

Abstract
Zika virus (ZIKV) is a mosquito-borne flavivirus that has emerged as a threat to global health. The family of adenosine deaminases acting on dsRNA (ADARs) are human host factors important for the genetic diversity and evolution of ZIKV. Here, we further investigated the role of ADAR1 in ZIKV replication by utilizing CRISPR/Cas9-based gene editing and RNAi-based gene knockdown techniques. Both ADAR1 knockout and knockdown significantly reduced ZIKV RNA synthesis, protein levels, and viral titers in several human cell lines. Trans-complementation with the full-length ADAR1 form p150 or the shorter form p110 lacking the Zα domain restored viral replication levels suppressed by the ADAR1 knockout. Moreover, we observed that the nuclear p110 form was redistributed to the cytoplasm in response to ZIKV infection. ADAR1 was not involved in viral entry but promoted viral protein translation by impairing ZIKV-induced activation of protein kinase regulated by dsRNA (PKR). Of note, the RNA-editing activity of ADAR1 was not required to promote ZIKV replication. We also found that the proviral role of ADAR1 was partially mediated through its ability to suppress IFN production and PKR activation. Our work identifies ADAR1 as a proviral factor involved in ZIKV replication, suggesting that ADAR1 could be a potential antiviral target.
Keywords: viral replication, interferon, flavivirus, RNA editing, host-pathogen interaction, post-transcriptional regulation, adenosine deaminase, host factor, positive stranded RNA virus, protein kinase regulated by dsRNA, Zika virus, arbovirus
Introduction
Zika virus (ZIKV)3 is an emerging member of the Flavivirus genus of the Flaviviridae family, which includes a number of medically important arboviruses such as dengue virus, Japanese encephalitis virus, and yellow fever virus (1). Although ZIKV was isolated in 1947 in the Zika forest of Uganda, the first outbreak of ZIKV took place until 2007 on Yap Island, Micronesia (2). The next significant outbreak occurred in 2015 in Brazil and rapidly spread throughout the Americas, infecting nearly 2 million people (3). ZIKV is transmitted to humans through the bites of mosquitoes (Aedes aegypti and Aedes albopictus). Unlike dengue virus and Japanese encephalitis virus, ZIKV can also be spread by blood-borne, transplacental, and sexual transmission (4). Although the infection of ZIKV often leads to mild and self-limiting febrile diseases such as fever, rash, conjunctivitis, and arthralgia, it is also associated with Guillain–Barré syndrome and severe birth defects, most notably fetal microcephaly (5–7). Nowadays, ZIKV has become a global pandemic threat due to its rapid spread and increased virulence.
ZIKV is a positive single-stranded RNA virus with a genomic size of 10.7 kb (8). ZIKV virion binds to host cell-surface receptors, leading to receptor-mediated endocytosis. Upon the viral genome, RNA is released into the cytoplasm, and is translated into a single polypeptide, which is subsequently cleaved by viral and host proteases into three structural (C, prM, and E) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins (9). The viral RNA is synthesized within the replication complex located on the surface of endoplasmic reticulum, followed by viral protein translation. Then progeny virions are assembled within the endoplasmic reticulum, become matured during transit through the Golgi, and are released into the extracellular environment (10).
ZIKV is divided into two major lineages, the Asian lineage and the African lineage (11). The Asian lineage has been evolved to modified strains that are responsible for neurological complications including Guillain–Barré syndrome and microcephaly (12). Recent reports have revealed that the genetic changes of ZIKV RNA genome during its evolution contribute to the virulence, pathogenesis, and epidemic of contemporary ZIKV strains (7). The bioinformatics analysis comparing available sequences of ZIKV strains showed that the guanine usage is growing in the ZIKV RNA-positive strand due to adenine to guanine transitions, which is mediated by the family of adenosine deaminases acting on dsRNA (ADARs) (13, 14). The ADAR family includes three members: ADAR1, ADAR2, and ADAR3 (15). ADAR1 and ADAR2 catalyze the hydrolytic C6 deamination of adenosine to produce inosine in dsRNA. The ADAR2 protein is constitutively expressed and mainly localizes to the nucleus (16). ADAR1, as the most prevalent form of ADARs, has two forms: a full-length p150 form and a shorter p110 form. The p150 form possesses two Z-DNA–binding domains (Zα and Zβ), three dsRNA-binding domains (dsRBD), and a deaminase catalytic domain, whereas the p110 form lacks the Zα domain (16). The p150 form is IFN-inducible and located in both cytoplasm and nucleus, whereas the p110 form is constitutively and ubiquitously expressed, localized predominantly to nucleus (17). Functionally, ADAR1 plays an essential role in the development, apoptosis, miRNA processing, etc. (18). The mutations of ADAR1 have been linked to the severe human autoimmune disease, Aicardi–Goutiéres syndrome (19).
