

Abstract
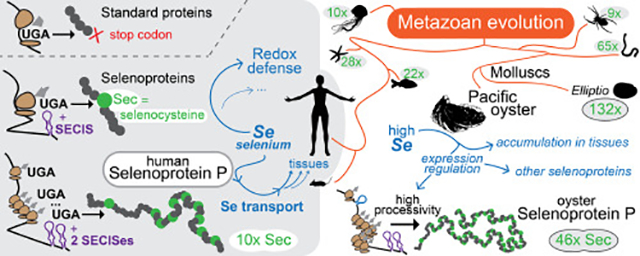
Selenoproteins typically contain a single selenocysteine, the 21st amino acid, encoded by a context redefined UGA. However, human Selenoprotein P (SelenoP) has a redox-functioning selenocysteine in its N-terminal domain and 9 selenium transporter-functioning selenocysteines in its C-terminal domain. Here we show that diverse SelenoP genes are present across metazoa with highly variable numbers of Sec-UGAs, ranging from a single UGA in certain insects, to 9 in common spider, and up to 132 in bivalve molluscs. SelenoP genes were shaped by a dynamic evolutionary process linked to selenium usage. Gene evolution featured modular expansions of an ancestral multi-Sec domain, which led to particularly Sec-rich SelenoP proteins in many aquatic organisms. We focused on molluscs, and chose Pacific oyster Magallana gigas as experimental model. We show that oyster SelenoP mRNA with 46 UGAs is translated full-length in vivo. Ribosome profiling indicates that selenocysteine specification occurs with ~5% efficiency at UGA1 and approaches 100% efficiency at distal 3’ UGAs. We report genetic elements relevant to its expression, including a leader ORF and an RNA structure overlapping the initiation codon that modulates ribosome progression in a selenium dependent manner. Unlike their mammalian counterparts, the two SECIS elements in oyster SelenoP (3’UTR recoding elements) do not show functional differentiation in vitro. Oysters can increase their tissue selenium level up to 50-fold upon supplementation, which also results in extensive changes in selenoprotein expression.
Keywords: selenoprotein, selenocysteine, recoding, dynamic redefinition, evolution
Graphical Abstract
Introduction
Selenium is an essential trace element for humans and for many other organisms [1]. The major reason for its importance in living systems is its occurrence in catalytic sites of certain oxidoreductases: selenium mediated reactions are thought to be readily reversible and hence the presence of selenium in proteins and certain tRNAs enables them to resist permanent oxidation [2]. Facilitation of resistance to oxidative inactivation relates to its irreplaceable role in mammalian interneurons in preventing fatal epileptic seizures [3]. Selenium is also important in other aspects of human health such as male fertility [4,5]. The deleterious consequences of deviations from physiologically important environmental levels of selenium in mammals are well known, including extreme selenium deficiency (e.g. in some regions of China until dietary supplementation) and its toxic levels (e.g. in parts of the American West) [6,7]. However, less is known about the significance for human health of intermediate dietary levels such as those found in North American and European soils and derived food. Suboptimal selenium levels are thought to have been important in prior era mass extinction events [8].
The biological effects of selenium in mammals are largely mediated by its incorporation in specific proteins (selenoproteins) in the form of selenocysteine (Sec), the 21st encoded amino acid [9,10]. With a small number of interesting bacterial exceptions [11], Sec is encoded by UGA (due to frequent switching from RNA to DNA in this report, UGA will also be used for the DNA counterpart rather than TGA). While in standard decoding UGA specifies translation termination, its meaning is dynamically redefined to specify Sec in response to mRNA-specific recoding signals and multiple specialized accessory factors. In eukaryotes, the most important such mRNA signal is part of the 3’ UTR folded into a structure termed SECIS (SElenocysteine Insertion Sequence) [12,13]. SECIS elements have a quartet of non-Watson-Crick base pairs [14,15] to which the protein SECISBP2 binds, and ribosomal protein L30 is also relevant to the interaction [16,17]. SECISBP2 binds the specialized elongation factor (EF-sec) for selenocysteinyl-tRNA [18–23]. Additional trans-acting factors including eIF4a3 [24] and nucleolin [25] are important regulators of the Sec incorporation machinery. In some eukaryotic selenoprotein mRNAs, cis-acting structures known as Sec redefinition elements (SREs) are also found adjacent to the UGA [26–29].
Nearly all selenoproteins contain a single Sec which is often at the active site. One clear exception is Selenoprotein P (SelenoP [30]) that contains a single redox-active Sec in its N-terminal domain [31–33] and in addition, multiple Secs in its C-terminal domain [34–36]. Full-length SelenoP serves a selenium transport function of critical importance for the brain, testes and other tissues [4,37]. Apart from limited bioinformatic studies in amphioxus and sea urchins [38,39], SelenoP has been almost exclusively investigated to date in vertebrates. SelenoP mRNAs are unique among vertebrate selenoproteins in having two rather than one SECIS element in their 3’ UTRs [40] (though some isoforms of human SelenoP mRNA has only one SECIS [37]). More recently, in addition to SRE-like elements, the SelenoP RNA structure termed Initiation Stem Loop (ISL) was identified with proposed roles in translation initiation [41].
The majority of known selenoproteins have non-Sec-containing orthologs in other organisms wherein Sec is replaced by cysteine (Cys), suggesting that, for several functions, the advantage of selenolate- versus thiolate-based catalysis is not universal or that the use of Sec is also associated with deleterious consequences that can outweigh its catalytic advantage. The presence of orthologs with either Sec or Cys has been exploited for bioinformatics studies that have yielded extensive information on these proteins. For example, analyses of selenoproteomes have uncovered trends in Sec utilization across species [38] including massive independent selenoprotein losses in insects, higher plants, fungi and protists [42–44]. Aquatic organisms have been shown to have generally large selenoproteomes, in contrast to terrestrial organisms which have reduced them through losses of selenoprotein genes or Sec to Cys replacements [33]. For studies of genes encoding SelenoP, the issue is more complicated as it involves the use of multiple UGA codons. Such bioinformatic analysis of vertebrate SelenoP has revealed up to 22 in-frame UGA codons in some genes [45]. The largest number of UGAs identified bioinformatically so far in an invertebrate SelenoP mRNA is in the purple sea urchin Strongylocentrotus purpuratus, where 28 are present [33]. Here we carried out detailed analyses of SelenoP in metazoa revealing unexpected diversity of its forms and new regulatory aspects of multiple codon redefinition.
Results
SelenoP genes and selenocysteine codon locations in metazoa
The computational search of metazoan genomes and transcriptomes resulted in 3,464 predictions of SelenoP sequences, which after filtering yielded 1,228 high quality non-redundant genes (Methods). SelenoP was found in ~50% of metazoan genomes, clustering in specific lineages. SelenoP was missing from the genomes of tunicates, Platyhelminthes, all nematodes except Trichinella, and the great majority of arthropods (Fig 1). Computational “translations” revealed the presence of sequences encoding Sec-rich C-terminal domains in SelenoP in various metazoan lineages. Diverse C-terminal domains were observed in vertebrates, echinoderms, arachnids, cnidarians, and various members of Lophotrochozoa (lineage including Annelida, Nemertea and molluscs, among others). To investigate the evolution of SelenoP and the emergence of C-terminal domains, we reconstructed a gene tree derived from the sequence alignment of the N-terminal portion of SelenoP proteins (Supplementary Fig 1). This tree was used to define 14 phylogenetic clusters of encoded proteins. Integrated with the species tree (Fig 2), this was used as backbone to inspect the evolutionary conservation of various sequence fetures including the position of the first Sec-encoding UGA codon (UGA1), clustering of multiple UGAs in the distal segment, two SECIS and an exon intron boundary just 3’ of UGA1 (though this last information is available only for genomic predictions, with fewer gene sequences obtainable in invertebrates than in vertebrates). The alignment of representative sequences for each protein cluster is presented in Supplementary Fig 2.
Fig 1. SelenoP genes identified in metazoan species.

The figure shows a summary of the SelenoP genes found in metazoa. Genes are represented as rectangles colored by phylogenetic cluster (see Fig 2). Sec residues are indicated as vertical black lines, SECIS as circles. Note that this representation does not capture the remarkable diversity within each group, particularly in the number of Sec residues (Fig 2). In the species tree on the left, some lineages are indicated with abbreviations: deuterostomes, protostomes, Lophotrochozoa. Abbreviations were used also for Heterobranchia and Caenogastropoda (molluscs, gastropods; Supplementary Note 3).
Fig 2. Tree representation of SelenoP in diverse metazoan species.

Colour coding of individual genes displayed follows phylogenetic clustering (Methods). This representation (legend top left) displays the positions of putative Sec-UGAs as black lines, and introns as red lines (for predictions in genomes only). Next to each gene, predicted SECIS elements are shown in blue and brown circles. The inner circle shows the SelenoP gene with the highest number of UGAs per species; the outer circle shows the gene with the second highest amount, if present; in a few cases, more than two genes were identified, and they were omitted in this representation. Note that only genes passing all filters (Methods) are displayed here; genes apparently missing may have been filtered out for low quality sequence (e.g. many non-placental vertebrates appear to lack SelenoP1 or SelenoP2).
Vertebrates
Vertebrate genomes contain two main classes of SelenoP genes: 1) an ortholog of human SelenoP with two SECIS elements and a Sec-rich C-terminal domain, hereafter referred as SelenoP1 for clarity; and 2) a shorter, single-Sec codon paralog hereafter termed SelenoP2 (but also known as Sepp2, SelPb). As previously reported [45], SelenoP2 is present in all vertebrates except placental mammals, where it was lost. SelenoP1 and SelenoP2 share the same intron structure and form sister clades in the gene tree. This suggests that the two genes originated by gene duplication approximately at the root of vertebrates; likely, the two genes were retained after a whole genome duplication [46]. The Sec codon of SelenoP2 which corresponds to the first Sec codon of SelenoP1 (hereafter referred as UGA1) was converted to a Cys codon in two vertebrate lineages, Anura (group of Amphibia including frogs) and Galeomorphii sharks (cartilaginous fish) (Supplementary Note 1). The number of Sec codons in SelenoP1 varies considerably across vertebrates. Hystricomorph rodents (i.e. guinea pigs, mole rats) had the lowest number (5–7 codons). At the other end, 3 species of Seriola amberjack fish (S. dumerili, S. rivoliana, S. quinqueradiata) had 33–37 (S. lalandi had instead only 25). However, the highest number of putative Sec-UGAs in a vertebrate, 49, was observed in Oreobates cruralis. In this species of robber frog, SelenoP1 contains a repeated C-terminal region, so that its mRNA includes ~28 more UGAs than its closest relatives. However, since we observed this extension in a single species and sequence source, we cannot exclude that this is an artefact of transcriptome assembly.
Cephalochordates
The selenoproteome of cephalochordate Branchiostoma floridae (amphioxus) has been previously reported [39]. Its SelenoP mRNA has multiple Sec codons and two SECIS elements. However, instead of the encoded protein having a Sec-rich C-terminal tail like other species, amphioxus SelenoP is formed by tandem repetitions of the thioredoxin-like domain. In the mRNA each of the Sec codons is located in a homologous context to UGA1 of human SelenoP. Through our genomic searches, we found that this gene structure is conserved in the cephalochordate lineage.
Echinoderms
SelenoP was previously reported in the echinoderm S. purpuratus (purple sea urchin) [33] Indeed, our search identified SelenoP in all echinoderms, forming a single monophyletic cluster in the reconstructed tree. As in S. purpuratus, the SelenoP of these other species contains multiple Sec-UGAs and two SECIS elements with the highest number of UGAs, 43, being identified in the brittlestar Amphiura filiformis. However, several other species (e.g. sea cucumber Parastichopus parvimensis) contain a SelenoP gene with a single UGA, which is at the characteristic UGA1 position, and a single SECIS.
Molluscs
We identified SelenoP in all main classes of molluscs: bivalves, cephalopods, and gastropods. With few exceptions ascribed to imperfect assembly quality, all molluscan SelenoP genes encode a long Sec-rich C-terminal domain, with clear homology within molluscs. The highest number of putative Sec codons in any SelenoP sequence searched to date are in the freshwater mussel Elliptio complanata. With 131 in-frame UGAs, Sec is predicted to be the second most abundant amino acid in the encoded protein. We confirmed Sec-UGA numbers by RT-PCR and Sanger sequencing (Supplementary Note 2). This revealed one additional Cys codon converted to a Sec codon raising the number to 132; variants with increasing numbers of UGAs may be segregating in the population. While E. complanata constitutes an outstanding outlier, high Sec counts were present in most bivalves (e.g. 46 in Pacific oyster, M. gigas), and in many cephalopods (e.g. 66 in the golden cuttlefish Sepia esculenta) and gastropods (e.g. 45 in the veined rapa whelk Rapana venosa; 14 in the edible periwinkle Littorina littorea). Within gastropods, we observed UGA1 converted to Cys in the lineage comprising Heterobranchia and Caenogastropoda, although its precise phylogeny is difficult to solve (Supplementary Note 3). Interestingly, we did not detect more than one SECIS for SelenoP in this lineage, despite featuring multiple distal Sec-UGAs.
Other Lophotrochozoa
Molluscs belong to the phylum Lophotrochozoa, which also includes Platyhelminthes (flatworms), Nemertea (ribbon worms) and Annelida (segmented worms). While we did not find SelenoP in Platyhelminthes, the other two categories of worms have highly Sec codon rich SelenoP genes. For example, 65 in-frame UGAs are present in SelenoP of the bootlace worm Lineus longissimus (Nemertea), and 66 in Platynereis dumerilii (Annelida). Two SECIS elements were identified in most multi-Sec codon SelenoP genes in worms. In the reconstructed gene tree, worm sequences formed a single cluster that also included brachiopods (e.g. Lingula anatina). This cluster includes a few genes with a single UGA and one or no SECIS (although this could be due to poor assembly quality). The predicted C-terminal Sec-rich domain has homology between Mollusca, Nemertea and Annelida, supporting common inheritance of Sec codon-rich SelenoP within Lophotrochozoa.
Nematodes
SelenoP is missing in the great majority of nematodes, with the sole exception of the Trichinella genus (an early-branching lineage of parasites). Several Trichinella species have a distant SelenoP homolog that has a Cys codon in place of UGA1. While in some cases there are 2–3 in-frame UGAs at the end of the coding sequence (CDS), they are not conserved in this genus, and no SECIS candidate is detected in these genes (with a single possible exception). It is likely that SelenoP is not a selenoprotein in Trichinella, and that other nematodes either lost this gene entirely or it diverged beyond our recognition power.
Insects
We did not find SelenoP in the great majority of insects, including fruit flies, mosquitos and beetles. However, SelenoP was identified in various early branching insect lineages, including Zygentoma (silverfish, firebrats), Odonata (dragonflies, damselflies), Blattodea (cockroaches, termites), Phasmatodea (stick-insects), and Paraneoptera (lice). These genes typically feature a single SECIS and UGA1 (Sec) and form a single cluster in the reconstructed protein tree (Supplementary Fig 1). Beyond the N-terminal thioredoxin-like domain, insect SelenoP genes encode a ~550 residue C-terminal domain with no similarity to any characterized protein. All the above-mentioned insect lineages contain the Sec-machinery, which is not ubiquitous in this class: several branches of holometabolous insects (Endopterygota) lost it in independent events [42,47]. Consistently, we did not find SelenoP in holometabola, with the sole exception of some Hymenoptera (wasps). In these selenoproteinless species, UGA1 is not conserved and there is no detectable SECIS, indicating that this remote hymenopteran SelenoP homolog is not a selenoprotein.
Other arthropods
We observed a remarkable diversity in arthropods. Crustaceans appear not to contain SelenoP. However, the centipede Strigamia maritima has a SelenoP with a single UGA, situated at the characteristic UGA1 location. SelenoP genes from arachnida (spiders, ticks, mites) formed three phylogenetic clusters, likely representing a single orthologous group with diverse divergence rates (Supplementary Note 4). Interestingly, we did not find any SECIS elements in SelenoP genes of Acariformes (arachnids). This group included both genes with an in-frame UGA corresponding to UGA1, and genes with diverse codons in its place. Future research will clarify whether these genes encode selenoproteins, or perhaps if their mRNAs are translated through a distinct mechanism of UGA recoding.
Cnidaria
We found SelenoP in many Cnidaria (early branching lineage of marine metazoans). Upon protein tree reconstruction, cnidarian sequences formed two distinct phylogenetic clusters. The smallest group consisted of sequences from Scyphozoa (true jellyfish such as Aurelia aurita, which has paralogs in both clusters). Genes in this cluster have classic position UGA1 replaced with a Cys codon, a distal region with multiple UGAs, and apparently only one SECIS element. The other and largest cluster encompassed hydrozoans, box jellyfish, sea anemones and corals. The majority of genes in this cluster carry a single UGA at the characteristic UGA1 position, and either one or two SECIS elements. A few genes in this cluster had instead multiple UGAs, both at the UGA1 position and in the distal region (homologous to Scyphozoa), and two SECIS elements.
