

Abstract
Schizophrenia is a debilitating psychiatric disorder with approximately 1% lifetime risk globally. Large-scale schizophrenia genetic studies have reported primarily on European ancestry samples, potentially missing important biological insights. Here, we report the largest study to date of East Asian participants (22,778 schizophrenia cases and 35,362 controls), identifying 21 genome-wide significant associations in 19 genetic loci. Common genetic variants that confer risk for schizophrenia have highly similar effects between East Asian and European ancestries (rg = 0.98 ± 0.03), indicating that the genetic basis of schizophrenia and its biology are broadly shared across populations. A fixed-effect meta-analysis including individuals from East Asian and European ancestries identified 208 significant associations in 176 genetic loci (53 novel). Trans-ancestry fine-mapping reduced the sets of candidate causal variants in 44 loci. Polygenic risk scores had reduced performance when transferred across ancestries, highlighting the importance of including sufficient samples of major ancestral groups to ensure their generalizability across populations.
Editorial summary:
Genome-wide meta-analysis with individuals of East Asian or European ancestry identifies 176 loci associated with schizophrenia. Despite consistent genetic effects across populations, polygenic risk models trained in one population have reduced performance in the other population.
Schizophrenia is an often disabling psychiatric disorder that occurs worldwide with a lifetime risk of about 1%1. It is well established that genetic factors contribute to the susceptibility of schizophrenia. Recently, 145 genetic loci have been associated with schizophrenia in samples of primarily European ancestry2,3 (EUR), but this still represents the tip of the iceberg with respect to common variant liability to the disorder: the highly polygenic nature of common variation underlying this disorder predicts that there are hundreds more loci to be discovered4.
Most genetic studies of schizophrenia have been performed in EUR samples, with relatively few studies in other populations5-8. This is a substantial deficiency for multiple reasons, particularly as it greatly limits the discovery of biological clues about schizophrenia. For some causal variants, ancestry-related heterogeneity yields varying allele frequency and linkage disequilibrium (LD) patterns such that associations that can be detected in one population may not be readily detected in others. Examples include a nonsense variant in TBC1D4, which confers muscle insulin resistance and increases risk for type 2 diabetes, common in Greenland but rare or absent in other populations9, several Asian-specific coding variants that influence blood lipids10, a variant highly protective against alcoholism that is common in Asian populations but uncommon elsewhere11, and two loci associated with major depression12 that are more common in the Chinese populations than EUR12,13 (rs12415800: 45% versus 2%; rs35936514: 28% versus 6%).
Even if alleles have similar frequencies across populations, the effects of alleles on risk might be specific to certain populations if there are prominent but local contributions of clinical heterogeneity, gene-environment (GxE) or gene-gene (GxG) interactions. In addition, there have been debates about differences in prevalence, symptomatology, etiology, outcome, and course of illness across geographical regions14-19. Understanding the genetic architecture of schizophrenia across populations provides insights into whether any differences represent etiologic heterogeneity on the illness.
Finally, polygenic risk score (PRS) prediction is emerging as a useful tool for studying the effects of genetic liability, identifying more homogeneous phenotypes, and stratifying patients. However, previous studies have shown that prediction accuracy decays with increasing genetic divergence between the risk allele discovery and target datasets20,21. The risk predicted, measured as R2, was only 45% as accurate in EAS as in EUR individuals when computed from GWAS of Europeans22. These differences can be explained by ancestry-related differences in allele frequencies, LD, and other factors22. Importantly, the applicability of training data from EUR studies to those of non-European ancestry has not been fully assessed, leaving uncertainty as to the biological relevance of discoveries made in EUR samples for non-Europeans21.
Results
Schizophrenia genetic associations in East Asian populations.
To our knowledge, this is the first study to combine multiple samples with schizophrenia across East Asia (EAS) to systematically examine the genetic architecture of schizophrenia in individuals of EAS ancestry. We compiled 22,778 schizophrenia cases and 35,362 controls from 20 samples from East Asia (Supplementary Table 1). Individual-level genotypes were available from 16 sample collections (Supplementary Table 1), on which we performed quality control, imputation and association tests (Methods and Supplementary Table 2). Two sample collections (TAI-1 and TAI-2) were trio-based and pseudo-controls were used. Four sample collections made available summary statistics for 22K-31K selected variants (Methods) that had been analyzed in published studies7,8. Compared with the latest study using only Chinese individuals8, our study has about twice the sample size, and is much more diverse.
We used a two-stage study design (Supplementary Table 1a). Stage 1 included 13 sample collections for which we had individual genotype data (13,305 cases and 16,244 controls after quality control). Stage 2 incorporated the remaining 7 sample collections: full genotype data from 3 sample collections that arrived after the stage 1 data freeze, and summary statistics (for selected variants) from 4 sample collections (Supplementary Table 1). Meta-analyses across stage 1 samples and across all EAS samples were conducted using a fixed-effect model with inverse-variance weighting. QQ plots (Supplementary Fig. 1) showed no inflation of test statistics (indicating that ancestry effects have been well controlled) with λgc = 1.14, λ1000 = 1.01, and LD Score regression23 (LDSC) intercept = 1.0145 ± 0.011 using stage 1 samples.
Combining stages 1 and 2, we found 21 genome-wide significant associations at 19 loci (Table 1, Fig. 1, Supplementary Table 3, and Supplementary Data Sets 1 and 2), an additional 14 associations over the most recent schizophrenia genetic study of Chinese ancestry8. Most associations were characterized by marked differences in allele frequencies between the EAS and EUR samples: for 15 of 21 loci, the index variants had a higher minor allele frequencies (MAF) in EAS than EUR. The higher allele frequency potentially confers better power to detect associations in EAS. For example, we identified a locus (Supplementary Data Sets 1) with the top association (rs374528934) having strong evidence in EAS (P = 5 × 10−11) but not in EUR using the stage 1 samples. rs374528934 has MAF of 45% in EAS but only 0.7% in EUR. No other variant in this locus is significantly associated with schizophrenia in EUR. This locus contains CACNA2D2 (encoding the calcium channel α2δ-2 subunit) associated with childhood epilepsy24,25, and to which the anticonvulsant medication gabapentin binds, suggesting a path for further therapeutic investigation25. This finding also adds new evidence to the calcium signaling pathway suggested to be implicated in psychiatric disorders26,27.
Table 1 ∣.
Genome-wide significant loci in the East Asian populations.