Not surprisingly, as a dsRNA-binding and -editing protein, ADAR1 is involved in the infection of many viruses (16). Initially, ADAR1 was known as an antiviral factor regulated by IFNs, whereas accumulating literature has shown that ADAR1 acts as a proviral factor in many viral infections, such as HIV-1, measles virus, vesicular stomatitis virus (VSV), and hepatitis D virus (17, 20–22). The proviral role of ADAR1 could be exerted in either an editing-dependent or editing-independent manner (23). ADAR1 edits the viral RNA substrates at specific sites (such as the 5′-UTR of HIV) or at multiple nonspecific sites (hyperediting) to promote their replication (18). In an editing-independent manner, ADAR1 inhibits the antiviral IFN production through suppressing RIG-I RNA binding (24, 25) or inhibits the dsRNA-regulated protein kinase (PKR), a classic antiviral protein that blocks the viral protein synthesis by phosphorylating the eukaryotic translation initiation factor 2α (eIF2α) (26, 27).
Although the epidemiology studies indicated that the host editing mediated by ADAR might be a force leading to ZIKV evolution, whether ADAR is involved in ZIKV acute infection remains unknown. To probe the role of ADAR1 in the ZIKV infection, we utilized CRISPR/Cas9 and RNAi techniques to down-regulate the levels of endogenous ADAR1. We found that ADAR1 was a proviral factor for ZIKV replication through inhibiting the activation of PKR and then enhancing the viral protein synthesis. The N-terminal Z-DNA–binding domain and dsRNA-binding domains of ADAR1 were sufficient for its proviral effect. These findings demonstrated that ADAR1 plays an important role in the acute infection of ZIKV, suggesting ADAR1 as a potential target for the development of antiviral agents.
Results
ADAR1 promotes the replication of ZIKV
To test whether ADAR1 has an impact on the ZIKV replication, we utilized the CRISPR/Cas9 gene-editing technique to generate the ADAR1 knockout (KO) cells. Two independent ADAR1 KO (both p150 and p110 forms) cells were isolated, in which the disruption of the ADAR1 gene was confirmed by genomic DNA sequencing (data not shown). The Western blotting data showed that no ADAR1 protein was detected in ADAR1 KO1 and ADAR1 KO2 cells, even upon the stimulation of IFN-β (Fig. 1A). The cell viability and growth rate between the control cells and two ADAR1 KO cells were comparable (Fig. 1B). Then, the control cells and ADAR1 KO cells were infected with ZIKV at a multiplicity of infection (MOI) of 3. The quantitative reverse transcription PCR (qRT-PCR) data showed that the viral RNA levels at 6, 9, and 12 h postinfection (p.i.) in both ADAR1 KO cells were significantly lower than in the control cells (p < 0.01; Fig. 1C); similarly, the viral E protein levels in ADAR1 KO cells were largely reduced (Fig. 1D), and the percentages of E-positive staining cells measured by flow cytometry were significantly down-regulated (Fig. 1, E and F). The plaque assay data revealed that the viral titers in ADAR1 KO1 and KO2 cells were 87.9 and 88.3% lower than the control cells, respectively (all p < 0.001; Fig. 1G). To explore whether ADAR1 affects the replication and transmission of ZIKV, we performed a multistep virus growth assay. The cells were infected by ZIKV at MOI 0.01, and the viral supernatants were collected at 24, 48, and 72 h p.i. for the plaque assay. At 24 h p.i., the ZIKV titers in all three cells were barely detected; and at 48 and 72 h p.i., the ZIKV titers in the ADAR1 KO cells were significantly reduced (all p < 0.05; Fig. 1H). These observations indicated that ADAR1 promotes the ZIKV replication.
Figure 1.

The ZIKV replication was impaired in ADAR1 knockout cells. A, Western blotting to detect the levels of ADAR1. The ADAR1 KO A549 cells were generated by the CRISPR/Cas9 technique. Two different knockout cells (ADAR1 KO1 and ADAR1 KO2) were confirmed by genomic DNA sequencing and subjected to Western blotting. GAPDH was probed as an internal control. The ratios of p110/actin were quantified by Quantity One, as shown below the representative blot of three independent experiments. B, cell growth curve of ADAR1 KO cells. Cell numbers were counted by trypan blue staining. C–G, virus replication levels in the ADAR1 KO cells. Control cells or ADAR1 KO cells were infected with ZIKV at MOI 3. Cells were harvested at the indicated time points for viral RNA measurement (C) or at 24 h p.i. for Western blotting (D) or flow cytometry (E and F); the supernatants were collected at 24 h p.i. for the plaque assay (G). H, multistep virus growth assay. Control cells and ADAR1 KO cells were infected with ZIKV at MOI 0.01. At 24, 48, and 72 h p.i., the supernatants were collected for the plaque assay. Data are shown as mean ± S.D. (error bars) of at least three independent experiments. **, p < 0.01; ***, p < 0.001, unpaired, two-tailed Student's t test.
