three groups: human (purple), discarded due to low quality (orange), and fraction passing quality control and not being of human origin (green). (B) Fitted Nonpareil curves and estimated average coverage for each metagenome after human and low-quality reads where removed from each data set. The horizontal dashed lines indicate 100% (upper red line) and 95% (bottom red line) coverage. Empty circles indicate the size (x axis) and estimated average coverage (y axis) of the data sets, and the lines after that point are projections of the fitted model. The inset plot shows the distribution of estimated coverage values in randomly drawn subsets of 1,000 reads per library for each pathotype and control group. Note that samples where DAEC was isolated showed less diverse communities (higher coverage) than other groups, including control samples.
After removal of the human reads, between 221 Mbp and 3.2 Gbp of reads per metagenome remained for analysis (average = 1.78 Gbp) (Table S2). One ETEC sample (Q53) had only ∼1,200 reads after quality control and was therefore removed from further analysis. To evaluate the fraction of the total extracted DNA from the stool sample that was sequenced (i.e., determine the coverage of the microbial community by sequencing), we estimated the sequencing coverage using Nonpareil 3, a read redundancy-based algorithm (28). Although the sequencing coverage varied among samples and pathotype groups, generally ≥80% of the microbial community was covered in the majority of samples, except for one EPEC sample (R126) that had very low coverage and was therefore discarded from further analysis (Fig. 1B; Table S2). The coverage estimates also reflected differences in the complexity of the microbial communities among pathotypes, showing that the DAEC group (defined by the recovery of a DAEC isolate) contained, in general, metagenomes with simpler microbial communities compared to the two other pathotypes (Kruskal-Wallis rank sum test, χ2 = 9.07, df = 3, P = 0.02), whereas control samples had more complex (diverse) gut microbial communities compared to all diarrheal samples (Wilcoxon rank sum test, W = 278, P = 0.03). Overall, our community coverage results suggested that despite the high frequency of coeluting host DNA in some disease samples, our metagenomic sequencing effort was adequate to assess the microbial community disturbance during diarrhea and to detect and recover abundant microbial community members and the putative pathogen.
Differences in microbial community composition and diversity between diarrhea and control samples.
The overall microbial community compositions and diversities in diarrhea and control groups were assessed based on 16S rRNA gene amplicon sequencing. Comparison of the normalized relative abundance at the phylum level indicated that three major phyla dominated the gut microbial communities in both diarrheal and control individuals, namely, Bacteroidetes, Firmicutes, and Proteobacteria. In general, Bacteroidia and Clostridia were the two most abundant classes in all samples. In contrast to the control group, the majority of samples in the diarrhea group also exhibited a large proportion of sequences belonging to Gammaproteobacteria, a bacterial group comprising several enteric pathogens (Wilcoxon rank sum test, W = 1,231, P = 1.733e–05), which indicated a possible underlying bacterial infection in individuals with diarrhea (Fig. S2A). Alpha-diversity analysis based on Faith’s phylogenetic diversity revealed significant differences between case and control data sets. Consistent with several other recent studies (see, for example, references 15, 16, 17, 18, 19, and 29), control stool samples showed, in general, a significantly higher microbial diversity than the cases (Kruskal-Wallis test, H = 15.9, P = 6.06e–05) (Fig. S2B). Analysis of the overall community dissimilarity at the genus level also revealed significant differences between diarrhea and control groups (permutational multivariate analysis of variance [PERMANOVA], pseudo-F = 2.47, P = 0.002). Diarrhea samples clustered more closely together compared to control samples, although some overlap between several samples of the two groups was also observed (Fig. S2C).
Differences in the overall relative abundance of the OTU taxonomically assigned to E. coli in diarrhea compared to control samples, by about 1 order of magnitude (mean of 6.39% of total reads in cases versus 0.81% in control, Welch’s two-sample test, P = 0.009), were also detected. Few samples had as much as 30% of their 16S rRNA gene-containing read sequences assigned to E. coli, although other diarrheal samples had very low E. coli abundances, comparable to those in control samples (Fig. S2D). Conversely, three samples from the control group had as much as a 3 to 5% relative abundance of E. coli, which might indicate an asymptomatic E. coli infection and/or higher abundance of commensal E. coli. Therefore, we next sought to identify samples where pathogenic E. coli was most likely the causative agent of diarrhea by examining the companion shotgun metagenomic and epidemiological data.
Identification of disease samples where E. coli was most likely the causative agent of diarrhea.
Our observations of the overall community composition, diversity, and estimated taxon abundance in the diarrheal samples based on 16S rRNA gene amplicon data (see, e.g., Fig. S2D) indicated that E. coli was likely not the causative agent of disease in all diarrhea samples, even though an E. coli pathotype isolate was cultured from all diarrhea samples and at least one virulence marker gene was detected by PCR. In other words, isolation and PCR detection of E. coli pathotypes in stool samples from individuals with diarrhea might have reflected the recovery of a rare isolate in situ that was not involved in infection, or a stage of infection with E. coli but not necessarily diarrhea caused by E. coli. Therefore, we next aimed to determine the diarrheal cases that were most likely attributable to pathogenic E. coli. For this purpose, we assessed the metagenomic data sets, recovered metagenome-assembled population genomes (MAGs) from the metagenomes, and the genomes of isolates for five main lines of evidence.
(i) Metagenomic abundance. We estimated the in situ metagenomic abundance of the E. coli pathotype isolate (or the MAG that originated from the same sample as the isolate) and a reference commensal E. coli strain HS (NC_009800.1), using the diarrhea metagenomic data set from which the isolate was recovered. We expected that the estimated abundance of the pathogenic isolate would be higher than the commensal strain in the diarrhea metagenome, as well as in control metagenomes in a competitive, read-based search. Competitive mapping, e.g., mapping reads against a single combined data set containing both commensal and pathogenic genomes, was preferred over regular mapping to avoid double counting reads that map to very conserved regions that could potentially lead to overestimation of the calculated abundance.
(ii) Virulence factors. We evaluated the presence or absence of a large array of E. coli virulence factors, including pathotype-specific marker genes and enterotoxins in the metagenomic data sets of diarrhea and control samples, based on the sequencing coverage of the genes shown by metagenomic reads relative to the coverage of the rest of the genome (e.g., these factors sometimes did not assemble as part of the corresponding MAG; hence, a read-based approach was used; see Materials and Methods for details). We expected that the abundance of virulence factors would be higher in the diarrhea metagenomes than in the control metagenomes.