| Stage 1 | Stage 2 | Combined | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | BP | AL | P | OR | P | OR | P | OR |
| rs4660761 | 1 | 44440146 | A/G | 3.6E-06 | 0.91 | 3.53E-04 | 0.92 | 5.08E-09 | 0.91 |
| rs848293 | 2 | 58382490 | A/G | 3.7E-10 | 0.90 | 3.10E-09 | 0.87 | 9.87E-18 | 0.89 |
| rs17592552 | 2 | 201176071 | T/C | 8.4E-10 | 0.86 | 2.68E-05 | 0.89 | 1.50E-13 | 0.88 |
| rs2073499 | 3 | 50374293 | A/G | 1.1E-09 | 0.89 | 2.14E-05 | 0.91 | 1.33E-13 | 0.90 |
| rs76442143 | 3 | 51043599 | T/C | 6.9E-09 | 1.14 | 1.03E-02 | 1.08 | 6.40E-10 | 1.12 |
| rs10935182 | 3 | 136137422 | A/G | 1.3E-06 | 0.90 | 1.33E-04 | 0.90 | 7.08E-10 | 0.90 |
| rs4856763 | 3 | 161831675 | A/G | 3.9E-06 | 0.92 | 8.54E-06 | 0.91 | 1.73E-10 | 0.92 |
| rs13096176 | 3 | 180752138 | T/C | 3.1E-07 | 0.88 | 2.21E-03 | 0.90 | 3.35E-09 | 0.89 |
| rs6832165 | 4 | 24270210 | C/G | 3.7E-08 | 1.12 | 3.70E-01 | 1.08 | 2.79E-08 | 1.12 |
| rs13142920 | 4 | 176728614 | A/C | 9.5E-05 | 0.93 | 5.85E-06 | 0.89 | 4.85E-09 | 0.92 |
| rs4479913 | 6 | 165075210 | A/G | 3.6E-07 | 1.13 | 9.98E-05 | 1.12 | 1.53E-10 | 1.12 |
| rs320696 | 7 | 137047137 | A/C | 5.5E-08 | 0.90 | 1.07E-02 | 0.93 | 2.81E-09 | 0.91 |
| rs11986274 | 8 | 38259481 | T/C | 5.1E-04 | 1.07 | 2.73E-06 | 1.11 | 1.44E-08 | 1.08 |
| rs2612614 | 8 | 65310836 | A/G | 2.2E-08 | 1.14 | 4.51E-02 | 1.06 | 1.62E-08 | 1.11 |
| rs4147157 | 10 | 104536360 | A/G | 6.6E-10 | 0.90 | 3.87E-07 | 0.89 | 1.32E-15 | 0.89 |
| rs10861879 | 12 | 108609634 | A/G | 4.8E-07 | 1.09 | 5.00E-03 | 1.07 | 1.18E-08 | 1.08 |
| rs1984658 | 12 | 123483426 | A/G | 5.1E-11 | 0.89 | 2.14E-04 | 0.92 | 8.62E-14 | 0.90 |
| rs9567393 | 13 | 32763757 | A/G | 3.5E-08 | 1.11 | 4.37E-03 | 1.07 | 1.13E-09 | 1.09 |
| rs9890128 | 17 | 1273646 | T/C | 3.5E-08 | 0.90 | 2.44E-02 | 0.91 | 2.61E-09 | 0.90 |
| rs11665111 | 18 | 77622996 | T/C | 5.2E-06 | 1.08 | 6.89E-04 | 1.09 | 1.46E-08 | 1.09 |
| rs55642704 | 18 | 77688124 | T/C | 1.1E-06 | 1.09 | 7.11E-06 | 1.10 | 3.76E-11 | 1.09 |
BP, genomic position in HG19; AL, reference and non-reference alleles; OR, odds ratio; P, P-value. n (EAS stage 1) = 13,305 cases, 16,244 controls; n (EAS stage 1+2) = 22,778 cases, 35,362 controls. Fixed effect inverse variance meta-analysis was utilized to generate P-values.
Figure 1 ∣. Genetic associations in East Asian populations.
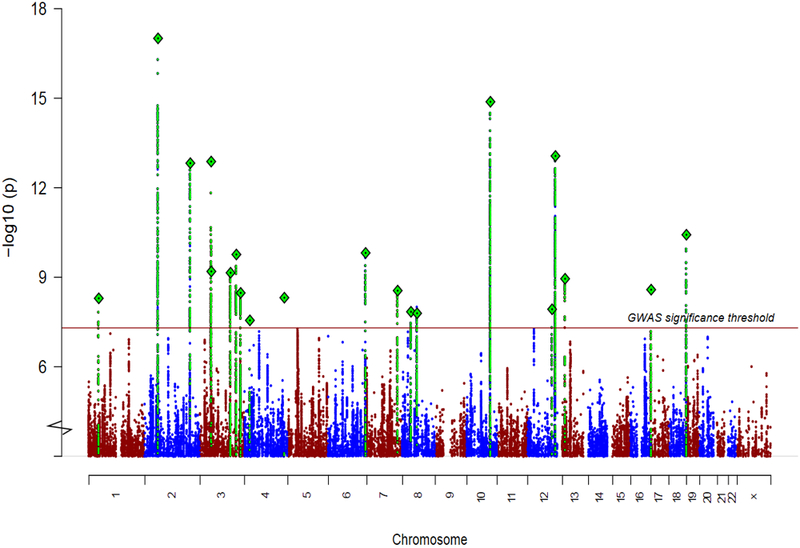
Manhattan plot for schizophrenia genetic associations using East Asian samples (stages 1 and 2; n = 22,778 cases; 35,362 controls).
Genetic effects are consistent across populations.
For causal variants, heterogeneity of genetic effects across populations could arise from clinical heterogeneity, differences in pathophysiology, environmental differences that change the genetic effects (GxE interaction), or interaction with other genetic factors that may differ in frequency across populations (GxG interaction). Heterogeneity in estimating genetic effect sizes may also be a consequence of differential correlation across genetic markers in a region, when investigating variants that are tagging the causal variant but do not exert any influence on the trait in question. Such heterogeneity does not reflect biological differences, but is rather statistical in nature. While it is assumed that biological pathways underlying complex human disorders are generally consistent across populations, genetic heterogeneity has been observed in other genetically complex disorders28. The large EAS sample allowed us to systematically explore the heterogeneity of genetic effects influencing liability to schizophrenia across two major world populations.
Using LDSC23 and common variants (MAF > 5%) outside of the MHC region, we found that the SNP-heritability of schizophrenia is very similar in EAS (0.23 ± 0.03) and EUR (0.24 ± 0.02) (Methods and Supplementary Fig. 2a). Using the same set of variants, we found that the genetic correlation for schizophrenia between EAS and EUR was indistinguishable from 1 (rg = 0.98 ± 0.03) (using POPCORN29, a method designed for cross-ancestry comparisons). This finding indicates that the common variant genetic architecture of schizophrenia outside of the MHC region is highly consistent across EAS and EUR.
Genetic correlations between schizophrenia and 11 other psychiatric disorders and behavior traits also showed no significant differences when estimated within EUR and across EAS-EUR (Supplementary Fig. 2b). In agreement with recent reports30-33, we observed significant positive genetic correlations for schizophrenia with bipolar disorder, major depressive disorder, anorexia nervosa, neuroticism, autism spectrum disorder, and educational attainment. We observed significant negative correlations with general intelligence, fluid intelligence score, prospective memory, and subjective well-being.
We used partitioned LDSC23 to look for heritability enrichment in diverse functional genomic annotations defined and used in previous publications34,35 (Methods and Supplementary Fig. 2c,d). Using EAS stage 1 samples, we observed significant enrichment (after Bonferroni correction) in regions conserved across 29 mammals (Conserved LindbladToh36). No other annotations were significantly enriched, and there were no significant differences between EUR-only and EAS-only enrichments (P = 0.16, two-sided paired t test).
We identified gene-sets that are enriched for schizophrenia genetic associations using MAGMA37 and gene-set definitions from a recent schizophrenia exome sequencing study38 (Methods). Despite large differences in sample size and genetic background, the gene-sets implicated in EAS and EUR samples were highly consistent: we observed no significant differences between gene-set ranks using the EAS samples from the ranks using EUR samples (P = 0.72, Wilcoxon test, two-sided). In addition, 9 of the top 10 gene-sets identified using the EAS samples are also among the top 10 gene-sets identified using EUR samples (Supplementary Fig. 3).
A study of EUR individuals suggested that common schizophrenia alleles are under strong background selection3. We performed two analyses and found that the natural selection signatures, including positive and background selections, are consistent in schizophrenia-associated loci across EAS and EUR populations. First, we compared the signatures in the top 100 associated loci in EAS to those in EUR. Among the selection signatures we calculated (Methods), none showed a significant difference across populations (Supplementary Fig. 4a, P > 0.05 for all panels, two-sided t test). We next asked whether the population differentiation drives schizophrenia variants to have different effects in different populations. Using 295 autosomal variants that are genome-wide significant in EAS, EUR or EAS-EUR combined samples, we did not observe a correlation (R2 = 0.003, Supplementary Fig. 4b) between the population differentiation (measured by Fst) and the heterogeneity of effect size (measured by log10P-value from the heterogeneity test across EAS and EUR).
As a further test, we examined whether the effect size estimates from EUR differ from those from EAS. We performed a heterogeneity test (Cochran's Q) for the most significant variants in the 108 published schizophrenia-associated loci2. Among them, 7 variants showed significant heterogeneity after Bonferroni correction (Supplementary Table 4). Postulating that this might in part be driven by the inflation of EUR estimates as a result of the winner’s curse, we applied a correction for the winner’s curse39, after which none of the variants showed evidence for significant heterogeneity, and the P-values from the heterogeneity test follow a uniform distribution (P = 0.10, Kolmogorov–Smirnov test, two-tailed).
