

Abstract
SMYD3, a SET and MYND domain containing lysine methyltransferase, catalyzes the transfer of the methyl group from a methyl donor onto the Nε group of a lysine residue in the substrate protein. Methylation of MAP3 kinase kinase (MAP3K2) by SMYD3 has been implicated in Ras-driven tumorigenesis. The crystal structure of SMYD3 in complex with MAP3K2 peptide reveals a shallow hydrophobic pocket (P-2), which accommodates the binding of a phenylalanine residue at the −2 position of the substrate (F258) is a crucial determinant of substrate specificity of SMYD3. To better understand the substrate preference of SMYD3 at the −2 position, molecular dynamics (MD) simulations and the MM/GBSA method were performed on the crystal structure of SMYD3-MAP3K2 complex (PDB: 5EX0) after substitution of F258 residue of MAP3K2 to each of the other 19 natural residues, respectively. Binding free energy calculations reveal that the P-2 pocket prefers an aromatic hydrophobic group and none of the substitutions behave better than the wild-type phenylalanine residue does. Furthermore, we investigated the structure–activity relationships (SAR) of a series of non-natural phenylalanine derivative substitutions at the −2 position and found that quite a few modifications on the sidechain of F258 residue could strengthen its binding to the P-2 pocket of SMYD3. These explorations provide insights into developing novel SMYD3 inhibitors with high potency and high selectivity against MAP3K2 and cancer.
1. Introduction
The lysine methylation is a principal regulatory mechanism that influences the protein activity, stability, and function.1 SMYDs (SET and MYND domain containing proteins) catalyze the transfer of the methyl group from S-adenosyl-l-methionine (SAM) onto the Nε group of a lysine residue in the target protein substrate. Both histones and nonhistone proteins have been shown to be the substrates of SMYD family proteins. Their reported histone targets include H3K4 (SMYD1 and SMYD3), H3K36 (SMYD2), and H4K5 (SMYD3),2−5 functioning in the regulation of chromatin structure and gene expression.6−11 Nonhistone substrates can also be methylated by SMYD proteins (Figure 1A). SMYD2 has a broad spectrum of substrates including tumor suppressor protein p53 (K370),12 retinoblastoma (Rb, K860),13 estrogen receptor α (ERα, K266),14 heat shock protein 90 (HSP90, K615),15 and period circadian protein homolog 2 (PER2, K798).16 In the case of SMYD3, vascular endothelial growth factor receptor 1 (VEGFR1, K831) and MAP3 kinase kinase (MAP3K2, K260) have been reported to be the substrates of SMYD3.17,18
Figure 1.
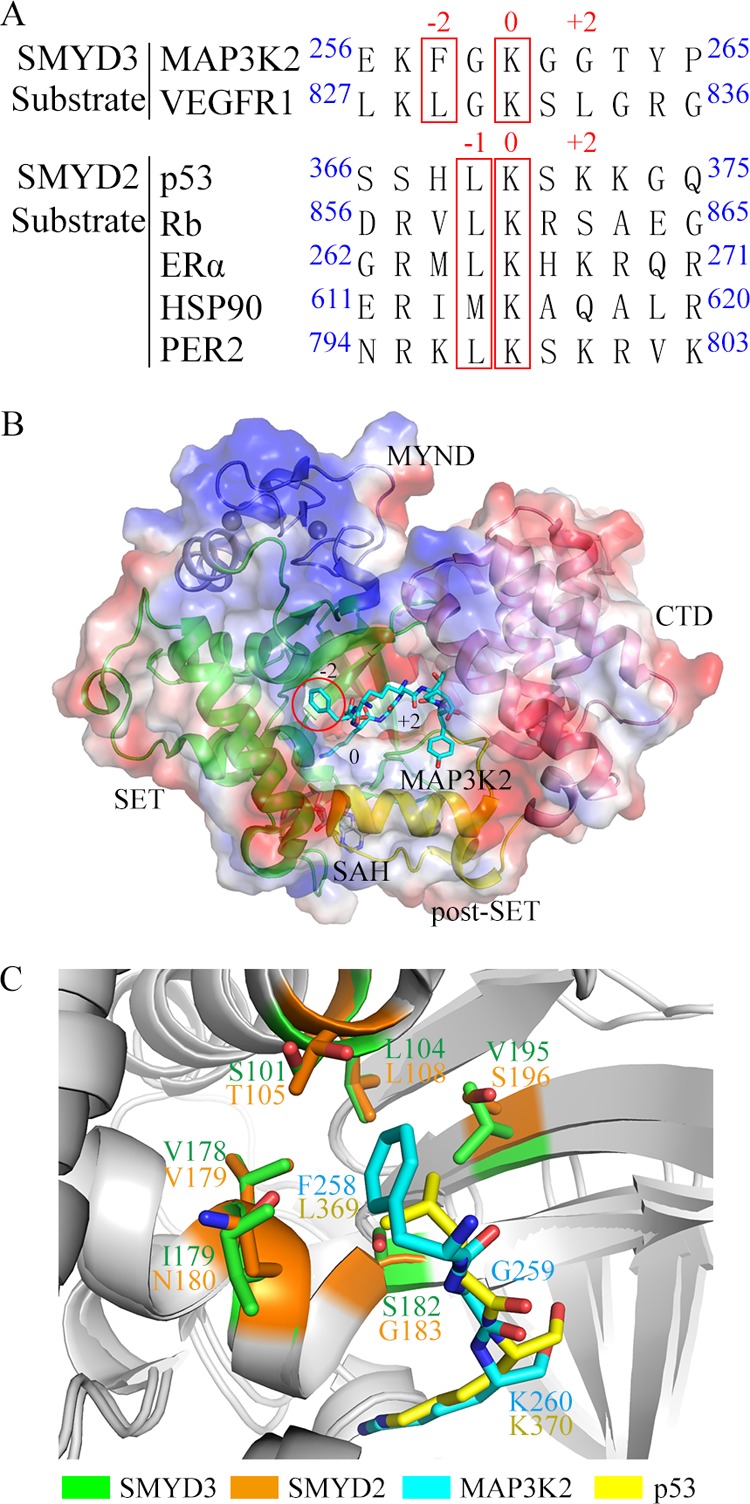
Comparison between the substrates of SMYD3 and SMYD2. (A) Amino acid sequence alignment of the reported substrates of SMYD3 and SMYD2. The −2- and 0-position residues in substrates of SMYD3 and the −1- and 0-position residues in substrates of SMYD2 are highlighted in the red boxes, respectively. (B) Electrostatic potential and substrate-binding cleft on the surface of SMYD3. The SET, MYND, post-SET, and C-terminal domains of SMYD3 are shown in green, blue, yellow, and pink, respectively. SAH and the MAP3K2 peptide are shown in a stick model (carbon, gray, and cyan, respectively; nitrogen, blue; oxygen, red). Zinc ions are shown as gray spheres. The −2-position F258 residue in MAP3K2 peptide is highlighted in the red circle. (C) Structural alignment of the shallow hydrophobic pocket of SMYD3 and SMYD2 with their substrates MAP3K2 and p53 peptide, respectively. The residues at the −2 position in SMYD3, at the −1 position in SMYD2, in MAP3K2 peptide, and in p53 peptide are shown in a stick model with carbon in green, orange, cyan, and yellow, respectively.