(iii) Intrapopulation diversity. We estimated the degree of E. coli intrapopulation diversity by calculating the average nucleotide identity of the metagenomic reads (defined as ANIr) mapping to the reference genome (pathotype isolate or reference commensal in competitive blast searches) with a percent identity between 90 and 100%, i.e., reads representing the total E. coli population in a sample. We expected that the degree of intrapopulation diversity (or clonality) of the pathogenic E. coli population would be lower (more clonal) compared to the E. coli population in control samples, as is often the case for active infection-associated pathogens (30).
(iv) Membership in disease-associated clonal complexes. We examined the phylogenetic clonal complex that the pathotype isolate was assigned to (or the MAG that originated from the same sample as the isolate). For the isolate of interest, we expected that other isolates within the same clonal complex would be more frequently associated with disease than control samples. For this purpose, we built a core-genome phylogeny based on ∼1,200 orthologous genes for a total of 263 E. coli isolates obtained from the EcoZUR project. Clonal complexes within the phylogenetic tree were identified based on their ANI values (31) using the PAM algorithm (partitioning around the medoids) with k medoids, where k was determined by the local gain in the average Silhouette width for each level of clustering (see Fig. S3) (32). Clonal complexes corresponded to sets of strains clustered together in the core genome phylogeny, typically with >99% ANI among them (versus <99% ANI between clonal complexes).
(v) Virulent E. coli MAGs. Finally, we performed binning of the assembled metagenomic contigs in order to recover high-quality E. coli MAGs (Table S2). In addition, we assessed the presence of virulence genes and enterotoxins in the recovered MAGs and built a phylogenetic tree using isolates and MAGs derived from the same sample to evaluate whether the isolates obtained in culture were good representatives of the indigenous population(s). We expected to recover complete, high-quality E. coli MAGs carrying canonical virulence genes.
Taking these five lines of evidence together, our expectation was that the putative E. coli pathogen (isolate and/or MAG) was likely the causative agent of disease in the sample that it was recovered from when (i) the E. coli population presented a higher estimated metagenomic abundance compared to the commensal E. coli strain HS; (ii) the E. coli population had more virulence genes than its counterparts from control samples, including the detection of key enterotoxins and pathotype-specific marker genes; (iii) the corresponding diarrheal metagenome harbored a more clonal population of E. coli compared to control metagenomes; (iv) the isolates were assigned to a clonal complex in the core genome phylogenetic tree that was enriched in isolates from other disease (as opposed to control) samples; and (v) we were able to recover E. coli MAGs harboring the typical pathotype-specific virulence genes. We tested these expectations for the three pathotype groups with the highest numbers of diarrhea cases in our data set: DAEC, ETEC, and EPEC.
DAEC as the causative agent of diarrhea.
Our results indicated that approximately 50% of the samples from which DAEC isolates were obtained (i.e., 8 samples of 16 total), showed metagenomic signatures consistent with the isolate being the causative agent (Fig. 2). These samples (Q51, Q196, E158, Q56, Q65, E230, E124, and E27) exhibited the following signatures: (i) higher abundance of the pathogenic isolate compared to the reference commensal strain or the total E. coli population in the control samples, i.e., 27.81% versus 0.6%, on average (Fig. 2A); (ii) detection of 40 or more virulence factors that were present in metagenomic contigs or were binned into MAGs at similar or higher sequence coverage than the MAG (Fig. 2B); (iii) reduced intrapopulation sequence diversity with ANIr values of ≥99% for the isolate and typically ANIr values of <99% for the reference commensal genome (Fig. 2C); (iv) the recovered pathotype isolate(s) was generally grouped in 11 phylogenetic clonal complexes composed of more isolates originating from cases of diarrhea than control samples (Fig. 2D); and finally, (v) the recovery of seven high-quality E. coli MAGs that encoded the pathotype (afaB) and other E. coli virulence factors. We also observed that the isolates within these clonal complexes were evenly distributed between rural and urban settings, which suggested that distinct DAEC genotypes were each associated with small-scale diarrheal outbreaks in northern Ecuador (see Fig. S3 in the supplemental material).
FIG 2.

Characteristics of samples where DAEC was most likely the causative agent. (A) Estimated metagenomic abundance of the reference commensal E. coli (strain HS, in light red) and the DAEC isolate (in red) recovered from the sample, along with the ELISA-based detection of rotavirus and bioinformatic detection of Adenovirus_F for each sample analyzed (rows). Samples where high-quality E. coli MAGs were recovered from the corresponding metagenome are denoted by a star. (B) Presence (detection) of four hallmark virulence factors in the metagenome, including the DAEC marker gene (afaB) and three enterotoxins, i.e., the hemolysin subunit B (hlyB), the heat-labile enterotoxin (eltA), and the secreted autotransporter toxin (sat). (C) Estimated E. coli intrapopulation diversity measured by ANIr of reads against the reference commensal strain HS (light orange) and the isolate obtained from the sample (dark brown). To avoid any potential bias by low in situ abundance, only samples where the average sequence depth of the reference genome was ≥1× were evaluated for ANIr. (D) Number of isolates that originated from cases of diarrhea (in red) versus control samples (in green) and were assigned in the same core-genome-based clonal complex as the isolate (epidemiology).
Although two DAEC-positive diarrhea samples showed ANIr values of >99% and the presence of the afaB gene (e.g., Q310 and Q49), they presented lower metagenomic abundances of the isolate compared to the control samples and/or other positive samples and relatively lower numbers or the absence of virulence genes. In addition, for these samples, no MAGs were recovered, a finding consistent with the relatively low abundance of E. coli. Therefore, these samples were not as conclusive with respect to whether the isolate was the etiological agent. Of particular interest was the observation that two of these samples (E27 and B89) were also positive for rotavirus and two (B274 and Q310) had a substantial number of reads mapping to adenovirus, a nonenveloped, double-stranded DNA virus causing acute gastroenteritis primarily in children (33). These results indicated that despite the PCR detection of a DAEC marker gene in these individuals, other viral pathogens, rather than E. coli, might have been responsible for the diarrhea phenotype.
To evaluate whether or not the DAEC MAGs were representative of the DAEC isolates recovered from the same samples, as well as the diversity of the population(s) present in the disease samples, we performed a phylogenetic reconstruction with MAGs and isolates. Core-gene phylogeny revealed that MAGs and isolate genomes clustered together in the majority of samples (Fig. 3A), with the average ANI between the pair of MAG and isolate genome originating from the same sample being 99.92%, except for sample Q196, where the estimated ANI was 97.29%. Further evaluation showed that the recovered MAG from sample Q196 was a high-quality genome, with 97.1% completeness, 1.85% contamination, and a 43.45 estimated index of strain heterogeneity (SH; scale between 0 and 100). This finding suggests that the isolate represented a minor member of the total E. coli population in sample Q196.
FIG 3.

