

Abstract
Oxidative modification of Cys residues by NO results in S-nitrosylation, a ubiquitous post-translational modification and a primary mediator of redox-based cellular signaling. Steady-state levels of S-nitrosylated proteins are largely determined by denitrosylase enzymes that couple NAD(P)H oxidation with reduction of S-nitrosothiols, including protein and low-molecular-weight (LMW) S-nitrosothiols (S-nitroso-GSH (GSNO) and S-nitroso-CoA (SNO-CoA)). SNO-CoA reductases require NADPH, whereas enzymatic reduction of GSNO can involve either NADH or NADPH. Notably, GSNO reductase (GSNOR, Adh5) accounts for most NADH-dependent GSNOR activity, whereas NADPH-dependent GSNOR activity is largely unaccounted for (CBR1 mediates a minor portion). Here, we de novo purified NADPH-coupled GSNOR activity from mammalian tissues and identified aldo-keto reductase family 1 member A1 (AKR1A1), the archetypal mammalian SNO-CoA reductase, as a primary mediator of NADPH-coupled GSNOR activity in these tissues. Kinetic analyses suggested an AKR1A1 substrate preference of SNO-CoA > GSNO. AKR1A1 deletion from murine tissues dramatically lowered NADPH-dependent GSNOR activity. Conversely, GSNOR-deficient mice had increased AKR1A1 activity, revealing potential cross-talk among GSNO-dependent denitrosylases. Molecular modeling and mutagenesis of AKR1A1 identified Arg-312 as a key residue mediating the specific interaction with GSNO; in contrast, substitution of the SNO-CoA–binding residue Lys-127 minimally affected the GSNO-reducing activity of AKR1A1. Together, these findings indicate that AKR1A1 is a multi-LMW-SNO reductase that can distinguish between and metabolize the two major LMW-SNO signaling molecules GSNO and SNO-CoA, allowing for wide-ranging control of protein S-nitrosylation under both physiological and pathological conditions.
Keywords: S-nitrosylation, nitric oxide, enzyme purification, enzyme kinetics, molecular modeling, mutagenesis, AKR1A1, denitrosylase, denitrosylation, GSNO reductase, S-nitroso-coenzyme A reductase
Introduction
Physiological NO-based signaling is transduced primarily by S-nitrosylation, the phylogenetically conserved post-translational modification of Cys thiols by NO to form S-nitrosothiols (SNOs)3 (1), with broad-ranging effects across health and disease (2, 3). Both the formation of S-nitroso-proteins (SNO-proteins) and the removal of NO from SNO-proteins are governed enzymatically. SNO formation entails the combined actions of nitric-oxide synthases, SNO synthases, and trans-nitrosylases (4), whereas denitrosylase enzymes terminate SNO-based signals. The activity of denitrosylases generally determines the steady-state levels of cellular S-nitrosylation (5).
Low-molecular-weight (LMW) thiols that carry SNO, including S-nitroso-GSH (GSNO) and S-nitroso-CoA (SNO-CoA), are central to protein S-nitrosylation through their ability to establish S-nitrosylation/denitrosylation equilibria with target SNO-proteins (5). Denitrosylation reactions involving LMW-SNO are generally coupled to the oxidation of NAD(P)H. In particular, SNO-CoA reductases (SCoRs) in yeast (Adh6) and mammals (AKR1A1) oxidize NADPH to reduce SNO-CoA, thereby regulating SNO-protein levels (6, 7); no appreciable NADH-dependent SCoR activity has been identified to date (6). By contrast, GSNO reductase (GSNOR, Adh5) metabolizes GSNO using reducing equivalents from NADH (8, 9). Such GSNOR-dependent regulation of S-nitrosylation widely impacts cellular function in both health and disease (5). However, whereas NADH/NAD+ ratios are often ≪1, the ratio of NADPH/NADP+ is typically ≫1; NADPH therefore serves as the general reducing agent in cells (5). Evidence for NADPH-coupled reduction of GSNO was found in the enzyme carbonyl reductase 1 (CBR1) (10); however, it is unclear to what extent CBR1 operates across cells and tissues. Therefore, we sought to identify additional enzymes by performing de novo purification and characterization of NADPH-dependent GSNO reductase activity from mammalian tissues.
Results
NADPH-dependent GSNO reductase activity is present across tissues
NADPH oxidation in the presence of GSNO was observed to varying degrees in lysates from mouse tissues (Fig. 1A), with the highest activity found in the kidneys. Previous reports suggested that the thioredoxin/thioredoxin reductase (Trx/TrxR) system can metabolize GSNO using NADPH as reducing equivalent (11); however, addition of the TrxR inhibitor auranofin to mouse tissue extracts had no effect on NADPH oxidation in the presence of GSNO (Fig. 1A). Further, GSNOR-deficiency increased GSNO-dependent NADPH oxidation in both kidneys and liver, but not in the lungs, heart, or spleen (Fig. 1A). This suggests a compensatory increase in NADPH-dependent GSNOR activity when NADH-dependent GSNOR activity is lost, particularly in tissues with high GSNOR activity (Fig. 1B). Acute treatment of kidney extracts from GSNOR−/− mice with GSNO increased SNO-protein levels (Fig. 1C, lanes 3 and 4), which was reversed by the co-addition of NADPH (Fig. 1C, fifth and sixth lanes). Treatment with auranofin did not prevent NADPH-dependent protein denitrosylation (Fig. 1C, fifth lane versus sixth lane), indicating that neither Trx/TrxR–mediated protein denitrosylation (12) nor GSNO metabolism are dominant in this system. Together, these results identify NADPH-dependent GSNO reductase activity across tissues to regulate protein S-nitrosylation.