Lastly, we evaluated the heterogeneity of schizophrenia genetic effects within EAS samples. None of the EAS associations showed significant heterogeneity across EAS samples (Supplementary Table 3). Using their principal components (PC), we further grouped the samples into the Northeast Asian, Southeast Asian and Indonesian subpopulations (Methods). We then performed a heterogeneity test (Cochran's Q) and found no significant heterogeneity among the three subpopulations (Supplementary Fig. 5).
Schizophrenia genetic associations from the meta-analysis of EAS and EUR.
As the genetic effects observed in EAS are largely consistent with those observed in EUR, we performed a meta-analysis including the EUR and EAS samples (stages 1 and 2) using a fixed-effect model with inverse-variance weighting40. The EUR + EAS samples in this analysis (56,418 cases and 78,818 controls) included all samples of EUR ancestry (33,640 cases and 43,456 controls) from the previous publication2 with the exclusion of three samples of EAS ancestry and the deCODE samples (1,513 cases and 66,236 controls), which only had summary statistics for selected variants. The three EAS samples (IMH-1, HNK-1 and JPN-1) excluded from EUR samples were included in our EAS stage 1.
We identified 208 independent (both in EAS and EUR) variants associated with schizophrenia across 176 genetic loci (Fig. 2 and Supplementary Tables 5 and 6), among which 53 loci were novel (not reported in refs. 2,3,7,8). Of the 108 schizophrenia-associated loci reported in the previous EUR study2, 89 remained significant in this study (Supplementary Table 4). Using simulations with a correction for winner’s curse39, we found that this is consistent with an expected over-estimation of the effect sizes due to the winner's curse in the previous study, rather than implying the 19 loci no longer significant in this study were false-positives (Supplementary Note). In addition, the deCODE samples (1,513 cases and 66,236 controls) were not included in the present study, causing the power for loci that had low MAF in EAS to drop.
Figure 2 ∣. Schizophrenia associations in EUR and EAS samples.
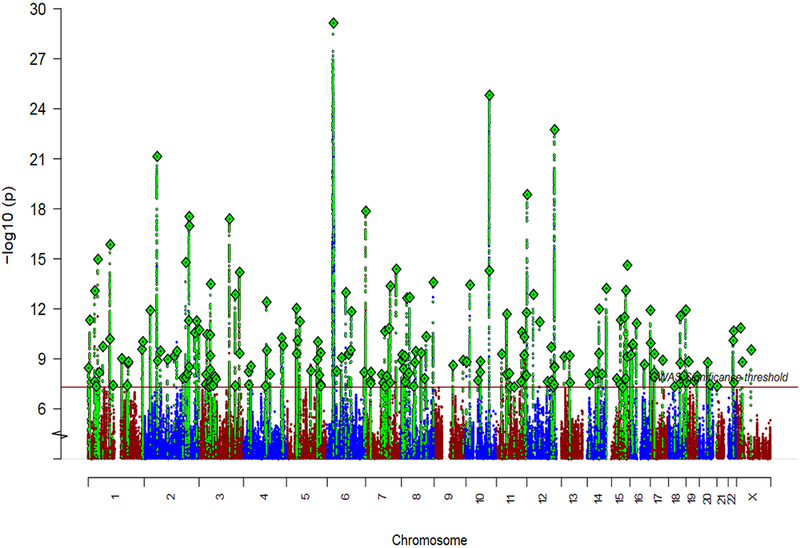
Manhattan plot for the schizophrenia genetic associations from the EAS (stages 1 and 2) + EUR meta-analysis (n = 56,418 cases; 78,818 controls).
Population diversity improves fine-mapping.
Causal variants in complex genetic disorders are defined as those that mechanistically contribute to the disorders, but this does not imply that the variant in isolation is likely to result in the disorder41,42. Due to LD, disease-associated loci from genome-wide association studies usually implicate genomic regions containing many associated variants. A number of approaches allow for the associated variants to be refined to a smaller set of the most plausible (or credible) candidate causal variants43-46. Loci implicated in psychiatric disorders usually have small effect sizes and as a result have generally poor performance using such approaches2,3.
Diversity in genetic background across populations can be used to improve fine-mapping resolution47. Here we demonstrate that resolution can be improved by exploiting differences in the patterns of LD between causal (directly associated) and LD (indirectly) associated variants. Based on the premise that genetic effects are highly consistent across populations, the causal variants will have consistent effects across populations, whereas non-causal variants can have inconsistent effects due to population-specific LD patterns. We therefore expect causal variants to have greater statistical significance and less heterogeneity in trans-ancestry meta-analysis compared to other alleles that are indirectly associated via LD (Supplementary Fig. 6). Using an algorithm based on this expectation (Methods), we fine-mapped 59 schizophrenia associations that reached genome-wide significance in the EUR and stage 1 EAS combined meta-analysis, had MAF > 0.01 in both EAS and EUR, and for which we had >95% coverage of common variants (MAF > 1%) with imputation INFO > 0.6 (Supplementary Table 7). The MHC region was excluded from the fine-mapping analysis due to its long range LD. Stage 2 EAS samples were excluded because not all had full genome coverage, which confounds the fine-mapping outcome (Methods).
Results from this EAS-EUR trans-ancestry approach improved upon those using only EUR, with 44 out of 59 loci mapped to a smaller number of candidate causal variants (Supplementary Table 7). For example, a locus on chromosome 1 (238.8-239.4 Mb), which initially contained 7 potentially causal variants based on a published fine-mapping method43 and EUR samples only, was resolved to a single variant, rs11587347, with 97.6% probability (Fig. 3a). This variant showed strong association in both populations, while the other 6 variants are equally associated in EUR but not in EAS (Fig. 3b,c). Over all associations, the median size of the 95% credible set, defined as the minimum list of variants that were >95% likely to contain the causal variant, dropped from 49 to 30, and the number of associations mapped to ≤5 variants increased from 2 to 7 (Fig. 3d). The number of associations mapped to a single variant with greater than 50% probability increased from 5 to 8, and median size of the genomic regions the associations mapped decreased from 154 kb to 94 kb.
Figure 3 ∣. Trans-ethnic fine-mapping improves resolution.

a, An association was mapped to a single variant (rs11587347) after adding EAS samples and using the trans-ancestry fine-mapping approach. Regional association plots were generated using http://locuszoom.org/ and LD from 1000 Genomes Project Phase 3 EUR subjects. b, LD with the lead variant (rs11587347). c, The lead variant (rs11587347) has strong association significance in both populations and low heterogeneity across populations. a-c, n (EAS stage 1) = 13,305 cases, 16,244 controls; n (EUR PGC2) = 33,640 cases, 43,456 controls. d, Number of variants in the 95% credible set using the trans-ancestry (EAS+EUR) and published fine-mapping approaches (EUR only).
Transferability of genetics across populations.
For genome-wide significant loci that individually explain >0.05% of the variance in schizophrenia liability in either ancestry, we compared the variance explained across EAS and EUR. Variance was approximated as 2f(1 – f)log(OR)2/(π2/3) (ref. 48) (Supplementary Fig. 7). Although these variants most often have comparable odds ratio across populations, their allele frequencies can differ. Variance explained, combining the effect size (OR) and prevalence of the risk allele (f), can be regarded as an approximate measure of the importance of a causal variant in a population. In our analysis, most of the trans-ancestry differences in variance explained is explained by allele frequency differences. One of the implications of this observation, as suggested in recent studies21,49,50, is that even if the risk alleles and effect sizes are primarily shared across populations, the disease predictive power of individual alleles, and of composite measures of those risk alleles such as PRS, may not be equivalent across populations.