Core genome-based phylogenetic and ANI relatedness among DAEC (A) and ETEC (B) isolates and MAGs recovered from the same sample of infectious diarrhea. Annotations in blue denote pairs of genomes (isolates and MAGs) that clustered closely in the phylogenetic reconstruction, while annotations in red denote more divergent pairs of genomes. The environmental E. coli strain TW158338 was used an outgroup.
Next, we evaluated whether any correlation existed between the percentage of human reads detected in the DAEC metagenomes and the estimated metagenomic abundance of the isolate for the set of eight samples that had strong evidence of DAEC-caused diarrhea. Our results showed a relatively strong positive linear correlation between the two variables (Pearson’s R = 0.65, P = 0.08) (Fig. 4). This observation indicated that the fraction of human reads observed in the metagenome might be directly related to the infection by pathogenic DAEC strains.
FIG 4.
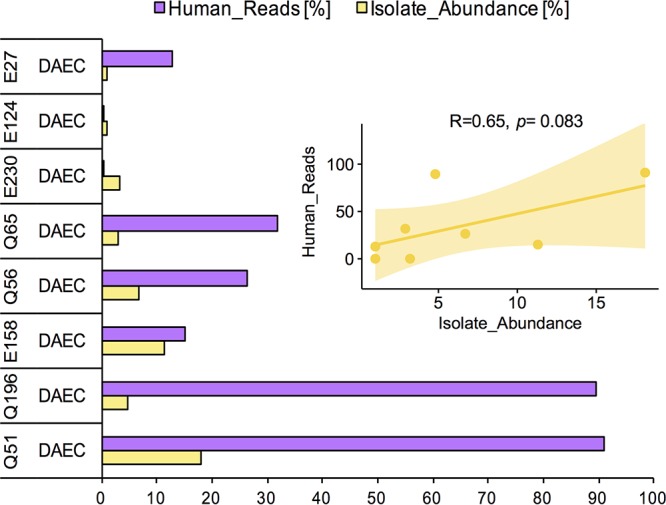
Correlation between recovered fraction of human metagenomic reads and DAEC pathogen abundance. The bar plot shows the observed percentage of the total metagenomic reads assigned to human (purple) and the estimated metagenomic abundance of the E. coli genome for the samples with strong evidence of DAEC infection. The inset plot shows the Pearson correlation analysis of the two variables, revealing a positive linear correlation.
ETEC and EPEC as causative agents of diarrhea.
A similar approach was applied to the samples that were positive for ETEC and EPEC by isolation and PCR to identify representative cases of infection (see Fig. S4 and S5 in the supplemental material). The signal of E. coli infection was, in general, more clear in ETEC (compared to DAEC) but much less clear in EPEC. Seven of ten samples (70%) had strong evidence of infection caused by the ETEC isolate (E184, Q294, B295, B45, B62, B244, and B255). In these seven metagenomic samples, at least one of the two ETEC marker genes, i.e., heat-labile (eltA) and/or heat-stable (sta) enterotoxins, was detected. The remaining three samples (i.e., B109, B68, and B200) did not show strong evidence of E. coli being the infectious agent (Fig. S4), since they presented a very low abundance of E. coli and/or the absence of at least one ETEC marker gene and/or relatively low clonality (ANIr < 99%). The recovery of high-quality ETEC MAGs was possible for six samples. All ETEC isolates and MAGs cluster together in the core genome phylogeny in only three clonal complexes, independent of the geographic origin of the genomes (Fig. S3 and Fig. 3B).
For EPEC-positive samples, the diagnostic marker gene (eaeA) was recovered in only two of the eight samples (E187 and E162). However, even for the latter two samples, the analysis in intrapopulation diversity revealed no clonal population. Recovery of high-quality E. coli MAGs was possible in only one sample (R135), but <50% of the total EPEC hallmark or other virulence genes were detected in the metagenomes or the MAG (Fig. S5). Given that no strong metagenomic signature of pathogenicity was observed in the majority of EPEC samples, we excluded this pathogroup from further analysis that focused on the detection of further pathotype-specific gut microbiome signatures.
Gut microbiome signatures of DAEC and ETEC infections.
The identified samples with strong evidence of DAEC or ETEC infection were analyzed further in order to elucidate differences between diarrhea and control groups on the gut microbiome such as gene content or microbial community composition alterations. In total, seven discriminative taxa between diarrhea and control samples were detected: Prevotella copri (Kruskal-Wallis test, P = 0.05), E. coli (P = 2.26e–5), Alloprevotella tannerae (P = 0.04), Campylobacter concisus (P = 0.01), Haemophilus haemolyticus (P = 0.05), Fusobacterium mortiferum (P = 0.02), and Bifidobacterium longum (P = 0.04) (Fig. S6A). P. copri was most strongly enriched in the control group, while E. coli was the phylogroup mostly differentiating the diarrhea group, revealing a negative correlation in the abundance pattern for these two species (Spearman’s rho = −0.39, P = 0.012) (Fig. S6C). In addition, a principal-component analysis (PCA) of species abundance indicated that microbial communities from DAEC versus ETEC infections clustered separately (Fig. S6B).
To explore whether pathotype-specific alterations (signatures) in the gut microbiome exist that discriminate DAEC versus ETEC infections, we evaluated whether or not differences in gene content and/or composition existed after removing reads assigned to E. coli from the metagenomic libraries and focusing on the samples with strong metagenomic evidence for infection identified above. The initial taxonomic characterization revealed at least four species that were discriminatory of DAEC versus ETEC infections. Specifically, Fusobacterium mortiferum (P = 0.025) and Campylobacter concisus (P = 0.011) were significantly more abundant in ETEC infections (Fig. 5A and B), while Bifidobacterium longum (P = 0.040) and Alloprevotella tannerae (P = 0.046) were significantly more enriched in DAEC infections (Fig. 5C and D). A PCA based on taxonomic composition at the species level also revealed that metagenomes associated with ETEC infections tended to be taxonomically more similar among themselves, whereas DAEC samples showed more diversity. Notably, one sample positive for DAEC (E124) also showed the eltA gene in the metagenome, which is associated with ETEC, and tended to be more dissimilar to any other DAEC or ETEC metagenome, indicating that this individual could have suffered from a coinfection with DAEC and ETEC strains. Further within-species diversity analysis based on single nucleotide polymorphism (SNP) patterns of conserved genes (strain level) suggested that at least two E. coli strains coexisted in the community rather than one strain harboring DAEC and ETEC virulence genes. One strain dominated the population with a ∼94% relative abundance, while the second strain represented ∼6% of the population. On the other hand, the metabolic gene profiling of the gut microbiome did not reveal substantial differences between infections by the two pathotypes. Therefore, our results revealed that significant, albeit rather narrow, taxonomic but not metabolic signatures might exist in the gut microbiome that differentiate DAEC from ETEC infections, which warrants further investigation.