Figure 1.

NADPH-dependent GSNO reductase activity in murine tissues. A and B, murine tissue extracts were incubated with 0.1 mm NADPH (A) or NADH (B) in the absence or presence of 0.2 mm GSNO, and NADPH consumption (A) or NADH consumption (B) (absorbance at 340 nm) was followed over time. Auranofin (2 μm) (A) and GSNOR−/− extracts (A and B) were used to rule out GSNO catabolizing activities by thioredoxin/thioredoxin reductase and GSNOR, respectively. A, the values are means ± S.D. for n = 3. **, p < 0.01 by one-way analysis of variance with Tukey's correction for multiple comparisons. B, in the heart, NADH-dependent GSNO reductase activity was not detected (n.d.) because NADH oxidase activity alone was greater than combined NADPH and GSNO consumption. The values are means ± S.D. for n = 3. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01 by t test using the Holm–Sidek method for determining significance. C, GSNO-coupled protein S-nitrosylation is regulated by novel GSNO reductase activity. Representative Coomassie-stained SDS-PAGE gel of SNO-proteins enriched by SNO-RAC following incubation of GSNOR−/− kidney lysates (0.5 mg/ml) for 10 min with 0.1 mm GSNO alone or in presence of 0.1 mm NADPH. The lysates were preincubated with 2 μm auranofin or vehicle (DMSO) for 30 min to inhibit thioredoxin/thioredoxin reductase-dependent SNO-protein denitrosylation. The results are representative of three independent experiments.
Purification and identification of AKR1A1 as novel NADPH-coupled GSNO reductase activity in mammalian tissues
NADPH-dependent GSNO reductase activity was purified to homogeneity from bovine kidney extract (Fig. 2, Table 1, Fig. S1, and Table S1) and identified as aldo-keto reductase 1A1 (AKR1A1). AKR1A1 is the founding member of the aldo-keto reductase superfamily of oxidoreductases, and orthologs are found throughout the vertebrate subphylum (13). Importantly, we previously identified AKR1A1 as the primary mammalian NADPH-dependent SNO-CoA reductase (6) and have described a renoprotective role for AKR1A1-dependent denitrosylation via metabolic reprogramming following acute kidney injury (7). Immunodepletion of AKR1A1 from kidney extracts of WT (C57BL/6) mice (Fig. 3A) markedly reduced NADPH-dependent GSNO reductase activity (Fig. 3B). Further, NADPH-dependent GSNO reductase activity is reduced by ∼50% in kidney extracts from AKR1A1+/− mice and ∼90% in kidney extracts from AKR1A1−/− mice (Fig. 3, C and D). Finally, kidney extracts from AKR1A1+/+ and AKR1A1−/− mice were treated with GSNO alone or together with NADPH, and total SNO-protein levels were assessed by SNO-RAC with Coomassie Blue staining (Fig. 3E). Treatment with GSNO increased SNO-protein levels in both AKR1A1+/+ and AKR1A1−/− kidney extracts (Fig. 3E, third and fourth lanes). SNO-protein levels were drastically reduced by co-addition of NADPH with GSNO in AKR1A1+/+ kidney extracts (Fig. 3E, fifth lane) but not in AKR1A1−/− extracts (Fig. 3E, sixth lane). Together, these data confirm that AKR1A1 is responsible for the majority of NADPH-dependent GSNO reductase activity in kidney tissues.
Figure 2.
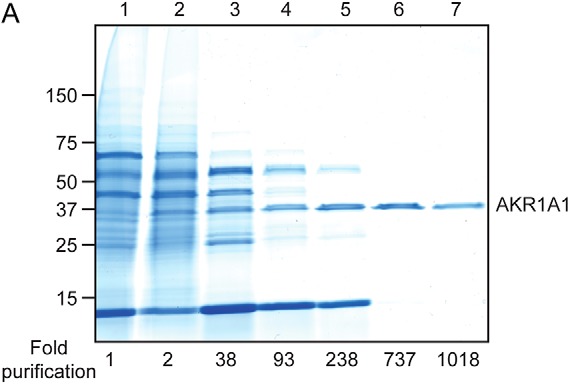
Purification of NADPH-dependent GSNO reductase activity. Shown is a representative Coomassie-stained SDS-PAGE gel corresponding to the chromatographic purification scheme of NADPH-dependent GSNO reductase activity (described in Table 1), which yielded a highly purified species from a crude kidney extract (lane 1) that was identified as aldo-keto reductase 1A1 (AKR1A1) (lane 7) (see also Fig. S1 and Table S1).
Table 1.
Purification of NADPH-dependent GSNO reductase activity from bovine kidney
| Purification step | Volume | Total protein | Total activity | Specific activity | Yield | Fold purification |
|---|---|---|---|---|---|---|
| ml | mg | units | units/mg | % | ||
| 1. Kidney soluble lysate | 105 | 5533.5 | 66.78 | 0.012 | 100 | 1 |
| 2. Ammonium sulfate (30–60%) fractionation | 34.5 | 2070.3 | 49.68 | 0.024 | 74.44 | 2 |
| 3. HiPrep Q fast flow | 22.5 | 91.8 | 41.35 | 0.456 | 61.94 | 37.68 |
| 4. HiPrep Phenyl fast flow | 23 | 15.7 | 17.7 | 1.13 | 26.52 | 93.38 |
| 5. Mono Q | 2.8 | 3.53 | 15.06 | 3.43 | 17.87 | 283.6 |
| 6. HiTrap Phenyl HP | 2.75 | 0.536 | 4.746 | 8.916 | 7.1 | 736.8 |
| 7. G-200 Superdex | 1.8 | 0.24 | 2.94 | 12.33 | 4.39 | 1019 |
Figure 3.