Here we evaluate this empirically. We assessed how much variation in schizophrenia risk can be explained in EAS using both EAS stage 1 and EUR training data. Using a standard clumping approach, we first computed PRS using a leave-one-out meta-analysis approach with EAS summary statistics (Methods), which explained ~3% of schizophrenia risk using genome-wide variants on the liability scale (R2 = 0.029 at P = 0.5). In contrast, when EUR summary statistics were used to calculate PRS in the EAS samples, a maximum of only ~2% of schizophrenia risk was explained (R2 = 0.022 at P = 0.1) despite a greater than 3-fold larger EUR effective sample size (Fig. 4 and Supplementary Fig. 8). The variance explained across various P-value thresholds provides a proxy for the signal-to-noise ratio, which differs by training population—relative to the EUR training data, variants from the EAS training data with more permissive P-values improve the EAS prediction accuracy. These results indicate that larger EAS studies will be needed to explain similar case/control variance as currently explained in EUR individuals. Further, although individual loci typically have the same direction and similar magnitude across populations, aggregating variants that differentially tag causal loci across populations for genetic risk prediction results in considerable variability in prediction accuracy.
Figure 4 ∣. Genetic risk prediction accuracy in EAS from EAS or EUR training data.
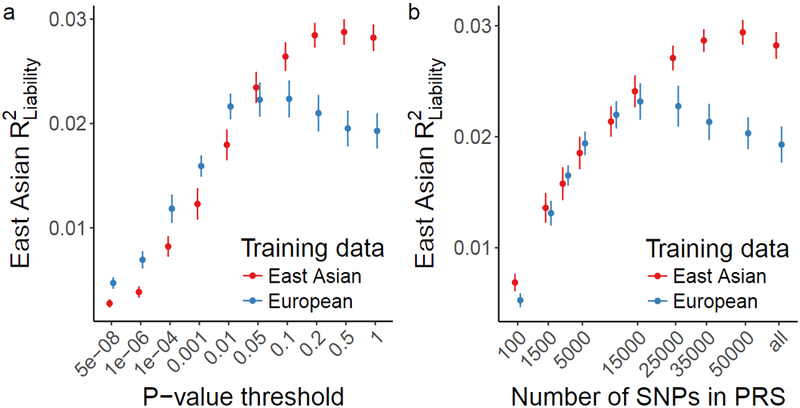
Polygenic risk scores were computed with GWAS summary statistics from EAS and EUR populations as training sets. EAS risk alleles and weights were computed with a leave-one-out meta-analysis approach across the 13 stage 1 samples. Error bars indicate the 95% confidence interval. LD panel for clumping is from EUR and EAS 1000 Genomes Phase 3 samples. a, Case/control variance explained in EAS samples by variants from EAS and EUR training data with a P-value more significant than the threshold. b, Case/control variance explained by the n most significant independent variants. a-b, For EAS stage 1: 13,305 cases and 16,244 controls; For EUR 33,640 cases and 43,456 controls.
Discussion
To date, most large-scale psychiatric genetics studies have been based on samples of primarily EUR ancestry6. To increase global coverage, we compiled the largest non-European psychiatric genetics cohort to date and leveraged its size and diversity to provide new insights into the genetic architecture of schizophrenia. This study includes all available major genotyped schizophrenia samples of East Asia ancestry, and presents analyses that have not previously been performed with sufficient power in psychiatric genetics. Although the first schizophrenia genetic associations from two much smaller studies of Chinese ancestry51,52 were not genome-wide significant in the present EAS analysis, several loci from their subsequent better powered studies7,8 reached genome-wide significance. Consistent with a study using EUR samples3, we note that this is consistent with the expected inflation of effect size from small studies rather than suggesting loci in previous studies are false positives.
When a single population is used to identify the disease-associated loci, the discovery is skewed towards disease-associated variants that have greater allele frequency in that population (Supplementary Fig. 9). When multiple populations are used, disease-associated variants are equally represented across the allele frequency spectrum in these populations (Supplementary Fig. 9). This demonstrates that including global samples improves power to find disease associations for which the power varies across populations. In this study, for example, more EUR than EAS samples would be required to detect around half of the new loci, as the MAF is higher in EAS than in EUR in these loci.
For traits like body mass index and autoimmune diseases, we observed heterogeneity across populations in genetic effects28,53, which may point to interactions between genetic associations and environment factors and/or other genetic loci. In contrast, for schizophrenia, we did not find significant heterogeneity across EAS and EUR ancestries. Analyses of genetic heritability, genetic correlation, gene-set enrichment and natural selection signatures converge on the conclusion that the schizophrenia biology is substantially shared across EAS and EUR ancestries (with MHC as a potential exception, discussed later). This remarkable genetic correlation (rg = 0.98) demonstrates that schizophrenia risk alleles operate consistently across different ethnic and cultural backgrounds, at least across EAS and EUR ancestries. Given that the main putative environmental risk factors (migration, urbanicity and substance misuse) differ across populations, this finding also suggests any specific genetic liability to schizophrenia acting via these routes is minimal.
We note that a direct comparison of the effect sizes estimated in EAS with those estimated in EUR has reduced accuracy as we do not know the exact schizophrenia causal variants. This is further complicated by inflation in effect size estimates due to the winner’s curse, which are of different magnitudes due to the sample size. Increasing the sample size, especially in those of non-European ancestries, will reduce the bias and enable a better isolation of causal variants, leading to a more precise comparison of the genetic effect size across populations.
The major histocompatibility complex (MHC) hosts the strongest schizophrenia association in EUR54. In this study, we did not find a significant schizophrenia association in MHC in EAS. An earlier EUR study55 mapped the MHC associations to a set of variants (in LD) at both distal ends of the extended MHC (lead variant: rs13194504) and the complement component 4 (C4). None of these associations was significant in EAS in this study, which is consistent with previous studies of the Chinese ancestry7,8,51,52. This, however, does not necessarily suggest population heterogeneity in their pathophysiological effect, as we attribute the disappearance of MHC signals partially to low frequencies. rs13194504 has MAF < 1% in EAS compared with 9% in EUR, and the C4-BS allele is extremely uncommon in samples from China and Korea56,57. Another reason may be the EUR-specific LD. In EUR, multiple protective alleles that contribute to the MHC associations are all on the same haplotype across about 6 Mb, due to an extremely long and EUR-specific haplotype that generates LD patterns at 5-Mb scale. This may also be the reason that association signals span so many Mb of genome, and the aggregate association signal (at variants that are in partial LD to multiple signals) is stronger than the signals at the individual associations.
Two recent studies using much smaller samples with individuals of Chinese ancestries7,8 reported variants in MHC significantly associated with schizophrenia (rs115070292 and rs111782145, respectively). The two studies did not replicate each other’s findings as the reported risk alleles are in very weak LD (r2 = 0.07) and are not in LD with the EUR MHC associations. rs115070292, from Yu et al.7, is more frequent in EAS (12%) than in EUR (2%) with P = 10−9 using 4,384 cases and 5,770 controls of Chinese ancestry. This variant was not significantly associated in our study (P = 0.44) even though some samples from that earlier study were included in the current study (BJM-1, 1,312 cases and 1,987 controls). The OR estimated from these shared samples marginally differs from that estimated using all EAS samples (P = 0.018), and this association showed marginally significant heterogeneity across all EAS samples (P = 0.039). Similarly, we did not replicate the association at rs111782145 from Li et al.8 (P = 0.47), again despite sample overlap (2,555 cases and 3,952 controls).
The lack of replication across all these studies reflects the complexity of the MHC region and the limited power for the MHC signals in EAS. As demonstrated in previous studies of complex disorders, it is still possible that when sample size increases for the EAS, genome-wide association within the MHC region could emerge. A study designed for the MHC region, such as in ref. 55, will be necessary to delineate the contribution of MHC to schizophrenia in EAS individuals.
Genetic associations usually implicate a large genomic region and thus it can be challenging to map their molecular functions. We designed a novel algorithm to leverage the population diversity to fine-map schizophrenia associations to precise sets of variants. Using this algorithm, we reduced the number of candidate variants associated with schizophrenia and facilitated the functional interpretation of these associations. Our algorithm only maps the primary association signals in a locus because the power to fine-map signals beyond that, especially in the EAS samples, is still limited at the current sample size for schizophrenia. We also made an assumption that there is only one causal variant driving the primary association signal. In the scenario that there is a haplotypic effect driven by multiple variants in strong LD, our approach will split the posterior probability among these variants. We expect the causal variants to have non-trivial probability so that they will still be reported in the credible set for future studies. Imputation quality plays a key role in fine-mapping as the power to map the causal variant decreases if it is poorly imputed. We restricted our study to genetic associations that have MAF > 1% in both EAS and EUR populations to ensure the imputation quality. For these associations, we found no major change in the size of the credible sets when the EUR samples were imputed using the more powerful Haplotype Reference Consortium (HRC) panel58. However, the HRC reference panel, with its much larger sample size and better characterization of low frequency and rare variants, could improve fine-mapping resolution for variants with MAF ≤ 1%59.