Protein homology and modularity in SelenoP
Our analyses highlighted a number of differences between SelenoP genes of vertebrates and invertebrates. We thus proceeded to a comprehensive computational characterization aimed to elucidate their evolution. All SelenoP proteins identified have a similar N-terminal thioredoxin-like domain. Yet, Sec-rich tails have no obvious homology across distant metazoan groups: Blastp does not detect significant homology (e-value<0.01) between the human SelenoP tail and that of any other phylogenetic cluster representatives. Aiming to clarify the evolution of SelenoP, we developed an improved procedure to detect local homology in protein sequences (Methods, Supplementary Fig 3). Our method can detect both inter- and intra-sequence similarities, which is relevant because various multi-Sec tails features obvious modularity, with similar motifs repeated in tandem (Supplementary Fig 2). Our results are shown in Fig 3. All SelenoP proteins match each other for the first ~200 residues, corresponding to the thioredoxin-like domain. At the C-terminal domain, several proteins contained shifted matches with themselves, indicating modularity. Besides the aforementioned thioredoxin-like repetition in cephalochordata, the simplest case was in spider (arachnida). Here, Sec residues located in the C-terminal region, formed by four repetitions of a 2-Sec motif, are separated from the thioredoxin-like domain by a region that also featured self-similarity. The sequences of molluscs, echinoderms and marine worms presented outstanding modularity, with high counts of matches both with themselves and with each other. Importantly, we detected local homology between some of these repetitions and the C-terminal domain of human SelenoP1 (Fig 3; expanded in Supplementary Fig 3C). These matches span the entire length of the human multi-Sec tail, from the Sec encoded by UGA2 to the end of the protein. Even with our conservative e-value cut-off, it is possible that some of these alignments are spurious and do not represent true homology, since SelenoP distal sequences have a very skewed C/U-rich composition. However, our finding of a perfectly conserved TESCQU motif between human and bootlace worm L. longissimus (Supplementary Fig 3C) is a strong indication that genuine homology is present. Notably, the alignments between the human and invertebrate sequences contained the binding site of ApoER2 in vertebrates, with its key motif ZQZ (Z representing Sec or Cys). Human SelenoP has two nearby ZQZ sites, the second of which was shown to be essential for ApoER2 binding [32]. Pacific oyster and bootlace worm have 14 and 29 ZQZ motifs, respectively, most of which have a similar sequence context to the second site in human. Among the rest of selected SelenoP representatives, only purple sea urchin S. purpuratus has ZQZ; a single occurrence is located at the very end of its sequence, but its context does not resemble its human counterpart. The significance of these results for the evolution of SelenoP is presented in the Discussion.
Fig 3. Homology and modularity in SelenoP protein sequences.

The plot represents local homology matches identified between SelenoP representatives (Methods). The central panel is used to illustrate the plot meaning. Each panel consists of a dot-matrix-like plot, with each diagonal line representing a match between a query (e.g. arachnida; P. tepidariorum) and a target (either the query itself or any other representative), color coded by target sequence. At the top of each panel, a horizontal line shows the query length and the positions of Sec residues. In arachnida, the C-terminal domain with 8-Sec consists of 4 repetitions of a 2-Sec module, resulting in multiple matches of this sequence with itself. Some C-terminal domains show high modularity (intra-sequence matches), and also homology with other representatives (inter-sequence matches). We identified matches of vertebrate SelenoP tail (human; top-left panel) with regions of invertebrate Sec-rich C-termini; these are shown in Supplementary Fig 3C.
Invertebrate SelenoP translation in supplemented rabbit reticulocyte lysates
In order to monitor Sec-incorporation with T7-transcribed SelenoP mRNA, we used co-translational 75Se labelling in rabbit reticulocyte lysate (RRL) supplemented with rat C-terminal domain of SECISBP2 (CT-SECISBP2) [22,27,48,49]. This system was previously shown to support translation of vertebrate full-length SelenoP [50], but has never been tested with invertebrate SelenoP. We selected a few SelenoP representative sequences for tests of in vitro translatability. The house spider (Parasteatoda tepidariorum; arthropod, arachnid; 9 Sec-UGAs and two SECIS) mRNA yielded a product of ~70 kDa, whose synthesis was dependent on the presence of CT-SECISBP2 (Fig 4A, green asterisk). Interestingly, its migration on gels was slower than its predicted full-length molecular weight (ca. 50 kDa). This may be due to its proline-rich amino acid composition (Supplementary Fig 2) or may indicate the presence of post-translational modifications. The owl limpet (Lottia gigantea; mollusc, aquatic gastropod; Sec-UGAs and one bona fide SECIS) mRNA yielded no detectable radiolabelled product (not shown). However, Pacific oyster (Magallana gigas, formerly known as Crassostrea gigas [51]; mollusc, bivalve; 46 Sec-UGAs, two SECIS) mRNA yielded a labelled product of ~30 kDa, conditional on the addition of CT-SECISBP2 (Fig 4B, red asterisk). The estimated molecular weight of this product approximates that expected from decoding the mRNA up to the fourth UGA codon. In addition, a CT-SECISBP2 dependent diffuse band was also observed (Fig 4B, red bracket). Since UGA positions in the distal region of oyster SelenoP are in very close proximity, the diffuse band could indicate premature termination within the UGA-rich distal segment.
Fig 4. Invertebrate SelenoP mRNA translation in CT-SECISBP2 supplemented RRL.

On top, the structures of SelenoP mRNA of (A) house spider, P. tepidariorum and (B) Pacific oyster, M. gigas, are displayed, including the location of Sec-UGAs within the CDS and SECIS elements in the 3’UTR. Below, the results of their in vitro translation experiments are shown. We tested mRNAs with either their native initiation context or an added Kozak consensus, with or without supplemented CT-SECISBP2. The first lane shows the translation product of zebrafish SelenoP mRNA as control for processive Sec incorporation [56]. A low molecular weight product (~25 kDa) was observed across all experiments even in the absence of added mRNA (Supplementary Fig 5) and was ascribed to RRL background. Green asterisks (A) denote a radiolabeled product corresponding to a full-length spider SelenoP. Red asterisks (B) denote oyster SelenoP termination product at UGA 3 or 4, and red bracket highlights a diffuse product(s) indicative of possible termination between UGAs 4–46.
Effect of oyster SelenoP sequence elements on in vitro translation
A search in the CDS of SelenoP from six bivalves, each containing more than 45 UGAs, revealed the potential for formation of a stem loop structure close to the initiation codon. Because of the degree of conservation, presence of sequence covariation and analogy to a known vertebrate counterpart [41], we term this element ‘Initiation Stem Loop’ (ISL). In M. gigas the ISL spans CDS positions 40 to 160 from the annotated AUG (Supplementary Fig 4). The possibility that the ISL acts as a barrier and perhaps influences initiation efficiency at the AUG prompted further in vitro translation experiments with M. gigas SelenoP mRNA. In order to assess the effect of the ISL and other sequence features, we generated and tested several oyster SelenoP variants. In vitro transcribed mRNAs corresponding to each of these variants were translated in RRL labeled with 75Se and supplemented with CT-SECISBP2. First, we noticed that the M. gigas SelenoP open reading frame (ORF) began with the start codon in a weak Kozak context (c.GAUGcG), which differs from the oyster transcriptome consensus (AaaAUGgc, closely similar to that identified in mammals). We thus replaced this with a strong Kozak context AcCAUGGca. There was no product difference, except perhaps a slight increase in the amount of the termination product (Fig 4B lane 4, Supplementary Fig 5A, lane 4). Second, we identified a small 5’ Leader ORF (16 codons, beginning 73 nts 5’ of SelenoP start codon), whose AUG is in a modest Kozak context (c.GAUGAA) which is expected to cause 30 to 40% of the scanning subunits to initiate translation [52–54]. Upon removal of the entire native 5’ leader (yet retaining 110 nt of vector sequence as 5’UTR), there was a modest reduction in the amount of the termination product (Supplementary Fig 5A, lane 5). Third, the disruption of sequence complementarity in the ISL through synonymous codon mutations resulted in the absence of detectable 75Se product (Supplementary Fig 5A, lane 6; note that the product due to ribosomes terminating at UGA1 would not have been detected by 75Se labelling). An additional experiment using 35SMet labelling (Supplementary Fig 5B, lane 7) detected the presence of significantly reduced early termination product at UGA1 upon ISL mutation, suggesting that initiation efficiency is reduced but not entirely abolished. Fourth, sequence inspection revealed that M. gigas SelenoP mRNA has an RNA structure homologous to vertebrate Sec redefinition element 2/3 (SRE2/3). It is located in CDS positions 1585–1705, spanning UGAs 31–35 in M. gigas (Supplementary Fig 4). Deleting the sequence from UGA11 to UGA46 yielded no SelenoP-specific radiolabeled product (Supplementary Fig 5A, lane 7). While this deletion removes the SRE 2/3, the occurrence of initiation (Supplementary Fig 5B, lane 9) means that the lack of 75Se labelled product likely reflects long range mRNA folding being relevant to Sec-specification.
Lastly, we focused on SECIS elements. M. gigas SelenoP mRNA contains two SECIS in its 3’UTR with signature conserved motifs also found in vertebrates (Supplementary Fig 4). As in vertebrates, the two oyster SelenoP SECIS elements also differ by an additional structural element, found in SECIS 1 (type II) but not SECIS 2 (type I). Oyster SECIS elements function to permit full length 75Se labelled product from zebrafish SelenoP CDS (Supplementary Fig 5A, lane 8). Appending oyster SECIS 1 or oyster SECIS 2 to the zebrafish CDS yielded full-length product with similar efficiency (Supplementary Fig 5A, lanes 9 and 10) suggesting that each oyster individual SECIS element has high affinity to rat SECISBP2, to a similar degree as the zebrafish combined SECIS elements (Supplementary Fig 6). The absence of functional distinction between oyster SECIS 1 and 2 in this heterologous system contrasts with their vertebrate counterparts. The “Berry model” for vertebrate SelenoP translation asserts that SECIS 2 functions to reprogram UGA1 for Sec insertion, while SECIS 1 acts for processive Sec insertion at the distal UGAs. This model was not proposed in absolute terms and much evidence from vertebrate in vitro protein synthesis, cell culture and mice with specific mutants of the endogenous gene, supports the proposed preferential action of the two SECIS elements [37,55–57].
Attempting to improve in vitro translation of oyster SelenoP, we considered using its native Sec recoding machinery. We identified a single homolog of human SECISBP2 in the M. gigas genome, consistent with earlier findings in various invertebrates. We refer to this gene as SECISBP2, though it is also homologous to SECISBP2L (Supplementary Note 5) [58,59]. We purified a recombinant full-length M. gigas SECISBP2 (Supplementary Fig 7C). In RRL supplemented with oyster SECISBP2, instead of rat SECISBP2, oyster SelenoP mRNA did not yield detectable 75Se labelled product (not shown). In the same system, translation of rat SelenoP mRNA did result in UGA specification of Sec but likely only by UGA1 (Supplementary Fig 7D). Our results suggest partial incompatibility between the translation machinery of oysters and vertebrates, and/or unknown oyster trans-acting components lacking in RRL, necessary for processive Sec incorporation. While meaningful for mechanistic insights, our in vitro experiments fell short of definitive evidence of full-length oyster SelenoP synthesis. In quest of it, we turned to a different experimental model.
Selenium biology of the oyster M. gigas
As our analyses revealed highly Sec-rich SelenoP genes in molluscs, and bivalves in particular, we performed in vivo experiments aimed to characterize the role of selenium in mollusc biology. Pacific oyster M. gigas was chosen for its widespread importance in the human food industry, and the availability of genome and transcriptome sequences [60,61]. We identified thirty-two selenoprotein genes in the genome sequence of M. gigas, along with a complete set of factors required for selenoprotein synthesis (tRNA-Sec, PSTK, SEPSECS, SECISBP2, EEFSec and SEPHS2) (Supplementary Table 1). All of these genes were also identified in a comprehensive transcriptome assembly [61], providing evidence for their expression. Additionally, a transcript encoding a SelenoW protein (SELENOW.1) was identified in the transcriptome. The sequence was supported by both RNA-seq and riboseq, but was missing in the genome assembly. Oyster selenoproteins belonged to 22 distinct families. These encompassed all selenoprotein families previously described in invertebrate metazoans [39], including three families not found in vertebrates: AHPC, MSRA and DSBA. In addition, we identified a Sec-containing radical S-adenosyl methionine (RSAM) gene (Radical SAM/Cys-rich domain; Interpro IPR026351) in M. gigas, including a SECIS downstream of CDS. RSAM selenoproteins had never been observed in metazoa but had been previously reported in bacteria [62] and in a single-cell eukaryote, the harmful bloom alga Aureococcus anophagefferens (UniProt F0XY08) [63]. Sequence searches revealed numerous RSAM selenoproteins in bivalves and other metazoan lineages (Supplementary Fig 8). Crustaceans, bivalves and most cnidarian species contain Sec in their RSAM protein, while echinoderms possess a Cys homolog. We identified two genes belonging to the SelenoP family in M. gigas. The second gene, here referred to as oyster SelenoPb, also contains two SECIS elements, but it is shorter and has fewer UGAs than its paralog SelenoP (Supplementary Note 6). Our analysis suggests that oyster SelenoPb appeared by tandem duplication in the Crassostrea lineage, which includes M. gigas.
Selenium uptake and accumulation in oyster tissues
Oysters feed on natural phytoplankton by filter-feeding. They are known for their capacity to filter microscopic food sources from water as well as their ability to bioaccumulate sediments, nutrients and even pollutants from their environment [64]. To investigate the effects of selenium concentration on the M. gigas selenoproteome, and particularly the translation of SelenoP, we carried out an oyster selenium supplementation where the microalgae food source (Tetraselmis sp.) was grown in Se-rich medium by the addition of 28.9 μM (5mg/L) final concentration of sodium selenite. This concentration was previously suggested to allow optimal Se uptake without inducing toxicity to algal cells [65,66]. Guided by preliminary analyses, we chose to focus on adult male oysters (Supplementary Note 7). Two groups of 10 individuals were distributed to separate tanks and fed weekly for 6 weeks with equal amounts of micro-algae pre-grown with or without Sesupplementation, and were then analyzed by ICP-MS (Methods). Total selenium levels in tissues of selenium supplemented oysters increased 20-fold on average compared to non-supplemented controls (Table 1), whose levels were also compared to those in farmed marine mussels, Mytilus edulis (Supplementary Table 2). Remarkably, individual oysters accumulated 50-fold higher selenium levels than in the control group without mortality. Other material from the supplemented tank, including oyster non cytoplasmic debris, sea water, and the algae that served as food source, also showed elevated selenium levels (Supplementary Table 3).
Table 1. Selenium accumulation in oyster tissues.
Total Se levels in whole body soft tissue from 7 males fed a Se enriched diet (A-G) and 5 from an unsupplemented control group (A-E), individually determined by ICP-MS, are shown together with their Standard Deviation (SD) and Relative Standard Deviation (RSD). Values for certified reference materials are also indicated.
| Individual Oyster Tissue | Non-supplemented | Supplemented | ||||
|---|---|---|---|---|---|---|
| Av. Se per individual (μg/g) | SD | RSD | Av. Se per individual (μg/g) | SD | RSD | |
| A | 3.47 | 0.27 | 7.5 | 80.2 | 0.8 | 1.1 |
| B | 3.3 | 0.2 | 6.15 | 48.4 | 0.34 | 0.69 |
| C | 3.12 | 0.25 | 7.95 | 179 | 0.28 | 0.16 |
| D | 4.72 | 0.23 | 4.94 | 36.8 | 0.43 | 0.17 |
| E | 2.9 | 0.18 | 6.1 | 19.9 | 0.32 | 1.59 |
| F | 52.1 | 0.52 | 0.98 | |||
| G | 13 | 0.2 | 1.56 | |||
| Average Se concentration per group (μg/g of tissue) | 3.502 | 61.3 | ||||
|
Certified Reference Materials NRCC, National Research Council of Canada NRCC Reference Material Site |
Av. Se μg/g | SD | RSD | |||
| DORM-1* | 1.46 | 0.03 | 2.27 | |||
| TORT-2** | 5.64 | 0.09 | 1.68 | |||
certified value: 1.62 ± 0.12 μg/g (dogfish muscle)
certified value: 5.63 ± 0.6 μg/g (Lobster hepatopancreas)
Effects of selenium supplementation on selenoproteome expression
To assess the effects of selenium concentration on selenoprotein gene expression, we performed RNA sequencing (RNA-seq) and ribosome profiling (riboseq) of oysters with and without selenium supplementation. While RNA-seq characterizes overall transcript levels, riboseq can elucidate ribosome occupancy and potential regulation at specific regions, by mapping ribosome-protected fragments (RPFs) to mRNAs. For each group, we selected whole body tissue from two oysters that had similar levels of total measured selenium (average 3.4 μg/g and 17 μg/g for non-supplemented and supplemented, respectively). The soft tissues of each pair were pooled and prepared for sequencing. A preliminary assessment of our ribosome profiling data was performed through metagene analysis (Supplementary Note 8). Despite a noticeable difference between supplementation groups, our data featured considerable triplet periodicity and quality suitable for downstream analysis. We thus quantified expression of Sec machinery and selenoprotein genes and compared them with the whole oyster transcriptome (Supplementary Fig 9). Transcripts with low coverage were discarded (Methods).
Our results show a general trend of an increase in selenoprotein mRNA abundance (Fig 5, red) and RPFs (Fig 5, blue) upon selenium supplementation, consistent with previous reports of ribosome profiling on selenium supplemented mice [67,68]. Translation efficiency (TE), estimated as RPF abundance relative to mRNA abundance, was actually reduced in oyster selenoproteins upon supplementation (Supplementary Fig 9C), because the transcript level increase (up to a 7-fold in individual selenoproteins) was greater than the RPF increase (about 3-fold).The observations in oyster differ from reports in mice wherein Se has a greater effect at protein level than at transcript level [67]. As for Sec machinery genes, we found no selenium-dependent regulation on EEFSEC, while SEPHS2 was upregulated and SEPSECS, surprisingly, downregulated (Fig 5).