SMYD3 has been implicated in a variety of cancers including liver, colon, and breast cancer related to its methyltransferase activity on nonhistone targets.19−22 Methylation of VEGFR1 K831 by SMYD3 enhances its autophosphorylation and kinase activity in the cell and promotes carcinogenesis.23−25 Methylation of MAP3K2 at K260 in the cytoplasm activates the MAP kinase signaling module and promotes RAS-driven tumorigenesis.18,26 These results suggest that targeting the methyltransferase activity of SMYD3 would be a useful strategy in anticancer therapy.
Our previous work solved the crystal structure of SMYD3 in complex with MAP3K2 peptide.27 The structure reveals that there is an amphiphilic cleft for substrate binding on the surface of SMYD3. A shallow hydrophobic pocket (P-2), which accommodates the binding of a phenylalanine residue at the −2 position of the substrate (F258) is a crucial determinant of the substrate specificity of SMYD3 (Figure 1B). This P-2 pocket is composed of several hydrophobic residues including L104, V178, I179, and V195, with two serine residues, S101 and S182, standing at the perimeters of the pocket (Figure 1C). SMYD2 contains a similar shallow hydrophobic pocket to hold the leucine residue at −1 position of the substrate p53, with L108, V179, N180, S196, T105, and G183 taking the place of L104, V179, I179, V195, S101, and S182 of SMYD3, respectively (PDB IDs: 3TG5 and 5EX0, Figure 1C). It is interesting to find that a majority of the reported substrates of SMYD2 include a leucine residue at the −1 position12−16 (Figure 1A). And the importance of this hydrophobic pocket in substrate binding preference of SMYD2 has been proved by several experimental and computational studies.16,28−32
In the past decade, several computational protein design protocols have been reported to predict the specificity of protein–protein or protein–ligand interactions.33−36 Coupled protein docking and conformational ensemble methods37−39 were further developed to enhance the accuracy of prediction. For example, a multistate computational procedure combining backbone ensemble, energy minimization, amino acid substitution, and fitness calculation was developed to probe the substrate specificity of SMYD2.32 The result reveals that SMYD2 exhibits narrow specificity for substrates at the −1 and +2 positions. Besides these approaches, molecular dynamics (MD) simulations are valuable adjuncts to reveal substrate recognitions by conformational dynamics. The motions of substrate and protein induced by their mutual interactions could be directly observed through trajectories.40−42
To better understand the substrate preference of SMYD3 at the −2 position, different kinds of natural amino acid substitutions are first used as probes to explore the binding preference of the P-2 pocket in this work. Binding free energy calculations of MD simulations from the MM/GBSA method reveal that the P-2 pocket plays a crucial role in substrate binding with narrow specificity. It prefers an aromatic hydrophobic group and none of the substitutions behave better than the wild-type phenylalanine does. Furthermore, a series of non-natural phenylalanine derivative substitutions at the −2 position was investigated by similar MD simulations procedures. Results show that quite a few modifications on the sidechain of F258 residue could further stabilize the sidechain conformation, and thus strengthen its binding to the P-2 pocket of SMYD3. These results provide insights into developing novel SMYD3 inhibitors with high potency and high selectivity against MAP3K2 and cancer.
2. Results and Discussion
First, we performed a 100-ns MD simulation to observe the stability of SMYD3-MAP3K2 complex. SMYD3 is composed of three domains, an N-terminal catalytic domain (SET and post-SET), a MYND domain inserts into the SET domain, and a C-terminal domain (CTD) (Figure 1B). The crystal structure of SMYD3-MAP3K2 complex reveals that the SET and post-SET domains of SMYD3 contribute most of the binding force to substrate binding other than the MYND and CTD domains. root mean square fluctuations (RMSF) analyses of each residue of the complex after 100-ns MD simulation show that the MYND and CTD domains of SMYD3 are more flexible than the SET and post-SET domains (Figure 2A). In the case of MAP3K2 peptide, residues from −2 to +2 positions (F258 to G262) are more stable than the remaining N- and C-terminal residues (Figure 2B). Figure 2C exhibits time evolutions of root mean square deviations (RMSD) of the stable regions of SMYD3 and MAP3K2. The RMSD values of both SET and post-SET domains of SMYD3 and F258 to G262 residues of MAP3K2 reach a stable plateau after ∼10 ns of simulations and keep a position around an average value of 2.3 and 1.0 Å during the remaining MD simulations, respectively. These results indicate that a 50-ns MD simulation unit performed on the stable regions of SMYD3 and MAP3K2 in the following study is sufficient for detecting reliable binding free energies and conformational dynamics of the complex.
Figure 2.