We further validated the proviral effect of ADAR1 by silencing ADAR1 through an RNAi strategy. Cells were transfected with a negative control siRNA (siNC) or two different siADAR1s (siADAR1-1 or siADAR1-2) targeting the ADAR1 gene. The protein levels of ADAR1 p150 and p110 forms in the A549 cells transfected with siADAR1s were significantly lower than in the cells transfected with siNC (Fig. 2A), confirming that the knockdown of ADAR1 expression by these siRNAs was effective. The control cells and ADAR1 knockdown cells were infected with ZIKV at MOI 3 and harvested for detection of viral replication levels. The ADAR1 silencing led to a significant reduction of E protein accumulation (Fig. 2A) and viral RNA levels at 6, 9, and 12 h p.i. (all p < 0.01; Fig. 2B). The viral yields of ZIKV in the ADAR1 knockdown cells were decreased by 85.0 and 72.5% (all p < 0.01; Fig. 2C). In the multistep virus growth assay, the ADAR1 knockdown resulted in significantly lower viral yields at 48 h and 72 h p.i. (all p < 0.05; Fig. 2D).
Figure 2.

Effect of ADAR1 knockdown on the ZIKV replication. A–C, virus replication levels. A549 cells were transfected with siRNAs for 48 h, followed by ZIKV infection at MOI 3. Cells were harvested at 24 h p.i. for real-time PCR to measure the viral RNA level (A) or for Western blotting to measure the viral protein E level (B); the supernatants were collected at 24 h p.i. for the plaque assay (C). D, multistep virus growth assay. siRNA-transfected cells were infected with ZIKV at MOI 0.01. At 24, 48, and 72 h p.i., the supernatants were collected for the plaque assay. E and F, viral replication levels in SNB19 cells. The SNB19 cells were transfected with siRNA, followed by ZIKV infection at MOI 3. Cells and supernatants were harvested at 24 h, and viral protein levels (E) and viral yields (F) were measured as above. Data are shown as mean ± S.D. (error bars) of at least three independent experiments. **, p < 0.01; ***, p < 0.001, unpaired, two-tailed Student's t test.
As ZIKV infection is associated with severe neurological diseases (7), we examined the role of ADAR1 in the ZIKV replication in human glioblastoma SNB19 cells. Transfection of siADAR1-1 or siADAR1-2 in SNB19 cells led to more than 80% reduction of ADAR1 protein levels (Fig. 2E). As expected, the viral E protein levels and viral titers in the ADAR1 knockdown cells were significantly reduced in SNB19 cells (Fig. 2, F and G), suggesting that the proviral effect of ADAR1 was not cell-specific.
Both p110 and p150 forms of ADAR1 confer the proviral activity
To analyze which form of ADAR1, p110 or p150, mediates its proviral effect, we introduced the WT p110 or p150 genes into the ADAR1 KO cells by lentivirus-mediated transduction. The p110– or p150–trans-complemented cells were sorted by flow cytometry. It is noteworthy that a p110 protein band was found in the p150-complemented cells, even the ATG codon of p110 was mutated (28). The control cells, the p110–trans-complemented cells, and the p150–trans-complemented cells were infected with ZIKV at MOI 3, and the viral replication levels were measured at 24 h p.i. Both the p110 or p150 trans-complementation largely restored the E protein levels in the ADAR1 KO cells (Fig. 3A). Consistently, the ZIKV titers in the p110- or p150-expressing cells were enhanced by 4.2- or 5.8-fold, respectively (all p < 0.01; Fig. 3B), suggesting that either the p110 or p150 form of ADAR1 was sufficient to support the replication of ZIKV.
Figure 3.
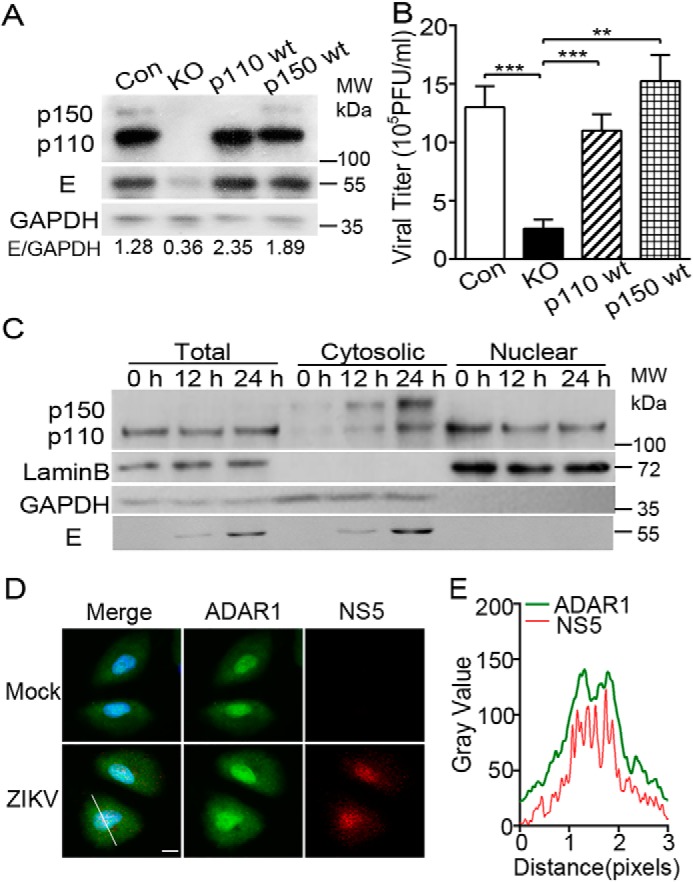
Role of ADAR1 p110 and p150 forms in the ZIKV replication. A and B, replication levels of ZIKV virus in p110- and p150-complemented cells. The ADAR1 KO cells were transduced with p110- and p150-expressing lentiviral constructs and sorted by flow cytometry. Cells were infected with ZIKV at MOI 3. Cells and supernatants were collected at 24 h p.i. for Western blotting (A) and plaque assay (B). Western blots were probed with antibodies against ADAR1, E, or β-actin. Data are shown as mean ± S.D. (error bars) of three independent experiments. **, p < 0.01; ***, p < 0.001, unpaired, two-tailed Student's t test. C, A549 cells were infected with mock or ZIKV. Cells were harvested at 0, 12, and 24 h p.i. for protein extraction. Proteins derived from cytoplasmic fraction and nuclear fraction of the cells were subjected to Western blotting. GAPDH and LaminB served as the loading control for cytoplasmic fraction and nuclear fraction. NS5 served as the control for ZIKV infection. D, A549 cells were infected with mock or ZIKV (MOI 3). Cells were fixed at 24 h p.i. and processed for immunofluorescence microscopy detecting ADAR1 (green), ZIKV NS5 (red), and DAPI (blue). Scale bar, 10 μm. E, co-localization analysis was performed by ImageJ.