Fig 5. Selenium supplementation effects on expression of oyster selenoprotein and Sec-decoding “machinery” genes.

The gene expression response to selenium supplementation is expressed as the fold change in mRNA abundance from RNA-seq (red, RPKM) or ribosome footprints from riboseq (blue, RPFKM) between Se-supplemented and non-supplemented samples. Absolute expression values are shown in Supplementary Fig 9. The bottom panel shows how selenoprotein and Sec machinery genes compare to the distribution of the whole oyster transcriptome. Supplementary Table 1 indicates the correspondence between oyster gene identifiers used here and human genes. Selenoproteins DIO.2, SELENOH.1 and Sec machinery PSTK.2, SBP2.117 were omitted from this analysis for low coverage.
Analogously to previous research [67,69], we obtained a surrogate estimate of Sec-UGA redefinition efficiency (URE) calculated as the ratio of RPF density downstream to upstream of UGA (Methods). URE represents the proportion of ribosomes that translated past the Sec-UGA codon compared to those that initiated translation. It cannot be calculated for selenoproteins with UGA-Sec near to the very 5’ or 3’ end of CDS. Consistent with previous reports of Sec-UGA redefinition in mice, we observed that the majority of selenoproteins exhibited a low URE, although there were several exceptions (Supplementary Table 4). The analysis of ribosome profiling for oyster SelenoP is presented hereafter.
In vivo translation of 46 Sec-UGA SelenoP
Ribosome coverage of oyster SelenoP mRNA
Ribosome profiling allowed us to monitor oyster SelenoP translation in vivo. RNA-seq showed full coverage along its mRNA, including its 5’ 110 nt Leader, CDS and 3’ UTR, while riboseq was mostly limited to the CDS as expected (Fig 6A). Notably, some RPFs mapped to a specific 61nt region spanning the previously identified 5’ Leader ORF, supporting that it is translated. Ribosome coverage was only moderately greater in the sample derived from selenium supplemented tissue. In the non-supplemented sample, the most abundant fragments were mapped from 15 nts 5’ of the CDS start and sharply dropped 20 nts 3’ of it (Fig 6A green arrow). The ISL RNA structure was found 20 nt 3’ of the coverage drop (ISL at positions 37–157 from main ORF AUG). The amount of these fragments was greatly reduced upon selenium supplementation. Comparing the RPF coverage map for the two conditions, the fragments from the non-supplemented material were somewhat less abundant approaching UGA1 and some of their boundaries moderately shifted. Inferences from such complex patterns in profiling data merit caution, but the data showed a broad RPF accumulation prior to UGA1 that was also present in mammalian SelenoP ribosome profiling data [41,69]. We computed URE for UGA1 by comparing RPF density in the CDS 5’ of UGA1 to the density in the region between UGA1 and UGA2. The URE for the non-supplemented group was 5.6% whereas that for the supplemented group was 4.7% (Supplementary Table 4). Thus, selenium level does not exert a strong influence on UGA1 redefinition in oyster, unlike its mammalian counterpart [67,69]. The role of the pause site is considered in the Discussion.
Fig 6. Monitoring Pacific oyster SelenoP translation by ribosome profiling.

(A) RNA-seq reads (grey) and riboseq RPFs (red for non-supplemented sample, blue for selenium supplemented) aligned to SelenoP mRNA positions. The coverage corresponds to full reads with no P-site offset, normalized to the total number of reads mapping to the mRNA. The RNA-seq reads were merged for the supplemented and non-supplemented. The 5’ leader ORF spans −55 to −22 nt positions (orange) whereas the main ORF is depicted as starting at position 0 (brown). Conserved RNA structures are shown in the gene model in grey. A selenium-regulated pause at initiation is indicated in the plot. (B, C) Immunoblot of the oyster tissue lysates used for library preparation using SelenoP custom antibodies targeting N-terminal (B) and C-terminal (C) portions of the protein. Red arrow indicates putative full-length protein detected at around 68 kDa. A densitometry graph (whiskers indicate SD of two technical replicates per sample) depicts the change in full-length band intensity in the non-supplemented vs Se-supplemented sample from each antibody; band intensity was normalized against a Ponceau-S loading control.
As introduced earlier (Supplementary Note 6), a second gene, termed oyster SelenoPb (SELENOP.1) was identified in M. gigas exhibiting high homology to SelenoP, 5’ of UGA1. However, we detected minimal RPF coverage 3’ of UGA1 in SelenoPb where the sequences are less homologous, suggesting that its expression levels were significantly lower, if not almost undetectable, compared to SelenoP. We therefore deduce that reads obtained in the presented SelenoP coverage map should accurately represent translation of its mRNA. The oyster RPFs map to the full length of the predicted CDS up to the UAG termination codon (Fig 6A). This result strongly supports full-length 46 Sec-UGA SelenoP translation in M. gigas. Translation of UGAs 2–46 appeared to be continuous and processive as indicated by the absence of ribosomal pausing and approximately equal RPF coverage across the UGA-rich segment, comparable to that found for its mammalian counterpart translation, both in vitro [57] and in mice [67].
Detection of full-length oyster SelenoP by immunoblot
Portions of the lysates used for ribosome profiling were further employed for immunoblot analysis. Custom M. gigas SelenoP antibodies were raised against the N-terminal region encoded upstream of UGA1 (anti-NT SelenoP) and the C-terminal region encoded after UGA43 (anti-CT SelenoP). The antibodies were tested for peptide affinity and specificity (Supplementary Fig 10). Both antibodies detected an immunoreactive product that migrated on SDS-PAGE at an apparent molecular weight of 68 kDa, matching the predicted molecular weight of full-length oyster SelenoP (Fig 6B, red arrow). Full-length product appeared to increase upon Se supplementation, consistent with the observed increase in RPF abundance (Fig 6B, C). Anti-CT SelenoP also recognized a very prominent product with an apparent molecular weight below 37 kDa. Since this was not recognized by the N-terminal specific antibody, it likely represents a non-specific protein (see below), although we cannot exclude possible proteolytic cleavage products.
75Selenium labelling of oyster larvae
Human SelenoP contains 10 in-frame UGA codons, which can also specify Cys instead of Sec to some degree dependent on the selenium supply level [70]. Our attempts to immunoprecipitate endogenous SelenoP from oyster tissues using the custom antibodies were unsuccessful. Consequently, to monitor in vivo Sec incorporation in oyster SelenoP, proteins were extracted from 75Se labeled free-swimming 7- and 14-day larvae and analyzed by autoradiography (Fig 7A) (preliminary experiments guided the choice of larval age; see Methods). A parallel Western blot with custom SelenoP antibodies detected a product at ~68 kDa, similar to that of the most intensely 75Se labelled protein. It corresponds to the predicted full-length M. gigas SelenoP. This is strong evidence for multiple Sec-incorporation in-vivo, and that SelenoP is the most selenium-rich protein in the M. gigas proteome. These results do not preclude the possibility of some level of specification of Cys at UGA sites, but if it occurs at all, it is very likely at a low level (also since some decoding of UGA as Cys in human cells is only detectable at low selenium [70]). The presence of lower molecular weight products detected by both autoradiography and Anti-NT SelenoP (Fig 7, blue arrows), suggests the presence of SelenoP termination products of 27 and 42 kDa derived from termination at UGA 2 and UGA 10–12 respectively. The additional radiolabeled products, not detected by our antibodies, should correspond to the other abundant selenoproteins. A counterpart of the <37 kDa product described in the last section was not detected.
Fig 7. 75Se labelled proteins in M. gigas larvae.

(A) Autoradiography of radiolabeled proteins in 7-day and 14-day old oyster larvae. Each protein preparation was loaded in two lanes as technical replicates. (B) Anti-N-terminal and (C) Anti-C-terminal SelenoP immunoblots of oyster larvae proteins. Red arrows indicate a radiolabeled and immunoreactive bands at an apparent molecular weight of 68 kDa, which is approximately the predicted size of full-length SelenoP. Blue arrows point to possible early SelenoP termination products detected by both autoradiography and parallel Anti-NT SelenoP probing.
Discussion
Evolution of Sec in SelenoP
Our characterization of SelenoP genes across metazoa brings a conundrum. We observed numerous short SelenoP2-like genes, encoding the thioredoxin-like domain, and long SelenoP1-like genes, encoding the thioredoxin-like domain and an additional C-terminal domain containing multiple Sec residues. There are two main evolutionary scenarios to explain these observations. The first involves common ancestry, wherein the occurrence of the multi-Sec sequence predates the split of these groups (i.e., SelenoP1-like is the ancestral state). In this scenario, the tail diverged extensively across groups, unlike the N-terminal thioredoxin-like domain, and also, the sequence encoding the tail was lost in SelenoP2-like genes. The alternative scenario involves convergent elongation: the various metazoan groups inherited a short SelenoP gene, which underwent independent elongations to acquire non-homologous multi-UGA distal sequences (i.e, SelenoP2like is the ancestral state). Based on the apparent lack of homology between the SelenoP tails of distant metazoans, we previously hypothesized that vertebrate SelenoP1 originated by duplication of SelenoP2, implying that convergent elongation had occurred in other lineages [37,41]. However, our improved procedure for finding local homology allowed us to detect significant matches between the Sec-rich tail of human SelenoP and that of various invertebrates (Fig 3). These findings support common ancestry as most likely scenario. To challenge this hypothesis, we reconstructed a phylogenetic tree of SECIS elements (Methods). If common ancestry is the correct explanation, human SECIS 1 should cluster with SECIS 1 of other species, with all SECIS 2 forming their own phylogenetic cluster. If instead convergent elongation occurred, the SECIS 1 and SECIS 2 sequences corresponding to each independent elongation should cluster together. Our reconstructed tree (Supplementary Fig 11) features low bootstrap values (i.e., topology reliability), reflecting the challenge of phylogenetic reconstruction of RNA structures. Yet, we observed that SECIS 1 from vertebrates, marine worms, molluscs and some cnidaria cluster together, while their SECIS 2 form a distinct cluster, therefore further supporting the common ancestry scenario.
The multi-Sec tail of SelenoP shows obvious modularity in several lineages. The repeated motifs in any modular SelenoP are more similar to other occurrences within the same protein than to the sequence encoded by SelenoP in other metazoan groups. This suggests that these repetitions propagated in relatively recent times, after the evolutionary split between the various metazoan groups. Our observations indicate that, even in the context of common ancestry, independent events of elongation occurred to extend the C-terminal tail by repetition of a Sec-containing module. In other lineages, the tail was instead shortened or lost entirely. Altogether, our analyses point to a very dynamic process acting on the C-terminal tail, characterized by a fast rate of divergence, extension and contraction.
We believe that this process was driven by the environmental availability of selenium across biological niches, as well as the mutable reliance on selenium utilization by the various metazoan lineages. As a fundamental actor in selenium homeostasis of metazoa, SelenoP is tightly linked with selenium supply. At the level of individuals, SelenoP in plasma is a biomarker of selenium concentration that has been adopted as the reference standard in several European countries [71,72]. At the species level, the number of Sec residues in SelenoP significantly correlates with selenoproteome size, and thus with the degree of selenium utilization as previously reported [33] and confirmed by our analysis (Supplementary Fig 12). It was previously hypothesized that selenium utilization is particularly important for aquatic life [38]. While the large selenoproteome of vertebrates apparently constitutes an exception, it was recently discovered that terrestrial vertebrates have relaxed evolutionary constraints on selenoprotein genes compared to fish [73]. Sec residues in the tail of SelenoP appear to follow a nearly neutral process of conversion to Cys. This may suggest that the large selenoproteome of mammals could be a legacy from our past in aquatic environments, slowly decreasing during our relatively slow evolution. Our own analyses of metazoan SelenoP are very much consistent with the aquatic hypothesis. While there are some exceptions (e.g. crustaceans lacking SelenoP), the proteins richest in Sec residues were all found in aquatic species. In particular, SelenoP evolution led to a truly remarkable number of Sec residues in this protein in molluscs and marine worms, with freshwater mussel E. complanata topping the list with 131/132.
Selenium biology in bivalves
Fish and shellfish are known for their high content of selenium, with the mussel Mytilus edulis (blue mussel) scoring the highest selenium concentration in a recent survey across various marine organisms [74]. Yet, the role of selenium in the biology of these animals is virtually unexplored. Here, we characterized the oyster selenoproteome in detail, and provided insights into selenium effects on gene expression regulation in-vivo. We identified 32 selenoproteins in oyster, more than in mammals (25 selenoproteins) [45]. We provide the first report of an animal Sec-containing RSAM protein in oysters and found that it is also present in other bivalves, cnidarian and crustacean species (while echinoderms possess a Cys homolog).
Supplementation studies revealed that the oysters can accumulate extremely high levels of Se in their tissues (Table 1), with an increase of up to 50-fold per individual. We further analyzed the effect of selenium supplementation on selenoprotein mRNA expression, translation and UGA redefinition efficiency, using RNA-seq and riboseq. For these analyses, we used samples with around 6-fold increase in total Se levels. While more replicates and conditions would be necessary for precise quantitative assessments, our results show clear qualitative trends. In response to selenium, we observed a general upregulation of RPF counts, reflective of protein product abundance, with individual selenoproteins exhibiting up to a 3-fold increase. For SelenoP, RPF abundance correlated with protein abundance observed by immunodetection. At the transcript level, the trend of increased expression of selenoproteins was also evident, and featured an even stronger response (Fig 5, Supplementary Fig 9). This contrasts with earlier experiments in mice, where selenium supplementation caused a greater effect on translation, and mRNA levels were relatively unchanged upon selenium increase [67]. This discrepancy could be attributed to different rates of mRNA turnover, stability or susceptibility to nonsense mediated decay (NMD) between organisms. Selenoproteins that presented the highest selenium response in mRNA levels in the Howard et al. study are known to be regulated during transcription, e.g. Gpx1 which is a target for NMD [75,76] and SelenoW which exhibits a high rate of mRNA turnover [77]. The difference observed in oysters might indicate the presence of an alternative regulatory system, perhaps a dedicated transcription factor sensing selenium levels. Further indications of translation playing a lesser role in selenium-response in oysters than in mammals come from the analysis of recoding efficiency. Oyster response to selenium was quite diverse across selenoproteins: not all selenoproteins exhibited enhanced URE or any strong 3’RPF increase, contrary to findings in mice [67].
Our analysis highlighted interesting differences in the regulation of the oyster selenoproteome by dietary Se compared to mammals. These are not surprising, given their long phylogenetic distance and many differences in physiology. It has been previously reported that, in oysters, accumulated minerals are compartmentalized resulting in a higher mineral turnover rate. Further, subcellular compartmentalization of metals in bivalves may contribute to detoxification and may explain how they can circumvent toxicity as a result of bioaccumulation [64]. Oysters also possess an open circulatory system compared to a closed system in mammals. Such biological difference raises the question of whether the selenoproteins identified in oysters exhibit the same function. For instance, SelenoP in mammals has been strongly implicated in Se transport, as a hierarchy of selenium supplementation to tissues is achieved by preferential delivery of long SelenoP isoforms to brain and testis [32]. In oysters, we cannot exclude a possible role of SelenoP as a Se-storage protein due to the high Sec content in its C-terminal domain. Selenium and SelenoP have also been implicated in mercury chelation [78]. The high Sec content of SelenoP could be a possible adaptation to filter-feeding lifestyle, enabling oyster to tolerate high accumulation of toxic metals [79].
Upon supplementation, the remarkable increase in total selenium in tissues was accompanied by a more modest upregulation of selenoprotein levels. Thus, the levels of selenium in tissues cannot be accounted by just protein production, contrary to the situation in mammals where most increase in Se tissue level is directed to incorporation into selenoproteins [80,81]. This suggests the presence of additional molecular mechanisms for selenium accumulation, which may involve specialized protein machinery or non-protein components that bind Se, e.g. low molecular weight selenocompounds or selenosugars.
Altogether, our work paves the way for investigating selenium biology in oysters and other molluscs. Considering the importance of these species in the food industry and their high selenium content, we expect that our research will prompt further studies to elucidate the function of selenoproteins and other aspects of selenium metabolism in these species.
Insights into the genetic decoding mechanism of oyster SelenoP
We identified a stem loop in oyster SelenoP mRNA, termed ISL, that is 37–157 nt 3’ of the main ORF start and 15 nt 5’ of UGA1. Its mammalian counterpart was shown to modulate translation initiation in vitro; analogous stem loops were characterized in non-selenoprotein mRNAs in bacteria [82]. In oyster, the function of ISL is selenium responsive. The oyster ribosome profiling shows an accumulation of abundant RPFs on the 5’ side of the ISL, indicative of blockage of ribosome progression. The peak of RPFs is observed only in non-supplemented samples, suggesting a regulatory mechanism sensing selenium level. Whether the ISL functions directly as a potential selenium riboswitch is outside the scope of the present work. Does it merely act as a “gate” for ribosome progression to avoid wasteful downstream translation? Alternatively, is it part of a mechanism for programming ribosomes at initiation so that they later decode UGA as Sec? In one model, a ribosome stalled at ISL would lead to its following ribosome having increased initiation potential at the main ORF start codon. Leader ORF translation may be relevant, but extensive work outside the scope of the present study, is needed to assess significance. Disruption of the block when selenium (or perhaps toxins) became plentiful, could lead to a burst of downstream translation and potentially SelenoELENOP synthesis. However, the ribosome profiling of living oysters shows only a 1.5-fold increase of main ORF translation under conditions of a 6-fold total selenium increase, and Western blot analysis on tissues with more elevated selenium also does not show a dramatic increase (Supplementary Fig 13). Also, there is not a substantial increase of leader ORF protected fragments on selenium supplementation. While future studies involving selenium depletion are desirable, a paradoxical result was obtained with the heterologous in vitro translation experiments: 5’UTR deletion and synonymous codon substitution to disrupt the pairing involved in the ISL led to reduced levels of initiation in vitro (Supplementary Fig 5B, lanes 5 and 6, respectively).