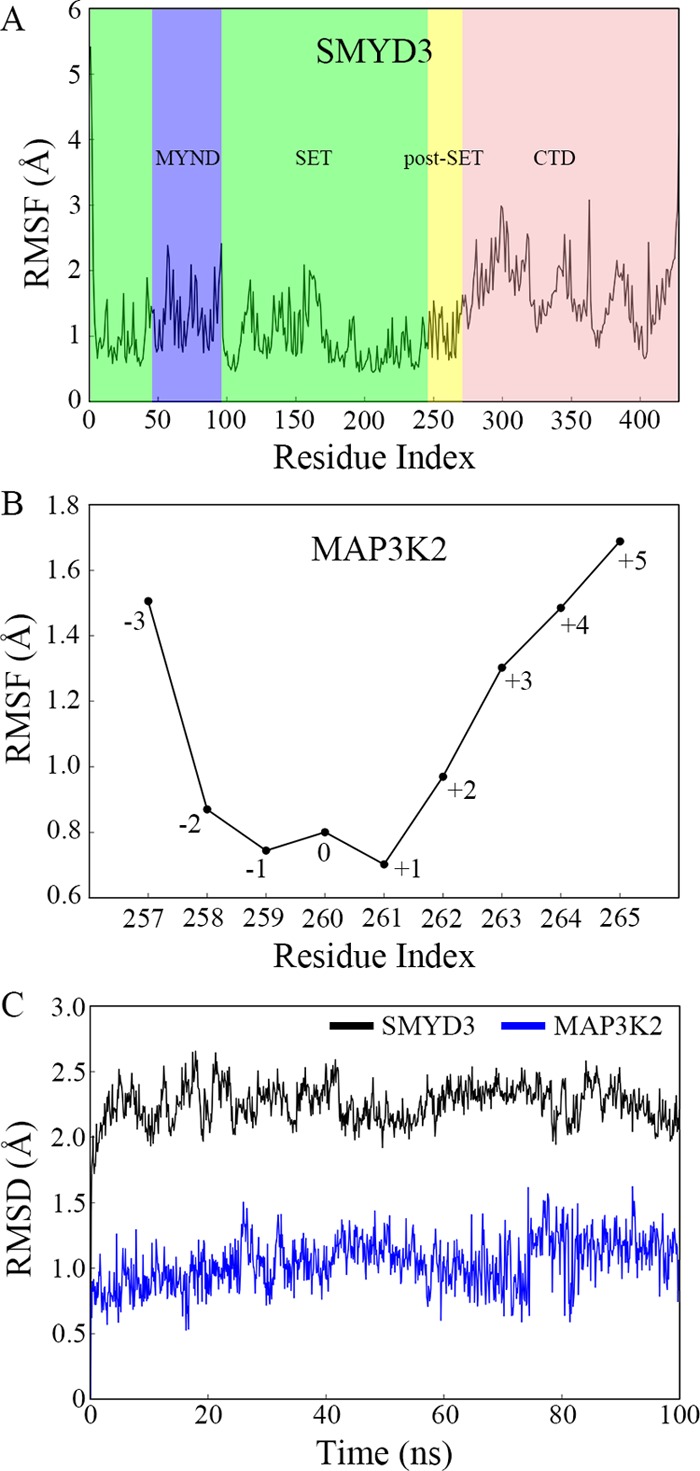
Structural deviation and flexibility in the 100-ns MD simulation. (A) RMSF as a function of the residue index of SMYD3. The SET, MYND, post-SET, and CTD domains are shaded in green, blue, yellow, and pink, respectively. (B) RMSF as a function of the residue index of MAP3K2 peptide. (C) Time evolutions of RMSD of the SET and post-SET domains in SMYD3 and the residues F258–G262 in MAP3K2 peptide.
2.1. Conformational Dynamics of F258 and Substitutions at the −2 Position
Binding preference at the −2 position was detected by the conformational dynamics of the F258 residue and each of its substitutions. A 50-ns MD simulation was performed three times. Averaged binding free energy of SMYD3 to F258 residue or its substitution from the last 2-ns was calculated, as shown in Figure 3A and Table 1, and time evolutions of its binding free energy in each repetition is depicted in Figure S1. The binding free energy tends to convergence, which means the complex has reached a relatively stable state during MD simulations. The results show that wild-type F258 of MAP3K2 has a most negative binding free energy of −9.64 kcal/mol toward SMYD3, indicating the most favorable substrate–protein recognition. Replacement of F258 to a leucine or a methionine residue results in an increased binding free energy of −5.46 and −5.31 kcal/mol, respectively. Other hydrophobic residue substitutions like I, V, C, and Y show tolerable but much weaker binding energy toward SMYD3. F258Y introduces an additional hydroxyl group to the phenyl ring, which lowers its binding affinity to the P-2 hydrophobic pocket. While in the case of the remaining F258 substitutions, the calculated binding free energy may be too high for efficient recognition. These results are in consistence with our previous experimental data27 and similar to the substrate specificity of SMYD2 at −1 position proved by computational and experimental approaches.41
Figure 3.

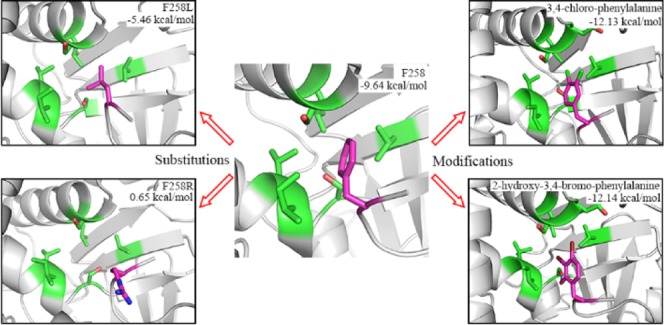
Conformational dynamics of F258 and substitutions at the −2 position. (A) The binding free energies between SMYD3 and F258 or its substitutions. The averaged value and standard deviation of three repetitions in each system are depicted in the histogram, respectively. The same scheme is used in the following figures unless explicitly specified. (B) The initial conformation in the crystal structure. F258 in MAP3K2 peptide and the residues around F258 in SMYD3 are shown in a stick model. The carbon atoms are shown in magenta and green, respectively. The main chain of the residue is hidden for brevity if not necessary. The same scheme is used in the following figures unless explicitly specified. The distances between F258 and the marked residues in SMYD3 are shown in the color map. (C) Conformational alignment of F258 after MD simulation with carbon in magenta and F258 in the crystal structure with carbon in cyan. (D–H) The conformations of other representative substitutions after MD simulations.
Table 1. Averaged Binding Free Energies and Standard Deviations between SMYD3 and F258 or Its Substitutions from Three Times-MD Simulations, Respectivelya.
| repetition |
|||||
|---|---|---|---|---|---|
| residue type | average | std. dev. | 1 | 2 | 3 |
| PHE | –9.64 | 0.13 | –9.64 | –9.52 | –9.77 |
| LEU | –5.46 | 0.95 | –5.47 | –6.41 | –4.50 |
| MET | –5.31 | 1.19 | –4.44 | –6.66 | –4.82 |
| ILE | –4.32 | 0.98 | –5.42 | –3.52 | –4.02 |
| VAL | –3.99 | 0.32 | –3.95 | –3.69 | –4.33 |
| CYS | –3.89 | 1.50 | –3.49 | –2.62 | –5.55 |
| TYR | –3.88 | 0.74 | –3.64 | –3.28 | –4.71 |
| PRO | –2.46 | 0.80 | –2.40 | –1.70 | –3.29 |
| THR | –2.14 | 1.10 | –2.13 | –3.25 | –1.06 |
| HIS | –1.92 | 0.75 | –2.74 | –1.26 | –1.77 |
| ALA | –1.74 | 0.87 | –2.46 | –1.98 | –0.77 |
| TRP | –0.85 | 1.13 | 0.44 | –1.42 | –1.58 |
| ASN | –0.04 | 1.41 | 0.70 | –1.66 | 0.85 |
| GLY | –0.03 | 0.42 | –0.01 | –0.46 | 0.38 |
| GLN | 0.03 | 1.18 | 1.05 | 0.32 | –1.27 |
| SER | 0.22 | 0.79 | 0.16 | 1.04 | –0.54 |
| ARG | 0.65 | 0.84 | –0.24 | 1.42 | 0.78 |
| GLU | 1.03 | 0.91 | 1.99 | 0.89 | 0.19 |
| LYS | 1.98 | 0.56 | 2.62 | 1.68 | 1.64 |
| ASP | 2.37 | 0.84 | 1.91 | 3.35 | 1.86 |
All binding free energies are in kcal/mol.