FIG 5.

Differentially abundant (diagnostic) taxa between DAEC and ETEC infections. Differentially abundant species were reported if they had a corrected P value of ≤0.05 and an effect size (the magnitude of the difference between groups) of ≥0.8. (A and B) Proportions of metagenomic sequences assigned to Fusobacterium mortiferum and Campylobacter concisus, respectively. (C and D) Proportions of sequences assigned to Bifidobacterium longum and Alloprevotella tannerae, respectively. (E) PCA plot based on the taxonomic composition of each metagenome (annotated at the species level using clade-specific marker genes with MetaPhlAn2) after removal of human and E. coli reads from the libraries.
DISCUSSION
Diagnostic testing for diarrheal pathogens has relied for decades on culture-based techniques that do not provide a quantitative estimate of the pathogen or the response of the gut microbiome to the infection. Consequently, a significant fraction of diarrhea episodes remains undiagnosed, and others are spuriously associated with infections of putative pathogens, due to the challenges associated with the accurate detection of the etiological agent using traditional techniques. The signatures of pathogen-specific disturbances in the ecology of the healthy gut microbiome also remain uncharacterized. Here, we provide a novel metagenome-based methodology that employs pathogen population in situ abundance, the level of intrapopulation diversity, and virulence gene content (see, for example, Fig. 2) to study diarrheal cases and to provide diagnostic resolution that is usually not attainable by traditional methods.
Application of our methodology to child diarrheal samples and age-matched controls collected in northern Ecuador showed that DAEC was likely the causative agent of several diarrheal cases, and the DAEC isolates recovered from these samples were assigned to 11 distinct clonal complexes. These complexes were apparently associated with small-scale diarrheal outbreaks given that the great majority of isolates in the complexes (>75% of total) were recovered from disease as opposed to control samples (despite the similar number of case and control samples and isolates in our data set), and the subjects that provided the disease samples were from both rural and urban settings.
Even though DAEC is not thought to be a highly virulent E. coli pathotype (24, 34), DAEC infections were found to be accompanied by coelution of large amounts of human DNA and conferred small (in terms of number of taxa affected) but significant shifts in the composition of the gut microbiome relative to the control or infections caused by other pathotypes (e.g., ETEC) according to our study. These findings echoed the findings by Huang et al. (16), who applied shotgun metagenomic to stool samples collected from two geographically isolated foodborne outbreaks in the United States, where the etiologic agent was identified by culture-dependent methods as distinct strains of Salmonella enterica subsp. enterica serovar Heidelberg. Similar to our study, the acute Salmonella infections described by the Huang et al. study were accompanied by a high frequency of coeluting human DNA sequences, significant shifts in the gut microbiome composition and diversity relative to healthy control samples, signatures of high abundance of the pathogen in the metagenomic diarrheal sample, and reduced intrapopulation diversity. Hence, it appears that the DAEC infections identified by our metagenomic analysis were likely caused by a DAEC strain.
Although the number of samples analyzed here was limited and thus the compositional shifts identified should be considered preliminary results only, our results do indicate that there may be consistent signatures in the gut microbiome that could provide reliable additional diagnostic features to distinguish different etiologic agents of diarrhea. The negative correlation in abundance between E. coli and P. copri was a notable such signature that should be explored in future work in order to elucidate the underlying mechanism(s) for the observed anticorrelation pattern. Of particular interest also was the observation that, in general, ETEC samples presented lower average metagenomic abundance of the pathogen (5.1%) than DAEC samples (27.8%) by ∼5-fold, on average, for the cases with clear evidence of E. coli pathotype infection. This observation suggests that ETEC infection may require a lower pathogen load in order to elicit disease than does DAEC infection, providing potentially an additional diagnostic metagenomic feature of ETEC versus DAEC infections. Consistent with the latter results, ETEC pathogens are thought to cause infection by the production of enterotoxins (22, 35) that alter the concentration of important cellular messengers such as cyclic AMP, cyclic GMP, and Ca2+, while DAEC is strongly attached to the cell surface, where it induces a cytopathic effect characterized by the development of long cellular extensions (24). These differences in mechanisms of pathogenicity might explain, at least in part, the differences in pathogen abundance and associated changes in the gut microbiome (see, for example, Fig. 5) that were observed in ETEC- and DAEC-dominated metagenomes.
A critical question in diagnostic testing for enteric pathogens is how often conventional culture-dependent techniques, such as those implemented for isolating E. coli from stool samples, readily capture the targeted pathogen as opposed to transient or dormant/inactive populations. Our metagenomic and/or epidemiological data showed that from the total set of diarrheal DAEC (n = 16) samples analyzed, the isolate was highly similar to the recovered MAG in only ∼30% of the cases (n = 5). The same results for ETEC-positive and EPEC-positive samples (by isolation and PCR) were 70 and 0%, respectively, for a total average of roughly about 50%. Thus, it appeared that in about half of the diarrheal samples the isolate likely was not the causative agent. This may help explain the results of many studies that have observed high rates of enteropathogen infection in individuals without diarrhea symptoms (36).
Beyond pathogen detection, our metagenomic study also generated complete or nearly complete pathogen MAGs directly from the samples, which allowed a more comprehensive characterization of the virulence and diversity of the infectious E. coli population. In addition to application in diagnostic testing, population genome binning can also be used to recover pathogenic genotypes and, when coupled to epidemiological information, to identify person-to-person transmission events and outbreak dynamics, which represent important tasks in public health investigations.
Although a small number of samples were excluded from the pathogen-specific signatures of the infection based on the relatively low coverage observed in comparison to other positive samples or to controls, it is important to highlight that finding a 0.05- to 0.1-fold coverage, i.e., the minimum threshold required for robust detection of a target genome in a complex gut metagenome (37), still translates to a relatively large number of cells in situ. The average E. coli genome size is 5 Mbp, and our metagenome libraries were, on average, ∼1.78 Gbp in size after removing contaminant host DNA and low-quality reads. Finding 0.05-fold coverage for an E. coli genome (∼5% of the estimated genome size) would require 0.25 Mbp of E. coli sequenced reads or ∼0.014% of the total metagenome size. Our extracted DNA came from an average of 1.26 × 106 cells in total based on the normalization of the average extracted DNA concentrations (6.45 ng/μl) with the estimated molecular weight of an E. coli bacterium 5.14 × 10−6 ng (38–40). Therefore, 0.014% of 1.26 × 106 would be 1.77 × 102 in total (i.e., >100 cells), which is still a large number of cells that could potentially cause a disease. Thus, the limit of detection of metagenomics, as applied here, was not low enough to detect relatively low-abundance microorganisms and should be combined with methods that offer a lower detection limit for this purpose, such as qPCR and isolation, for a more comprehensive assessment of enteric infections. Furthermore, sample heterogeneity could also account for the lack of metagenomic evidence of E. coli infection for the ∼50% of disease samples identified above as false-positive calls by isolation and PCR. Our typical sample size was ∼0.5 to 0.8 g, which represents a small portion of the total stool, and we might therefore have undersampled the (potential) E. coli population present in the gut. Multiple replicate samples and high sampling volumes (i.e., 2 to 5 g or more) should be used to avoid such sample heterogeneity issues in the future. Regardless of the potential effects of sample heterogeneity, our findings collectively highlight the potential of metagenomics as a diagnostic tool for infectious diseases, the strengths of combining traditional culture-based and PCR techniques with shotgun metagenomics, and the applicability of our bioinformatic framework to the study of enteric pathogens.