Our heterologous in vitro translation experiments revealed some degree of interchangeability in the Sec incorporation factors and SelenoP mRNA elements between invertebrates, fish and mammals. For instance, full-length translation of spider SelenoP with 9 Sec residues was achieved in rabbit reticulocyte lysates (RRL) with reconstituted rat CT-SECISBP2 indicating that all the factors necessary for its translation are present in this system. In contrast, while oyster SelenoP mRNA also yielded Se labelled product, it was not full length and only a product corresponding to termination at ~ UGAs 3 or 4. Replacement of the SECIS elements in zebrafish SelenoP mRNA with either both or single, oyster counterparts and translation in reconstituted RRL showed that either oyster SECIS was equally able to support full-length translation (Supplementary Fig 5A, lanes 8, 9 and 10). With caution in extrapolating from this heterologous system, this may mean that invertebrate SelenoP SECIS 1 and 2 are more functionally interchangeable than their mammalian counterparts. Another example of likely divergence involves SECISBP2. Invertebrates have a single gene in this family and vertebrates have two, SECISBP2 and SECISBP2L. The former is a primary binder of SECIS elements and mouse conditional deletions gene revealed a significant reduction in mRNA-levels with retention of Sec specification [69,83]. The function of SECISBP2L is still unclear. Further research will be necessary to untangle the functions of the two paralogs and also the differences with the oyster ortholog seen in our heterologous experiments.
Ribosome programming for UGA redefinition to Sec is a complex process where much is left to be understood. What features may be relevant for SelenoP, where translation is further complicated by progressive recoding of multiple UGAs? In standard decoding, UGA as well as other stop codons (UAG and UAA) are decoded by protein release factors and this is a slow process compared to the decoding of sense codons by cognate aminoacyl-tRNA. The relatively lower rate of competing release factor-mediated termination may be significant for UGA1 specification of Sec. However, while the critical feature(s) for the SECIS protein complex programming of ribosomes remain unclear, multiple features may be facilitatory. SRE1, an mRNA structure close to UGA1 is not required but is relevant to Sec specification efficiency [41]. The deleterious effect found in the work reported here, of deleting from UGA 11–16, is consistent with the possibility that particular long-range mRNA folding is relevant to bringing a SECIS complex in proximity to a ribosome close to UGA1. Prior results also pointed to a relevant long-range mRNA structure [41,84,85]. The ISL is also a candidate for playing a role in ribosome programming, but this requires further exploration. Another possible facilitator involves interactions between the exon junction protein complex (EJC) and SECIS elements. A likely constituent of the complex, eIF4a3 is a selective negative regulator of selenoprotein synthesis and binds type I, but not type II SECIS elements in mammals [24,86]. Notably, there is a conserved exon/intron junction 26 nts 3’ of SelenoP UGA1 (of potential NMD relevance, this distance is smaller than 50 nt from UGA1; all the remaining UGAs are in the last exon). In one model, binding of eIF4a3 in the EJC to SelenoP SECIS 2 (type I), which is involved in redefinition of UGA1, may serve to facilitate localization of SECIS 2 for later replacement association with the oncoming ribosome. Such a potential mechanism cannot be obligatory since UGA1 can be recoded from mRNA generated from constructs lacking the intron, yet the high level of termination observed with those constructs [55] suggests that it may be relevant for improved efficiency. We analysed the conservation of the exon/intron junctions in metazoan SelenoP genes, focusing on our set of predictions from genome sequences (Supplementary Fig 14). Our analyses highlighted 7 cases in which the first intron was lost. Three such cases were represented by UGA1-containing SECIS-lacking SelenoP genes identified in Acariformes, possibly translated by a Sec-independent mechanism (Supplementary Note 4). Notably, three other cases of intron loss were concomitant with substitution of UGA1 by a standard codon. It is tempting to interpret this observation as supportive of a facilitatory role for the EJC, as UGA1 substitution and loss of SECIS may have led to a lack of selective pressure to retain an EJC complex close to 3’ to at the former site of UGA1. In mice with deletions of endogenous SECIS 2, SECIS 1 can mediate some UGA 1 redefinition [37,41] and in part because of this there remains the possibility that the exon intron junction location is relevant in a subsidiary manner to the redefinition of UGA1 in WT conditions.
UGA is inferred to be an efficient terminator for standard decoding since it is the terminator for approximately 22% of M. gigas genes. Oyster ribosome profiling revealed inefficient (<5%) redefinition of SelenoP UGA1 but approximating to 100% efficiency for 3’ UGAs. In addition to the ribosome profiling results, the corresponding full-length translation of oyster SelenoP was demonstrated by metabolic Sec-incorporation into oyster SelenoP with 75Se labelling of oyster larvae, along with a parallel Western blot with anti-NT and anti-CT SelenoP antibodies. Given the larger numbers of distal UGAs in many cases and the proximity of these UGAs to each other (there are several occurrences of just one codon separating two UGA codons), we consider models involving the SECIS complex tracking with, or acting as a once off ribosome “switch” [87], to be more appealing than separate contacts with a SECIS complex at each UGA. While there is good evidence from zebrafish to mammals for the general validity of the Berry model (UGA1 redefinition mainly enabled by SECIS 2, and redefinition of 3’ UGAs by SECIS 1), we have no experimental data relevant to whether this model is applicable for invertebrates. However, our experiments showed that both SECIS elements of oyster SelenoP supported progressive Sec incorporation in vitro of a zebrafish construct, suggesting that such SECIS specialization is not present in oyster. While there has been substantial debate about whether there are subsets of ribosomes free of mRNA that are specialized for translating particular mRNAs, decoding multiple UGAs in SelenoP as Sec is the most striking example of mRNA-linked ribosome specialization. Elucidation of the mechanisms involved in this mRNA-linked specialization, including by structural studies, will be a difficult but rewarding challenge.
Methods
SelenoP gene finding, phylogeny, filtering
Gene prediction was carried out with the program Selenoprofiles v3.5c [88], which employs a protein alignment profile to scan nucleotide databases and find genes belonging to the same family. Due to its peculiar encoded C-terminal Sec-rich domain, SelenoP is particularly difficult to predict. This domain contains various stretches of repetitive sequences, resulting in lots of spurious hits in non-homologous repetitive regions of genomes during the very first step of Selenoprofiles (Blast search). We thus forced Selenoprofiles to perform the initial Blast search using only the conserved N-terminal domain. A second challenge derives from the C-terminal domain of SelenoP having no apparent similarity across various lineages, which hinders their discovery by homology. The 3’ portion of SelenoP genes can be found by looking downstream of regions with similarity to the N-terminal domain; in genomes, however, this is hampered by the potential presence of intervening introns. To remedy this, we searched abundant transcriptomic data (listed below), and used results to build a profile alignment containing representative SelenoP sequences across diverse lineages. This permitted the finding in genomes of homologous sequences encoding C-terminal domains. The newly created profile is now included in the Selenoprofiles package. In all our searches, we completed gene structures by extending homologous matches upstream (to the farthest AUG before any stop codon) and downstream (allowing UGA codons, but not any other stop). In total, we searched 1,159 NCBI genomes, 1,375 NCBI Transcriptome Shotgun Assemblies (TSA; all those available from metazoa excluding mammals and holometabolous insects), and 70 transcriptomes [89] from diverse lineages of lophotrochozoa. Our searches resulted in 3,464 SelenoP gene predictions in total across all sources. Upon their inspection, we noticed this set was highly redundant, with nearly identical multiple entries for the same genes, and included poor quality sequences, with incomplete gene structures and predictions from putative contaminations. We thus proceeded to filter and process these predictions through a number of analyses, described below, in order to obtain a bona-fide SelenoP set.
We aligned peptide sequences with ClustalO v.1.2.1 [90] then we processed them to merge those coming from the same genes, with the following procedure. First, for each species we detected clusters of predictions referring to the same transcript, defined as presenting at least 96% identity (excluding terminal gaps) in their CDS. Only one representative was kept for each cluster. Often, such predictions complemented each other, in that they shared a common region, but each individually lacked the N-terminal or C-terminal encoding sections; in these cases, a new consensus sequence was produced to span the full gene sequence. Next, we processed the resulting alignment to keep only one isoform per gene. We detected clusters of predictions from the same species sharing a stretch of at least 120 nearly identical (96%) nucleotides in their CDS; we selected the longest prediction for each group, and we dismissed the rest. Throughout this procedure, we reduced our SelenoP set down to 1,917 predictions. Next, we split the alignment in N- and C-terminal encoding sections, the former ending with sequence encoding the KDDFLIYDRCG motif in human, roughly corresponding to the end of SelenoP conservation across metazoan lineages. We thus built a protein tree using the N-terminal section, employing the routine ‘standard_trimmed_raxml’ in ETE3 [91,92] and we employed it as backbone for a number of analyses, including a ‘phylogenetic filtering’ procedure (explained below). Proteins from vertebrates formed a clear monophyletic cluster in the tree; thus, we split this portion of the tree from the rest for visualization purposes.
We proceeded to filter our raw prediction set using the resulting phylogenetic tree topology. The first phylogenetic filtering step was motivated by the observation of a number of SelenoP predictions clustering with species very distant from their annotated source organism. For example, the transcriptomes of Crassostrea hongkongensis and angulata (bivalves) contained SelenoP sequences nearly identical to Sus scrofa. These genes cluster together with all mammalian SelenoPs in the protein tree and are very distant from other sequences from bivalves. Both transcriptomes were deposited by the same researchers. We concluded that these genes are not actually present in Crassostrea, and that they derived from sequence contamination from pig. The inspection of the protein tree highlighted numerous analogous cases. We thus defined a score for each gene, expressing the consistency between the protein tree and the taxonomy of source organisms. We considered 10 ‘phylogenetic neighbours’ for each protein, defined as those with shortest distance in the reconstructed tree, and their annotated NCBI taxonomy (e.g. for Crassostrea hongkongensis this would consist of the following 15 units: ‘cellular organisms; Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Lophotrochozoa; Mollusca; Bivalvia; Pteriomorphia; Ostreoida; Ostreoidea; Ostreidae; Crassostrea’). For each gene, we thus computed the average proportion of its taxonomy units shared with its phylogenetic neighbours. This score was further normalized by the average value in the neighbours. The resulting taxonomy consistency score approaches 1.0 for nodes whose position in the protein tree is consistent with the species tree, and it is lower for putative contaminant sequences; we thus filtered out 64 genes with a taxonomy consistency score below 0.75. The second step of our phylogenetic filter was motivated by the presence of partial gene sequences (fragments), which we attributed to the imperfect quality of genomes and transcriptomes. These predictions can be readily detected by inspecting the sequence length across the protein tree: fragments are isolated cases with shorter lengths compared to their phylogenetic neighbours. We thus computed a length consistency score per prediction, defined as its CDS length divided by the average length of its 10 phylogenetic neighbours. We thus filtered out 474 genes with a score below 0.75 (i.e., those at least 25% shorter than their most similar sequences). Lastly, we also filtered out 139 predictions with pseudogene features (frameshifts or in-frame stops other than UGA) and 299 predictions without a (non-UGA) stop codon downstream (mostly in short transcriptomic fragments). Our final bona-fide set consisted of 1,228 SelenoP predictions. The N-terminal protein tree, cleared of filtered out predictions, is shown in Supplementary Fig 1.
Homology and modularity in SelenoP sequences
We set up a computational procedure to detect local homology both inter- and intra-sequences. We initially attempted to use Blastp v2.2.26 with permissive parameters (word size 2, max evalue 1000, query filtering turned off), but we realized that its algorithm by design does not allow detection of intra-sequence matches, thus precluding investigation of modularity. We finally employed Exonerate v2.4.0, run in ‘affine:local’ mode with permissive parameters (score threshold 30, word length 2), which instead returns both inter- and intra-sequence matches (Supplementary Fig 3A). Exonerate assigns a score to each match, but it does not provide measures of statistical significance (e.g. e-values). We thus ran both programs on the protein sequences of the representatives of all phylogenetic clusters, in all-against-all fashion. We exploited the presence of many identical alignments in the outputs of Exonerate and Blast to calibrate an ‘Exonerate score to log e-value’ ratio, thus allowing to assign ‘inferred e-values’ to Exonerate hits (Supplementary Fig 3B). We considered all Exonerate matches with ‘inferred e-value < 0.01, and we visualized them with R as dot-matrix-like plots (Fig 3). While we inspected results of all sequences, to reduce the complexity of plots we finally decided to represent only a subset of all sequence representatives per phylogenetic cluster, removing those providing redundant information (e.g. a single representative for arachnid SelenoP was retained).
SECIS prediction and phylogeny
We used the program SECISearch3 [93], to detect potential SECIS elements in SelenoP sequences. We considered regions of 3000 nucleotides downstream of each CDS (or shorter if reaching over the contig end), and we scanned them using all three methods implemented in SECISearch3. During our complex filtering procedure (explained above), some gene predictions were combined to form consensus entries that were longer than their individual components. In those cases, we assigned to each new consensus entry the SECIS set with the highest number of predicted SECIS elements, among the ones in its merged components. To predict the phylogenetic tree of SECIS elements, we produced a structural alignment using the program Cmalign from the Infernal package v1.1.1, using the SECIS model at the core of SECISearch3 as template. Then, to reduce the number of sequences for phylogenetic reconstruction, we trimmed out the most abundant classes of genes: phylogenetic clusters vertebrate SelenoP1 and vertebrate SelenoP2. We retained SECIS elements found in 40 genes selected randomly in each of these clusters and removed the rest from the alignment. We thus ran phylogenetic reconstruction using ETE3 [91] with ‘pmodeltest_soft_ultrafast’ for evolutionary model selection and ‘phyml_default_bootstrap’ for tree reconstruction. The resulting tree is shown in Supplementary Fig 11.
Tetraselmis sp. algal culture
For oyster feed, we cultured Tetraselmis sp. algae using artificial sea water (ASW) made with Instant Ocean Aquarium sea salts adjusted to a salinity of 35 ppt. Algae were diluted in 80 ml of algae media (80% ASW and 20% F2 Guillards media (Sigma G0154), either supplemented or not with 5 mg/L of sodium selenite (28.9 μM) (Sigma- 214485) diluted in artificial sea water. Algae were grown for 3 days with constant oxygenation and a 12h light/dark cycle to allow Se incorporation. On the day of feeding, algal cells were equalized by counting cell numbers and equal amounts were fed to each corresponding oyster tank weekly for 6 weeks.
Aquaculture, selenium supplementation and histology of Magallana gigas
Twenty diploid, two year old Pacific oysters obtained from an oyster cultivation farm in Co. Clare, Ireland were distributed equally into two tanks containing artificial seawater with salinity adjusted to 25 ppt and kept at constant temperature 16 °C. Denitrifying bacteria were added to aid in acclimation and break down of toxic waste. 24 h after feeding (to allow complete filtering of algal feed), oyster tissues were cross-sectioned and prepared for histology for gonad development and sex determination.
Cross-sections of diploid oysters were fixed in 90% fixative (Davidson Solution-Sigma) and 10% acetic acid for 24–48 h, dehydrated through graduated ethanol dilutions and paraffin-wax embedded. 7-μm-thick sections were cut and stained with hematoxylin-Eosin. The slides were analysed using a Nikon Eclipse 80i microscope with a 40X objective, images were captured with a Nikon DXM 1200-C camera, and processed using Andor IQ acquisition software (Andor Technology Ltd. Belfast, Northern Ireland). Sex and gonad development stage were analysed using previously published criteria [94,95]. Only male oysters with developed gonads were subjected to total selenium determination and ribosome profiling library preparation.
Inductively Coupled Mass Spectrometry (ICP-MS): Total selenium
Whole-body male oyster tissues were prepared for ICP-MS analysis by liquid nitrogen grinding. Independently, the algae-feed, tank water, and oyster cytoplasmic extract preparations were also analyzed. DigiPrep (SCP Science, Courtaboeuf, France) was used to heat the sample during acid digestion. The samples were weighed and left overnight in 0.2 to 0.5 mL of HNO3 (depending on quantity of the sample available). One mL of H2O2 was added and the sample was digested in a DigiPrep (the digestion program: 0–30 min at room temperature, 30–240 min at 65 °C). The digests were diluted to reach the HNO3 concentration of 4% and analyzed by ICP MS using the conditions optimized daily (nebulizer gas flow, RF power, lens voltage and collision gas flow) [96]. We carried out external calibration at 6 levels, adding selenium to a blank sample (mixture of nitric acid, hydrogen peroxide and water) to reach Se concentrations of 0.25, 0.5, 1, 2.5, 5 and 10 ppm. The ICP MS instrument used was an Agilent 7700 (Tokyo, Japan) fitted with a collision cell and a Meinhard nebulizer (Glass Expansion, Romainmotier, Switzerland).