Finally, this large-scale EAS sample allowed us to empirically evaluate the congruence of the genetic basis of schizophrenia between EAS and EUR. In spite of a cross-population common variant genetic correlation being highly consistent, we found that polygenic risk models trained in one population have reduced performance in the other population due to different allele frequency distributions and LD structures. This highlights the importance of including all major ancestral groups in genomic studies both as a strategy to improve power to find disease associations and to ensure the findings have maximum relevance for all populations.
METHODS
Overview of samples
EAS samples, full-genome.
Genome-wide genotype data was obtained from 16 samples from East Asia (Supplementary Table 1). Two of these samples (TAI-1 and TAI-2) had parent-offspring trios and were processed as case/pseudo-controls. DSM-IV was used for diagnosing all schizophrenia cases in these samples, except for the trios (TAI-1 and TAI-2), for which DIGS was used. All samples were processed according to quality control (QC) procedures reported in ref. 2, with details reported in following sections. After QC, genotypes were phased and imputed against the 1000 Genomes Project Phase 3 reference panel6. Principal component analysis (PCA) was conducted across samples via imputed best guess genotypes to identify and remove overlapping samples across datasets, cryptic related samples and population outliers. Eight PCs that were associated to case-control status were included in univariate logistic regression to control for the population stratification in each sample.
EAS samples, selected variants.
Summary statistics were obtained for a set of variants from four EAS samples (BJM-2, BJM-3, BJM-4, BIX-5) that had been analyzed in published studies7,8. The summary statistics included odds ratio, standard error, reference and tested alleles for variants that have P < 10−5 in either stage 1 or the meta-analysis combining stage 1 and EUR samples. Between 22,156 and 31,626 variants were available after the exclusion of strand ambiguous60 variants (Supplementary Table 2).
EUR samples.
Genotypes for EUR schizophrenia patients and controls were obtained from the Psychiatric Genomics Consortium as reported in ref. 2. All samples of EUR ancestry were included in this study except for the deCODE samples (1,513 cases and 66,236 controls). We also note that three sample collections of EAS ancestry reported in ref. 2 were not included in the EUR samples in our analysis but were included in the EAS samples (IMH-1, HNK-1 and JPN-1). The same procedures used in processing EAS samples were applied to the EUR samples.
EAS subpopulations.
To investigate the heterogeneity of schizophrenia genetics effects within EAS, we grouped the samples based on their principal components. Other than Indonesians (UWA-1), which fall into their own subpopulation, samples were grouped into Northeast Asian subpopulation if their average PC2 was significantly greater than 0 (BIX-2, BJM-1, XJU-1, JPN-1, KOR-1) and into Southeast Asian subpopulation if their average PC2 was significantly less than 0 (TAI-1, TAI-2, IMH-1, IMH-2, HNK-1, BIX-3). The remaining samples (UMC-1, SIX-1, BIX-1, BIX-4) were not included in subpopulations. The heterogeneity test (Cochran’s Q) across subpopulations, calculated pairwise and in three-way, was conducted using the RICOPILI pipeline61.
Quality control
Quality control procedures were carried out as part of the RICOPILI pipeline61 (https://sites.google.com/a/broadinstitute.org/ricopili/home) with the following steps and parameters: (1) excluding variants with call rate below 95%; (2) excluding subjects with call rate below 98%; (3) excluding monomorphic variants; (4) excluding subjects with inbred coefficient above 0.2 and below −0.2; (5) excluding subjects with mismatch in reported sex and chromosome X computed sex; (6) excluding variants with missing rate differences greater than 2% between cases and controls; (7) subsequent to step 6, excluding variants with call rate below 98%; and (8) excluding variants in violation of Hardy-Weinberg equilibrium (P < 10−6 for controls or P < 10−10 for cases). Numbers of variants or subjects removed in each step are reported in Supplementary Table 2.
Phasing and imputation
All datasets were phased using SHAPEIT62 and IMPUTE263 using regular steps and parameters. Additional processing for trios (TAI-1 and TAI-2) was carried out such that case/pseudo-controls were identified and imputed. All samples were imputed to the 1000 Genomes Project Phase 3 reference panel64 (2,504 subjects, including 504 EAS subjects). Imputation procedures resulted in dosage files and best guess genotypes in PLINK65 binary format. The former was used for subsequent association analysis, and the latter was used in the PCA and PRS analyses.
Sample overlaps, population outliers and population stratification
We used Eigenstrat66 to calculate the principal components for all the samples using the best guess genotypes from imputation (Supplementary Fig. 10b). We computed the identity-by-descent matrix to identify intra- and inter- dataset sample overlaps. Samples with pi-hat > 0.2 were extracted, followed by Fisher-Yates shuffle on all samples. The number of times with which each sample was related to another sample was tracked, and samples that were related to more than 25 samples were removed. When deciding which samples to retain, trios were preferred, followed by cases, and thereafter a random sample for each related pair was removed, resulting in removal of 704 individuals.
To identify population outliers, k-means clustering was conducted using the first 20 PCs from PCA and covariates representing each of the 13 stage 1 samples. Guided by results of k-means clustering and visual inspection of PCA plots, 46 individuals were identified as outliers and were excluded. Further population-level inspection was carried out by merging the 1000 Genomes Project Phase 1 reference samples with stage 1 samples and conducting PCA (Supplementary Fig. 10a). Using similar approaches reported above, no further samples were excluded as population outliers.
Eight PCs that were associated with case/control status with P < 0.2 were used as covariates for association analysis in each sample (PCs 1, 4, 5, 6, 8, 9, 15, and 19). QQ plots (Supplementary Fig. 1) showed that the population structure has been well controlled.
Association analysis and meta-analysis
Association analysis was carried out for each sample using PLINK65 and genotype dosage from imputation. Only variants having imputation INFO ≥ 0.6 and MAF ≥ 1% were included in the analysis. We performed logistic regression with PCs identified in the prior subsection as covariates to control for population stratification within each study. Fixed-effect meta-analysis67, weighted by inverse-variance, was then used to combine association results across samples. Meta-analysis for European samples were conducted in the same matter. In order to find independent schizophrenia associations in both EUR and EAS populations (Supplementary Table 5), we performed LD clumping twice using the 1000 Genomes Project Phase 3 EUR and EAS reference panels, respectively (with default parameters in RICOPILI).
Chromosome X analysis
Chromosome X genotypes were processed separately from autosomal variants. Quality control was conducted separately for males and females, using similar quality control parameters as above. Cases and pseudo-controls were built out of the trios. Phasing and imputation were then performed on males and females separately for each sample, followed by logistic regression with the same PCs, and meta-analysis combining samples (same parameters as the autosomal analyses). Results were generated for EAS stage 1 samples and EUR-EAS combined samples (excluding BIX-1, BIX-2 and BIX-3). EAS stage 2, BIX-1, BIX-2 and BIX-3 samples do not have chromosome X data and were therefore not analyzed.
Genetic correlation and heritability
Schizophrenia heritabilities in the observed scale for samples of EUR and EAS ancestry were estimated from their summary statistics using LDSC23. We converted the heritabilities in the observed scale to liability scale assuming the schizophrenia population prevalence at 1%. The LD scores were pre-computed from the 1000 Genomes Project Phase 3 reference panel in EUR and EAS respectively (https://github.com/bulik/ldsc). Only autosomal variants having MAF greater than 5% in their respective population were included in the analysis, and variants in the MHC region were not included due to the long-range LD.