MATERIALS AND METHODS
EcoZUR study design.
Surveys were carried out using Android devices and the Open Data kit program (http://opendatakit.org). Prior to enrollment, all participants signed a consent document approved by the Institutional Review Board of Emory University (IRB00065781) and the Universidad San Francisco de Quito (USFQ 2013-145M). The research protocol was also approved by the Ecuadorian Ministry of Health (MSP-DIS-2014-0055-O). Further details about the methods employed in the EcoZUR study can be found in Smith et al. (27).
E. coli isolation, pathotype identification, and rotavirus detection.
Fresh stool samples were incubated in E. coli-specific media and tested for different pathotypes based on the presence of specific virulence genes determined by PCR. For each stool sample, five lactose-positive colonies were isolated on MacConkey’s agar media (MKL) and non-lactose-fermenting isolates were further cultured and tested on Chromocult agar media (Merck, Darmstadt, Germany) for β-glucuronidase activity. We tested five colonies per stool sample, because this is a standard procedure for detecting the dominant E. coli population (>97% chance [41]); a higher number of colonies was also not practical for the large number of samples collected as part of the EcoZUR study (>1,000 samples collected). Colonies unable to ferment lactose were identified by biochemical tests as Shigella or E. coli using the API 20E test (bioMérieux, Marcy l’Etoile, France); we focused on E. coli isolates only for this study. The five colonies were pooled, resuspended in 300 μl of sterile-distilled water, boiled for 10 min to release the DNA. Identification of E. coli pathotypes was performed by PCR screening for the following target virulence genes (see also Table 1 for primer sequence information): bfp for typical EPEC, lt and sta for ETEC, ipaH for EIEC and shigellae, aggR for EAEC, eaeA for atypical EPEC, and afaB for DAEC. Positive pools for eaeA were subsequently tested for stx1 and stx2 genes for the differentiation of potential EHEC infections. If a pooled sample tested positive for any virulence factor, then each of the five isolates were retested individually to identify the specific isolate carrying the virulence gene. In addition, fresh stool samples were also tested for rotavirus antigens using a RIDA Quick Rotavirus test (r-Biopharm, Darmstadt, Germany).
DNA extraction, library preparation, and sequencing.
DNA from E. coli isolates was extracted using the Wizard genomic DNA purification kit (Promega). DNA for stool metagenomes was extracted from a homogenized stool mix using the Mo Bio PowerSoil DNA isolation kit. In both cases, the purity and concentration of the DNA was estimated using a NanoDrop spectrophotometer (Thermo Scientific) and a Qubit 2.0 dsDNA high-sensitivity assay (Invitrogen, Carlsbad, CA). For isolates and metagenome DNA sequencing, libraries were prepared using an Illumina Nextera XT DNA library preparation kit according to the manufacturer’s instructions except that the protocol was terminated after isolation of cleaned double-stranded libraries. After this, libraries were quantified using the Qubit 1X dsDNA HS assay kit (ThermoFisher) and run on a high-sensitivity DNA chip using a Bioanalyzer 2100 instrument (Agilent) to determine library insert sizes. An equimolar mixture of the isolates libraries (final loading concentration of 10 pM) was sequenced on an Illumina MiSeq instrument (School of Biological Sciences, Georgia Institute of Technology) using a MiSeq reagent v2 kit for 500 cycles (2 × 250-bp paired end run; Illumina, Inc., San Diego, CA). Metagenomic libraries were sequenced in the Illumina HiSeq 2500 instrument in the rapid run mode for 300 cycles (150-bp, paired-end mode).
Libraries for 16S rRNA gene amplicon sequencing were amplified and sequenced using 16S rRNA primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′), which target the V4 region of the gene (∼254 bp) as previously described (42). PCR amplifications were performed in duplicates to a final volume of 20 μl containing 0.5 U of AccuPrime Pfx polymerase, 1× AccuPrime reaction mix, 200 nM concentrations of each primer, and 1 μl of template DNA. Amplification conditions included an initial denaturation of 2 min at 95°C, followed by 25 cycles of 95°C for 20 s, 55°C for 30 s, and 72°C for 30 s, and a single final extension step at 72°C for 6 min. Specific amplification was verified by agarose gel electrophoresis and duplicate samples were pooled and purified using DNA purification SPRI (solid-phase reversible immobilization) magnetic beads (Applied Biological Materials) according to the manufacturer’s instructions. Purified amplicons were then pooled in an equimolar concentration and sequenced on the Illumina MiSeq instrument (2 × 250-bp paired end run) as recommended by the manufacturer for low-diversity sequencing. The resulting sequencing data set supporting the results reported here were submitted to the National Center for Biotechnology Information (NCBI) under BioProject PRJNA486009. The metadata associated with the sequencing data generated in this study is available as supporting information.
Read quality control, assembly, and taxonomic annotation.
Raw FASTQ reads from both E. coli isolates and stool metagenomes were quality trimmed using the SolexaQA++ package (43). Specifically, the scripts dynamicTrim.pl and LengthSort.pl were used to trim individual reads to the longest continuous segment for which phred quality score was ≥20 (Q ≥ 20, which represents ∼99% accuracy per nucleotide position). Reads shorter than 50 bp were discarded. Both isolate and metagenome libraries were processed using MiGA (Microbial Genomes Atlas), a recently developed system for data management and processing of microbial genomes and metagenomes (http://microbial-genomes.org/) (44). Quality-filtered reads were de novo assembled using IDBA-UD with precorrections (45), and protein-coding sequences were predicted using MetaGeneMark (46). In addition, MiGA reports the percentage of contamination and completeness for genome sequences based on recovery of lineage-specific marker genes, as well as the likely taxonomic origin of windows of 10 genes across the query genome using the MyTaxa engine (47), which allows the user to manually inspect the genomes for possible contamination through the so-called “MyTaxaPlots” barplots.
16S rRNA gene amplicon data processing.