AKR1A1 is an NADPH-dependent GSNO reductase in mammals. A, representative Western blotting analysis following immunodepletion of AKR1A1 from WT mouse kidney extracts. IP, immunoprecipitate. B, relative NADPH-dependent GSNO reductase activity in IgG or AKR1A1 immunodepleted kidney extracts from A. The bars represent means ± S.D. for n = 3. ****, p < 0.0001 by Student's t test. C, representative Western blotting analysis of kidney extracts from AKR1A1+/+, AKR1A1+/−, and AKR1A1−/− mice. The example shown is representative of three independent experiments. D, relative NADPH-dependent GSNO reductase activity in kidney extracts from AKR1A1+/+, AKR1A1+/−, and AKR1A1−/− mice. The bars represent means ± S.D. for n = 3. ****, p < 0.0001 by one-way analysis of variance with Tukey's correction for multiple comparisons. E, representative Coomassie-stained SDS-PAGE gel illustrating SNO-proteins isolated by SNO-RAC following treatment of kidney extracts from AKR1A1+/+ and AKR1A1−/− mice with 0.1 mm GNSO (in the presence or absence of 0.1 mm NADPH). The results are representative of three independent experiments. F, NADPH-dependent GSNO reductase activity across various tissues from AKR1A1+/+ and AKR1A1−/− mice. The extracts were incubated with 0.2 mm GSNO and 0.1 mm NADPH. The bars represent means ± S.D. for n = 3. **, p < 0.01; ***, p < 0.001 by Student's t test. G, representative Western blotting analysis of the indicated tissue extracts from AKR1A1+/+ and AKR1A1−/− mice. The example is representative of three independent experiments. H, Western blotting analysis for AKR1A1 expression in liver extracts from GSNOR+/+ or GSNOR−/− mice. I, quantification of AKR1A1 expression in liver extracts from GSNOR+/+ or GSNOR−/− mice (H). Bands (n = 5) were quantified using ImageJ. ****, p < 0.0001 by Student's t test.
AKR1A1-dependent GSNO reductase activity was assessed across multiple tissues, namely the liver, lungs, heart, and spleen (Fig. 3, F and G). AKR1A1 accounts for a significant portion of NADPH-dependent GSNO reductase activity in the liver, lungs, and spleen, but not in the heart (Fig. 3F), corresponding to the relative expression of AKR1A1 in these tissues (Fig. 3G). This suggests that the role of AKR1A1 as a GSNO reductase will be most prominent in tissues with higher expression levels (7) and also that additional reductases may be present to account for remaining activity in AKR1A1−/− tissues.
As demonstrated in Fig. 1A, NADPH-dependent GSNO reductase activity is increased in both the kidneys and liver from GSNOR−/− mice. To determine whether AKR1A1 could account for this increased GSNO metabolizing activity, we performed Western blotting analysis for AKR1A1 in liver extracts from GSNOR+/+ and GSNOR−/− mice. AKR1A1 protein level was increased in liver extract from GSNOR−/− mice (Fig. 3, H and I), suggesting that AKR1A1 expression is up-regulated in the absence of GSNOR, the principal NADH-dependent GSNO reductase, and providing evidence for a physiological role of AKR1A1-mediated GSNO reduction. Additionally, quantitative RT-PCR across a variety of tissues (Fig. S2) suggests that basal AKR1A1 activity may be higher than that of either GSNOR (Adh5) or CBR1; however, definitive analyses require mRNA copy numbers and enzyme activity measurements. Taken together, these data indicate that AKR1A1 likely serves as a physiological GSNO reductase across tissues.
Enzymatic properties of AKR1A1 using GSNO as substrate
Product analysis by MS revealed GSH sulfinamide (Fig. S3) to be the major product of GSNO reduction by AKR1A1 in the presence of NADPH, suggesting a hydride transfer reaction mechanism (Fig. 4A) similar to both the NADPH-dependent reduction of SNO-CoA by AKR1A1 (6, 14) and the NADH-dependent reduction of GSNO by GSNOR (8, 15). The hydride transfer mechanism common to aldo-keto reductases requires an active site tyrosine (Tyr-50 in AKR1A1) to act as a proton donor for the reaction (16). To confirm the requirement of this residue for the reduction of GSNO by AKR1A1, we generated and purified catalytically dead AKR1A1Y50A and assessed its ability to reduce GSNO. Reaction of AKR1A1Y50A with GSNO failed to consume NADPH, compared with AKR1A1WT (Fig. 4B). Together, these data confirm that reduction of GSNO by AKR1A1 follows the canonical AKR reaction scheme, specifically hydride transfer from NADPH to the nitrogen atom of the SNO moiety and protonation of the oxygen atom by the active site Tyr-50 to generate the S-(N-hydroxy) intermediate that rearranges to form GSH sulfinamide (8). Serial addition of limiting amounts of NADPH in the presence of excess GSNO demonstrated a 1:1 GSNO:NADPH stoichiometry (Fig. 4C). Kinetic analysis of purified bovine AKR1A1 revealed Km of 87.8 ± 5.1 μm and Kcat of 471 ± 8 min−1 (Fig. 4D), whereas analysis of purified recombinant human AKR1A1 gave Km of 184.1 ± 8.1 μm and Kcat of 948 ± 14 min−1 (Fig. 4E).
Figure 4.