We computed the genetic correlations between schizophrenia and other traits within EUR and across EUR and EAS. EUR and EAS (stage 1 only) summary statistics for autosomal variants from this study were used as schizophrenia genetic association inputs for their respective populations. Traits tested included schizophrenia2, bipolar68, major depression69, anorexia nervosa70, neuroticism & subjective well-being (SWB)71, autism spectrum disorder (PGC 2015 release, available at http://www.med.unc.edu/pgc), attention deficit hyperactivity disorder (with samples of non-European ancestry removed, available at http://www.med.unc.edu/pgc)72, education attainment73, general intelligence74, fluid intelligence score and prospective memory result (using individuals from UK Biobank; http://www.nealelab.is/uk-biobank). Only variants having MAF greater than 5% were available and included. Variants in the MHC region were excluded from the analysis. Genetic correlations within EUR were computed using LDSC with LD scores pre-computed on the 1000 Genomes Project Phase 3 reference panel (503 EUR subjects). Genetic correlations across EUR and EAS were computed using POPCORN29. POPCORN uses a Bayesian approach which assumes that genotypes are drawn separately from each population and effect sizes follow the infinitesimal model. The inflation of z scores could then be modelled and a weighted likelihood function which was maximized to find heritability and genetic correlation. Genetic correlations in POPCORN were computed in the “genetic effect” mode, which estimates the correlation based on the LD covariance scores and effect sizes from summary statistics.
Partitioned heritability
Partitioned LDSC34 was conducted to look for heritability enrichment in diverse annotations using EAS (stage 1) and EUR autosomal variants (summary statistics), respectively. LD scores for each annotation were computed using a combination of PLINK65 and LDSC23 using the 1000 Genomes Project EAS and EUR subjects, respectively. We used baseline annotations34 and additional annotations including chromatin accessibility in brain dorso-lateral prefrontal cortex through the Assay for Transposase-Accessible Chromatin using sequencing peaks (ATAC Bryois)35, conserved regions located in “ATAC Bryois” (ATAC Bryois & Conserved LindbladToh)35, and introgressed regions from Neanderthal (Neanderthal Vernot)75. Variants can be included in multiple annotations. Multi-allelic variants were removed.
Gene-set analysis
We performed gene and gene-set based tests using MAGMA37. Genome-wide summary statistics for autosomal variants from EAS, EUR and EAS+EUR meta-analyses were used in this analysis. Variant-to-gene annotation was performed using RefSeq NCBI37.3 with a window of 5 kb upstream and 1.5 kb downstream. LD was taken from 1000 Genomes Project EAS, EUR and EUR-EAS panels, respectively. The gene-based P-values were computed using F-test and multivariate linear model, and competitive tests were used for gene-set analysis. Seventy gene-sets were selected and tested in this study (Supplementary Table 8), including those from the Molecular Signatures Database76, related to psychiatric diseases38,77,78 and from ‘gwaspipeline’(https://github.com/freeseek/gwaspipeline/blob/master/makegenes.sh). Gene-sets were ranked for EUR, EAS and EAS+EUR analyses, respectively. The top-ranking gene-sets were compared across analyses to identify common schizophrenia pathways. Additionally, Wilcoxon sign rank tests was conducted to compare the ranking of gene-sets between the EUR and EAS datasets.
Natural selection analysis
We used the CHB and CEU panels from the 1000 Genomes Project Phase 3 to investigate the natural selection signatures in schizophrenia-associated loci for EAS and EUR populations, respectively. We used the following selection signatures, with their sensitivity to timeframes discussed in ref 3. Integrated Haplotype Score (iHS): iHS captures the haplotype homozygosity at a given variant. We calculated iHS using the R rehh package79. Genetic distance between variants was determined using HapMap phase II genetic map. Ancestral and derived alleles were obtained from the 1000 Genome project, which inferred the ancestral state using six primates on the EPO (Enredo-Pecan-Ortheus) pipeline. Only biallelic variants that have MAF ≥ 5% were included in the analysis. Cross Population Extended Haplotype Homozygosity (XPEHH)80: XPEHH detects variants under selection in one population but not the other. We used CEU as the reference panel when calculating XPEHH for CHB and vice versa. Fixation index (Fst): Fst measures the population differentiation due to genetic structure. We estimated Fst using the Weir and Cockerham approach81, which is robust to sample size effects. Absolute derived allele frequency difference (∣ΔDAF∣): ∣ΔDAF∣ measures population differentiation between CHB and CEU populations. Composite of Multiple signals (CMS)82-85: CMS combines iHS, XPEHH, Fst and ∣ΔDAF∣. As a result, CMS potentially has better power to detect the selection signature. For each variant, , in which pi is the rank of the variant using method i, sorted by increasing P-values, divided by the total number of variants. B statistic: B statistic measures the background selection. We calculated the B statistic as in ref. 84.
Trans-ethnicity fine-mapping
For a disease-associated genetic locus, fine-mapping defines a “credible set” of variants that contains the causal variant with certain probability (e.g., 99% or 95%). Bayesian fine-mapping approaches2,43,86,87 have been widely used for studies of a single ancestry. Here, we extended a Bayesian fine-mapping approach85 (Defining credible sets, Methods) to studies of more than one ancestry. Intuitively, the extension was achieved through a prior calculated from the heterogeneity across ancestries, such that variants that have different odds ratio across populations will have a smaller prior probability to be the causal variant.
As in several previous studies2,86, we restricted our fine-mapping analysis to the primary association signal in each locus. This is done by taking P variants that are in LD with the lead variant (the variant having the most significant P-value) with r2 > 0.1 in EUR or EAS. Assume D represents the data including the genotype matrix X for the P variants and disease Y for N individuals, and β represents a collection of model parameters. We define the model, denoted by A, as the causal status for the P variants in locus: A ≡ {aj}, in which aj is the causal status for variant j. aj = 1 if the variant j is causal, and aj = 0 if it is not. For the primary association signal and under the presumption that the causal variant is the same across all ancestries, one and only one of the P variants is causal: ∑jaj = 1. For convenience, we define Aj as the model in which only variant j is causal, and A0 as the model in which no variant is causal (null model). The probability of model Aj (where variant j is the only causal variant in the locus) given the data (D) can be calculated using Bayes’s rule:
With the steepest descent approximation, the assumption of a flat prior on the model parameters (β), and the assumption of one causal variant per locus (equation 2 in ref. 86), Pr(Aj∣D) can be approximated as:
| (1) |
in which N is the sample size. We denote as the χ2 test statistic for variant j, which can be calculated from the P-value from the meta-analysis combining EAS and EUR samples. Using equation 3 in ref. 86, we have:
| (2) |
Pr(Aj) is the prior probability that variant j is causal. We have shown that schizophrenia causal variants have consistent genetic effect across populations. Therefore, we model the prior probability as a function of the heterogeneity measured in I2:
| (3) |
Using equations 2 and 3, Pr(Aj∣D) in equation 1 can be calculated as
We only use stage 1 samples in fine-mapping so the variants have the same sample size (assuming all variants have good imputation quality). Therefore, N−1/2, Pr(D) and can be regarded as constants,
The normalized causal probability for variant j is then
And the 95% credible set of variants is defined as the smallest set of variants, S, such that
Polygenic risk score analysis
We constructed PRS using a pruning and thresholding approach in a study set of EAS individuals with training summary statistics from either EUR or EAS individuals. In the former case, we used summary statistics from all EUR individuals in this study; in the latter case, we used a leave-one-out meta-analysis approach across the 13 stage 1 samples to build PRS.