16S rRNA gene libraries were processed using the default parameters in Qiime2 2018.11 pipeline (https://qiime2.org/) (48). In brief, 16S rRNA gene sequences were first denoised and quality filtered using Dada2, an algorithm that uses a statistical model for correcting errors introduced in the Illumina amplicon sequencing and infers underlying sample sequences clustering highly similar amplicon sequence variants (ASVs) (49). After quality control, a feature table of ASVs that are 100% identical was generated. Taxonomy was assigned to ASVs using the q2-feature-classifier (50) classify‐sklearn naive Bayes taxonomy classifier against the Greengenes 13_8 99% OTU reference sequences (51). Low abundance ASVs (those ASVs with a total count fraction lower than 0.005% of the total) were removed from the final feature table, as suggested previously (52). Alpha-diversity estimates were computed in rarefied libraries to avoid bias in the detection of differentially abundant taxa given the underlying differences in library size. ASV richness was estimated using Faith’s phylogenetic diversity (53). Beta-diversity analyses were performed using different distance metrics, including Bray-Curtis, Jaccard, and weighted and unweighted UNIFRAC (54). The resulting distance matrices were visualized through principal coordinate analysis plots. The same distance matrices were then used to conduct a statistical test of dissimilarities, i.e., a permutational multivariate nonparametric analysis of dissimilarities PERMANOVA (55), performed using the R’s package Vegan (56). Differentially abundant OTUs (or ASVs) between diarrhea and control samples were identified using STAMP, a data analysis metagenomic software that reports differentially abundant features using effect sizes and confidence intervals (57).
Population genome binning and in situ metagenome abundance.
MaxBin2 (58) was used to bin previously assembled contigs into metagenome-assembled genomes (MAGs) for the recovery of E. coli population genome with a minimum contig length threshold of 2,000 bp. Prior to binning, Bowtie 2 (59) was used to align short-read sequences to assembled contigs, and SAMtools (60) was used to sort and convert SAM files to the BAM format. Sorted BAM files were then used to calculate the coverage (mean representation) of each contig in each sample metagenome. The quality of each resulting MAG was evaluated with MiGA (see above) and CheckM v1.0.3 (61) using taxonomy-specific workflow for “Escherichia coli.” Only E. coli MAGs with a higher quality score than 60, calculated as the estimated completeness minus five times the estimated contamination (62), were retained (Table S3). The taxonomic affiliation of each MAG was then confirmed with MiGA, which uses a combination of the genome-aggregate average nucleotide identity concept, or ANI, and the average amino acid identity, AAI, to taxonomically classify a query genomic sequence against its reference genome databases and find the closest match with P < 0.05.
The in situ abundance of isolates (sequencing depth), MAGs, and reference commensal E. coli strain HS in a sample was calculated by using the number of metagenomic reads competitively mapping on each genome above a cutoff nucleotide identity of ≥95% and a query sequence coverage by the alignment of ≥50% using Bowtie and SAMtools normalized by the estimated genome size by the MiGA analysis (genome completeness). The abundance of RpoB genes (sequencing depth) was calculated by using the total number of reads identified as RpoB genes by the ROCker pipeline as described previously (63), normalized by the average length of the RpoB genes. The genome equivalent, that is, the total number of cells sampled representing the genome of interest (or carrying the gene of interest), was estimated from the ratio of the abundance of the query genome (or gene) to that of the RpoB genes.
Read-based detection of virulence factors and estimation of E. coli intrapopulation diversity.
Genome and metagenome virulence profiling was examined using the Virulence Factors Database (VFDB; http://www.mgc.ac.cn/VFs/) (64) filtered for E. coli specifically. Metagenomic reads were mapped against the VFDB database, and gene presence or absence was determined by the number of reads recruited by the VF genes (≥1×) and the length of the gene that was covered by reads (≥70%; lower gene abundance or coverage was considered gene absence). E. coli intrapopulation diversity was estimated by calculating the average nucleotide identity of the metagenomic reads (defined as ANIr) mapping to the reference genome (pathotype isolate or reference commensal) with a percent identity between 90 and 100%, i.e., reads representing the total E. coli population in a sample, based on competitive mapping. For this task, the function enve.recplot2.ANIr of the R package enveomics.R v1.3 (65) was used as described previously (https://github.com/lmrodriguezr/enveomics/tree/master/enveomics.R).
Phylogenetic analysis of genomes of isolates and MAGs.
Orthologous genes of isolates, MAGs and reference E. coli strains were identified using reciprocal best matches with protocols detailed as described previously (66). Sequences of orthologous genes present in all the genomes (core genes) were extracted and aligned using MUSCLE v3.8.35 (67). The resulting alignment was concatenated and trimmed with Gblocks 0.91b (68) to remove noisy and/or uninformative regions. Phylogenetic reconstructions were estimated using FastTree v2.1.7 (69) with 1,000 bootstrap replicates and the GTR-GAMMA substitution model for nucleotide sequences and visualized in iTOL (70).
Supplementary Material
ACKNOWLEDGMENTS
We thank to all participants from Ecuador (Quito, Esmeraldas, Borbón, and the rural villages) who agreed to participate in the EcoZUR study and donated their specimens for the successful completion of this study. We thank the local authorities of the Ecuadorian Ministry of Public Health for providing access to the facilities in their hospitals. We also thank Denys Tenorio, Mauricio Ayoví, Xavier Sanchez, Edison Puebla, and Kate Bohnert for assistance in carrying out the field portions of the study.
Funding for this study was provided by National Institutes of Allergy and Infectious Diseases grant 1K01AI103544 and by Colciencias through a doctoral fellowship to A.P.-G. The content of this manuscript is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
We declare that we have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01820-19.