Besides binding free energies, binding modes at the P-2 pocket could be traced after MD simulations. The initial conformation of F258 in the crystal structure is shown in the left panel of Figure 3B. Distances of sidechain-centroids between F258 and each of the six residues forming the P-2 pocket of SMYD3 (numbered 1–6) are shown in a graduated color map at the right panel, from blue (closest) to red (farthest). Moreover, the averaged distance in the color map would help to estimate the occupancy rate of F258 in the P-2 pocket. Average conformations of F258 and its 19 replacements in the P-2 pocket during the last 2-ns MD simulations are shown in Figures 3C–H and S2. From the results, we can see that the conformation of F258 is rather stable, and its averaged distance to the P-2 pocket residues changed from 6.37 Å to a little bit more favorable 6.24 Å after simulations (Figure 3C). Among the 19 substitutions of F258, only leucine and methionine residues insert into and fully occupy the P-2 pocket like the phenylalanine residue does, with a slightly larger distances of 6.57 and 6.92 Å, respectively (Figure 3D,E). In the case of other hydrophobic substitutions like I, V, C, A, and W, their sidechains are not symmetrically presented (I), too short (V, C, A), or too large (W) to fully insert into the P-2 pocket (Figures 3F,G and S2). Other residues with hydrophilic or charged sidechains like arginine could not be stably located inside the pocket and the unfavorable bindings lead to their sidechains flipping out of the pocket during MD simulations (Figures 3H and S2).
The P-2 pocket of SMYD3 is a shallow hydrophobic pit mainly formed by L104, V178, I179, and V195; its binding preference is associated with the hydrophobic property and the sidechain volume of the interacting residue. From our calculation results, −2 position residue with aromatic hydrophobic sidechain shows a much negative binding free energy to the P-2 pocket than residues with small or hydrophilic sidechains. Meanwhile, nonpolar sidechains occupy the −2 pocket better and demonstrate a much favorable binding mode than polar or charged ones. These are consistent with the size and hydrophobic property of the P-2 pocket.
Our previous work showed that the catalytic activity of SMYD3 toward MAP3K2 peptide decreased ∼6-fold when F258 was mutated to a leucine residue, and no methylation activity was detected when an F258R mutation was introduced to the substrate.27 Combined with binding free energy calculations and structural analyses, this work provides good explanations of the experimental results. Compared with the wild-type phenylalanine residue, a leucine residue has a 4.2 kcal/mol higher binding free energy to the P-2 pocket, indicating a lower binding capacity (−5.46 to −9.64 kcal/mol). MD simulations demonstrate that a leucine residue can occupy the P-2 pocket well but has a lower occupancy rate than phenylalanine (6.57–6.24 Å). These may be the major reasons causing a lower activity of SMYD3 on F258L mutant of the substrate. This can be further proved by the substitution of the −2 residue of VEGFR1, another reported substrate of SMYD3. VEGFR1 happens to contain a leucine residue at the −2 position (L829, Figure 1A), and the activity of SMYD3 on VEGFR1 is lower than that on MAP3K2.27 When L829 is substituted by a phenylalanine residue, the binding free energy of F829 to the P-2 pocket decreases from −4.49 to −8.52 kcal/mol (Table S1), which confirms the importance of −2 phenylalanine to the activity of SMYD3.
To further prove that the binding affinity is related to the catalytic activity directly, in other words, is not affected by the change of the catalytic property of SMYD3 when catalyzing on mutated substrates, we first measured Kcat value of SMYD3 on wild-type MAP3K2 peptide and F258L mutant by in vitro methyltransferase assay. Results show similar Kcat values of SMYD3 on wild-type MAP3K2 (5.51 ± 0.85 h–1) compared with that on F258L mutant (5.13 ± 0.58 h–1), indicating a constant catalytic property of SMYD3 (Table S2). In addition, we demonstrate computationally that the active site structures remain the same for different mutants during MD simulations by calculating the averaged RMSD value of the key residues both in the active site and in the P-2 pocket of SMYD3 (Table S3). Results show small RMSDs (from 0.58 to 1.21 Å) with reasonable standard deviations (0.08–0.36 Å), indicating that the structures of the active site of SMYD3 remain the same for all of the 19 mutated substrates during MD simulations.
In the case of F258R mutation, the positively charged large sidechain of an arginine residue can hardly fit into the P-2 pocket after the MD simulations, and binding free energies rose to an even positive charged value with big deviations among paralleled simulations (Table 1). The failure of recognition at the −2 position may disturb substrate binding, hindering the insertion of the target lysine residue into the catalytic pocket. The improper distance or uncomfortable circumstance between the Nε group of lysine and the methyl group on SAM will prevent an efficient transfer of the methyl group.
2.2. SARs of Phenylalanine Derivatives at the −2 Position
Our research reveals that the P-2 pocket has a high binding preference for phenylalanine residue. The structure–activity relationship (SAR) studies of modifications on the −2 phenylalanine would be important for future inhibitor design targeting the P-2 pocket. For this propose, a series of substitutions of −2 residue by non-natural phenylalanine derivatives was investigated by similar MD simulation procedures. A total of 51 phenylalanine derivatives or mimics was chosen from the SwissSidechain database,43 which provides a unified web resource for hundreds of non-natural sidechains with biochemical parameters. Time evolutions of binding free energy (Figure S3) show that each complex has reached a relatively stable state during MD simulations. Binding free energy calculations show that quite a few modifications on the phenyl ring and two cycloalkyl mimics have more negative values than phenylalanine, indicating a more favorable binding to the P-2 pocket (Figure 4A, Table S4).
Figure 4.

Conformational dynamics of F258 modifications at the −2 position. (A) The binding free energies between SMYD3 and F258 modifications. (B–P) The conformations of the representative F258 modifications. The residue labels of SMYD3 are shown in (B) and omitted in the other figures for brevity. Hydrogen bond is depicted as dash line if it exists.