Product and kinetic analysis of AKR1A1 GSNO reductase activity. A, GSH sulfinamide was identified by MS as the major stable product of GSNO reduction by purified AKR1A1 (see Fig. S3 for product analysis). B, NADPH consumption (as measured by change in absorbance at 340-nm wavelength) by purified AKR1A1WT or AKR1A1Y50A (catalytically dead mutant) over 10 min in the presence of 0.1 mm GSNO and 0.1 mm NADPH. C, stoichiometry of NADPH:GSNO in AKR1A1-catalyzed GSNO reduction. Sequential additions of 0.084 mm NADPH to an excess of GSNO led to a mean consumption of 0.073 ± 0.0018 mm of GSNO, demonstrating a stoichiometry near 1:1. The results shown are representative of two independent experiments. D and E, kinetic analysis of GSNO reductase activity by purified native bovine AKR1A1 (D) and purified recombinant human AKR1A1 (E). Enzyme assays were performed in duplicate (D) or triplicate (E). Kinetic values were calculated using GraphPad Prism 7.
The results presented thus far identify AKR1A1 as a major NADPH-dependent GSNO reductase in mammalian tissues; AKR1A1 is also the major SNO-CoA reductase across tissues (6). Thus, AKR1A1 represents the first LMW-SNO reductase that metabolizes two major SNO signaling molecules. We sought to further understand the basis of molecular recognition of GSNO versus SNO-CoA by AKR1A1 (14). Modeling of GSNO with the active site of AKR1A1 produced a catalytically competent binding mode with the sulfur–nitrogen–oxygen group oriented toward the catalytic Tyr-50 and co-factor NADPH (Fig. 5A). The active site of AKR1A1 is surrounded by multiple charged and aromatic amino acids (Fig. 5A, highlighted in red) that could mediate the GSNO–AKR1A1 interaction, and the predicted binding mode identified Trp-22 and Arg-312 as potential mediators of this interaction. To test the role of these amino acids and other active site residues in binding of GSNO, we generated and purified AKR1A1 mutants (14) and tested the effect of mutation on in vitro reduction of GSNO. Mutations of AKR1A1 had varying effects on the catalytic efficiency of AKR1A1 for GSNO (Fig. 5B, Table 2, and Table S2), with some mutations increasing catalytic efficiency but others reducing catalytic efficiency. Most notably, AKR1A1R312A drastically reduced catalytic efficiency for GSNO (Fig. 5B). Arg-312 was predicted to form a hydrogen bond with the α-carboxylic acid group of the glutamic acid component of GSNO (Fig. 5A). Accordingly, mutation of Arg-312 to alanine led to an ∼5-fold increase in Km (Fig. 5C and Table 2), suggesting that binding of GSNO is dramatically altered in this mutant. Catalytic efficiency was also reduced because of a ∼60% reduction in Kcat (Fig. 5C and Table 2). In contrast, AKR1A1R312A increases Km for SNO-CoA by ∼2-fold, indicating a degree of specificity in the Arg-312–GSNO interaction (Table 2) (14). We previously identified Lys-127 as the key residue mediating the SNO-CoA–AKR1A1 interaction (14), where mutation of this residue to alanine increases Km for SNO-CoA ∼8-fold and reduces the ability of AKR1A1 to bind SNO-CoA-bound beads. Lys-127 was not predicted to interact with GSNO (Fig. 5A), and accordingly, AKR1A1K127A only very slightly increased Km for GSNO (Fig. 5D and Table 2) with no effect on Kcat. These results indicate that different residues are driving the interaction between AKR1A1 and GSNO versus SNO-CoA, with Arg-312 mediating the GSNO–AKR1A1 interaction and Lys-127 mediating the SNO-CoA–AKR1A1 interaction.
Figure 5.

Molecular modeling and mutagenic analysis of AKR1A1 GSNO reductase activity. A, binding model of GSNO in the AKR1A1 active site. Putative interacting residues and the SNO-CoA binding residue (Lys-127) are highlighted in red. NADPH carbons are colored green. B, catalytic efficiency (Kcat/Km) of AKR1A1 mutants expressed relative to AKR1A1WT. Kcat and Km were determined from two independent purifications (calculated using GraphPad Prism 7) and used to generate an average catalytic efficiency for each enzyme. C, kinetic analysis of GSNO reductase activity by AKR1A1WT and AKR1A1R312A. Enzyme assays were performed in triplicate. The AKR1A1WT curve is replotted from Fig. 4E for comparison. The kinetic values are listed in Table 2. D, kinetic analysis of GSNO reductase activity by AKR1A1WT and AKR1A1K127A. Enzyme assays were performed in triplicate. The AKR1A1WT curve is replotted from Fig. 4E for comparison. Kinetic values are listed in Table 2. E, relative GSNO reductase activity in the presence of increasing concentrations of GSH or CoA (CoASH). Increasing amounts of GSH and CoASH were added to a reaction mix of 100 μm GSNO/NADPH and 20 nm AKR1A1WT. The assays were performed in duplicate. The IC50 values were calculated in GraphPad Prism 7. n.d., not detected.
Table 2.
Kinetic comparison of GSNO and SNO-CoA reductase activities for wildtype and mutant human AKR1A1 enzymes
| Enzyme | Substrate | Kma | Kcata |
|---|---|---|---|
| μm | min−1 | ||
| WT | GSNO | 184 ± 8 | 948 |
| SNO-CoAb | 58 ± 4a | 959a | |
| K127A | GSNO | 336 ± 24 | 953 |
| SNO-CoAb | 410 ± 27a | 750a | |
| R312A | GSNO | 943 ± 104 | 383 |
| SNO-CoAb | 132 ± 11a | 410a |
a Km and Vmax were determined from Michaelis–Menten curves using GraphPad Prism 7. Kcat was calculated by dividing Vmax by the enzyme concentration in each assay. Enzyme assays were performed in triplicate.