For the EUR training data, we extracted EUR individuals (FIN, GBR, CEU, IBS, TSI) from 1000 Genomes Project64 Phase 3 as an LD reference panel to greedily clump variants. For the EAS LD reference panel, we created two panels: (1) an analogous EAS panel (CDX, CHB, CHS, JPT, KHV) from 1000 Genome Project64 Phase 3 (Fig. 4 and Supplementary Fig. 8c,d), and (2) an LD panel from best guess genotypes from each cohort in the study (Supplementary Fig. 8a,b,e,f). For both EAS and EUR prediction sets, we filtered to variants with a MAF greater than 1% in each respective population and removed indels and strand ambiguous variants. We subset each list of variants to those in the summary statistics with an imputation INFO > 0.9. We then selected approximately independent loci at varying P-value thresholds or top-ranking n variants using an LD threshold of r2 ≤ 0.1 in a window of 500 kilobase pairs in PLINK65 with the --clump flag. We treated the MHC with additional caution to minimize overfitting in this region, selecting only the most significant variant from the HLA region. To profile variants, we multiplied the log odds ratio for selected variants by genotypes and summed these values across the genome in PLINK65 using the --score flag for each of the 13 EAS stage 1 samples. We assessed case/control variance explained by computing Nagelkerke’s and a liability-scale pseudo-R2 as in Lee et al.88 by comparing a full model with the PRS and 10 principal components with a model excluding the PRS. Results of PRS were presented in two ways the first we selected SNP based on GWAS P-value thresholds (PT) (i.e. 5e-8, 1e-6, 1e-4, 0.001, 0.01, 0.05, 0.1, 0.2, 0.5, 1) and P-value ranks. In the latter, top ranked SNPs that exist between both EUR and EAS summary statistics were selected based on the SNP rank thresholds (i.e. top 100, 1,500, 5,000, 15,000, 25,000, 35,000, 50,000, all).
Ethics.
The study protocols were approved by the institutional review board at each center involved with recruitment. Informed consent and permission to share the data were obtained from all subjects, in compliance with the guidelines specified by the recruiting centre’s institutional review board. Samples recruited in mainland China were processed and analyzed in a Chinese server to comply with the Interim Measures for the Administration of Human Genetic Resources (regulation from the Ministry of Science and Technology of the People’s Republic of China). We set up the computer codes on the Chinese server so that analyses performed on these samples were exactly the same as other samples. Summary statistics from these Chinese samples, with no individual-level data, were then shared and combined with the rest of EAS samples.
Data availability.
Genome-wide summary statistics from EAS samples, EUR samples (“49 EUR samples”) and all samples (EAS and EUR combined) in this study can be downloaded from https://www.med.unc.edu/pgc/results-and-downloads/. Individual-level genotype data for EAS samples are available upon request from the contact authors (Supplementary Note). Alternately, requests can be made to the Psychiatric Genomics Consortium (PGC). In this case, access to individual-level genotypes from samples recruited outside of mainland China will go through the PGC “fast-track” approval. Access to individual-level genotypes from samples recruited within mainland China has to be approved by the individual Chinese contact authors (Supplementary Note), and are subject to the policies and approvals from the Human Genetic Resource Administration, Ministry of Science and Technology of the People’s Republic of China. Individual-level genotypes from samples recruited within mainland China have been stored and kept in a server physically located in mainland China. Analyses were performed on these samples using the same computer codes as those used for other EAS and EUR samples, which are available in the Code availability section.
Code availability.
Computer code used in this manuscript: RICOPILI (quality control, principal component analysis, pre-phasing, imputation, association test and meta-analysis) https://github.com/Nealelab/ricopili/wiki; embedded within RICOPILI (Eigenstrat https://github.com/DReichLab/EIG/tree/master/EIGENSTRAT; SHAPEIT https://mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html; EAGLE https://github.com/poruloh/Eagle; IMPUTE https://mathgen.stats.ox.ac.uk/impute/impute_v2.html; Minimac https://genome.sph.umich.edu/wiki/Minimac; POPCORN (trans-ancestry genetic correlation): https://github.com/brielin/Popcorn; LDSC (heritability, partitioned heritability and within-ancestry genetic correlation): https://github.com/bulik/ldsc; MAGMA (pathway analysis): https://ctg.cncr.nl/software/magma; Fine-mapping (Fine-mapping and PAINTOR): https://github.com/hailianghuang/FM-summary, https://github.com/gkichaev/PAINTOR_V3.0; REHH (selection): https://cran.r-project.org/web/packages/rehh/index.html; B score (background selection): http://www.phrap.org/othersoftware.html; PRS analyses: https://github.com/armartin/pgc_scz_asia
Supplementary Material
Acknowledgements
We thank Kenneth Kendler, John McGrath, James Walters, Douglas Levinson, and Michael Owen for helpful discussions. We thank Swapnil Awathi, Vassily Trubetskoy, and Georgia Panagiotaropoulou for their support with the RICOPILI analysis pipeline. We thank SURFsara and Digital China Health for computing infrastructure for this study. M.L. acknowledges National Medical Research Council Research Training Fellowship award (Grant No.: MH095:003/008-1014); A.R.M. acknowledges funding from K99MH117229; B.B. acknowledges funding support from the National Health and Medical Research Council (NHMRC Funding No. 1084417 and 1079583); L.G. acknowledges National Key Research and Development Program of the Ministry of Science and Technology of China (2016YFC1306802); M.I., Y.K. and N.I. acknowledge Strategic Research Program for Brain Sciences (SRPBS: Grant Numbers JP19dm0107097) and GRIFIN of P3GM (Grant Numbers JP19km0405201 and JP19km0405208) from the Japan Agency for Medical Research and Development (AMED); Y.K., M.K. and A.T. acknowledge the BioBank Japan Project from the Ministry of Education, Culture, Sports, and Technology (MEXT) of Japan; S-W.K. acknowledges Grant of the Korean Mental Health Technology R&D Project (HM15C1140); W.J.C. acknowledges Ministry of Education, Taiwan (‘Aim for the Top University Project’ to National Taiwan University, 2011-2017); Ministry of Science and Technology, Taiwan (MOST 103-2325-B-002-025); National Health Research Institutes, Taiwan (NHRI-EX104-10432PI); NIH/NHGRI grant U54HG003067; NIMH grant R01 MH085521; and NIMH grant R01 MH085560; S.J.G. acknowledges R01MH08552; B.J.M. acknowledges Australian NHMRC grant 496698. H-G.H. acknowledges Ministry of Education, Taiwan (‘Aim for the Top University Project’ to National Taiwan University, 2011-2017); MOST, Taiwan (MOST103-2325-B-002-025); NIH/NHGRI grant U54HG003067; NIMH grants R01 MH085521, R01 MH085560; and NHRI, Taiwan (NHRI-EX104-10432PI); P.Sk. acknowledges U01MH109536; B.J.M acknowledges Australian NHMRC grant 496698; J.L. acknowledges National Medical Research Council Translational and Clinical Research Flagship Programme (Grant No.: NMRC/TCR/003/2008) and National Medical Research Council under the Centre Grant Programme (Grant No.: NMRC/CG/004/2013); P.H. acknowledges funding support from the Medical Research Council MR/L010305/1; S.X. acknowledges funding support from the Strategic Priority Research Program (XDB13040100) and Key Research Program of Frontier Sciences (QYZDJ-SSW-SYS009) of the Chinese Academy of Sciences (CAS), the National Natural Science Foundation of China (NSFC) grant (91731303, 31525014, and 31771388), the UK Royal Society-Newton Advanced Fellowship (NAF\R1\191094), the Program of Shanghai Academic Research Leader (16XD1404700), the National Key Research and Development Program (2016YFC0906403), and the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01); P.F.S. acknowledges the PGC funding from U01 MH109528 and U01 MH1095320; M.J.D. acknowledges NIH/NIMH 5U01MH109539-02; M.C.O’D acknowledges funding from MRC (UK) G0800509 and NIMH ref 1U01MH109514-01; S.Q. acknowledges National Key Research and Development Program of China (2016YFC0905000, 2016YFC0905002); and Shanghai Key Laboratory of Psychotic Disorders (13dz2260500); K.S.H. acknowledges Grant of the National Research Foundation of Korea (2015R1A2A2A01002699), funded by the Ministry of Science, ICT and Future Planning; D.B.W and S.G.S acknowledge funding support from the National Health and Medical Research Council (NHMRC 513861); W.Y. acknowledges National Key R&D Program of China (2016YFC1307000); National Natural Science Foundation of China (81571313); Peking University Clinical Scientist Program supported by "the Fundamental Research Funds for the Central Universities" (BMU2019LCKXJ012); M.T. acknowledges R01MH085560: Expanding Rapid Ascertainment Networks of Schizophrenia Families in Taiwan; J.J.L. acknowledges funding support from Agency for Science, Technology and Research (A*STAR), Singapore; XC.M. acknowledges the National NSFC Surface project (81471374; PI: XC Ma); Y.S. acknowledges National Key R&D Program of China (2016YFC0903402); NSFC (31325014, 81130022, 81421061); and the 973 Program (2015CB559100). H.H. acknowledges support from NIH K01DK114379, NIH R21AI139012, Zhengxu & Ying He Foundation and Stanley Center for Psychiatric Research.