Among these beneficial modifications, halogen substitutions prove to be good candidates. And the less hydrophilic substitution results in a more favorable recognition as exampled by monosubstitution at the para position of the phenyl ring by different halogens. Binding free energy results show that substitutions by I (PHI, −10.53 kcal/mol), Br (4BF, −10.49 kcal/mol), and Cl (F20, −10.04 kcal/mol) are more suitable to the pocket than wild-type F258 (−9.64 kcal/mol), but F (PFF, −9.51 kcal/mol) is not a good candidate of substitution (Table 2, Figure 4B–E). These results correspond with the hydrophobic property of the P-2 pocket. Electronegativity of iodine is very close to that of carbon, and thus C–I bond is less polarized than other carbon–halogen bonds, which makes iodine substitution the most favorable. On the contrary, the electronegativity of fluorine is much higher than that of carbon, so the C–F bond is strongly polarized, which results in the unfavorable binding of fluorine substitution. Furthermore, monosubstitution by halogen at the para position of the phenyl ring seems to be more effective than that at the ortho or meta position (Table 2), which may due to the spatial locations of residues forming the P-2 pocket. As shown in Figure 3B, the distance between −2-position F258 and L104 residue of SMYD3 leaves adequate space to allow suitable modifications at the para position on the phenyl ring. In contrast, the sidechain hydroxyl group of S182 at the periphery of the P-2 pocket is rather close (∼4 Å) to the ortho and meta sites on one side of the phenyl ring, and thus it may hinder further modifications at those positions, especially for big hydrophobic halogen atoms. However, monosubstitution by the hydroxyl group at the ortho position is better than that at the meta or para position for recognition because its hydrophilic property is more suitable for interacting with S182 residue (Table 2). Double substitutions by halogens (except fluorine) at both para and meta sites result in a more favorable binding to the P-2 pocket as both of the halogen atoms insert into the pocket in good conformations (I34, B34, and CP3 in Table 3, Figure 4F–H). However, the para–ortho-site double modifications (CP2) is not as good as the para–meta one (CP3) because of the rather outer position and more hydrophilic property of the ortho site compared to the meta site in the pocket, though it is still better than a monosubstitution at the para site (F20) (Table 3, Figure 4I).
Table 2. Averaged Binding Free Energies between SMYD3 and the Representative Monosubstituted F258 Modificationsa.
| binding
free energy |
|||
|---|---|---|---|
| substituent group | ortho | meta | para |
| –I | –9.69 | –9.82 | –10.53 |
| –Br | –9.52 | –9.66 | –10.49 |
| –Cl | –8.96 | –9.46 | –10.04 |
| –F | –7.63 | –8.65 | –9.51 |
| –CH3 | –9.45 | –9.88 | –10.17 |
| –OH | –8.91 | –6.70 | –3.88 |
All binding free energies are in kcal/mol.
Table 3. Averaged Binding Free Energies between SMYD3 and the Representative Multisubstituted F258 Modifications or Cycloalkyl Derivativesa.
| substituent group/position |
||||||
|---|---|---|---|---|---|---|
| name | 2 | 3 | 4 | 5 | 6 | binding free energy |
| I34 | –I | –I | –12.79 | |||
| B34 | –Br | –Br | –12.29 | |||
| CP3 | –Cl | –Cl | –12.13 | |||
| CP2 | –Cl | –Cl | –9.97 | |||
| BOH | –OH | –Br | –Br | –12.14 | ||
| BO2 | –OH | –Br | –Br | –11.49 | ||
| BMO | –Br | –OH | –Br | –10.40 | ||
| DBY | –Br | –OH | –Br | –9.67 | ||
| ALC | cyclohexyl sidechain | –12.64 | ||||
| CPA | cyclopentyl sidechain | –10.82 | ||||
All binding free energies are in kcal/mol.
Substitution by the methyl group seems to be an alternative strategy that the mono-meta site (APD, −9.88 kcal/mol), mono-para site (4PH, −10.17 kcal/mol), and double-meta–para-site substitution (MP3, −9.80 kcal/mol) show a slightly lower binding free energy than F258 (−9.64 kcal/mol), respectively. A mono-ortho-site methyl modification is not as good as the para or meta substitution somehow (MPH, −9.45 kcal/mol) because the CH3 group at the ortho position may have space clashes with the aforementioned S182 residue of the P-2 pocket (Figures 4J and S4). Larger methyl group derivatives like the C(CH3)3 group behaves better than the methyl group at the para position (−10.46 to −10.17 kcal/mol) because of its greater hydrophobic property (Table S4, Figure 4K).
As one can predict, none of the hydroxyl group substitutions are suitable for the P-2 pocket, though the ortho substitution seems to be better than meta or para ones (Table 2). However, triple substitution by the hydroxyl group and halogen seems to be a good option as exampled by hydroxyl and halogen combinations. Substitutions by two iodine or bromine atoms and one hydroxyl group exhibit good binding free energies to the P-2 pocket and in the meantime attenuate the strong hydrophobic property introduced by the two halogen atoms (Table 3). Among these, BOH and BO2 with para-, meta-double bromine and ortho-hydroxyl group substitutions show pretty good binding free energy to the P-2 pocket as two bromine atoms insert into the pocket in good conformations, though BOH with a hydroxyl group positioned in the same side of the phenyl ring to bromine atoms behaves better than the opposite-side BO2 does (Figure 4L,M). If the hydroxyl group is positioned at the meta (BMO) or para (DBY) position, the binding free energy is higher and the hydrogen bond is formed with D100 instead of S182 residue, requiring a big conformational change in D100 (Table 3, Figures 4N and S4). Iodine may not be a good choice in these mixed triple substitutions because of its big size, though the binding free energy is rather outstanding (Table S4 and Figure S4). These findings throw light on future inhibitor designs by using substrate mimics at the −2 position, since the solubility of the small inhibitor can be increased by introducing a hydrophilic group but not at the expense of losing too much binding affinity.
Furthermore, substitutions of the whole phenyl ring by cycloalkyl mimics also show rather good results. This may be connected with the flexibility of naphthene that it could fit into the P-2 pocket better than a more rigid phenyl ring. Cyclohexyl ring is better than the cyclopentyl ring as it resembles the phenyl ring more in shape (Table 3, Figure 4O,P).
3. Conclusions
In this work, we use MD simulations and the MM/GBSA method to study the substrate preference of methyltransferase SMYD3. The P-2 pocket of SMYD3 is a well-defined shallow hydrophobic subpocket adjacent to the catalytic pocket. The −2-position F258 residue of MAP3K2 was substituted to each of the other 19 natural residues followed by a 50-ns MD simulation repeated three times, respectively. The energetic and structural results show that the P-2 pocket prefers an average-sized hydrophobic group. None of the substitutions behave better than the wild-type phenylalanine residue.