The SNO moiety gives selective preference for AKR1A1 to interact with SNO-CoA versus reduced or derivatized CoA (14), so that the SNO-CoA reductase activity of AKR1A1 is not inhibited by cytosolic concentrations of free CoA or acetyl-CoA. We tested whether this principle applied to the interaction of GSNO with AKR1A1. Competitive inhibition assays using increasing concentrations of GSH or CoA with fixed (0.1 mm) GSNO demonstrated that GSH and CoA are weak inhibitors of GSNO reduction by AKR1A1. GSH was unable to reach 50% inhibition even at supraphysiological concentrations, whereas CoA had an IC50 of ∼6.5 mm (Fig. 5E). These results again suggest that molecular recognition of SNO substrates by enzymes is facilitated by both the R group (GSH or CoA) and the SNO moiety, creating a strong preference for the SNO-modified substrate.
Discussion
GSNO and SNO-CoA play central roles in cellular signaling by stabilizing NO bioactivity and targeting proteins for S-nitrosylation (5). Accordingly, the enzymatic denitrosylases controlling GSNO and SNO-CoA levels are paramount to proper regulation of SNO-based signaling. Here we used de novo purification to identify AKR1A1 as a novel NADPH-dependent GSNO reductase, responsible for this activity in kidneys, liver, lungs, and spleen; AKR1A1-mediated GSNO reductase activity is also likely to play an important role in other tissues where AKR1A1 is highly expressed, including brain, intestines, stomach, testes, and white adipose tissue (7). With the discovery of AKR1A1, there are now three identified GSNO reductases (GSNORs 1–3): NADH-dependent GSNOR (Adh5), NADPH-dependent CBR1 (GSNOR2), and NADPH-dependent AKR1A1/SCoR (GSNOR3); together, these enzymes likely account for a majority of GSNO reductase activity across tissues.
AKR1A1 and CBR1 provide the first example of mammalian denitrosylases sharing substrate (GSNO) and co-factor (NADPH). Human AKR1A1 has a Km of 184 μm and Kcat of 948 min−1 for GSNO; CBR1 has a Km of 30 μm and Kcat of 450 min−1 for GSNO (10), giving CBR1 an ∼3-fold higher catalytic efficiency (Kcat/Km) for GSNO. Differences in tissue expression (Fig. S2), response to cellular stimuli, and regulation by post-translational modification are likely to determine which enzyme predominates in a given tissue, compartment, and condition. Careful comparisons of the SNO-proteomes regulated by each enzyme, particularly in tissues where all three GSNORs are expressed (e.g. liver, kidney, lungs, etc.), may help delineate shared versus specialized roles of these enzymes in regulating S-nitrosylation.
The ability of AKR1A1/SCoR to reduce GSNO identifies this enzyme with activity toward two major SNO signaling molecules (GSNO and SNO-CoA). Human AKR1A1 metabolizes SNO-CoA with a Km of 58 μm and Kcat of 959 min−1 (14) (Table 2); it metabolizes GSNO with a Km of 184 μm and Kcat of 948 min−1. Thus, AKR1A1 has an ∼3-fold higher catalytic efficiency (Kcat/Km) for SNO-CoA driven by an ∼3-fold lower Km for SNO-CoA, identifying SNO-CoA as the preferred SNO substrate for AKR1A1. However, up-regulation of AKR1A1 upon genetic deletion of GSNOR from mouse liver indicates that the GSNO reductase function of AKR1A1 is likely operative endogenously. More generally, although cytosolic CoA concentrations are lower than GSH levels, we do not believe thiol concentrations are limiting in formation of GSNO versus SNO-CoA or that absolute GSNO and SNO-CoA concentrations, per se, represent primary determinants of SNO-protein levels. Rather, we favor a model in which GSH/GSNO and CoA/SNO-CoA may subcompartmentalize and/or AKR1A1 may interact with proteins that preferentially bind GSNO or SNO-CoA to generate layers of specificity. Identifying proteins whose SNO levels are regulated by the distinct SNO reductase functions of AKR1A1 versus alternative denitrosylases will help clarify these issues and represent areas of ongoing work. Thus, proteins regulated by the GSNO reductase activity of AKR1A1 may be found by comparing the SNO proteomes and interactomes of AKR1A1 with those of GSNOR and CBR1. Further, comparison of the SNO-proteome controlled by the SNO-CoA–binding mutant AKR1A1K127A with the SNO-proteome controlled by AKR1A1WT would be informative.
The inability of GSH and CoA to effectively inhibit GSNO reduction by AKR1A1 is consistent with our previous findings with regards to the inability of CoA and acetyl-CoA to inhibit SNO-CoA reduction by this enzyme (14) and further support molecular recognition of the SNO group by target enzymes. This provides enzymes with a means to selectively interact with SNO substrates within the cellular milieu where GSNO and SNO-CoA are likely minor populations compared with GSH and CoA (and other derivatives thereof). Whether this concept applies more generally to other LMW-SNO denitrosylases or to protein denitrosylases such as thioredoxin-related proteins remains to be explored. However, it is most likely that both nitrosylases and denitrosylases exploit recognition of the stable SNO moiety to propagate and regulate SNO-based signaling broadly.