Footnotes
Competing interests
B.M.N. is a member of Deep Genomics Scientific Advisory Board. He also serves as a consultant for Camp4 Therapeutics Corporation, Takeda Pharmaceutical and Biogen. M.J.D. is a founder of Maze Therapeutics. The remaining authors declare no competing interests.
References
- 1.Stilo SA & Murray RM The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin. Neurosci. 12, 305–315 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardiñas AF et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visscher PM et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li YR & Keating BJ Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 6, 91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popejoy AB & Fullerton SM Genomics is failing on diversity. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu H et al. Common variants on 2p16.1, 6p22.1 and 10q24.32 are associated with schizophrenia in Han Chinese population. Mol. Psychiatry 22, 954–960 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Li Z et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Moltke I et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512, 190–193 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Tang CS et al. Exome-wide association analysis reveals novel coding sequence variants associated with lipid traits in Chinese. Nat. Commun. 6, 10206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke T-K et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112 117). Mol. Psychiatry 22, 1376–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.CONVERGE consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wray NR et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.World Health Organization. Report of the International Pilot Study of Schizophrenia. (1973).
- 15.Carpenter WT Jr, Strauss JS & Bartko JJ Flexible system for the diagnosis of schizophrenia: report from the WHO International Pilot Study of Schizophrenia. Science 182, 1275–1278 (1973). [DOI] [PubMed] [Google Scholar]
- 16.Jablensky A et al. Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten-country study. Psychol. Med. Monogr. Suppl. 20, 1–97 (1992). [DOI] [PubMed] [Google Scholar]
- 17.McGrath JJ Variations in the incidence of schizophrenia: data versus dogma. Schizophr. Bull 32, 195–197 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haro JM et al. Cross-national clinical and functional remission rates: Worldwide Schizophrenia Outpatient Health Outcomes (W-SOHO) study. Br. J. Psychiatry 199, 194–201 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Gureje O & Cohen A Differential outcome of schizophrenia: where we are and where we would like to be. Br. J. Psychiatry 199, 173–175 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Scutari M, Mackay I & Balding D Using genetic distance to infer the accuracy of genomic prediction. PLoS Genet. 12, e1006288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin AR et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100, 635–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin AR et al. Hidden ‘risk’ in polygenic scores: clinical use today could exacerbate health disparities. Nat. Genet. 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bulik-Sullivan B et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geisler S, Schöpf CL & Obermair GJ Emerging evidence for specific neuronal functions of auxiliary calcium channel α2δ subunits. Gen. Physiol. Biophys. 34, 105–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heyes S et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 134, 36–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gulsuner S et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown BC, Asian Genetic Epidemiology Network Type 2 Diabetes Consortium, Ye, C. J., Price, A. L. & Zaitlen, N. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okbay A et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies G et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N= 112 151). Mol. Psychiatry 21, 758–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cross-Disorder Group of the Psychiatric Genomics Consortium et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anttila V et al. Analysis of shared heritability in common disorders of the brain. Science 360, pii: eaap8757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryois J et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 9, 3121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genovese G et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, 1433–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowden J & Dudbridge F Unbiased estimation of odds ratios: combining genomewide association scans with replication studies. Genet. Epidemiol. 33, 406–418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borenstein Michael, Hedges Larry V., Higgins Julian P. T., Rothstein Hannah R.. Introduction to Meta-Analysis. (John Wiley & Sons, Ltd, 2009). [Google Scholar]
- 41.Spain SL & Barrett JC Strategies for fine-mapping complex traits. Hum. Mol. Genet. 24, R111–R119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaid DJ, Chen W & Larson NB From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swaminathan B et al. Fine mapping and functional analysis of the multiple sclerosis risk gene CD6. PLoS One 8, e62376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaulton KJ et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet. 47, 1415–1425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kichaev G & Pasaniuc B Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet. 97, 260–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawitan Y, Seng KC & Magnusson PKE How many genetic variants remain to be discovered? PLoS One 4, e7969 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Márquez-Luna C, Loh P-R, South Asian Type 2 Diabetes (SAT2D) Consortium, SIGMA Type 2 Diabetes Consortium & Price, A. L. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 41, 811–823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C-Y, Han J, Hunter DJ, Kraft P & Price AL Explicit modeling of ancestry improves polygenic risk scores and BLUP prediction. Genet. Epidemiol. 39, 427–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yue W-H et al. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nat. Genet. 43, 1228–1231 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Shi Y et al. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet. 43, 1224–1227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salinas YD, Wang L & DeWan AT Multiethnic genome-wide association study identifies ethnic-specific associations with body mass index in Hispanics and African Americans. BMC Genet. 17, 78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.International Schizophrenia Consortium et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen JY et al. Effects of complement C4 gene copy number variations, size dichotomy, and C4A deficiency on genetic risk and clinical presentation of systemic lupus erythematosus in East Asian populations. Arthritis Rheumatol. 68, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong GH et al. Association of complement C4 and HLA-DR alleles with systemic lupus erythematosus in Koreans. J. Rheumatol. 21, 442–447 (1994). [PubMed] [Google Scholar]
- 58.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sand PG A lesson not learned: allele misassignment. Behav. Brain Funct. 3, 65 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lam M et al. RICOPILI: Rapid Imputation for Consortias PipeLIne. bioRxiv. doi: 10.1101/587196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Connell J et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Howie B, Fuchsberger C, Stephens M, Marchini J & Abecasis GR Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 18, 497–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boraska V et al. A genome-wide association study of anorexia nervosa. Mol. Psychiatry 19, 1085–1094 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okbay A et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 48, 624–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okbay A et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sniekers S et al. Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence. Nat. Genet. 49, 1107–1112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Juric I, Aeschbacher S & Coop G The strength of selection against Neanderthal introgression. PLoS Genet. 12, e1006340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner TN et al. denovo-db: a compendium of human de novo variants. Nucleic Acids Res. 45, D804–D811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pirooznia M et al. High-throughput sequencing of the synaptome in major depressive disorder. Mol. Psychiatry 21, 650–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gautier M & Vitalis R rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177 (2012). [DOI] [PubMed] [Google Scholar]
- 80.Sabeti PC et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weir BS & Cockerham CC Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984). [DOI] [PubMed] [Google Scholar]
- 82.Grossman SR et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327, 883–886 (2010). [DOI] [PubMed] [Google Scholar]
- 83.Grossman SR et al. Identifying recent adaptations in large-scale genomic data. Cell 152, 703–713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma Y et al. Properties of different selection signature statistics and a new strategy for combining them. Heredity 115, 426–436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McVicker G, Gordon D, Davis C & Green P Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 5, e1000471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gormley P et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 48, 856–866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wellcome Trust Case Control Consortium et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 44, 1294–1301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee SH, Goddard ME, Wray NR & Visscher PM A better coefficient of determination for genetic profile analysis. Genet. Epidemiol. 36, 214–224 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome-wide summary statistics from EAS samples, EUR samples (“49 EUR samples”) and all samples (EAS and EUR combined) in this study can be downloaded from https://www.med.unc.edu/pgc/results-and-downloads/. Individual-level genotype data for EAS samples are available upon request from the contact authors (Supplementary Note). Alternately, requests can be made to the Psychiatric Genomics Consortium (PGC). In this case, access to individual-level genotypes from samples recruited outside of mainland China will go through the PGC “fast-track” approval. Access to individual-level genotypes from samples recruited within mainland China has to be approved by the individual Chinese contact authors (Supplementary Note), and are subject to the policies and approvals from the Human Genetic Resource Administration, Ministry of Science and Technology of the People’s Republic of China. Individual-level genotypes from samples recruited within mainland China have been stored and kept in a server physically located in mainland China. Analyses were performed on these samples using the same computer codes as those used for other EAS and EUR samples, which are available in the Code availability section.