As the P-2 pocket plays a significant role in substrate binding, we investigated the SARs of a series of non-natural phenylalanine derivative substitutions at the −2 position. Results show that the P-2 pocket has adequate space to allow hydrophobic modifications at the para positions on the phenyl ring, especially for halogen atoms like bromine and chlorine. However, the hydrophilic S182 residue in the P-2 pocket confines the size and hydrophobicity of the substitution at the ortho or meta position of the ring. Multiple substitutions by hydroxyl group and halogen combinations seem to be a good guidance for future inhibitor design to achieve both solubility and affinity.
Recently, EPZ030456 was developed as an SMYD3 inhibitor with a nanomolar inhibition concentration.44 However, the crystal structure shows that except for the headgroup that inserts into the lysine channel, the compound makes few specific interactions with SMYD3. To our knowledge, the P-2 pocket has not been shown as a target pocket in developing SMYD3 inhibitors. Investigations of binding preference of the P-2 pocket, as well as the SARs of modifications on the −2-position phenyl ring in this study, will provide insights into the rational drug design to develop novel SMYD3 inhibitors with high potency and selectivity.
4. Materials and Methods
4.1. Systems for MD Simulations
The previously solved crystal structure of SMYD3 and MAP3K2 complex (PDB: 5EX0) was used as the starting structure. F258 and G262 at the −2 and +2 positions of the MAP3K2 peptide were substituted to each of the other 19 natural residues, respectively. F258 was also substituted, respectively, to several non-natural residues chosen from the SwissSidechain database,43 which provides a unified web resource for hundreds of non-natural sidechains with biochemical parameters. The selection of non-natural residue types follows the rules: (1) Sidechains were mainly chosen from the set of phenylalanine derivatives. (2) Sidechains with large substituted groups on the phenyl ring such as phenyl group, phosphate group, nitro group, cyan group, and guanidine group were not considered in case of steric conflicts with the P-2 pocket. (3) Sidechains with modifications on the CB atom such as hydroxyl- or carbonyl substitutions were not considered to simplify the system of comparison. (4) There is no special requirement for the position (ortho, meta, or para) or the number (mono or multi) of the substituted group. (5) Two alanine derivatives were selected with cycloalkyl sidechains to resemble the phenyl ring. Names of these non-natural residues were shown by three-letter abbreviations. (6) Eleven phenylalanine derivatives with halogen-substitutions, which are not included in the database, were also employed in the SARs study. They are I34, B34, BOH, IMO, IO2, BO2, BMO, MIF, MBF, OIF, and OBF. Their structures were modified from 4-bromo-phenylalanine (4BF) in the database by changing the substituted groups manually. Finally, 51 non-natural sidechains were investigated in MD simulations.
4.2. Parameters in the MD Simulations
The LEaP module of the AMBER18 package45 was used for residue substitution and adding hydrogen atoms for each complex. The cofactor S-adenosyl-l-homocysteine (SAH) and three zinc ions in the crystal structure were reserved in the MD simulations. The chelating cysteine and histidine residues were deprotonated. The amber FF14SB force field46 was used for protein and natural residues. The force field parameters of non-natural residues and SAH were derived from the SwissSidechain database43 and the general Amber force field (GAFF),47 respectively. TIP3P water model48 was used to solvate the complex in a hexagonal explicit water box under the periodic boundary condition. The distance between the edges of the box and the closet atoms of the complex was 12 Å. Na+ was added as a counterion to neutralize each system.
For each solvated system, 5000-step energy minimization was performed, followed by a combined equilibration process with a 500-ps constant volume ensemble to heat the system from 0 to 300 K and a 500-ps constant pressure ensemble at a constant pressure of 1 bar. The Langevin thermostat49 and Berendsen barostat50 were used for temperature and pressure control, respectively. During equilibration, a force constant of 10 kcal mol–1 Å–2 as a harmonic constraint was applied. Then, 50-ns MD simulation of each system was performed in constant pressure ensembles at 300 K with the constraint released. The time step was set to 2 fs. The SHAKE algorithm51 was used to restrain all of the bond lengths involving hydrogen atoms. The particle mesh Ewald (PME) method52 was used to calculate the long-range electrostatic contributions. The cut-off value of the van der Waals interactions was set to 10 Å. Each system was performed three times under the same conditions to confirm the results. Besides, an extra 100-ns MD simulation of SMYD3 in complex with wild-type MAP3K2 peptide was performed to investigate their stability. The AMBER18 software package45 was employed to perform MD simulations. Coordinates were saved every 10 ps.
4.3. Analysis of the Simulations
The interactions between SMYD3 and the focused residues were calculated by the MM/GBSA method, which combines molecular mechanics (MM), the Generalized Born (GB) equations, and surface accessible (SA) calculations to calculate the binding free energy.53−55 The calculated binding free energy is given by
| 1 |
where ΔGbind, vac is the free energy of the complex in the gas phase, including nonbonded and bonded potentials calculated by the MM method, and ΔGcom, sol, ΔGrec, sol, and ΔGlig, sol are the free energies of the complex, receptor, and ligand in the aqueous phase, respectively. The solvated free energies include the polar and nonpolar free energies, which are computed by the GB and SA methods, respectively. In this work, the binding free energy between SMYD3 and the residue in the peptide substrate was calculated by a total of 200 snapshots extracted from the final 2 ns trajectory of each system. All of the parameters were set as default values in the calculations. In each system, the residue in the MAP3K2 peptide to be calculated and the whole protein with SAH and zinc ions were considered as ligand and receptor, respectively. Since we are mainly interested in the differences of the binding free energies, entropy was ignored in the calculations.
The cpptraj module56 of AmberTools18 was used to calculate the averaged structure, the averaged distance, root mean square fluctuations (RMSF), and root mean square deviations (RMSD). The illustrated representative conformation was averaged from the last 2-ns MD simulation and derived from the first repetition of each system. The distance was measured by sidechain-centroids between residues unless explicitly specified.
Acknowledgments
We thank grant supports from the Natural Science Foundation of China and the Chinese Ministry of Science and Technology (grant nos. 31622020, 2018YFA0107004 and 2019YFA0508902), Tianjin Funds for Distinguished Young Scientists (17JCJQJC45900), Beijing Municipal Science and Technology Project (Z171100000417001), and Project funded by China Postdoctoral Science Foundation (2018M641631).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01842.