Experimental procedures
NADPH-dependent GSNO reductase activity in murine tissues
Tissues (kidney, liver, lung, heart, and spleen) were harvested from 10–12-week-old C57BL/6 or AKR1A1+/+ mice and GSNOR−/− or AKR1A1−/−. Mice were used under a protocol consistent with the Guide for the Care and Use of Laboratory Animals (17) and approved by the Case Western Reserve University Institutional Animal Care and Use Committee. Tissues were homogenized in lysis buffer (50 mm phosphate buffer, pH 7.0, 150 mm NaCl, 0.1 mm EDTA, 0.1 mm diethylene triamine pentaacetic acid (DTPA), 1 mm PMSF, and a mixture of protease inhibitors (Roche)). The extracts were subsequently clarified by centrifugation (twice at 20,000 × g at 4 °C for 45 min), and the protein concentration was determined using the BCA assay. The activities were measured as described previously (6). Briefly, enzyme assays were performed in 50 mm phosphate buffer, pH 7.0 (containing 0.1 mm EDTA and DTPA), and contained 0.2 mm GSNO and 0.1 mm NADPH or NADH. The reactions were initiated by the addition of lysate and allowed to proceed for 1 min. All assays were performed in duplicate or triplicate using 1-cm-path length cuvettes, and the slope was used to calculate the activity. For some samples, lysates were treated with 2 μm auranofin (prepared in DMSO) prior to performing the assays to rule out the NADPH-dependent GSNO breakdown mediated by the thioredoxin system.
Purification of NADPH-dependent GSNO reductase activity
Bovine kidneys were obtained from Rockland Immunochemicals Inc. and classified as waste (no Institutional Animal Care and Use Committee approval required). Bovine kidney tissue (∼80 g) was suspended in 100 ml of lysis buffer (50 mm phosphate buffer, pH 7.0, 150 mm NaCl, 1 mm PMSF, and protease inhibitor mixture (Roche)). Initial lysis was performed in a blender using short pulses (five to six times), followed by homogenization with 20–30 strokes with a Dounce homogenizer (Wheaton). Following centrifugation twice at 60,000 × g for 45 min, the supernatant was taken as the starting material for assessment of enrichment of GSNO metabolizing activity. At this and all subsequent stages, enzyme activity was assessed with 0.2 mm GSNO, 0.1 mm NADPH in 50 mm phosphate buffer (pH 7) containing 0.1 mm EDTA and DTPA. The supernatant was precipitated with 30% ammonium sulfate followed by centrifugation at 20,000 × g, and the resultant supernatant was reprecipitated with 60% ammonium sulfate and pelleted at 20,000 × g. The second pellet was resuspended and dialyzed against Tris buffer, pH 8.0, at 4 °C. The dialyzed extract was applied at 1 ml/min onto a High Prep Q Fast Flow 16 × 10-mm column equilibrated with 20 mm Tris buffer, pH 8.0. The NADPH-dependent GSNO reductase activity was eluted with a linear 0–0.3 m NaCl gradient in 20 mm Tris buffer, pH 8.0. Active fractions were pooled and ammonium sulfate added to a final concentration of 1 m. Sample was then loaded onto a HiPrep Phenyl FF 16/10 column equilibrated with 20 mm Tris, pH 8.0, containing 1 m ammonium sulfate. Elution was achieved using a linear gradient of 0.7–0.4 m ammonium sulfate in Tris pH 8.0 buffer. Active fractions were pooled, and overnight dialyses were performed using 20 mm Tris, pH 8.0. Dialyzed sample was then loaded onto a MonoQ GL 5 × 50-mm column at a flow rate of 1 ml/min, and elution was carried out with a linear 0–0.3 m NaCl gradient in 20 mm Tris buffer, pH 8.0. Active fractions were pooled and ammonium sulfate added to final concentration of 1 m. Sample was then loaded onto a HiTrap Phenyl HP column equilibrated with 20 mm Tris, pH 8.0, containing 1 m ammonium sulfate at a flow rate of 0.5 ml/min. Finally, active fractions were concentrated to less than 200 μl of volume and then loaded onto a Superdex 200 Increase 10/300 column. Purification was assessed by running the various protein fractions on an SDS-PAGE gel and visualized by Brilliant Blue–G Colloidal Stain (Sigma). Protein bands were identified using MALDI-TOF/TOF MS/MS internally calibrated with trypsin autoproteolysis peaks. The MS spectrum was searched against the National Center for Biotechnology Information database, using the online version of Protein Prospector. MS analysis was carried out at Michael Hooker Proteomics and Mass Spectrometry Facility at the University of North Carolina.
Western blotting analysis
Western blotting analyses were performed using standard methods (7, 14). Antibodies used were AKR1A1 (Abnova, H00010327-D01P for Western blotting and immunoprecipitation) or AKR1A1 (Proteintech, 15054-1-AP for Western blotting); β-actin (Sigma–Aldrich, A1978); GSNOR (Proteintech, 11051-1-AP); and p97 (Fitzgerald, 10R-P104A).
Kinetic analysis of bovine and recombinant AKR1A1
Recombinant WT and mutant human AKR1A1 was purified as previously described (14). Enzyme assays were performed in 50 mm sodium phosphate (pH 7.0) containing 100 μm EDTA and DTPA, 100 μm NADPH, and varying concentrations of GSNO. GSNO was prepared freshly by reacting equal volumes of 0.1 m GSH and 0.1 m NaNO2 in MilliQ water with 0.1 m EDTA and 0.1 m DTPA. The reactions were performed in triplicate. Initial rates were calculated from the absorbance decrease (340 nm) using a combined extinction coefficient of 7.06 mm−1 cm−1 for GSNO and NADPH. Kinetic parameters (Km and Vmax) were determined in GraphPad Prism 7, and Kcat was calculated from Vmax and enzyme concentration. For NADPH consumption curves, reactions (100 μm GSNO, 100 μm NADPH, and 20 nm recombinant human AKR1A1) were monitored for 10 min while measuring absorbance at 340 nm. GSH and CoA competitive inhibition assays were performed in duplicate in the presence of varying concentrations of GSH or CoA (dissolved in 50 mm sodium phosphate buffer (pH 7.0) with 100 μm EDTA/DTPA) with a static concentration of 100 μm GSNO and 20 nm recombinant human AKR1A1. IC50 was determined using GraphPad Prism 7.