As the p110 form of ADAR1 predominantly resides in the nucleus whereas ZIKV replicates in the cytoplasm, we examined the localization of ADAR1 p150 and p110 forms in the mock-infected and ZIKV-infected cells. The cells were collected at 0, 12, and 24 h p.i. for preparation of whole-cell extraction or subcellular fractionation to separate the cytoplasmic and nuclear fractions. In response to ZIKV infection, the levels of both cytoplasmic p150 and p110 forms were increased, whereas the nuclear p110 levels were decreased. The increase of the p150 form could be partially mediated by the IFNs, whereas more p110 protein presented in the cytoplasm upon viral infection implied that the p110 might be recruited by ZIKV for its replication (Fig. 3C). As a control, the viral E protein levels increased as time elapsed, validating that the cells were infected.
Furthermore, the mock- and ZIKV-infected A549 cells were collected for the immunofluorescence microscopy assay to monitor the localization of ADAR1 and viral NS5 protein. In the ZIKV-infected cells, the ADAR1 protein and NS5 protein were both diffusely distributed in the cytoplasm and nuclei (Fig. 3D). Co-location analysis performed by ImageJ showed that the ADAR1 was co-localized with viral NS5 protein (Fig. 3E).
ADAR1 promotes the replication of ZIKV at the viral protein synthesis step
To identify which step of the viral life cycle ADAR1 acts at, we first examined ADAR1's role in the viral entry step. The control cells and ADAR1 KO cells were inoculated with the ZIKV virions at 4 °C for 1 h, in which condition the virions could bind the receptors but not be internalized. Total RNA was extracted for qRT-PCR measurement. We did not observe a difference of viral RNA levels between the control and ADAR1 KO cells (p > 0.05; Fig. 4A), indicating that ADAR1 did not affect the viral attachment. Then we incubated the ZIKV virions at 37 °C for 1 h to allow them to attach and internalize, followed by total RNA extraction and qRT-PCR. The viral RNA levels in ADAR1 KO cells were comparable with the control cells (p > 0.05; Fig. 4A), suggesting that ADAR1 is not involved in the viral endocytosis step.
Figure 4.

ADAR1 acted at the ZIKV protein translation step. A, role of ADAR1 in the viral entry. Control cells or ADAR1 KO cells were inoculated with ZIKV at MOI 3, followed by incubation at 4 °C or 37 °C for 1 h. Cells were extensively washed with PBS and harvested for RNA extraction and qRT-PCR. B, replicon assay. A549 cells were transfected with WT or GDD mutant viral RNAs. The cells were harvested at the indicated time points for the luciferase assay. Data are shown as mean ± S.D. of at least three independent experiments. C, Western blot analysis. Vero cells were transfected with siNC or siADAR1-2 for 24 h, followed by ZIKV infection at MOI 3. Cells were harvested at 24 h p.i. for Western blotting to measure the siRNA efficiency and the viral protein E levels. D, replicon assay. Vero cells were transfected with siNC or siADAR1-2 for 24 h, followed by transfection with WT or GDD mutant viral RNAs. The cells were harvested at the indicated time points for the luciferase assay. Data are shown as mean ± S.D. (error bars) of at least three independent experiments.