Ribosome Profiling and RNA sequencing library preparation
For ribosome profiling, soft tissues from two individual whole oysters, with similar determined levels of selenium from each supplementation group, were flash frozen in liquid nitrogen and subsequently pulverized by grinding using a ceramic pestle and mortar, pooled and lysed in 2 mL polysome lysis buffer (10 mM Tris HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1% sodium deoxycholate, 1% Triton X100, 100μg/ml cycloheximide, 2 mM DTT and 2μl Superase·IN RNase inhibitor). Lysates were centrifuged at 16,000 × g for 10 minutes at 4°C. One ml of supernatant was treated with 100U of RNAse I at 25°C for 45 minutes. RNAse I was deactivated with Superase·In at 65°C for 5 minutes. Monosome fractions were isolated by loading RNAse I-treated lysates on a 10%−60% continuous sucrose gradient and centrifuged at 35,000 RPM for 3 hrs at 4°C in a SW41Ti rotor in a Beckman ultracentrifuge. Gradients were chased with caesium chloride and fractions containing the monosomes were identified by OD reading at a wavelength of 256 nm. Ribosome-protected fragments (RPFs) were isolated from tightly defined monosome fractions by TRIzol extraction.
For RNA sequencing, polyA mRNA were isolated from the same samples using the Poly(A) Purist Mag kit (Ambion) and randomly fragmented using alkaline fragmentation buffer (2mM EDTA, 10 mM Na2CO3, 90 mM NaHCO3). Both RPFs and randomly fragmented PolyA were prepared following Ingolia’s ribosome profiling library preparation protocol [97], omitting the experimental ribosomal RNA (rRNA) depletion step. The prepared cDNA libraries were sequenced by BGI Genomics Co., Ltd. Shenzhen, China (www.bgi.com). The samples were pooled in one SE50 multiplex lane where 3GB of data were allocated for each ribosome profiling sample and 0.6 GB were allocated for each RNA-sequencing sample.
Bioinformatic analysis of RNA-seq and riboseq
Using Ribogalaxy [98], raw sequencing reads were pre-processed before alignment to the transcriptome. Oyster ribosomal RNA sequences 5.8S (ENSRNA022717831), 5S (ENSRNA02271792), 28S (AB102757.1) and 18S (ENSRNA022718259) were identified in silico using Bowtie version 1.1.1 [99] and removed. The remaining reads were mapped with Bowtie to the oyster transcriptome [61] using the same parameters as trips-viz [100]. Metagene analysis was performed to assess triplet periodicity and P-site offset as a quality control (Supplementary Note 8). For metagene analysis, start and stop codons in each transcript were obtained from the transcriptome assembly ORF annotation [61]. Previously, we had identified transcripts corresponding to selenoproteins using Selenoprofiles [88] and manually selected a single transcript as a representative for each selenoprotein gene (Supplementary Table 1). Read counts per selenoprotein gene were normalized by transcript length (kbp) per million mapped reads (RPKMs and RPFKMs, for RNA-seq and riboseq respectively) using the programs cuffquant and cuffnorm from the package cufflinks version 2.2.1 [101]. Genes with RPKM or RPFKM below 1.0 in either selenium supplementation group (DIO.2, SELENOH.1, PSTK.2, SBP2.117) were discarded to avoid over interpretation in the differential expression analysis due to low coverage. Translation efficiency (TE) was computed as the ratio of RPFKM/RPKM for each gene. To calculate Sec UGA-redefinition efficiency (URE) [67,69], we defined the 5’ and 3’ regions for each selenoprotein CDS based on the position of the Sec-UGA codon (UGA1 in case of SelenoP). URE was expressed as the ratio of RPF density in the region 3’ of UGA to the region 5’ of UGA (3’RPFKM/5’RPFKM). Reads within 15 nt from AUG and 5 nt from stop codon were omitted to remove bias introduced by cycloheximide treatment. URE was not computed for selenoprotein genes in which the UGA codon was located near the 3’ end of the CDS (SELENOK.1, SELENOO.2, SELENOS.1, TR.1, TR.2) and near the AUG start (DSBA.1, MSRA.2, SELENOW.1, SELENOW.2). Aligned RNA-seq and riboseq reads were visualized with trips-viz for each individual selenoprotein mRNA, and plots were generated to compare supplemented and non-supplemented samples. Mapping of ambiguous reads were allowed for genes with more than one transcript isoform in the transcriptome. Minimum and maximum fragment length were set at 25 to 35 nt respectively for trips-viz visualization, corresponding to the expected size of the RPFs during translation.
In-vivo 75Se labelling of oyster larvae
Free-swimming larvae at 7-day and 14-day old stages were gifts from an oyster cultivation facility (Pacific Sea Foods, Quilcene WA, USA). Additional larvae were obtained from the Rutgers Haskin Shellfish laboratory for method development. After 24h acclimation in 10 L of artificial sea water and initial feeding with Instant Shellfish Diet1800, 100–200 oyster larvae (100 μm in size) were transferred into a 12 well plate with artificial sea water spiked with 1 μl of 100 μM 75Se diluted in 1% DMSO for metabolic selenium incorporation. After 24 h, oyster larvae were washed, harvested and mechanically lysed in RIP-A lysis buffer (50 mM Tris-HCl pH 8.0, Triton X-100, 150 mM NaCl, 0.5% sodium deoxycholate and protease inhibitors). Protein extracts were electrophoresed in 10% SDS-PAGE, fixed, dried and exposed to a Phosphorimager screen. Parallel Western blot probing was performed using SelenoP custom antibodies.
Immunoblot and Densitometry
Custom antibodies, anti-NT SelenoP against oyster SelenoP N-terminal region before Sec-UGA 1 (immunogenic peptide TADGTDPVKARVN) and anti-CT SelenoP against the C-terminal region after Sec-UGA 43 (immunogenic peptide YCRTGTYDDRAH) were obtained from Genscript. These antibodies were tested for affinity and specificity (Supplementary Fig 10). Immunoblot analysis using SelenoP custom antibodies were performed on oyster lysates used in ribosome profiling analysis. Two of the same samples (replicate=2) were equalized to 50 μg of protein per lane, resolved on 12% SDS PAGE and transferred to nitrocellulose membrane (Protran). The membrane was blocked with 5% milk in phosphate buffer saline-Tween20 (0.5% PBS-T) overnight in 4°C and probed with 1:2500 dilutions of rabbit primary antibodies (anti-NT-SelenoP and anti-CT-SelenoP) in 5% milkPBS-T for 1 hr at RT. The membrane was washed 3 times with PBS-T. Immunoreactive bands were detected with appropriate fluorescently labelled secondary antibodies and scanned using LI-COR Odyssey® Infrared Imaging Scanner (LI-COR Biosciences). Full-length band intensity was quantified across samples using Image Lite Studio. Values were normalized against Ponceau S loading control and plotted as an average of two technical replicates.
SelenoP plasmid construction and oyster mutant generation
Native cDNA sequences were used to obtain gene-blocks (DNA fragments) from Integrated DNA Technologies (IDT). An exception was made for the sequence of spider P. tepidariorum, obtained instead through gene synthesis (GenScript), due to its sequence complexity hindering gene-blocks generation. All SelenoP gene-block constructs were cloned into the pcDNA3.1 TOPO-TA vector cleaved by BamHI and AgeI-HF.
The plasmid with zebrafish SelenoP cDNA [56] was used as positive control. Mutant constructs were generated by either PCR or site directed mutagenesis. The Kozak context was added by Quick Change II kit (Agilent Technologies) site-directed mutagenesis following manufacturer’s protocol and with primer sequences indicated in Supplementary Table 5. Mutants of the oyster SelenoP mRNA (Supplementary Fig 5) were generated as follows. Del. 5’UTR mutant (Results) was generated by PCR amplification of native oyster sequence with primers indicated and cloned into double-digested pCDNA3.1 TOPO-TA vector. Syn. ISL mutant were introduced through PCR in 3 steps. Removal of sequence encoding the C-terminal Sec-rich domain (Del. C-Term mutant) was carried out by a two-step cloning procedure where the CDS was re-ligated with its 3’UTR after UGA 11–46 removal by PCR. Addition of oyster SECIS elements to zebrafish coding region was via Pac1 and Not1 double digest removal of zebrafish SECIS 1 and re-ligation of oyster SECIS amplified by PCR including restriction sites. Oyster SECIS element sequences were subcloned into the pcDNA3.1 TOPO-TA vector containing zebrafish CDS insert (mutants: Fus. 3’UTR, Fus. SECIS 1 and Fus SECIS 2). All restriction enzymes used for each insert are indicated in Table S5. Inserts digested with BglII/Age1-HF were inserted into a BamH1/Age1HF cut pCDNA 3.1 TOPO-TA vector while the same restriction enzymes were used to cut the vector for all the other mutants. Subsequent ligation, transformation and positive colony selection were by standard procedures. Plasmid DNA was purified using GeneJET Mini-Prep Plasmid Isolation Kit (Thermo Fisher). DNA sequencing was by Eurofins, Germany.
In-vitro transcription, reticulocyte lysate (RRL) translation and 75Se labelling
All cloned plasmid constructs were linearized by a single restriction enzyme digestion, in vitro transcribed by T7 polymerase and capped using mMessege mMachine kit (Ambion). A 110 nt vector sequence is present between the T7 promoter and the respective construct sequence. RRL (Promega) reactions were reconstituted with rat CT-SECISBP2 or oyster SECISBP2 and radiolabelled with 75Se as described [50]. RRL reactions were denatured in sample buffer at 95°C for 5 min, electrophoresed on 10% or 15% SDS-PAGE. The gel was fixed, dried and exposed for autoradiography.
Oyster SECISBP2 recombinant protein purification
Full-length oyster SECISBP2 (FLSBP2) CDS trimmed with Xba1 and Sac1HF digestion was cloned into bacterial vector (Pj307) [102] cleaved with Spe1 and Sac1HF. The ampicillin resistant empty vector encodes an N-terminal Glutathione Sepharose Transferase (GST) tag followed by sequence specifying an HRV-3C protease cleavage site and a 6XHis-tag. A clone containing the FLSBP2 insert was transformed into BL21 competent E. coli. A selected colony was grown in 5ml of LB-AMP media. Cells were then transferred to 3L of LB-AMP media, grown at 37°C for 2h and on reaching OD600 of 0.6 induced with 0.05 mM IPTG and grown overnight at 15°C. Cells were centrifuged (5000×g,10 min), washed with PBS and lysed in a lysozyme-containing lysis buffer. Lysates were sonicated, centrifuged and incubated overnight with glutathione sepharose beads (GE Healthcare Sigma 17–0756-01) at 4°C. The GST beads were then washed 3x (50 mM Tris-HCl pH8, 150 mM NaCl, 0.1% NP-40), and finally with 10 bead volumes of protease cleavage buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 0.1% NP-40, 1 mM DTT and 1 mM EDTA). GST beads were then resuspended in 1 bead volume of cleavage buffer and 40 μl PreScission Protease (GE Healthcare, 27–0843-01) to cleave off the GST-tag. Concentration ensued in Amicon ultra-centrifugal filter units with 30 kDa molecular weight cut off (MWCO) concentrators (Sigma). Following gel filtration using a Superdex200 column, the buffer was substituted with PBS. An aliquot was subjected to SDS-PAGE and analysed with Coomassie staining and Western blot.
RT-PCR and sequencing
Elliptio complanata mussels were a gift from Department of Natural Resources, C-2 Annapolis Maryland, USA. Total RNA was isolated from their tissue using Trizol (Invitrogen™). Reverse transcription (RT) was performed with oligo dT primer using SuperScript® III First-Strand Synthesis System (Invitrogen). PCR on the resulting cDNA was with Phusion polymerase (NEB) using the forward primer 5’ TAAACTGGCTGAGCCGCGGTGGCTC3’ and reverse primers 5’ ATATATAATATGCTAAGAGTGATCA 3’ (T1) and 5’ AAGTGACAAAACGCACAAGCAATGTTAAAATGCAC 3’ (T2). Amplified PCR product was gel purified by Zymoresearch DNA isolation kit as per manual and sent for Sanger sequencing using the following forward sequencing primers: 5’ TTGCATTCCCTGCTTTTGACATACTGTCG 3’ (S1), 5’ ACGTTGATTTTTGATGACGTCACTCAGAT 3’ (S2) and 5’ TCCATTTGATGAAAGAAAGCTAAAGAA3’ (S3).
Supplementary Material
Supplementary Fig 1: Reconstructed tree of SelenoP genes.
This tree is based on an alignment of the proximal region of SelenoP genes predicted across metazoa. All vertebrate sequences cluster together in this phylogenetic reconstruction; for visualization purposes, their tree is shown in Panel A; the remaining genes are shown in Panel B. Next to each node, a graphical representation of aligned protein sequence is shown (see also Figs 1,2), coloured according to encoded protein clusters defined based on this tree topology. Sec-UGAs are represented as vertical black lines. For the subset of predictions that come from genome sequences, red lines indicate intron positions. In the outmost section, SECIS presence is shown (the first SECIS per gene in dark blue, the second in brown; if unique, it is colored in grey).
Supplementary Table 3 Selenium levels in relevant material from the supplementation trial.
Total Se levels were determined for each sample from the appropriate supplementation group including the algal food source grown and collected before feeding day (Methods), artificial sea water collected after the supplementation trial, tissue cytoplasmic extracts isolated after tissue lysis and the remaining debris separated by centrifugation. Standard deviation values (SD) were computed per sample group.
Supplementary Table 4 UGA redefinition efficiency calculations for each selenoprotein.
Selenoprotein genes UGA redefinition efficiency (URE) were computed as the ratio of the RPF density 5’ of UGA to the RPF density 3’ of UGA (excluding the first 15 nt of CDS). For SelenoP, the URE for UGA1 was calculated, and considering only the sequence between UGA1 and UGA2 to estimate RPF density 3’ of UGA.
Supplementary Fig 2: Alignment of SelenoP sequence representatives.
The figure shows an alignment of sequence representatives chosen for each protein cluster. Sec residues (U) are highlighted.
Supplementary Table 5: Table of primers used to PCR amplify mutant oyster constructs for subsequent cloning
Supplementary Fig 3: Local homology in SelenoP sequences.
(A) Results of local homology searches using Blast (left) and Exonerate (right) using spider P. tepidarium as query and all sequence representatives as target. Note that Blast fails to report matches of the query with itself, so that we used Exonerate for this task. (B) We identified a number of identical alignments in the output of Blast and Exonerate for all queries and targets and verified that the scores of the two programs are highly correlated (left). We thus calibrated a log(e-value) to Exonerate score ratio (right), which allowed us to assign inferred e-values to all Exonerate hits, used to filter them (see Fig 3: inferred e-value < 0.01). (C) Local homology identified between the C-terminal domain of human SelenoP and that of two invertebrates (see Fig 3, top-left panel).
Supplementary Fig 4: Conserved RNA structures identified in SelenoP of Pacific oyster M. gigas.
(A) Top: predicted RNA structure of (left to right) Initiation Stem Loop (ISL) at positions 40–160 from AUG; Selenocysteine Recoding Elements (SRE2/3) spanning positions 1585–1705; and SECIS 1 and 2, 194–267 and 585–657 nts 3’ of the UAG stop codon. The conserved SECIS motifs, the core and the A repeats at the apical loop, are in green. Bottom: gene structure indicating the coding sequence, UGA positions (vertical lines), and sequences that form secondary structure elements (shaded in grey). (B) ISL alignment across 6 bivalves. (B & C) Shaded positions correspond to RNA pairs in the structure above. (C) Alignment of SRE2/3 across 6 bivalves. Matching colors indicate the two strands of a stem. The species used to predict the conserved structures show in the alignment are: M. gigas, C. virginica, Mytilus galloprovincialis, Bathymodiolus platifrons, Pinctada martensii and Mizuhopecten yessoensis.
Supplementary Fig 5: Oyster SelenoP mRNA context-specific mutants tested by RRL translation.
The top panel shows representations of each in vitro transcribed mRNA (Methods). The figure depicts: brown box for oyster CDS and green box for zebrafish CDS. Black lines are Sec-UGA and the ISL structure is in black before UGA 1. SECIS 1 is in blue and SECIS 2 is in red. Red ‘X’ denotes deletion and ‘SM’ denotes synonymous codon mutations to disrupt conserved pairings. Yellow asterisk refers to oyster early termination product while the green arrow indicates full-length zebrafish SelenoP. Lane 1: negative control no mRNA, lane 2: zebrafish WT control, (lanes-3–10): oyster mutant constructs, including native transcript (lane 3), Kozak consensus added (lane 4), native 5’UTR removed (lane 5), ISL complementarity disrupted (lane 6), zebrafish CDS fused to the entire oyster 3’UTR (lane 7), or to only oyster SECIS 1 (lane 8) or oyster SECIS 2 (lane 9). Note that particular differences in product intensity and resolution are observed in the positive controls (WT zebrafish SelenoP) in individual autoradiography gels which could be attributed to RRL batch variability. Also, the low molecular weight product at ~25 kDa was observed across all experiments even in the absence of added mRNA (panel A, lane 1) and thus attributed to RRL background.
To test whether the absence of radiolabeled product in the ISL and C-terminal deletion mutants were due to lack of initiation or Sec-decoding of UGA1, we performed 35[S]Met labelling in the presence or absence of the concentration of G418 antibiotic that promotes 20% readthrough at UGA codons [103,104]. Translation reactions were performed in the absence of rat CT-SBP2, with added ‘cold’ selenium and either supplemented or not supplemented by G418. The RRL products were resolved in a 15% SDS-PAGE gel to detect the presence of the early termination product (5–6 kDa) before recoding of UGA1. We observed that all the constructs produced a labelled readthrough product corresponding to termination at UGA2 except for the C-terminal deletion mutant in the presence of G418. Moreover, in the absence of G418, the early termination 5 kDa product of oyster SelenoP was reduced with 5’UTR deletion and more significantly with ISL mutation and C-terminal deletion. Note that we also detect a >37 kDa 35SMet labelled product in the native and Kozak context constructs similar to the 75Se experiment (yellow asterisks).