AKR1A1 immunodepletion
For immunodepletion, 10 μg of AKR1A1 Ab or control IgG Ab was bound to protein G–Sepharose beads (Amersham Biosciences) in dilution buffer (50 mm phosphate buffer, pH, 7.0, 10 mm NaCl, 0.1 mm EDTA, and DTPA), and the volume was brought to 1 ml. Coupling of the antibody to the beads was done at 4 °C for 1 h on an end-to-end rotator. Antibody-bound beads were washed three times with dilution buffer to remove unbound antibodies. Freshly prepared kidney extract (65 μg) from C57BL/6 mice (The Jackson Laboratory) was added to the beads, and the volume was brought to 1 ml with dilution buffer, followed by constant rotation overnight at 4 °C. Supernatants were removed and used for in vitro assays and Western blotting. 125 μl of supernatant were used to assay for NADPH-dependent GSNO reductase activity, as described above. To assess immunodepletion, a small volume of supernatant (25 μl) was combined with 10 μl of 4× Laemmli sample buffer (Invitrogen). To the beads, 10 μl of dye was added, and the eluant was used as immunoprecipitation control.
Denitrosylation assay
Kidney tissues were homogenized in lysis buffer (50 mm phosphate buffer, pH 7.0, 150 mm NaCl, 0.1 mm EDTA, 0.1 mm DTPA, 1 mm PMSF, and a mixture of protease inhibitors (Roche)). The extract was centrifuged twice at 20,000 × g at 4 °C for 45 min. Supernatant containing 1 mg of protein was diluted in the assay buffer (50 mm phosphate buffer, pH 7.0) to achieve a final volume of 2 ml. The samples were then treated with 100 μm GSNO alone or in combination with NADPH or NADH for 10 min at room temperature. The reactions were stopped by addition of 3 volumes (6 ml) of ice-cold acetone, and denitrosylation was detected by a SNO-RAC technique (18). In brief, acetone-precipitated protein samples were dissolved in 2 ml of blocking buffer (0.2% S-methylmethanethiosulfonate and 2.5% SDS in HEN buffer: 100 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, pH 8.0) and incubated in dark for 20 min at 55 °C with frequent vortexing. The proteins were then precipitated with 3 volumes of ice-cold acetone at −20 °C for 20 min. Residual S-methylmethanethiosulfonate was removed by resuspending the pellets in 2 ml of HENS buffer (HEN containing 1% SDS), followed by protein precipitation with ice-cold acetone (as described in the previous step). Protein pellets were finally resuspended in 2 ml of HENS buffer, and freshly prepared thiol beads (50 μl) and ascorbate (final concentration, 30 mm) were added. After incubation for 3.5 h, the beads were subsequently washed four times with HENS buffer and two times with HENS/10 (1:10 dilution of HEN buffer containing 1% SDS). Proteins were eluted with 20 μl of elution buffer (HENS/10 containing 10% β-mercaptoethanol) with constant shaking for 20 min at room temperature. Eluted samples were mixed with sample buffer, separated by SDS-PAGE, and visualized by Coomassie Blue staining.
Products of AKR1A1-catalyzed GSNO catabolism
Samples (1 ml) were prepared containing 20 mm ammonium bicarbonate buffer, 200 μm GNSO, and 200 μm NADPH at 25 °C. The reactions were initiated by the addition of purified bovine AKR1A1 and allowed to continue until absorbance at 340 nm indicated complete consumption of GSNO (∼1 h). The samples were then centrifuged through a 10-kDa cutoff ultrafiltration membrane, and the filtrate was stored at −80 °C until analyzed. For MS analysis, the samples were diluted 1:2 in HPLC-grade acetonitrile. Formic acid was added to the samples at a final concentration of 0.1% (v/v). The samples were injected into a Thermo LTQ Oribtrap XL at a flow rate of 1 μl/min. The ion at 339 m/z was isolated and fragmented using collision-induced dissociation with a normalized collision energy of 35 V and an isolation width of 3.0.
Quantitative real-time PCR analysis of GSNO reductases
Various mouse tissues (∼20 mg) were harvested from C57BL/6J mice and preserved in RNA Later stabilization reagent (Qiagen). Tissues were homogenization in PureZOL (Bio-Rad) on a Tissue Lyser (Qiagen) using stainless steel beads (Qiagen). Chloroform was used to extract the aqueous phase, and RNA from this phase was purified using the Aurum purification kit (Bio-Rad) following the manufacturer's instructions. For reverse transcriptase reaction, 1 μg of total RNA was transcribed to cDNA using iScriptTM reverse transcription supermix (Bio-Rad). Quantitative PCR was performed with the TaqMan method (using the Roche Universal Probe Library System) on an Applied Biosystems Step One Plus real-time PCR system. Relative expression of AKR1A1 versus ADH5 or CBR1 was calculated using the 2-(ΔCt) method with normalization to 18S rRNA. For murine AKR1A1, probe 99 (Roche) was used with the following primers: 5′-GGTATATTGTGCCCATGATTACG-3′ and 3′-GGGGAGTAGCAGGCAATG-5′. For murine ADH5, probe 88 was used with the following primers: 5′-ACAGGACGCACATGGAAAG-3′ and 3′-ACACCAGCTTTGGGACACTC-5′. For murine CBR1, probe 17 was used with the following primers: 5′-AGGTGACAATGAAAACGAACTTT-3′ and 3′-GGACACATTCACCACTCTGC-5′. For murine 18S rRNA, probe 48 was used with the following primers: 5′-GCAATTATTCCCCATGAACG-3′ and 3′-GGGACTTAATCAACGCAAGC-5′.