To distinguish between the ADAR1 impact on protein translation and RNA replication, we utilized two subgenomic ZIKV replicons, WT or the NS5GDD mutant ZIKV replicons, which encode seven NS proteins with a Renilla luciferase reporter that replaced the structural proteins (29). The NS5GDD mutant replicon was used as an indicator of protein translation as the RNA-dependent RNA polymerase activity of NS5 was abolished. Two peaks of luciferase activities at 6 and 48 h post-transfection were detected in WT replicon RNA-transfected Vero cells, which represented the protein translation and RNA synthesis, respectively (data not shown). In contrast, only one peak of luciferase activity at 6 h post-transfection was seen in the NS5GDD mutant RNA-transfected Vero cells (data not shown). Then we compared the luciferase activities of the WT and NS5GDD mutant replicons in the control and ADAR1 KO A549 cells. Unfortunately, A549 cells were refractory to RNA transfection, so most of the cells died at 48 h post-transfection (data not shown). Therefore, our data only showed the relative luciferase activities measured with the cell lysates harvested at 6, 12, and 24 h post-transfection. At 6 h post-transfection, the luciferase activities of WT ZIKV and NS5GDD mutant replicons in the ADAR1 KO cells were significantly lower than in the control cells (p < 0.05 and 0.001, respectively; Fig. 4B), indicating that ADAR1 is involved in the viral protein translation. Similarly, significantly lower luciferase activities were achieved in the ADAR1 KO cells at 24 h post-transfection (p < 0.05 and 0.01, respectively; Fig. 4B).
Moreover, we explored whether ADAR1 affects the viral protein translation in Vero cells. The cells were transfected with siNC or siADAR1-2, followed by ZIKV infection. Transfection of siADAR1-2 led to a significantly lower protein level of ADAR1 in Vero cells (Fig. 4C). The ZIKV E protein level in the ADAR1 knockdown cells was significantly reduced (Fig. 4C), indicative of a supportive role of ADAR1 in Vero cells. Then, the luciferase activities of the WT and NS5GDD mutant replicons were compared in the control and ADAR1 knockdown Vero cells. Similar to the observations in A549 cells, the absence of ADAR1 resulted in 1.9–2.1-fold lower luciferase activities of WT and NS5GDD replicon RNAs at 6 and 48 h post-transfection (p < 0.05; Fig. 4D). The luciferase activities at 48 h post-transfection in the ADAR1 knockdown cells did not decrease further than at 6 h post-transfection, suggesting that the viral RNA replication was not influenced by the loss of ADAR1.
The proviral role of ADAR1 is mediated by suppressing PKR phosphorylation
Given that the proviral effect of ADAR1 occurred in the early translation step of ZIKV, we proposed that ADAR1 functions through interfering with the cellular translation machinery. As ADAR1 has been reported to inhibit the activation of PKR (30), a major eIF2α kinase in the context of viral infection (31), we hypothesized that the promotion of ADAR1 on the ZIKV protein translation might be exerted through PKR. To verify this hypothesis, we measured the levels of phosphorylated PKR and eIF2α in the control cells and ADAR1 KO cells. The cells were infected with ZIKV at MOI 3 and harvested at 6 or 18 h p.i. for Western blotting. The phosphorylation levels of PKR and eIF2α in the ADAR1 KO cells at all time points were significantly higher than those in the control cells (Fig. 5A). Correspondingly, the levels of ZIKV E protein in the ADAR1 KO cells were lower than in the control cells (Fig. 5A).
Figure 5.

The proviral role of ADAR1 was mediated through PKR, but not RNase L. A, Western blot analysis. The control cells and ADAR1 KO cells were infected with mock or ZIKV at MOI 3. Cells were collected at the indicated time points for Western blotting using the indicated antibodies. B and C, viral replication levels. The control cells and stable PKR knockdown cells were transfected with siNC or siADAR1-2, followed by ZIKV infection at MOI 3. Cells and the supernatants were harvested at 24 h p.i. The viral protein levels (B) and viral yields (C) were measured by Western blotting and a plaque assay as above. The Western blotting bands were quantified by Quantity One as shown below the representative blot of three independent experiments. D, Western blot analysis. The control cells and ADAR1 KO cells were infected with mock or ZIKV at MOI 3. At 24 h p.i., cells were harvested for Western blotting using the indicated antibodies. E, RNA integrity assay. The control cells and ADAR1 KO cells were transfected with mock, poly(I:C), or WT replicon RNAs. Total RNAs were extracted at 6 h post-transfection, and the integrity of 28S and 18S rRNA were measured by RNA electrophoresis (F) or by Agilent 2100 BioAnalyzer to determine the 28S/18S and RNA integrity number. G and H, viral replication levels. The control cells and RNase LKO cells were transfected with siNC or siADAR1-2, followed by ZIKV infection at MOI 3. At 24 h, cells and supernatants were collected for Western blotting and plaque assay. Data are shown as mean ± S.D. (error bars) of at least three independent experiments. NS, not significant; **, p < 0.01; ***, p < 0.001, unpaired, two-tailed Student's t test.
To confirm that PKR confers an anti-ZIKV activity and that its inhibition by ADAR1 contributes to the proviral effect of ADAR1, we generated two stable PKR knockdown cell lines using two different shRNAs targeting different regions of PKR (shPKR90 and shPKR405) as reported previously (32). The PKR knockdown cells were transfected with siNC or siADAR1, followed by ZIKV infection. At 24 h p.i., the Western blotting and plaque assay were performed. The PKR levels in two PKR knockdown cells were effectively depleted (Fig. 5B). As a consequence, the levels of p-PKR and p-eIF2α in the PKR knockdown cells were dramatically reduced. In both PKR knockdown cells, the viral E proteins were markedly higher than in the control cells, demonstrating that PKR plays an antiviral role in ZIKV replication. As expected, transfection of siADAR1 led to elevated levels of p-PKR and p-eIF2α and lower E protein accumulation. However, ADAR1 knockdown did not result in a significant decrease of E protein level in two PKR knockdown cells (Fig. 5B). Consistently, the viral yields in two PKR knockdown cells were 3–5-fold higher than in the control cells (Fig. 5C). Knockdown of ADAR1 led to significantly lower viral yields in PKR-sufficient cells, whereas it slightly but not significantly reduced the viral titers in the absence of PKR (Fig. 5C), suggesting that the PKR was a major mediator of ADAR1 proviral function.