REFERENCES
- 1.Troeger C, Forouzanfar M, Rao PC, Khalil I, Brown A, Reiner RC, Fullman N, Thompson RL, Abajobir A, Ahmed M, Alemayohu MA, Alvis-Guzman N, Amare AT, Antonio CA, Asayesh H, Avokpaho E, Awasthi A, Bacha U, Barac A, Betsue BD, Beyene AS, Boneya DJ, Malta DC, Dandona L, Dandona R, Dubey M, Eshrati B, Fitchett JRA, Gebrehiwot TT, Hailu GB, Horino M, Hotez PJ, Jibat T, Jonas JB, Kasaeian A, Kissoon N, Kotloff K, Koyanagi A, Kumar GA, Rai RK, Lal A, El Razek HMA, Mengistie MA, Moe C, Patton G, Platts-Mills JA, Qorbani M, Ram U, Roba HS, Sanabria J, Sartorius B, Sawhney M, Shigematsu M, Sreeramareddy C, Swaminathan S, Tedla BA, Jagiellonian RT-M, Ukwaja K, Werdecker A, Widdowson M-A, Yonemoto N, El Sayed Zaki M, Lim SS, Naghavi M, Vos T, Hay SI, Murray CJL, Mokdad AH. 2017. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis 17:909–948. doi: 10.1016/S1473-3099(17)30276-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selendy JMH. 2011. Water and sanitation-related diseases and the environment: challenges, interventions, and preventive measures. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 3.Kotloff KL. 2017. The burden and etiology of diarrheal illness in developing countries. Pediatr Clin North Am 64:799–814. doi: 10.1016/j.pcl.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Devleesschauwer B, Haagsma JA, Mangen M-J, Lake RJ, Havelaar AH. 2018. The global burden of foodborne disease, p 107–122. In Food safety economics. Springer, New York, NY. [Google Scholar]
- 5.Kelly D, Khurram NA, Hickman RA, Pei Z. 2018. Quantitative approach in clinical microbiology: a paradigm shift toward culture-free methods, p 599–615. In Advanced techniques in diagnostic microbiology. Springer, New York, NY. [Google Scholar]
- 6.Forbes JD, Knox NC, Ronholm J, Pagotto F, Reimer A. 2017. Metagenomics: the next culture-independent game changer. Front Microbiol 8:1069. doi: 10.3389/fmicb.2017.01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller RR, Montoya V, Gardy JL, Patrick DM, Tang P. 2013. Metagenomics for pathogen detection in public health. Genome Med 5:81. doi: 10.1186/gm485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sleight SC, Wigginton NS, Lenski RE. 2006. Increased susceptibility to repeated freeze-thaw cycles in Escherichia coli following long-term evolution in a benign environment. BMC Evol Biol 6:104. doi: 10.1186/1471-2148-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States: major pathogens. Emerg Infect Dis 17:7. doi: 10.3201/eid1701.p11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frickmann H, Schwarz NG, Rakotozandrindrainy R, May J, Hagen RM. 2015. PCR for enteric pathogens in high-prevalence settings: what does a positive signal tell us? Infect Dis (Lond) 47:491–498. doi: 10.3109/23744235.2015.1022212. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Platts-Mills JA, Juma J, Kabir F, Nkeze J, Okoi C, Operario DJ, Uddin J, Ahmed S, Alonso PL, Antonio M, Becker SM, Blackwelder WC, Breiman RF, Faruque ASG, Fields B, Gratz J, Haque R, Hossain A, Hossain MJ, Jarju S, Qamar F, Iqbal NT, Kwambana B, Mandomando I, McMurry TL, Ochieng C, Ochieng JB, Ochieng M, Onyango C, Panchalingam S, Kalam A, Aziz F, Qureshi S, Ramamurthy T, Roberts JH, Saha D, Sow SO, Stroup SE, Sur D, Tamboura B, Taniuchi M, Tennant SM, Toema D, Wu Y, Zaidi A, Nataro JP, Kotloff KL, Levine MM, Houpt ER. 2016. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study. Lancet 388:1291–1301. doi: 10.1016/S0140-6736(16)31529-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acácio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 13.Conway T, Cohen PS. 2015. Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol Spectr 3:MBP-0006-2014. doi: 10.1128/microbiolspec.MBP-0006-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossi E, Cimdins A, Lüthje P, Brauner A, Sjöling Å, Landini P, Römling U. 2018. It’s a “gut feeling”: Escherichia coli biofilm formation in the gastrointestinal tract environment. Crit Rev Microbiol 44:1–30. doi: 10.1080/1040841X.2017.1303660. [DOI] [PubMed] [Google Scholar]
- 15.Nelson AM, Walk ST, Taube S, Taniuchi M, Houpt ER, Wobus CE, Young VB. 2012. Disruption of the human gut microbiota following norovirus infection. PLoS One 7:e48224. doi: 10.1371/journal.pone.0048224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang AD, Luo C, Pena-Gonzalez A, Weigand MR, Tarr CL, Konstantinidis KT. 2017. Metagenomics of two severe foodborne outbreaks provides diagnostic signatures and signs of coinfection not attainable by traditional methods. Appl Environ Microbiol 83:e02577-16. doi: 10.1128/AEM.02577-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pop M, Walker AW, Paulson J, Lindsay B, Antonio M, Hossain MA, Oundo J, Tamboura B, Mai V, Astrovskaya I, Corrada Bravo H, Rance R, Stares M, Levine MM, Panchalingam S, Kotloff K, Ikumapayi UN, Ebruke C, Adeyemi M, Ahmed D, Ahmed F, Alam MT, Amin R, Siddiqui S, Ochieng JB, Ouma E, Juma J, Mailu E, Omore R, Morris JG, Breiman RF, Saha D, Parkhill J, Nataro JP, Stine OC. 2014. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol 15:R76. doi: 10.1186/gb-2014-15-6-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monira S, Nakamura S, Gotoh K, Izutsu K, Watanabe H, Alam NH, Nakaya T, Horii T, Ali SI, Iida T, Alam M. 2013. Metagenomic profile of gut microbiota in children during cholera and recovery. Gut Pathog 5:1. doi: 10.1186/1757-4749-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis 197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 20.Youmans BP, Ajami NJ, Jiang Z-D, Campbell F, Wadsworth WD, Petrosino JF, DuPont HL, Highlander SK. 2015. Characterization of the human gut microbiome during travelers’ diarrhea. Gut Microbes 6:110–119. doi: 10.1080/19490976.2015.1019693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaper JB, Nataro JP, Mobley H. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 22.Fleckenstein JM. 2013. Enterotoxigenic Escherichia coli, p 183–213. In Escherichia coli. Elsevier, New York, NY. [Google Scholar]
- 23.Kaur P, Chakraborti A, Asea A. 2010. Enteroaggregative Escherichia coli: an emerging enteric food-borne pathogen. Interdiscip Perspect Infect Dis 2010:254159. doi: 10.1155/2010/254159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Servin AL. 2014. Pathogenesis of human diffusely adhering Escherichia coli expressing Afa/Dr adhesins (Afa/Dr DAEC): current insights and future challenges. Clin Microbiol Rev 27:823–869. doi: 10.1128/CMR.00036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson JS, Giogha C, Wong Fok Lung T, Hartland EL. 2016. The genetics of enteropathogenic Escherichia coli virulence. Annu Rev Genet 50:493–513. doi: 10.1146/annurev-genet-120215-035138. [DOI] [PubMed] [Google Scholar]