Supplementary Fig 6: Testing affinity to rat SECISBP2 of oyster SECIS-fusion constructs.
MgCl2 can change conformation and affinity of RNA structures with K-turn motifs [105,106]. SECIS affinity was tested by the addition of increasing concentrations of MgCl2 (mM) to RRL translation reactions of (A) wild-type zebrafish (B) zebrafish fusion with oyster 3’UTR (C) zebrafish fusion with oyster SECIS 1 and (D) zebrafish fusion with oyster SECIS 2. Full-length zebrafish SelenoP product runs at 42 kDa.
Supplementary Fig 7: Supplementing RRL translation reactions with oyster SECISBP2.
(A) The positions of the functional domains, SID (SECIS Interaction Domain) and RBD (Ribosome Binding Domain) [107,108], are shown within oyster SECISBP2. (B) Alignment and extent of conservation of oyster SECISBP2 with both human SECISBP2 and SECISBP2L are shown (C) E. coli expressed His-tagged oyster SECISBP2 analysis by SDS PAGE with Coomassie staining and Western blot probing (anti-His tag). The green arrow points to purified full-length oyster SECISBP2 following cleavage of a GST-tag used for purification. (D) Autoradiogram of 75Se labelled rat SelenoP translated in RRL supplemented with varying concentrations of oyster SECISBP2. Black arrow points to a 27 kDa band suggestive of a single or double Sec-incorporation.
Supplementary Fig 8: Radical S-adenosyl Methionine (RSAM) selenoprotein in M. gigas and other species.
Multiple amino acid sequence alignment of Radical SAM (RSAM) proteins, showing only the C-terminus including the Sec or Cys position, 497. The residue at this position is highlighted in red for Sec and in orange for Cys. The sequences were obtained from NCBI GenBank Transcriptome Shotgun Assemblies (TSA). The phylogenetic tree on the left was obtained from NCBI Taxonomy.
Supplementary Fig 10 SelenoP Antibody characterization.
(A) Protein map of SelenoP including Sec-UGAs (vertical white lines) and antibody recognition sites (grey). N-terminal Ab (Anti-NT-SelenoP) is located before Sec - UGA1 and C-terminal Ab (Anti-CT-SelenoP) is located after Sec-UGA 43. (B) Antibody immunogenic peptide insertion: NT- N-terminal peptide (yellow) and CT C-terminal peptide (green) into a GST-MBP-6XHis (GST-MBP-NT-6XHis or GST-MBP-CT-6XHis) tagged vector for bacterial production. (C) Immunoblot of bacterial protein produced using antibody against the GST tag (Anti-GST), N-terminal SelenoP (Anti-NT-SelenoP) and C-terminal SelenoP (Anti-CT-SelenoP). Anti-CT-SelenoP also detected the N-terminal peptide suggesting non-specific recognition.
Supplementary Fig 9: Effects of selenium supplementation on the expression of selenoprotein and selenoprotein machinery genes.
Expression levels of Sec machinery (top) and selenoprotein genes (bottom) assayed through riboseq (first column, expressed in RPFKM+1) and RNA-seq (second column, RPKM+1). The third column shows translation efficiency, calculated as RPFKM/RPKM ratio. All values were computed separately for Se-supplemented (orange) and non-supplemented samples (green). The bottom panel shows how Sec machinery and selenoprotein genes compare with the distribution of the whole oyster transcriptome.
Supplementary Fig 11: Tree of SelenoP SECIS elements.
The plots show the reconstructed phylogenetic tree based on a structural alignment of SelenoP SECIS elements (Methods). Outside of the circular tree in the middle, there are three layers. The first layer contains bars representing the distance of SECIS predictions from the CDS. This is color coded to distinguish multiple SECIS elements identified in the same gene: blue for SECIS 1, brown for SECIS 2, grey if a single SECIS was found. The second layer contains schemes of SelenoP genes for which each SECIS was identified, following the same representation as Figs 1–2. The third layer contains species names, color coded by lineage. Two major clusters of SECIS elements are highlighted in light and dark grey, roughly corresponding to the SECIS 1 (top) and SECIS 2 (bottom-left).
Supplementary Fig 12: Correlation between number of selenoproteins per species and Sec residues in SelenoP.
The plot shows the relationship between the number of selenoproteins found in transcriptomes or genomes and the number of putative Sec-UGAs in SelenoP genes. The plot contains one point per species, colored by phylogenetic cluster as in Fig 1. Our analysis shows significant correlation (Pearson’s r=0.161; p-value<1e-4), even when subsampling vertebrates (considering only 40 vertebrates and all non-vertebrate species: Pearson’s r=0.298; p-value<1e-4). Selenoproteins were predicted in nucleotide assemblies by Selenoprofiles (all predictions with label “selenocysteine”), and highly similar genes predicted in the same assembly were removed with the program Cdhit v4.6 [109]. When multiple assemblies were available for a given species, the one richest in selenoproteins was used, and when multiple homologous SelenoP genes were identified, the one with most Sec-UGAs was used. Only those species with predicted selenoproteins and Sec-containing SelenoP were plotted and considered for this analysis.
Supplementary Fig 13: Anti- SelenoP immunoblot of oyster tissues from with varying total Se concentrations determined.
Oyster tissues with determined total Se levels were probed with antibodies against N and C terminal portions of SelenoP. Two total protein dilutions were loaded on the gel (25 and 50μg) of each sample including one representative sample from the non-supplemented group (3 μg/g total Se) and two representative samples from the Se-supplemented group (13, 17 and 80 μg/g) with variable elevated total Se levels determined.
Supplementary Fig 14: Exon/intron junction conservation in metazoan SelenoP.
The plot displays the gene structures of SelenoP gene predictions obtained from genome assemblies, indicating the position of exon/intron junctions. Note that only one representative species, Falcus pellegrinus, is shown for vertebrate SelenoP1 and SelenoP2, since intron positions are conserved in this gene across all the vertebrate clade. Genes that have lost the intron 3’ of UGA1 are highlighted in various colors. Light blue indicates two cases in which intron loss was accompanied by UGA1 replacement by a standard codon, and SelenoP ceased to be a selenoprotein. Green indicates the case of sea snail Colubraria reticulata (gastropod) where intron loss was also concomitant to UGA1 replacement by a Cys codon, but SelenoP remained a selenoprotein due to its distal UGAs and SECIS element. Yellow indicates the case of sea anemone Exaiptasia pallida, where UGA1 was retained despite the absence of introns. Lastly, grey indicates the case of three Acariformes (arachnida) which possess UGA1 but lost its neighbouring intron. These do not present any SECIS element and may be translated through a different form of readthrough (see Results).
Supplementary Note 1: Sec to Cys conversions in vertebrate SelenoP2
Supplementary Note 2: Validation of 132 UGAs in SelenoP of Elliptio complanata
Supplementary Note 3: Sec to Cys conversion of SelenoP in gastropods
Supplementary Note 4: Divergence of SelenoP in arachnida
Supplementary Note 5: SECISBP2 orthology between vertebrates and oysters
Supplementary Note 6: SelenoP duplication in oysters
Supplementary Note 7: Oysters for selenium supplementation experiments
Supplementary Note 8: Assessment of ribosome profiling data
Supplementary Table 1 Selenoproteome of M. gigas
List of selenoprotein and machinery genes in M. gigas, indicating the corresponding transcript in the assembled transcriptome and the contig in the genome assembly where each gene was found. The last column indicates the homology with human genes; in case of complex orthology relationships, only the family is indicated.
Supplementary Table 2 Marine mussels, Mytilus edulis, total selenium measurement
Total selenium levels were determined for eight individual farmed marine mussels (Mytilus edulis) collected from Collorus, Co. Kerry, Ireland. Standard deviation values (SD) and Relative Standard Deviation (RSD) values are indicated.
Highlights.
SelenoP has 10 selenocysteine-UGAs in human, but what about other metazoa?
Remarkable diversity of SelenoP: from 1 Sec-UGA in insects to 132 in bivalves
Translation and in vivo Sec-incorporation of a 46-UGA SelenoP in Pacific oyster
Regulatory RNA elements in oyster SelenoP with roles in initiation and Sec-decoding
New insights into SelenoP evolution and processive translation of multiple Sec-UGAs
Acknowledgements
We thank Sumangala Shetty and Mark Pinkerton for advice, Iarfhlaith Connellan from Cartron Point Shellfish Ltd, Co. Clare for providing the diploid oysters and microalgae, Lizzie Nelson of Pacific Sea Foods, Quilcene, Washington for a gift of oyster larvae, Matt Ashton of Department of Natural Resources, Maryland for a gift Elliptio complanata fresh water mussels and Declan O’Sullivan of Collorus, Co. Kerry, Ireland for provision of marine farmed Mytilus edulis mussels, Ximing Guo for oyster larvae from the Rutgers Haskin Shellfish Laboratory. M.M. was supported by EMBO short-term fellowship ASTF 289 – 2014. The work was supported by the following grants: NIHDK117149, GM065204 and AG021518 (V.N.G.), BIO2014–57291-R from the Spanish Ministry of Economy and Competitiveness (R.G.), NIH GM077073 (P.R.C.), Science Foundation Ireland (13/IA/1853) and Irish Research Council (IRCLA/2019/74 n) (J.F.A).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Labunskyy VM, Hatfield DL, Gladyshev VN, Selenoproteins: molecular pathways and physiological roles., Physiol. Rev 94 (2014) 739–77. doi: 10.1152/physrev.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Maroney MJ, Hondal RJ, Selenium versus sulfur: Reversibility of chemical reactions and resistance to permanent oxidation in proteins and nucleic acids, Free Radic. Biol. Med 127 (2018) 228–237. doi: 10.1016/j.freeradbiomed.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Freitas F. Porto, Seibt T, Mehr L, Aichler M, Walch A, Lamp D, Jastroch M, Miyamoto S, Wurst W, Ursini F, Arnér ESJ, Fradejas-Villar N, Schweizer U, Zischka H, Friedmann Angeli JP, Conrad M, Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis, Cell. 172 (2018) 409–422.e21. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- [4].Burk RF, Hill KE, Regulation of Selenium Metabolism and Transport, Annu. Rev. Nutr 35 (2015) 109–134. doi: 10.1146/annurev-nutr-071714-034250. [DOI] [PubMed] [Google Scholar]
- [5].Schweizer U, Fradejas-Villar N, Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism, FASEB J. 30 (2016) 3669–3681. doi: 10.1096/fj.201600424. [DOI] [PubMed] [Google Scholar]
- [6].Xia YM, Hill KE, Burk RF, Biochemical studies of a selenium-deficient population in China: measurement of selenium, glutathione peroxidase and other oxidant defense indices in blood., J. Nutr 119 (1989) 1318–26. doi: 10.1093/jn/119.9.1318. [DOI] [PubMed] [Google Scholar]
- [7].Jackson ML, Selenium: geochemical distribution and associations with human heart and cancer death rates and longevity in China and the United States., Biol. Trace Elem. Res 15 (n.d.) 13–21. http://www.ncbi.nlm.nih.gov/pubmed/2484511 (accessed July 25, 2019). [DOI] [PubMed] [Google Scholar]
- [8].Long JA, Large RR, Lee MSY, Benton MJ, Danyushevsky LV, Chiappe LM, Halpin JA, Cantrill D, Lottermoser B, Severe selenium depletion in the Phanerozoic oceans as a factor in three global mass extinction events, Gondwana Res. 36 (2016) 209–218. doi: 10.1016/j.gr.2015.10.001. [DOI] [Google Scholar]
- [9].Böck A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F, Selenocysteine: the 21st amino acid., Mol. Microbiol 5 (1991) 515–20. http://www.ncbi.nlm.nih.gov/pubmed/1828528 (accessed August 19, 2013). [DOI] [PubMed] [Google Scholar]
- [10].Atkins JF, Gesteland RF, The twenty-first amino acid., Nature. 407 (2000) 463, 465. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- [11].Mukai T, Englert M, Tripp HJ, Miller C, Ivanova NN, Rubin EM, Kyrpides NC, Söll D, Facile Recoding of Selenocysteine in Nature, Angew. Chemie Int. Ed 55 (2016) 5337–5341. doi: 10.1002/anie.201511657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berry MJ, Banu L, Chen Y, Mandel SJ, Kieffer JD, Harney JW, Larsen PR, Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3’ untranslated region, Nature. 353 (1991) 273–276. http://www.nature.com/nature/journal/v353/n6341/abs/353273a0.html. [DOI] [PubMed] [Google Scholar]
- [13].Berry MJ, Banu L, Harney JW, Larsen PR, Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons., EMBO J. 12 (1993) 3315–22. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=413599&tool=pmcentrez&rendertype=abstract (accessed July 13, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Walczak R, Carbon P, Krol A, An essential non-Watson-Crick base pair motif in 3’UTR to mediate selenoprotein translation., RNA. 4 (1998) 74–84. http://www.ncbi.nlm.nih.gov/pubmed/9436910 (accessed July 25, 2019). [PMC free article] [PubMed] [Google Scholar]
- [15].Grundner-Culemann E, Martin GW, Harney JW, Berry MJ, Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes., RNA. 5 (1999) 625–35. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1369790&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM, A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs., EMBO J. 19 (2000) 306–14. doi: 10.1093/emboj/19.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chavatte L, Brown BA, Driscoll DM, Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes, Nat. Struct. \& Mol. Biol 12 (2005) 408–416. http://www.nature.com/nsmb/journal/v12/n5/abs/nsmb922.html. [DOI] [PubMed] [Google Scholar]
- [18].Bubenik JL, Miniard AC, Driscoll DM, Characterization of the UGA-recoding and SECIS-binding activities of SECIS-binding protein 2., RNA Biol. 11 (2014) 1402–13. doi: 10.1080/15476286.2014.996472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fagegaltier D, Hubert N, Yamada K, Mizutani T, Carbon P, Krol A, Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation., EMBO J. 19 (2000) 4796–805. doi: 10.1093/emboj/19.17.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tujebajeva RM, Copeland PR, Xu XM, Carlson BA, Harney JW, Driscoll DM, Hatfield DL, Berry MJ, Decoding apparatus for eukaryotic selenocysteine insertion., EMBO Rep. 1 (2000) 158–63. doi: 10.1038/sj.embor.embor604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zavacki AM, Mansell JB, Chung M, Klimovitsky B, Harney JW, Berry MJ, Coupled tRNA(Sec)-dependent assembly of the selenocysteine decoding apparatus., Mol. Cell 11 (2003) 773–81. http://www.ncbi.nlm.nih.gov/pubmed/12667458 (accessed July 4, 2014). [DOI] [PubMed] [Google Scholar]
- [22].Donovan J, Caban K, Ranaweera R, Gonzalez-Flores JN, Copeland PR, A novel protein domain induces high affinity selenocysteine insertion sequence binding and elongation factor recruitment, J. Biol. Chem 283 (2008) 35129 http://www.jbc.org/cgi/content/abstract/283/50/35129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carlson BA, Yoo M-H, Tsuji PA, Gladyshev VN, Hatfield DL, Mouse models targeting selenocysteine tRNA expression for elucidating the role of selenoproteins in health and development., Molecules. 14 (2009) 3509–27. doi: 10.3390/molecules14093509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Budiman ME, Bubenik JL, Miniard AC, Middleton LM, Gerber CA, Cash A, Driscoll DM, Eukaryotic Initiation Factor 4a3 Is a Selenium-Regulated RNA-Binding Protein that Selectively Inhibits Selenocysteine Incorporation, Mol. Cell 35 (2009) 479–489. doi: 10.1016/j.molcel.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miniard AC, Middleton LM, Budiman ME, Gerber CA, Driscoll DM, Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression., Nucleic Acids Res. (2010). doi: 10.1093/nar/gkq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Howard MT, Aggarwal G, Anderson CB, Khatri S, Flanigan KM, Atkins JF, Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons., EMBO J. 24 (2005) 1596–607. doi: 10.1038/sj.emboj.7600642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Howard MT, Moyle MW, Aggarwal G, Carlson BA, Anderson CB, A recoding element that stimulates decoding of UGA codons by Sec tRNA[Ser]Sec., RNA. 13 (2007) 912–20. doi: 10.1261/rna.473907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maiti B, Arbogast S, Allamand V, Moyle MW, Anderson CB, Richard P, Guicheney P, Ferreiro A, Flanigan KM, Howard MT, A mutation in the SEPN1 selenocysteine redefinition element (SRE) reduces selenocysteine incorporation and leads to SEPN1-related myopathy., Hum. Mutat 30 (2009) 411–6. doi: 10.1002/humu.20879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bubenik JL, Miniard AC, Driscoll DM, Alternative transcripts and 3’UTR elements govern the incorporation of selenocysteine into selenoprotein S., PLoS One. 8 (2013) e62102. doi: 10.1371/journal.pone.0062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gladyshev VN, Arnér ES, Berry MJ, Brigelius-Flohé R, Bruford EA, Burk RF, Carlson BA, Castellano S, Chavatte L, Conrad M, Copeland PR, Diamond AM, Driscoll DM, Ferreiro A, Flohé L, Green FR, Guigó R, Handy DE, Hatfield DL, Hesketh J, Hoffmann PR, Holmgren A, Hondal RJ, Howard MT, Huang K, Kim H-Y, Kim IY, Köhrle J, Krol A, Kryukov GV, Lee BJ, Lee BC, Lei XG, Liu Q, Lescure A, Lobanov AV, Loscalzo J, Maiorino M, Mariotti M, Prabhu KS, Rayman MP, Rozovsky S, Salinas G, Schmidt EE, Schomburg L, Schweizer U, Simonović M, Sunde RA, Tsuji PA, Tweedie S, Ursini F, Whanger PD, Zhang Y, Selenoprotein Gene Nomenclature., J. Biol. Chem (2016). doi: 10.1074/jbc.M116.756155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kryukov G, Gladyshev V, Selenium metabolism in zebrafish: multiplicity of selenoprotein genes and expression of a protein containing 17 selenocysteine residues, Genes to Cells. 5 (2000) 1049 http://www.genestocellsonline.org/cgi/content/abstract/5/12/1049. [DOI] [PubMed] [Google Scholar]
- [32].Kurokawa S, Eriksson S, Rose KL, Wu S, Motley AK, Hill S, Winfrey VP, McDonald WH, Capecchi MR, Atkins JF, Arnér ESJ, Hill KE, Burk RF, Sepp1(UF) forms are N-terminal selenoprotein P truncations that have peroxidase activity when coupled with thioredoxin reductase-1., Free Radic. Biol. Med 69 (2014) 67–76. doi: 10.1016/j.freeradbiomed.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lobanov AV, Hatfield DL, Gladyshev VN, Reduced reliance on the trace element selenium during evolution of mammals., Genome Biol. 9 (2008) R62. doi: 10.1186/gb-2008-93-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hill KE, Lloyd RS, Yang JG, Read R, Burk RF, The cDNA for rat selenoprotein P contains 10 TGA codons in the open reading frame., J. Biol. Chem 266 (1991) 10050–3. http://www.ncbi.nlm.nih.gov/pubmed/2037562 (accessed July 25, 2019). [PubMed] [Google Scholar]
- [35].Himeno S, Chittum HS, Burk RF, Isoforms of Selenoprotein P in Rat Plasma, J. Biol. Chem 271 (1996) 15769–15775. doi: 10.1074/jbc.271.26.15769. [DOI] [PubMed] [Google Scholar]