As ADAR1 has been reported to regulate the activation of RNase L, which degrades the RNA and confers an antiviral activity against some viruses (33), we next examined whether RNase L is responsible for the proviral effect of ADAR1. The RNase L protein level in ADAR1 KO cells was slightly higher than in the mock-infected cells, but the RNase L levels in two ZIKV-infected cells were similar (Fig. 5D). Next, we examined the RNA integrity by RNA electrophoresis and an rRNA cleavage assay. Control cells and ADAR1 KO cells were transfected with poly(I:C) or ZIKV replicon RNAs and harvested at 6 h post-transfection. The RNA gel showed that three major rRNA bands (28S, 18S, and 5S) remained intact in both control cells and ADAR1 KO cells upon transfection with ZIKV replicon RNA (Fig. 5E). In contrast, rRNA degradation was observed in the poly(I:C)-stimulated cells (Fig. 5E). Further rRNA cleavage assay data showed that no significant difference of 28S/18S and RNA integrity number was observed among these samples (Fig. 5F). These observations demonstrated that the RNA stability was not affected by ADAR1 KO at 6 h post-transfection of replicon RNA. To probe the role of RNase L in the ZIKV replication, we generated RNase L KO cells by the CRISPR/Cas9 technique. The RNase L KO cells were transfected with siNC or siADAR1, followed by ZIKV infection. At 24 h p.i., Western blotting and plaque assay were performed. Our data showed that the E protein accumulation (Fig. 5G) and the viral yields (Fig. 5H, white bars) in the RNase L KO cells were comparable with the control cells, consistent with recent work reporting that ZIKV production is resistant to RNase L antiviral activity (34). Of note, the ADAR1 knockdown in the control cells and RNase L KO cells resulted in similar reductions of E protein levels and viral yields (Fig. 5, G and H), indicating that the proviral effect of ADAR1 was not exerted through RNase L.
The editing activity of ADAR1 is not required for its proviral function
To pinpoint whether the RNA deaminase activity is required for ADAR1 to promote ZIKV replication, we introduced two point mutations in the deaminase domain (H910Q and E912A) of p110 and p150 forms (28), generating two plasmids expressing editing-deficient p110 and p150 mutants (p110C and p150C). The p110C and p150C genes were transduced into ADAR1 KO cells by lentivirus-mediated transduction, and cells were sorted by flow cytometry. The control cells, ADAR1 KO cells, and transduced cells expressing WT p110, p110C, WT 150, and p150C were infected with ZIKV at MOI 3. At 24 h p.i., the cells and supernatants were collected for Western blotting (Fig. 6A) and plaque assay, respectively (Fig. 6B). In all of the WT and mutant ADAR1–trans-complemented cells, the phosphorylation levels of PKR and eIF2α were significantly lower than in the ADAR1 KO cells. We did not notice a significant difference of p-PKR and p-eIF2α levels between the cells expressing WT (p110 and p150) and mutant ADAR1 (p110C and p150C) (Fig. 6A). Consistently, the viral E protein levels were restored by the trans-complementation of either WT or mutant ADAR1 (Fig. 6A). As expected, the viral yields in the p110C- or p150C-expressing cells were comparable with those in the p110- or p150-expressing cells, around 3.6- and 4.3-fold higher than the ADAR1 KO cells (p < 0.01; Fig. 6B).
Figure 6.

The deaminase activity of ADAR1 was not required to promote viral replication. The ADAR1 KO cells were transduced with the lentiviral constructs expressing WT p110 or p150 form or their mutants lacking the deaminase activity (H910Q/E912A) (p110 C and p150 C). The transduced cells were sorted by flow cytometry. Cells were infected by ZIKV at MOI 3. Cells and supernatants were collected at 24 h p.i. for Western blotting (A) and plaque assay (B). Western blots were probed with the indicated antibodies. C and D, replicon assay. Cells were transfected with pFK-SGR (C) or pFK-SGR-GDD (D) RNAs. The cells were harvested at the indicated time points for the luciferase assay. E and F, viral replication levels. The control cells and ADAR1 KO, p110-expressing, and p110N-expressing cells were infected with ZIKV. The viral E protein levels and viral titers were measured by Western blotting (E) or plaque assay (F) as above. Data are shown as means ± S.D. (error bars) of three independent experiments. **, p < 0.01; ***, p < 0.001, unpaired, two-tailed Student's t test.