Conformational dynamics of F258 and its natural and non-natural substitutions at the −2 position; structure of F258 modifications; and in vitro methyltransferase activity assay (PDF)
Author Contributions
This article was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang J.; Berger S. L. The Emerging Field of Dynamic Lysine Methylation of Non-Histone Proteins. Curr. Opin. Genet. Dev. 2008, 18, 152–158. 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Tan X.; Rotllant J.; Li H.; De Deyne P.; Du S. J. SmyD1, a Histone Methyltransferase, Is Required for Myofibril Organization and Muscle Contraction in Zebrafish Embryos. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2713–2718. 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. A.; Sims R. J.; Gottlieb P. D.; Tucker P. W. Identification and Characterization of Smyd2: A Split SET/MYND Domain-Containing Histone H3 Lysine 36-Specific Methyltransferase That Interacts with the Sin3 Histone Deacetylase Complex. Mol. Cancer 2006, 5, 26 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R.; Furukawa Y.; Morita M.; Iimura Y.; Silva F. P.; Li M.; Yagyu R.; Nakamura Y. SMYD3 Encodes a Histone Methyltransferase Involved in the Proliferation of Cancer Cells. Nat. Cell Biol. 2004, 6, 731–740. 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- Van Aller G. S.; Reynoird N.; Barbash O.; Huddleston M.; Liu S.; Zmoos A.-F.; McDevitt P.; Sinnamon R.; Le B.; Mas G.; Annan R.; Sage J.; Garcia B. A.; Tummino P. J.; Gozani O.; Kruger R. G. Smyd3 Regulates Cancer Cell Phenotypes and Catalyzes Histone H4 Lysine 5 Methylation. Epigenetics 2012, 7, 340–343. 10.4161/epi.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M.; Jenuwein T. The Many Faces of Histone Lysine Methylation. Curr. Opin. Cell Biol. 2002, 14, 286–298. 10.1016/S0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- Grewal S. I.; Elgin S. C. R. Heterochromatin: New Possibilities for the Inheritance of Structure. Curr. Opin. Genet. Dev. 2002, 12, 178–187. 10.1016/S0959-437X(02)00284-8. [DOI] [PubMed] [Google Scholar]
- Wu J.; Grunstein M. 25 Years after the Nucleosome Model: Chromatin Modifications. Trends Biochem. Sci. 2000, 25, 619–623. 10.1016/S0968-0004(00)01718-7. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Reinberg D. Transcription Regulation by Histone Methylation: Interplay between Different Covalent Modifications of the Core Histone Tails. Genes Dev. 2001, 15, 2343–2360. 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- Spellmon N.; Holcomb J.; Trescott L.; Sirinupong N.; Yang Z. Structure and Function of SET and MYND Domain-Containing Proteins. Int. J. Mol. Sci. 2015, 16, 1406–1428. 10.3390/ijms16011406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Perez-Burgos L.; Placek B. J.; Sengupta R.; Richter M.; Dorsey J. A.; Kubicek S.; Opravil S.; Jenuwein T.; Berger S. L. Repression of P53 Activity by Smyd2-Mediated Methylation. Nature 2006, 444, 629–632. 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Saddic L. A.; West L. E.; Aslanian A.; Yates J. R.; Rubin S. M.; Gozani O.; Sage J. Methylation of the Retinoblastoma Tumor Suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Tanaka K.; Yan J.; Li J.; Peng D.; Jiang Y.; Yang Z.; Barton M. C.; Wen H.; Shi X. Regulation of Estrogen Receptor by Histone Methyltransferase SMYD2-Mediated Protein Methylation. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17284–17289. 10.1073/pnas.1307959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M.; Lanouette S.; Elisma F.; Tremblay V.; Butson J.; Figeys D.; Couture J.-F. Proteomic Analyses of the SMYD Family Interactomes Identify HSP90 as a Novel Target for SMYD2. J. Mol. Cell Biol. 2011, 3, 301–308. 10.1093/jmcb/mjr025. [DOI] [PubMed] [Google Scholar]
- Cornett E. M.; Dickson B. M.; Krajewski K.; Spellmon N.; Umstead A.; Vaughan R. M.; Shaw K. M.; Versluis P. P.; Cowles M. W.; Brunzelle J.; Yang Z.; Vega I. E.; Sun Z.; Rothbart S. B. A Functional Proteomics Platform to Reveal the Sequence Determinants of Lysine Methyltransferase Substrate Selectivity. Sci. Adv. 2018, 4, eaav2623 10.1126/sciadv.aav2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunizaki M.; Hamamoto R.; Silva F. P.; Yamaguchi K.; Nagayasu T.; Shibuya M.; Nakamura Y.; Furukawa Y. The Lysine 831 of Vascular Endothelial Growth Factor Receptor 1 Is a Novel Target of Methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- Mazur P. K.; Reynoird N.; Khatri P.; Jansen P. W. T. C.; Wilkinson A. W.; Liu S.; Barbash O.; Van Aller G. S.; Huddleston M.; Dhanak D.; Tummino P. J.; Kruger R. G.; Garcia B. A.; Butte A. J.; Vermeulen M.; Sage J.; Gozani O. SMYD3 Links Lysine Methylation of MAP3K2 to Ras-Driven Cancer. Nature 2014, 510, 283–287. 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva F. P.; Hamamoto R.; Kunizaki M.; Tsuge M.; Nakamura Y.; Furukawa Y. Enhanced Methyltransferase Activity of SMYD3 by the Cleavage of Its N-Terminal Region in Human Cancer Cells. Oncogene 2008, 27, 2686–2692. 10.1038/sj.onc.1210929. [DOI] [PubMed] [Google Scholar]
- Sarris M. E.; Giakountis A.; Moulos P.; Talianidis I.; Hatzis P. Smyd3-Associated Regulatory Pathways in Cancer. Semin. Cancer Biol. 2017, 42, 70–80. 10.1016/j.semcancer.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Sarris M. E.; Moulos P.; Haroniti A.; Giakountis A.; Talianidis I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Ma S.-J.; Liu Y.-M.; Zhang Y.-L.; Chen M.-W.; Cao W. Correlations of EZH2 and SMYD3 Gene Polymorphisms with Breast Cancer Susceptibility and Prognosis. Biosci. Rep. 2018, 38, BSR20170656 10.1042/BSR20170656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A. D.; Camp E. R.; Fan F.; Shen L.; Gray M. J.; Liu W.; Somcio R.; Bauer T. W.; Wu Y.; Hicklin D. J.; Ellis L. M. Vascular Endothelial Growth Factor Receptor-1 Activation Mediates Epithelial to Mesenchymal Transition in Human Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 46–51. 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- Starovasnik M. A.; Christinger H. W.; Wiesmann C.; Champe M. A.; de Vos A. M.; Skelton N. J. Solution Structure of the VEGF-Binding Domain of Flt-1: Comparison of Its Free and Bound States 1 1 Edited by P. E. Wright. J. Mol. Biol. 1999, 293, 531–544. 10.1006/jmbi.1999.3134. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Hooper A. T.; Zhong Z.; Witte L.; Bohlen P.; Rafii S.; Hicklin D. J. The Vascular Endothelial Growth Factor Receptor (VEGFR-1) Supports Growth and Survival of Human Breast Carcinoma. Int. J. Cancer 2006, 119, 1519–1529. 10.1002/ijc.21865. [DOI] [PubMed] [Google Scholar]