- [36].Ma S, Hill KE, Caprioli RM, Burk RF, Mass spectrometric characterization of full-length rat selenoprotein P and three isoforms shortened at the C terminus. Evidence that three UGA codons in the mRNA open reading frame have alternative functions of specifying selenocysteine insertion or translation termination., J. Biol. Chem 277 (2002) 12749–54. doi: 10.1074/jbc.M111462200. [DOI] [PubMed] [Google Scholar]
- [37].Wu S, Mariotti M, Santesmasses D, Hill KE, Baclaocos J, Aparicio-Prat E, Li S, Mackrill J, Wu Y, Howard MT, Capecchi M, Guigó R, Burk RF, Atkins JF, Human selenoprotein P and S variant mRNAs with different numbers of SECIS elements and inferences from mutant mice of the roles of multiple SECIS elements., Open Biol. 6 (2016) 160241. doi: 10.1098/rsob.160241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lobanov AV, Fomenko DE, Zhang Y, Sengupta A, Hatfield DL, Gladyshev VN, Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life., Genome Biol. 8 (2007) R198. doi: 10.1186/gb2007-8-9-r198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jiang L, Ni J, Liu Q, Evolution of selenoproteins in the metazoan, BMC Genomics. 13 (2012) 446. doi: 10.1186/1471-2164-13-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hill KE, Lloyd RS, Burk RF, Conserved nucleotide sequences in the open reading frame and 3’ untranslated region of selenoprotein P mRNA., Proc. Natl. Acad. Sci. U. S. A 90 (1993) 537–41. doi: 10.1073/pnas.90.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mariotti M, Shetty S, Baird L, Wu S, Loughran G, Copeland PR, Atkins JF, Howard MT, Multiple RNA structures affect translation initiation and UGA redefinition efficiency during synthesis of selenoprotein P., Nucleic Acids Res. 45 (2017) 13004–13015. doi: 10.1093/nar/gkx982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chapple CE, Guigó R, Relaxation of selective constraints causes independent selenoprotein extinction in insect genomes, PLoS One. 3 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mariotti M, Santesmasses D, Capella-Gutierrez S, Mateo A, Arnan C, Johnson R, D’Aniello S, Yim SH, Gladyshev VN, Serras F, Corominas M, Gabaldón T, Guigó R, Evolution of selenophosphate synthetases: emergence and relocation of function through independent duplications and recurrent subfunctionalization., Genome Res. 25 (2015) 1256–67. doi: 10.1101/gr.190538.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mariotti M, Salinas G, Gabaldón T, Gladyshev VN, Utilization of selenocysteine in early-branching fungal phyla, Nat. Microbiol (2019). doi: 10.1038/s41564-018-0354-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mariotti M, Ridge PG, Zhang Y, Lobanov AV, Pringle TH, Guigo R, Hatfield DL, Gladyshev VN, Composition and evolution of the vertebrate and mammalian selenoproteomes., PLoS One. 7 (2012) e33066. doi: 10.1371/journal.pone.0033066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dehal P, Boore JL, Two rounds of whole genome duplication in the ancestral vertebrate., PLoS Biol. 3 (2005) e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mariotti M, Selenocysteine Extinctions in Insects, in: Raman C, Goldsmith M, Agunbiade T (Eds.), Short Views Insect Genomics Proteomics. Entomol. Focus Vol 4, Springer, Cham, 2016: pp. 113–140. [Google Scholar]
- [48].Mehta A, Rebsch CM, Kinzy SA, Fletcher JE, Copeland PR, Efficiency of mammalian selenocysteine incorporation., J. Biol. Chem 279 (2004) 37852–9. doi: 10.1074/jbc.M404639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Latrèche L, Jean-Jean O, Driscoll DM, Chavatte L, Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine., Nucleic Acids Res. 37 (2009) 5868–80. doi: 10.1093/nar/gkp635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shetty SP, Shah R, Copeland PR, Regulation of Selenocysteine Incorporation into the Selenium Transport Protein, Selenoprotein P, J. Biol. Chem 289 (2014) 25317–25326. doi: 10.1074/jbc.M114.590430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Salvi D, Mariottini P, Molecular taxonomy in 2D: a novel ITS2 rRNA sequence-structure approach guides the description of the oysters’ subfamily Saccostreinae and the genus Magallana (Bivalvia: Ostreidae), Zool. J. Linn. Soc 179 (2016) 263–276. doi: 10.1111/zoj.12455. [DOI] [Google Scholar]
- [52].Iacono M, Mignone F, Pesole G, uAUG and uORFs in human and rodent 5’untranslated mRNAs., Gene. 349 (2005) 97–105. doi: 10.1016/j.gene.2004.11.041. [DOI] [PubMed] [Google Scholar]
- [53].Churbanov A, Rogozin IB, Babenko VN, Ali H, Koonin EV, Evolutionary conservation suggests a regulatory function of AUG triplets in 5’-UTRs of eukaryotic genes., Nucleic Acids Res. 33 (2005) 5512–20. doi: 10.1093/nar/gki847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hinnebusch AG, Ivanov IP, Sonenberg N, Translational control by 5’-untranslated regions of eukaryotic mRNAs, Science (80-. ). 352 (2016) 1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stoytcheva Z, Tujebajeva RM, Harney JW, Berry MJ, Efficient incorporation of multiple selenocysteines involves an inefficient decoding step serving as a potential translational checkpoint and ribosome bottleneck., Mol. Cell. Biol 26 (2006) 9177–84. doi: 10.1128/MCB.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shetty SP, Sturts R, Vetick M, Copeland PR, Processive incorporation of multiple selenocysteine residues is driven by a novel feature of the selenocysteine insertion sequence, J. Biol. Chem 293 (2018) 19377–19386. doi: 10.1074/jbc.RA118.005211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fixsen SM, Howard MT, Processive selenocysteine incorporation during synthesis of eukaryotic selenoproteins., J. Mol. Biol 399 (2010) 385–96. doi: 10.1016/j.jmb.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Donovan J, Copeland PR, Evolutionary history of selenocysteine incorporation from the perspective of SECIS binding proteins., BMC Evol. Biol 9 (2009) 229. doi: 10.1186/14712148-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Donovan J, Copeland PR, Selenocysteine Insertion Sequence Binding Protein 2L Is Implicated as a Novel Post-Transcriptional Regulator of Selenoprotein Expression., PLoS One. 7 (2012) e35581. doi: 10.1371/journal.pone.0035581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H, Xiong Z, Que H, Xie Y, Holland PWH, Paps J, Zhu Y, Wu F, Chen Y, Wang J, Peng C, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z, Zhu Q, Feng Y, Mount A, Hedgecock D, Xu Z, Liu Y, Domazet-Lošo T, Du Y, Sun X, Zhang S, Liu B, Cheng P, Jiang X, Li J, Fan D, Wang W, Fu W, Wang T, Wang B, Zhang J, Peng Z, Li Y, Li N, Wang J, Chen M, He Y, Tan F, Song X, Zheng Q, Huang R, Yang H, Du X, Chen L, Yang M, Gaffney PM, Wang S, Luo L, She Z, Ming Y, Huang W, Zhang S, Huang B, Zhang Y, Qu T, Ni P, Miao G, Wang J, Wang Q, Steinberg CEW, Wang H, Li N, Qian L, Zhang G, Li Y, Yang H, Liu X, Wang J, Yin Y, Wang J, The oyster genome reveals stress adaptation and complexity of shell formation., Nature. 490 (2012) 49–54. doi: 10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- [61].Riviere G, Klopp C, Ibouniyamine N, Huvet A, Boudry P, Favrel P, GigaTON: an extensive publicly searchable database providing a new reference transcriptome in the pacific oyster Crassostrea gigas, BMC Bioinformatics. 16 (2015) 401. doi: 10.1186/s12859-0150833-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang Y, Fomenko DE, Gladyshev VN, The microbial selenoproteome of the Sargasso Sea., Genome Biol. 6 (2005) R37. doi: 10.1186/gb-2005-6-4-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gobler CJ, Lobanov AV, Tang Y-Z, Turanov AA, Zhang Y, Doblin M, Taylor GT, Sañudo-Wilhelmy SA, Grigoriev IV, Gladyshev VN, The central role of selenium in the biochemistry and ecology of the harmful pelagophyte, Aureococcus anophagefferens., ISME J. 7 (2013) 1333–43. doi: 10.1038/ismej.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rainbow PS, Smith BD, Trophic transfer of trace metals: Subcellular compartmentalisation in bivalve prey and comparative assimilation efficiencies of two invertebrate predators, J. Exp. Mar. Bio. Ecol 390 (2010) 143–148. doi: 10.1016/J.JEMBE.2010.05.002. [DOI] [Google Scholar]
- [65].Umysová D, Vítová M, Dousková I, Bisová K, Hlavová M, Cízková M, Machát J, Doucha J, Zachleder V, Bioaccumulation and toxicity of selenium compounds in the green alga Scenedesmus quadricauda., BMC Plant Biol. 9 (2009) 58. doi: 10.1186/1471-2229-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun X, Zhong Y, Huang Z, Yang Y, Selenium accumulation in unicellular green alga Chlorella vulgaris and its effects on antioxidant enzymes and content of photosynthetic pigments., PLoS One. 9 (2014) e112270. doi: 10.1371/journal.pone.0112270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Howard MT, Carlson BA, Anderson CB, Hatfield DL, Translational Redefinition of UGA Codons Is Regulated by Selenium Availability, J. Biol. Chem. 288 (2013) 19401–19413. doi: 10.1074/jbc.M113.481051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tsuji PA, Carlson BA, Anderson CB, Seifried HE, Hatfield DL, Howard MT, Dietary Selenium Levels Affect Selenoprotein Expression and Support the Interferon-γ and IL-6 Immune Response Pathways in Mice., Nutrients. 7 (2015) 6529–49. doi: 10.3390/nu7085297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fradejas-Villar N, Seeher S, Anderson CB, Doengi M, Carlson BA, Hatfield DL, Schweizer U, Howard MT, The RNA-binding protein Secisbp2 differentially modulates UGA codon reassignment and RNA decay., Nucleic Acids Res. 45 (2017) 4094–4107. doi: 10.1093/nar/gkw1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Turanov AA, Everley RA, Hybsier S, Renko K, Schomburg L, Gygi SP, Hatfield DL, Gladyshev VN, Regulation of Selenocysteine Content of Human Selenoprotein P by Dietary Selenium and Insertion of Cysteine in Place of Selenocysteine, PLoS One. 10 (2015) e0140353. doi: 10.1371/journal.pone.0140353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xia Y, Hill KE, Byrne DW, Xu J, Burk RF, Effectiveness of selenium supplements in a low-selenium area of China, Am. J. Clin. Nutr 81 (2005) 829–834. doi: 10.1093/ajcn/81.4.829. [DOI] [PubMed] [Google Scholar]
- [72].Kipp AP, Strohm D, Brigelius-Flohé R, Schomburg L, Bechthold A, Leschik-Bonnet E, Heseker H, German Nutrition Society (DGE), Revised reference values for selenium intake., J. Trace Elem. Med. Biol 32 (2015) 195–9. doi: 10.1016/j.jtemb.2015.07.005. [DOI] [PubMed] [Google Scholar]
- [73].Sarangi GK, Romagné F, Castellano S, Distinct Patterns of Selection in Selenium-Dependent Genes between Land and Aquatic Vertebrates, Mol. Biol. Evol 35 (2018) 1744–1756. doi: 10.1093/molbev/msy070. [DOI] [PubMed] [Google Scholar]
- [74].Bryszewska MA, Måge A, Determination of selenium and its compounds in marine organisms, J. Trace Elem. Med. Biol 29 (2015) 91–98. doi: 10.1016/j.jtemb.2014.10.004. [DOI] [PubMed] [Google Scholar]
- [75].Moriarty PM, Reddy CC, Maquat LE, Selenium Deficiency Reduces the Abundance of mRNA for Se-Dependent Glutathione Peroxidase 1 by a UGA-Dependent Mechanism Likely To Be Nonsense Codon-Mediated Decay of Cytoplasmic mRNA, Mol. Cell. Biol 18 (1998) 2932–2939. doi: 10.1128/MCB.18.5.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sunde RA, Raines AM, Selenium regulation of the selenoprotein and nonselenoprotein transcriptomes in rodents., Adv. Nutr 2 (2011) 138–50. doi: 10.3945/an.110.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gu QP, Ream W, Whanger PD, Selenoprotein W gene regulation by selenium in L8 cells., Biometals. 15 (2002) 411–20. http://www.ncbi.nlm.nih.gov/pubmed/12405536 (accessed July 25, 2019). [DOI] [PubMed] [Google Scholar]
- [78].Spiller HA, Rethinking mercury: the role of selenium in the pathophysiology of mercury toxicity., Clin. Toxicol. (Phila) 56 (2018) 313–326. doi: 10.1080/15563650.2017.1400555. [DOI] [PubMed] [Google Scholar]
- [79].Hedge LH, Knott NA, Johnston EL, Dredging related metal bioaccumulation in oysters., Mar. Pollut. Bull 58 (2009) 832–40. doi: 10.1016/j.marpolbul.2009.01.020. [DOI] [PubMed] [Google Scholar]
- [80].Schomburg L, Schweizer U, Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium, BBA - Gen. Subj (2009). doi: 10.1016/j.bbagen.2009.03.015. [DOI] [PubMed] [Google Scholar]
- [81].Kurokawa S, Berry MJ, Selenium. Role of the Essential Metalloid in Health, in: 2013: pp. 499–534. doi: 10.1007/978-94-007-7500-8_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jagodnik J, Chiaruttini C, Guillier M, Stem-Loop Structures within mRNA Coding Sequences Activate Translation Initiation and Mediate Control by Small Regulatory RNAs., Mol. Cell 68 (2017) 158–170.e3. doi: 10.1016/j.molcel.2017.08.015. [DOI] [PubMed] [Google Scholar]
- [83].Seeher S, Schweizer U, Targeted deletion of Secisbp2 reduces, but does not abrogate, selenoprotein expression and leads to striatal interneuron loss, Free Radic. Biol. Med 75 (2014) S9. doi: 10.1016/j.freeradbiomed.2014.10.849. [DOI] [PubMed] [Google Scholar]
- [84].Turanov AA, Lobanov AV, Fomenko DE, Morrison HG, Sogin ML, Klobutcher LA, Hatfield DL, Gladyshev VN, Genetic Code Supports Targeted Insertion of Two Amino Acids by One Codon, Science (80-. ). 323 (2009) 259. doi: 10.1126/science.1164748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shetty SP, Copeland PR, The Selenium Transport Protein, Selenoprotein P, Requires Coding Sequence Determinants to Promote Efficient Selenocysteine Incorporation, J. Mol. Biol 430 (2018) 5217–5232. doi: 10.1016/j.jmb.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Budiman ME, Bubenik JL, Driscoll DM, Identification of a signature motif for the eIF4a3–SECIS interaction, Nucleic Acids Res. 39 (2011) 7730–7739. doi: 10.1093/nar/gkr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Atkins JF, Bock A, Matsufuji S, Gesteland RF, Dynamics of the Genetic Code, in: Gesteland RF, Cech TR, Atkins JF (Eds.), RNA World, 2nd ed., Press, 1999: pp. 637–673. [Google Scholar]
- [88].Mariotti M, Guigó R, Selenoprofiles: profile-based scanning of eukaryotic genome sequences for selenoprotein genes., Bioinformatics. 26 (2010) 2656–63. doi: 10.1093/bioinformatics/btq516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kocot KM, Struck TH, Merkel J, Waits DS, Todt C, Brannock PM, Weese DA, Cannon JT, Moroz LL, Lieb B, Halanych KM, Phylogenomics of lophotrochozoa with consideration of systematic error, Syst. Biol 66 (2017) 256–282. doi: 10.1093/sysbio/syw079. [DOI] [PubMed] [Google Scholar]
- [90].Sievers F, Higgins DG, Clustal Omega for making accurate alignments of many protein sequences, Protein Sci. 27 (2018) 135–145. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Huerta-Cepas J, Serra F, Bork P, ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data., Mol. Biol. Evol 33 (2016) 1635–8. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Stamatakis A, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics. 30 (2014) 1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mariotti M, Lobanov AV, Guigo R, Gladyshev VN, SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins., Nucleic Acids Res. 41 (2013) e149. doi: 10.1093/nar/gkt550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jouaux A, Heude-Berthelin C, Sourdaine P, Mathieu M, Kellner K, Gametogenic stages in triploid oysters Crassostrea gigas: Irregular locking of gonial proliferation and subsequent reproductive effort, J. Exp. Mar. Bio. Ecol 395 (2010) 162–170. doi: 10.1016/J.JEMBE.2010.08.030. [DOI] [Google Scholar]
- [95].Li Q, Liu W, Shirasu K, Chen W, Jiang S, Reproductive cycle and biochemical composition of the Zhe oyster Crassostrea plicatula Gmelin in an eastern coastal bay of China, Aquaculture. 261 (2006) 752–759. doi: 10.1016/J.AQUACULTURE.2006.08.023. [DOI] [Google Scholar]
- [96].Ogra Y, Ishiwata K, Encinar J. Ruiz, Łobiński R, Suzuki KT, Speciation of selenium in selenium-enriched shiitake mushroom, Lentinula edodes., Anal. Bioanal. Chem 379 (2004) 861–6. doi: 10.1007/s00216-004-2670-6. [DOI] [PubMed] [Google Scholar]
- [97].Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS, The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments., Nat. Protoc 7 (2012) 1534–50. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Michel AM, Mullan JPA, Velayudhan V, O’Connor PBF, Donohue CA, Baranov PV, RiboGalaxy: A browser based platform for the alignment, analysis and visualization of ribosome profiling data., RNA Biol. 13 (2016) 316–9. doi: 10.1080/15476286.2016.1141862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome., Genome Biol. 10 (2009) R25. doi: 10.1186/gb2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kiniry SJ, O’Connor PBF, Michel AM, Baranov PV, Trips-Viz: a transcriptome browser for exploring Ribo-Seq data, Nucleic Acids Res. 47 (2019) D847–D852. doi: 10.1093/nar/gky842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks., Nat. Protoc 7 (2012) 562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Antonov I, Coakley A, Atkins JF, Baranov PV, Borodovsky M, Identification of the nature of reading frame transitions observed in prokaryotic genomes., Nucleic Acids Res. 41 (2013) 6514–30. doi: 10.1093/nar/gkt274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Gupta N, DeMong LW, Banda S, Copeland PR, Reconstitution of selenocysteine incorporation reveals intrinsic regulation by SECIS elements., J. Mol. Biol 425 (2013) 2415–22. doi: 10.1016/j.jmb.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF, Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy., Ann. Neurol. 48 (2000) 164–9. http://www.ncbi.nlm.nih.gov/pubmed/10939566 (accessed July 25, 2019). [PubMed] [Google Scholar]
- [105].Ashraf S, Huang L, Lilley DMJ, Sequence determinants of the folding properties of box C/D kink-turns in RNA, RNA. 23 (2017) 1927–1935. doi: 10.1261/rna.063453.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Goody TA, Melcher SE, Norman DG, Lilley DMJ, The kink-turn motif in RNA is dimorphic, and metal ion-dependent., RNA. 10 (2004) 254–64. doi: 10.1261/rna.5176604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Fletcher JE, Copeland PR, Driscoll DM, Krol A, The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2., RNA. 7 (2001) 1442–53. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1370188&tool=pmcentrez&rendertype=abstract (accessed September 25, 2013). [PMC free article] [PubMed] [Google Scholar]
- [108].Caban K, Kinzy SA, Copeland PR, The L7Ae RNA Binding Motif Is a Multifunctional Domain Required for the Ribosome-Dependent Sec Incorporation Activity of Sec Insertion Sequence Binding Protein 2, Mol. Cell. Biol. 27 (2007) 6350–6360. doi: 10.1128/MCB.0063207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Fu L, Niu B, Zhu Z, Wu S, Li W, CD-HIT: Accelerated for clustering the next-generation sequencing data, Bioinformatics. 28 (2012) 3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1: Reconstructed tree of SelenoP genes.