To further validate the role of ADAR1-editing activity in the viral replication, we compared the luciferase activities of the ZIKV replicons between the WT ADAR1 and the editing-deficient mutant-expressing cells. The control cells, ADAR1 KO cells, ADAR1-complemented cells (p110 and p150), and ADAR1 mutant–complemented cells (p110C and p150C) were transfected with ZIKV replicon RNAs and harvested at 6 h post-transfection. The luciferase activities of WT replicon RNA in the WT p110, p110C, WT p150, and p150C cells were increased by 1.6-, 1.7-, 1.7-, and 1.6-fold compared with the ADAR1 KO cells, respectively (all p < 0.01; Fig. 6C); similarly, the trans-complementation of WT or mutant ADAR1 largely restored the luciferase activities of NS5GDD mutant replicon (all p < 0.01; Fig. 6D).
To examine the involvement of the ADAR1 N-terminal domain in the ZIKV replication, we constructed the plasmid expressing a truncated p110 protein, which contains its Zβ-DNA–binding domain and three dsRNA-binding domains (p110N) but not the catalytic domain. The p110N-expressing cells were established by transducing and sorting. The viral replication levels in control cells, ADAR1 KO cells, and p110- or p110N-expressing cells were compared by Western blotting and plaque assay. The phosphorylation of PKR and eIF2α induced by ZIKV infection were largely impaired in the p110N-expressing cells (Fig. 6E). As expected, the viral E protein level of ZIKV was completely restored by p110N trans-complementation (Fig. 6E). In addition, the viral yield in the p110N-expressing cells was fully rescued, similar to the p110-expressing cells (p < 0.001; Fig. 6F).
The above data demonstrated that the Z-DNA–binding domain and dsRNA-binding domains of ADAR1, instead of its catalytic domain, are important for its proviral role.
The proviral role of ADAR1 is partially mediated by suppressing the IFN production
As ADAR1 has been implicated to suppress the IFN production (25, 35–37) and ZIKV is sensitive to IFN action (10, 38), we further tested whether ADAR1 is involved in the IFN production induced by ZIKV. The mRNA levels of IFN-β in the control and ADAR1 KO cells were compared by qRT-PCR. As expected, the ADAR1 knockout led to an increased level of IFN-β mRNA (all p < 0.01; Fig. 7A). To investigate whether the elevated IFNs produced in the ADAR1 KO cells has an influence on the viral replication, we utilized the IFNAR1 knockout cells (IFNAR1KO) (39) and STAT1 knockout cells (STAT1KO), in which the IFNAR1 or STAT1 gene was ablated by CRISPR/Cas9-based gene editing. The level of ISG protein PKR in the control cells was enhanced by more than 2-fold upon IFN-β stimulation, but not altered in the IFNAR1KO and STAT1KO cells (Fig. 7B), confirming that the knockout of IFNAR1 or STAT1 effectively blocked the IFN signaling pathway.
Figure 7.

The ZIKV-induced IFN level was regulated by ADAR1 and partially contributed to the ADAR1 proviral effect. A, real-time PCR to measure the IFN-β mRNA levels. Control cells and ADAR1 KO cells were mock-infected or infected with ZIKV at MOI 3. Cells were collected at the indicated time points for RNA extraction and qRT-PCR. B, Western blot analysis. IFNAR1KO and STAT1KO cells were stimulated by IFN-β. At 24 h, cells were harvested for Western blotting using the indicated antibodies. GAPDH was probed as an internal control. C and D, viral replication levels. Control cells, IFNAR1KO, and STAT1KO cells were transfected with siNC or siADAR1. At 48 h post-transfection, cells were infected with ZIKV at MOI 3. At 24 h p.i., the cells were collected for Western blotting using antibodies against ADAR1, p-PKR, PKR, p-eIF2α, eIF2α, E, or GAPDH (C), and the supernatants were collected for the plaque assay (D). Data are shown as means ± S.D. (error bars) of three independent experiments. **, p < 0.01; ***, p < 0.001.
Next, we transfected the siADAR1-2 into the control cells, IFNAR1KO cells, and STAT1KO cells, followed by ZIKV infection at 48 h post-transfection. At 24 h p.i., the cells and supernatants were collected for Western blotting and plaque assay. As shown in Fig. 7C, the loss of IFNAR1 and STAT1 decreased the levels of PKR, and the phosphorylation forms of PKR and eIF2α, leading to higher E protein levels (Fig. 7C, lanes 3 and 5 versus lane 1). The ADAR1 knockdown in all three cells increased the levels of p-PKR and p-eIF2α and reduced the viral E protein levels (Fig. 7C, lane 1 versus lane 2, lane 3 versus lane 4, and lane 5 versus lane 6). Notably, in the siADAR1-transfected IFNAR1KO or STAT1KO cells, the levels of p-PKR and p-eIF2α were much lower than in the ADAR1-silenced control cells, and higher E protein levels were observed when IFN signaling was blocked (Fig. 7C, lanes 4 and 6 versus lane 2). Consistently, the viral yields in the IFNAR1KO and STAT1KO cells were higher than in the control cells (Fig. 7D). The ADAR1 knockdown led to significantly lower viral yields in all three cells (p < 0.01 and 0.001; Fig. 7D). However, the -fold reduction mediated by ADAR1 knockdown was only 1.9–2.0-fold in the IFNAR1KO and STAT1KO cells, which was lower than in the control cells (p < 0.001 and 0.01; Fig. 7D). These data suggested that ADAR1 regulates the phosphorylation of PKR and eIF2α in both an IFN-dependent and IFN-independent manner, and its inhibitory effect on the IFN production partially contributes to its proviral effect.