Molecular modeling
Static protein/flexible ligand modeling of the interaction of GSNO with AKR1A1 was performed using Maestro 9.9 software. The AKR1A1 crystal structure (Protein Data Bank code 3H4G) was prepared by removal of H2O and fidarestat from the Protein Data Bank file. Original hydrogens were removed in Maestro and replaced, bond orders were assigned, and the structure was minimized. The docking grid was prepared around the active site at X = −2.099, Y = −24.407, Z = −6.508. The GSH structure was obtained from PubChem, and the GSNO structure was created from the GSH structure in Maestro. GSNO was prepared for docking in Maestro using the ligand preparation function. GSNO was docked to the active site grid using XP Glide Docking with postdocking minimization.
Author contributions
C. T. S., P. A., and J. S. S. conceptualization; C. T. S. and P. A. data curation; C. T. S. and P. A. formal analysis; C. T. S. and P. A. validation; C. T. S., P. A., N. M. V., and H.-L. Z. investigation; C. T. S. and P. A. visualization; C. T. S., P. A., A. H., and J. S. S. methodology; C. T. S. and P. A. writing-original draft; C. T. S., P. A., R. T. P., and J. S. S. writing-review and editing; R. T. P. and J. S. S. supervision; J. S. S. funding acquisition.
Supplementary Material
Acknowledgment
We thank Precious McLaughlin for technical assistance.
This work was supported by National Institutes of Health Grants R01DK119506, R01HL126900, P01HL075443, and P01HL128192 (to J. S. S.). This work was also supported in part by National Institutes of Health Grant T32GM007250 (to C. T. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S3.
- SNO
- S-nitrosothiol
- CoA
- coenzyme A
- GSNO
- S-nitrosoglutathione
- GSNOR
- GSNO reductase
- SCoR
- SNO-CoA reductase
- SNO-CoA
- S-nitroso-coenzyme A
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- LMW
- low-molecular-weight
- PMSF
- phenylmethylsulfonyl fluoride
- SNO-RAC
- S-nitrosothiol resin-assisted capture
- DTPA
- diethylene triamine pentaacetic acid.
References
- 1. Hess D. T., Matsumoto A., Kim S.-O., Marshall H. E., and Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 10.1038/nrm1569 [DOI] [PubMed] [Google Scholar]
- 2. Anand P., and Stamler J. S. (2012) Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. 90, 233–244 10.1007/s00109-012-0878-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foster M. W., Hess D. T., and Stamler J. S. (2009) Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 15, 391–404 10.1016/j.molmed.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seth D., Hess D. T., Hausladen A., Wang L., Wang Y.-J., and Stamler J. S. (2018) A multiplex enzymatic machinery for cellular protein S-nitrosylation. Mol. Cell 69, 451–464.e6 10.1016/j.molcel.2017.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stomberski C. T., Hess D. T., and Stamler J. S. (2019) Protein S-nitrosylation: determinants of specificity and enzymatic regulation of S-nitrosothiol–based signaling. Antioxid. Redox Signal. 30, 1331–1351 10.1089/ars.2017.7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anand P., Hausladen A., Wang Y.-J., Zhang G.-F., Stomberski C., Brunengraber H., Hess D. T., and Stamler J. S. (2014) Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 111, 18572–18577 10.1073/pnas.1417816112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou H.-L., Zhang R., Anand P., Stomberski C. T., Qian Z., Hausladen A., Wang L., Rhee E. P., Parikh S. M., Karumanchi S. A., and Stamler J. S. (2019) Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 565, 96–100 10.1038/s41586-018-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jensen D. E., Belka G. K., and Du Bois G. C. (1998) S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 331, 659–668 10.1042/bj3310659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu L., Hausladen A., Zeng M., Que L., Heitman J., and Stamler J. S. (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–494 10.1038/35068596 [DOI] [PubMed] [Google Scholar]
- 10. Bateman R. L., Rauh D., Tavshanjian B., and Shokat K. M. (2008) Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J. Biol. Chem. 283, 35756–35762 10.1074/jbc.M807125200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nikitovic D., and Holmgren A. (1996) S-Nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J. Biol. Chem. 271, 19180–19185 10.1074/jbc.271.32.19180 [DOI] [PubMed] [Google Scholar]
- 12. Benhar M., Forrester M. T., Hess D. T., and Stamler J. S. (2008) Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320, 1050–1054 10.1126/science.1158265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mindnich R. D., and Penning T. M. (2009) Aldo-keto reductase (AKR) superfamily: genomics and annotation. Hum. Genomics 3, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stomberski C. T., Zhou H.-L., Wang L., van den Akker F., and Stamler J. S. (2019) Molecular recognition of S-nitrosothiol substrate by its cognate protein denitrosylase. J. Biol. Chem. 294, 1568–1578 10.1074/jbc.RA118.004947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hedberg J. J., Griffiths W. J., Nilsson S. J., and Höög J. O. (2003) Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electrospray mass spectrometry. Eur. J. Biochem. 270, 1249–1256 10.1046/j.1432-1033.2003.03486.x [DOI] [PubMed] [Google Scholar]
- 16. Jin Y., and Penning T. M. (2007) Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 47, 263–292 10.1146/annurev.pharmtox.47.120505.105337 [DOI] [PubMed] [Google Scholar]
- 17. Committee for the Update of the Guide for the Care and Use of Laboratory Animals. (2011) Guide for the Care and Use of Laboratory Animals, National Academies Press, Washington, D.C. [Google Scholar]
- 18. Forrester M. T., Thompson J. W., Foster M. W., Nogueira L., Moseley M. A., and Stamler J. S. (2009) Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 27, 557–559 10.1038/nbt.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.