This tree is based on an alignment of the proximal region of SelenoP genes predicted across metazoa. All vertebrate sequences cluster together in this phylogenetic reconstruction; for visualization purposes, their tree is shown in Panel A; the remaining genes are shown in Panel B. Next to each node, a graphical representation of aligned protein sequence is shown (see also Figs 1,2), coloured according to encoded protein clusters defined based on this tree topology. Sec-UGAs are represented as vertical black lines. For the subset of predictions that come from genome sequences, red lines indicate intron positions. In the outmost section, SECIS presence is shown (the first SECIS per gene in dark blue, the second in brown; if unique, it is colored in grey).
Supplementary Table 3 Selenium levels in relevant material from the supplementation trial.
Total Se levels were determined for each sample from the appropriate supplementation group including the algal food source grown and collected before feeding day (Methods), artificial sea water collected after the supplementation trial, tissue cytoplasmic extracts isolated after tissue lysis and the remaining debris separated by centrifugation. Standard deviation values (SD) were computed per sample group.
Supplementary Table 4 UGA redefinition efficiency calculations for each selenoprotein.
Selenoprotein genes UGA redefinition efficiency (URE) were computed as the ratio of the RPF density 5’ of UGA to the RPF density 3’ of UGA (excluding the first 15 nt of CDS). For SelenoP, the URE for UGA1 was calculated, and considering only the sequence between UGA1 and UGA2 to estimate RPF density 3’ of UGA.
Supplementary Fig 2: Alignment of SelenoP sequence representatives.
The figure shows an alignment of sequence representatives chosen for each protein cluster. Sec residues (U) are highlighted.
Supplementary Table 5: Table of primers used to PCR amplify mutant oyster constructs for subsequent cloning
Supplementary Fig 3: Local homology in SelenoP sequences.
(A) Results of local homology searches using Blast (left) and Exonerate (right) using spider P. tepidarium as query and all sequence representatives as target. Note that Blast fails to report matches of the query with itself, so that we used Exonerate for this task. (B) We identified a number of identical alignments in the output of Blast and Exonerate for all queries and targets and verified that the scores of the two programs are highly correlated (left). We thus calibrated a log(e-value) to Exonerate score ratio (right), which allowed us to assign inferred e-values to all Exonerate hits, used to filter them (see Fig 3: inferred e-value < 0.01). (C) Local homology identified between the C-terminal domain of human SelenoP and that of two invertebrates (see Fig 3, top-left panel).
Supplementary Fig 4: Conserved RNA structures identified in SelenoP of Pacific oyster M. gigas.
(A) Top: predicted RNA structure of (left to right) Initiation Stem Loop (ISL) at positions 40–160 from AUG; Selenocysteine Recoding Elements (SRE2/3) spanning positions 1585–1705; and SECIS 1 and 2, 194–267 and 585–657 nts 3’ of the UAG stop codon. The conserved SECIS motifs, the core and the A repeats at the apical loop, are in green. Bottom: gene structure indicating the coding sequence, UGA positions (vertical lines), and sequences that form secondary structure elements (shaded in grey). (B) ISL alignment across 6 bivalves. (B & C) Shaded positions correspond to RNA pairs in the structure above. (C) Alignment of SRE2/3 across 6 bivalves. Matching colors indicate the two strands of a stem. The species used to predict the conserved structures show in the alignment are: M. gigas, C. virginica, Mytilus galloprovincialis, Bathymodiolus platifrons, Pinctada martensii and Mizuhopecten yessoensis.
Supplementary Fig 5: Oyster SelenoP mRNA context-specific mutants tested by RRL translation.
The top panel shows representations of each in vitro transcribed mRNA (Methods). The figure depicts: brown box for oyster CDS and green box for zebrafish CDS. Black lines are Sec-UGA and the ISL structure is in black before UGA 1. SECIS 1 is in blue and SECIS 2 is in red. Red ‘X’ denotes deletion and ‘SM’ denotes synonymous codon mutations to disrupt conserved pairings. Yellow asterisk refers to oyster early termination product while the green arrow indicates full-length zebrafish SelenoP. Lane 1: negative control no mRNA, lane 2: zebrafish WT control, (lanes-3–10): oyster mutant constructs, including native transcript (lane 3), Kozak consensus added (lane 4), native 5’UTR removed (lane 5), ISL complementarity disrupted (lane 6), zebrafish CDS fused to the entire oyster 3’UTR (lane 7), or to only oyster SECIS 1 (lane 8) or oyster SECIS 2 (lane 9). Note that particular differences in product intensity and resolution are observed in the positive controls (WT zebrafish SelenoP) in individual autoradiography gels which could be attributed to RRL batch variability. Also, the low molecular weight product at ~25 kDa was observed across all experiments even in the absence of added mRNA (panel A, lane 1) and thus attributed to RRL background.
To test whether the absence of radiolabeled product in the ISL and C-terminal deletion mutants were due to lack of initiation or Sec-decoding of UGA1, we performed 35[S]Met labelling in the presence or absence of the concentration of G418 antibiotic that promotes 20% readthrough at UGA codons [103,104]. Translation reactions were performed in the absence of rat CT-SBP2, with added ‘cold’ selenium and either supplemented or not supplemented by G418. The RRL products were resolved in a 15% SDS-PAGE gel to detect the presence of the early termination product (5–6 kDa) before recoding of UGA1. We observed that all the constructs produced a labelled readthrough product corresponding to termination at UGA2 except for the C-terminal deletion mutant in the presence of G418. Moreover, in the absence of G418, the early termination 5 kDa product of oyster SelenoP was reduced with 5’UTR deletion and more significantly with ISL mutation and C-terminal deletion. Note that we also detect a >37 kDa 35SMet labelled product in the native and Kozak context constructs similar to the 75Se experiment (yellow asterisks).
Supplementary Fig 6: Testing affinity to rat SECISBP2 of oyster SECIS-fusion constructs.
MgCl2 can change conformation and affinity of RNA structures with K-turn motifs [105,106]. SECIS affinity was tested by the addition of increasing concentrations of MgCl2 (mM) to RRL translation reactions of (A) wild-type zebrafish (B) zebrafish fusion with oyster 3’UTR (C) zebrafish fusion with oyster SECIS 1 and (D) zebrafish fusion with oyster SECIS 2. Full-length zebrafish SelenoP product runs at 42 kDa.
Supplementary Fig 7: Supplementing RRL translation reactions with oyster SECISBP2.
(A) The positions of the functional domains, SID (SECIS Interaction Domain) and RBD (Ribosome Binding Domain) [107,108], are shown within oyster SECISBP2. (B) Alignment and extent of conservation of oyster SECISBP2 with both human SECISBP2 and SECISBP2L are shown (C) E. coli expressed His-tagged oyster SECISBP2 analysis by SDS PAGE with Coomassie staining and Western blot probing (anti-His tag). The green arrow points to purified full-length oyster SECISBP2 following cleavage of a GST-tag used for purification. (D) Autoradiogram of 75Se labelled rat SelenoP translated in RRL supplemented with varying concentrations of oyster SECISBP2. Black arrow points to a 27 kDa band suggestive of a single or double Sec-incorporation.
Supplementary Fig 8: Radical S-adenosyl Methionine (RSAM) selenoprotein in M. gigas and other species.
Multiple amino acid sequence alignment of Radical SAM (RSAM) proteins, showing only the C-terminus including the Sec or Cys position, 497. The residue at this position is highlighted in red for Sec and in orange for Cys. The sequences were obtained from NCBI GenBank Transcriptome Shotgun Assemblies (TSA). The phylogenetic tree on the left was obtained from NCBI Taxonomy.
Supplementary Fig 10 SelenoP Antibody characterization.
(A) Protein map of SelenoP including Sec-UGAs (vertical white lines) and antibody recognition sites (grey). N-terminal Ab (Anti-NT-SelenoP) is located before Sec - UGA1 and C-terminal Ab (Anti-CT-SelenoP) is located after Sec-UGA 43. (B) Antibody immunogenic peptide insertion: NT- N-terminal peptide (yellow) and CT C-terminal peptide (green) into a GST-MBP-6XHis (GST-MBP-NT-6XHis or GST-MBP-CT-6XHis) tagged vector for bacterial production. (C) Immunoblot of bacterial protein produced using antibody against the GST tag (Anti-GST), N-terminal SelenoP (Anti-NT-SelenoP) and C-terminal SelenoP (Anti-CT-SelenoP). Anti-CT-SelenoP also detected the N-terminal peptide suggesting non-specific recognition.
Supplementary Fig 9: Effects of selenium supplementation on the expression of selenoprotein and selenoprotein machinery genes.
Expression levels of Sec machinery (top) and selenoprotein genes (bottom) assayed through riboseq (first column, expressed in RPFKM+1) and RNA-seq (second column, RPKM+1). The third column shows translation efficiency, calculated as RPFKM/RPKM ratio. All values were computed separately for Se-supplemented (orange) and non-supplemented samples (green). The bottom panel shows how Sec machinery and selenoprotein genes compare with the distribution of the whole oyster transcriptome.
Supplementary Fig 11: Tree of SelenoP SECIS elements.
The plots show the reconstructed phylogenetic tree based on a structural alignment of SelenoP SECIS elements (Methods). Outside of the circular tree in the middle, there are three layers. The first layer contains bars representing the distance of SECIS predictions from the CDS. This is color coded to distinguish multiple SECIS elements identified in the same gene: blue for SECIS 1, brown for SECIS 2, grey if a single SECIS was found. The second layer contains schemes of SelenoP genes for which each SECIS was identified, following the same representation as Figs 1–2. The third layer contains species names, color coded by lineage. Two major clusters of SECIS elements are highlighted in light and dark grey, roughly corresponding to the SECIS 1 (top) and SECIS 2 (bottom-left).
Supplementary Fig 12: Correlation between number of selenoproteins per species and Sec residues in SelenoP.
The plot shows the relationship between the number of selenoproteins found in transcriptomes or genomes and the number of putative Sec-UGAs in SelenoP genes. The plot contains one point per species, colored by phylogenetic cluster as in Fig 1. Our analysis shows significant correlation (Pearson’s r=0.161; p-value<1e-4), even when subsampling vertebrates (considering only 40 vertebrates and all non-vertebrate species: Pearson’s r=0.298; p-value<1e-4). Selenoproteins were predicted in nucleotide assemblies by Selenoprofiles (all predictions with label “selenocysteine”), and highly similar genes predicted in the same assembly were removed with the program Cdhit v4.6 [109]. When multiple assemblies were available for a given species, the one richest in selenoproteins was used, and when multiple homologous SelenoP genes were identified, the one with most Sec-UGAs was used. Only those species with predicted selenoproteins and Sec-containing SelenoP were plotted and considered for this analysis.
Supplementary Fig 13: Anti- SelenoP immunoblot of oyster tissues from with varying total Se concentrations determined.
Oyster tissues with determined total Se levels were probed with antibodies against N and C terminal portions of SelenoP. Two total protein dilutions were loaded on the gel (25 and 50μg) of each sample including one representative sample from the non-supplemented group (3 μg/g total Se) and two representative samples from the Se-supplemented group (13, 17 and 80 μg/g) with variable elevated total Se levels determined.
Supplementary Fig 14: Exon/intron junction conservation in metazoan SelenoP.
The plot displays the gene structures of SelenoP gene predictions obtained from genome assemblies, indicating the position of exon/intron junctions. Note that only one representative species, Falcus pellegrinus, is shown for vertebrate SelenoP1 and SelenoP2, since intron positions are conserved in this gene across all the vertebrate clade. Genes that have lost the intron 3’ of UGA1 are highlighted in various colors. Light blue indicates two cases in which intron loss was accompanied by UGA1 replacement by a standard codon, and SelenoP ceased to be a selenoprotein. Green indicates the case of sea snail Colubraria reticulata (gastropod) where intron loss was also concomitant to UGA1 replacement by a Cys codon, but SelenoP remained a selenoprotein due to its distal UGAs and SECIS element. Yellow indicates the case of sea anemone Exaiptasia pallida, where UGA1 was retained despite the absence of introns. Lastly, grey indicates the case of three Acariformes (arachnida) which possess UGA1 but lost its neighbouring intron. These do not present any SECIS element and may be translated through a different form of readthrough (see Results).
Supplementary Note 1: Sec to Cys conversions in vertebrate SelenoP2
Supplementary Note 2: Validation of 132 UGAs in SelenoP of Elliptio complanata
Supplementary Note 3: Sec to Cys conversion of SelenoP in gastropods
Supplementary Note 4: Divergence of SelenoP in arachnida
Supplementary Note 5: SECISBP2 orthology between vertebrates and oysters
Supplementary Note 6: SelenoP duplication in oysters
Supplementary Note 7: Oysters for selenium supplementation experiments
Supplementary Note 8: Assessment of ribosome profiling data
Supplementary Table 1 Selenoproteome of M. gigas
List of selenoprotein and machinery genes in M. gigas, indicating the corresponding transcript in the assembled transcriptome and the contig in the genome assembly where each gene was found. The last column indicates the homology with human genes; in case of complex orthology relationships, only the family is indicated.
Supplementary Table 2 Marine mussels, Mytilus edulis, total selenium measurement
Total selenium levels were determined for eight individual farmed marine mussels (Mytilus edulis) collected from Collorus, Co. Kerry, Ireland. Standard deviation values (SD) and Relative Standard Deviation (RSD) values are indicated.