- Colón-Bolea P.; Crespo P. Lysine Methylation in Cancer: SMYD3-MAP3K2 Teaches Us New Lessons in the Ras-ERK Pathway. BioEssays 2014, 36, 1162–1169. 10.1002/bies.201400120. [DOI] [PubMed] [Google Scholar]
- Fu W.; Liu N.; Qiao Q.; Wang M.; Min J.; Zhu B.; Xu R. M.; Yang N. Structural Basis for Substrate Preference of SMYD3, a SET Domain-Containing Protein Lysine Methyltransferase. J. Biol. Chem. 2016, 291, 9173–9180. 10.1074/jbc.M115.709832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Li L.; Zhang H.; Luo X.; Dai J.; Zhou S.; Gu J.; Zhu J.; Atadja P.; Lu C.; Li E.; Zhao K. Structure of Human SMYD2 Protein Reveals the Basis of P53 Tumor Suppressor Methylation. J. Biol. Chem. 2011, 286, 38725–38737. 10.1074/jbc.M111.262410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Sirinupong N.; Brunzelle J.; Yang Z. Crystal Structures of Histone and P53 Methyltransferase SmyD2 Reveal a Conformational Flexibility of the Autoinhibitory C-Terminal Domain. PLoS One 2011, 6, e21640 10.1371/journal.pone.0021640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Trescott L.; Holcomb J.; Zhang X.; Brunzelle J.; Sirinupong N.; Shi X.; Yang Z. Structural Insights into Estrogen Receptor α Methylation by Histone Methyltransferase SMYD2, a Cellular Event Implicated in Estrogen Signaling Regulation. J. Mol. Biol. 2014, 426, 3413–3425. 10.1016/j.jmb.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Larsen N. A.; Howard T.; Pollard H.; Green I.; Grande C.; Cheung T.; Garcia-Arenas R.; Cowen S.; Wu J.; Godin R.; Chen H.; Keen N. Structural Basis of Substrate Methylation and Inhibition of SMYD2. Structure 2011, 19, 1262–1273. 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Lanouette S.; Davey J. A.; Elisma F.; Ning Z.; Figeys D.; Chica R. A.; Couture J.-F. Discovery of Substrates for a SET Domain Lysine Methyltransferase Predicted by Multistate Computational Protein Design. Structure 2015, 23, 206–215. 10.1016/j.str.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Sammond D. W.; Eletr Z. M.; Purbeck C.; Kimple R. J.; Siderovski D. P.; Kuhlman B. Structure-Based Protocol for Identifying Mutations That Enhance Protein–Protein Binding Affinities. J. Mol. Biol. 2007, 371, 1392–1404. 10.1016/j.jmb.2007.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell D. J.; Kortemme T. Computer-Aided Design of Functional Protein Interactions. Nat. Chem. Biol. 2009, 5, 797–807. 10.1038/nchembio.251. [DOI] [PubMed] [Google Scholar]
- Karanicolas J.; Kuhlman B. Computational Design of Affinity and Specificity at Protein–Protein Interfaces. Curr. Opin. Struct. Biol. 2009, 19, 458–463. 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvizo O.; Mittal S.; Mayo S. L.; Schiffer C. A. Structural, Kinetic, and Thermodynamic Studies of Specificity Designed HIV-1 Protease. Protein Sci. 2012, 21, 1029–1041. 10.1002/pro.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.-S.; Love J. J.; Mayo S. L. A de Novo Designed Protein-Protein Interface. Protein Sci. 2007, 16, 2770–2774. 10.1110/ps.073125207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha R. K.; Leaver-Fay A.; Yin S.; Wu Y.; Butterfoss G. L.; Szyperski T.; Dokholyan N. V.; Kuhlman B. Computational Design of a PAK1 Binding Protein. J. Mol. Biol. 2010, 400, 257–270. 10.1016/j.jmb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Kortemme T. Backrub-Like Backbone Simulation Recapitulates Natural Protein Conformational Variability and Improves Mutant Side-Chain Prediction. J. Mol. Biol. 2008, 380, 742–756. 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair J. M. A.; Bavro V. N.; Ricci V.; Modi N.; Cacciotto P.; Kleinekathöfer U.; Ruggerone P.; Vargiu A. V.; Baylay A. J.; Smith H. E.; Brandon Y.; Galloway D.; Piddock L. J. V. AcrB Drug-Binding Pocket Substitution Confers Clinically Relevant Resistance and Altered Substrate Specificity. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 3511–3516. 10.1073/pnas.1419939112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi U.; Holliday M. J.; Eisenmesser E. Z.; Hamelberg D. Dynamical Network of Residue–Residue Contacts Reveals Coupled Allosteric Effects in Recognition, Catalysis, and Mutation. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 4735–4740. 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gfeller D.; Michielin O.; Zoete V. SwissSidechain: A Molecular and Structural Database of Non-Natural Sidechains. Nucleic Acids Res. 2012, 41, D327–D332. 10.1093/nar/gks991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; Raimondi A.; Jacques S. L.; West K.; Foley M.; Waters N. J.; Kuntz K. W.; Wigle T. J.; Scott M. P.; Copeland R. A.; Smith J. J.; Chesworth R. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D. A., Case, Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham T. E. III, Cruzeiro V. W. D., Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., Izadi S., Kovalenko A., Kurtzman T., Lee T. S., LeGra S., Y D. M., K P. A. Amber 18, 2018.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Pastor R. W.; Brooks B. R.; Szabo A. An Analysis of the Accuracy of Langevin and Molecular Dynamics Algorithms. Mol. Phys. 1988, 65, 1409–1419. 10.1080/00268978800101881. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald: An N ·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Kollman P. Free-Energy Calculations - Applications to Chemical and Biochemical Phenomena. Chem. Rev. 1993, 93, 2395–2417. 10.1021/cr00023a004. [DOI] [Google Scholar]
- Chong L. T.; Duan Y.; Wang L.; Massova I.; Kollman Pa. Molecular Dynamics and Free-Energy Calculations Applied to Affinity Maturation in Antibody 48G7. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14330–14335. 10.1073/pnas.96.25.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman P.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D.; Cheatham T. E. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.