- 26.Pasqua M, Michelacci V, Di Martino ML, Tozzoli R, Grossi M, Colonna B, Morabito S, Prosseda G. 2017. The intriguing evolutionary journey of enteroinvasive Escherichia coli (EIEC) toward pathogenicity. Front Microbiol 8:2390. doi: 10.3389/fmicb.2017.02390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith SM, Montero L, Paez M, Ortega E, Hall E, Bohnert K, Sanchez X, Puebla E, Endara P, Cevallos W, Trueba G, Levy K. 2019. Locals get travellers’ diarrhoea too: risk factors for diarrhoeal illness and pathogenic Escherichia coli infection across an urban-rural gradient in Ecuador. Trop Med Int Health 24:205–219. doi: 10.1111/tmi.13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez-R LM, Gunturu S, Tiedje JM, Cole JR, Konstantinidis KT. 2018. Nonpareil 3: fast estimation of metagenomic coverage and sequence diversity. mSystems 3:e00039-18. doi: 10.1128/mSystems.00039-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The HC, Florez de Sessions P, Jie S, Pham Thanh D, Thompson CN, Nguyen Ngoc Minh C, Chu CW, Tran T-A, Thomson NR, Thwaites GE, Rabaa MA, Hibberd M, Baker S. 2018. Assessing gut microbiota perturbations during the early phase of infectious diarrhea in Vietnamese children. Gut Microbes 9:38–54. doi: 10.1080/19490976.2017.1361093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richter TKS, Michalski JM, Zanetti L, Tennant SM, Chen WH, Rasko DA. 2018. Responses of the human gut Escherichia coli population to pathogen and antibiotic disturbances. mSystems 3:e00047-18. doi: 10.1128/mSystems.00047-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 32.Kaufman L, Rousseeuw PJ. 2009. Finding groups in data: an introduction to cluster analysis, vol 344 John Wiley & Sons, New York, NY. [Google Scholar]
- 33.Qiu F-Z, Shen X-X, Li G-X, Zhao L, Chen C, Duan S-X, Guo J-Y, Zhao M-C, Yan T-F, Qi J-J, Wang L, Feng Z-S, Ma X-J. 2018. Adenovirus associated with acute diarrhea: a case-control study. BMC Infect Dis 18:450. doi: 10.1186/s12879-018-3340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Bouguénec C, Servin AL. 2006. Diffusely adherent Escherichia coli strains expressing Afa/Dr adhesins (Afa/Dr DAEC): hitherto unrecognized pathogens. FEMS Microbiol Lett 256:185–194. doi: 10.1111/j.1574-6968.2006.00144.x. [DOI] [PubMed] [Google Scholar]
- 35.Mirhoseini A, Amani J, Nazarian S. 2018. Review on pathogenicity mechanism of enterotoxigenic Escherichia coli and vaccines against it. Microb Pathog 117:162–169. doi: 10.1016/j.micpath.2018.02.032. [DOI] [PubMed] [Google Scholar]
- 36.Levine MM, Robins-Browne RM. 2012. Factors that explain excretion of enteric pathogens by persons without diarrhea. Clin Infect Dis 55:S303–S311. doi: 10.1093/cid/cis789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castro JC, Rodriguez-R LM, Harvey WT, Weigand MR, Hatt JK, Carter MQ, Konstantinidis KT. 2018. imGLAD: accurate detection and quantification of target organisms in metagenomes. PeerJ 6:e5882. doi: 10.7717/peerj.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massie HR, Zimm BH. 1965. Molecular weight of the DNA in the chromosomes of E. coli and B. subtilis. Proc Natl Acad Sci U S A 54:1636. doi: 10.1073/pnas.54.6.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri MC, Tassani S, Piva F, Perez-Amodio S, Strippoli P, Canaider S. 2013. An estimation of the number of cells in the human body. Ann Hum Biol 40:463–471. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 41.Lautenbach E, Bilker WB, Tolomeo P, Maslow JN. 2008. Impact of diversity of colonizing strains on strategies for sampling Escherichia coli from fecal specimens. J Clin Microbiol 46:3094–3096. doi: 10.1128/JCM.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-R LM, Gunturu S, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, Konstantinidis KT. 2018. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res 46:W282–W288. doi: 10.1093/nar/gky467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng Y, Leung HCM, Yiu S-M, Chin F. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 46.Zhu W, Lomsadze A, Borodovsky M. 2010. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo C, Rodriguez-R LM, Konstantinidis KT. 2014. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. doi: 10.1093/nar/gku169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navas-Molina JA, Peralta-Sánchez JM, González A, McMurdie PJ, Vázquez-Baeza Y, Xu Z, Ursell LK, Lauber C, Zhou H, Song SJ. 2013. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol 531:371–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faith DP, Baker AM. 2006. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinform Online 2:117693430600200.117693430600200007. doi: 10.1177/117693430600200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lozupone CA, Hamady M, Kelley ST, Knight R. 2007. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clarke KR. 1993. Non‐parametric multivariate analyses of changes in community structure. Austral Ecol 18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 56.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. Package “vegan”: community ecology package, v2. https://cran.r-project.org/web/packages/vegan/vegan.pdf.
- 57.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y-W, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 59.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramirez-Gonzalez RH, Bonnal R, Caccamo M, MacLean D. 2012. Bio-SAMtools: ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Source Code Biol Med 7:6. doi: 10.1186/1751-0473-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. 2017. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol 2:1533. doi: 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 63.Orellana LH, Rodriguez-R LM, Konstantinidis KT. 2016. ROCker: accurate detection and quantification of target genes in short-read metagenomic data sets by modeling sliding-window bitscores. Nucleic Acids Res 45:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. 2005. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33:D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1. [DOI] [Google Scholar]
- 66.Pena-Gonzalez A, Rodriguez-R LM, Marston CK, Gee JE, Gulvik CA, Kolton CB, Saile E, Frace M, Hoffmaster AR, Konstantinidis KT. 2018. Genomic characterization and copy number variation of Bacillus anthracis plasmids pXO1 and pXO2 in a historical collection of 412 strains. mSystems 3:e00065-18. doi: 10.1128/mSystems.00065-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 69.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toma C, Lu Y, Higa N, Nakasone N, Chinen I, Baschkier A, Rivas M, Iwanaga M. 2003. Multiplex PCR assay for identification of human diarrheagenic Escherichia coli. J Clin Microbiol 41:2669–2671. doi: 10.1128/jcm.41.6.2669-2671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tornieporth NG, John J, Salgado K, de Jesus P, Latham E, Melo MC, Gunzburg ST, Riley LW. 1995. Differentiation of pathogenic Escherichia coli strains in Brazilian children by PCR. J Clin Microbiol 33:1371–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paton AW, Paton JC. 1998. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J Clin Microbiol 36:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le Bouguenec C, Archambaud M, Labigne A. 1992. Rapid and specific detection of the pap, afa, and sfa adhesin-encoding operons in uropathogenic Escherichia coli strains by polymerase chain reaction. J Clin Microbiol 30:1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.