Discussion
ADAR1, a classic IFN-inducible protein, has been indicated to positively or negatively regulate the replication of many RNA viruses in an editing-dependent or editing-independent manner (18, 23). Our study demonstrated that in addition to its role in the ZIKV evolution, ADAR1 is involved in the ZIKV life cycle in in vitro cultured cells in an editing-independent manner.
Our study presented several pieces of evidence to demonstrate that ADAR1 plays a proviral role in the ZIKV replication. First, the disruption of the ADAR1 gene in two independent A549 cell clones by the CRISPR/Cas9 technique resulted in a dramatic decrease of viral replication. Second, the siRNA-mediated ADAR1 knockdown in both A549 cells and SNB19 cells led to significant reductions of viral replication levels, indicating that the involvement of ADAR1 in ZIKV replication was not cell-specific. Third, the trans-complementation of either the p110 or p150 form of ADAR1 in the ADAR1 KO cells largely restored the replication levels of ZIKV, illustrating that both p110 and p150 forms of ADAR1 confer a proviral activity. Interestingly, more of the p110 form of ADAR1 was cytoplasmic upon ZIKV infection, providing an additional piece of evidence that the p110 is utilized by ZIKV.
To be noted, the ADAR1 KO A549 cells were successfully generated, and no difference of cell growth between WT cells and ADAR1 KO cells was observed in our study. In contrast, Li et al. (33) previously reported that the ADAR1 deletion in A549 cells led to a cell-lethal phenotype, which could be rescued by an ablation of RNase L. The exact reasons behind these contradictory observations are currently unknown but may relate to different single guide RNAs (sgRNAs) that were used in these studies. Our study agreed with two recent reports showing that knockout of ADAR1 in 293T cells, human embryonic stem cells, and A549 cells was successful, despite the fact that ADAR1 is essential for survival of a subset of cancer cell lines, including HCC366, NCI-H196, and NCI-H1650 cells (40, 41).
We found that ADAR1 functions at the protein translation in the ZIKV life cycle. In the control and ADAR1 KO cells, the RNA levels of the virions attached to the cells or internalized were comparable, indicating that the ADAR1 was not required for the viral entry. The observations that both the early RNA and protein levels were dramatically reduced in the ADAR1 KO cells promoted us to deduce that ADAR1 acts at an early stage of viral replication. Our hypothesis was supported by the ZIKV reporter replicon assay data, showing that the protein translation levels of both WT ZIKV and NS5GDD mutant replicon RNAs were significantly impaired in the ADAR1-depleted A549 cells and Vero cells. As in the ADAR1 knockdown cells, the reductions of luciferase activity at 6 and 48 h were comparable, so we excluded a role of ADAR1 in the viral RNA synthesis. The finding that phosphorylation levels of PKR and eIF2α were increased in the absence of ADAR1 suggested that ADAR1 facilitates the viral protein translation via inhibiting PKR (42). This finding was consistent with the studies with VSV and measles virus (17, 43). On the other hand, we found that the RNase L is not involved in the ADAR1 proviral effect, in keeping with recent work showing that the RNase L does not affect the viral yield of ZIKV (34).
Previously, Chung et al. (40) reported that in response to IFN stimulation, the expression of ADAR1 editing–defective mutant impairs the levels of p-PKR at 24 h post-treatment, but the inhibition is lost at 48 h post-treatment. They proposed that ADAR1 plays both editing- and nonediting-dependent roles in PKR phosphorylation induced by IFNs. In contrast, our study showed that the trans-complementation of all WT and mutant ADAR1 forms, even the truncated p110N protein, was able to inhibit the phosphorylation levels of PKR and eIF2α and to restore the viral replication levels. Our finding agreed with the report by Nie et al. (26) that the N-terminal Z-DNA-binding domains and dsRBDs, but not the C-terminal deaminase domain of ADAR1, are important to promote the VSV infection. It is worth pointing out that, although the deaminase activity of ADAR1 was not essential during viral acute infection, its editing activity of the viral RNA genome still has an impact on the ZIKV evolution, contributing to the sequence diversity and the change of virulence and pathogenesis (13, 14).
Finally, our study found that the ADAR1 negatively regulates the IFN levels induced by ZIKV, as seen in other virus studies (3, 36, 37). The viral replication levels of ZIKV in the IFNAR1KO and STAT1KO cells significantly decreased upon ADAR1 knockdown, indicating that at least part of the ADAR1 proviral effect is independent of IFN production. On the other hand, we noticed that the viral replication levels in the ADAR1-depleted IFNAR1KO and STAT1KO cells were substantially higher than in the ADAR1 knockdown control cells, implying that the proviral role of ADAR1 is also partially mediated by the IFN signaling pathway. This finding is not unexpected, because the PKR is an ISG protein, whose activity is regulated by IFNs. Our conclusion agreed with the study on ADAR1 and HCV, showing that the ADAR1 knockdown up-regulated the RIG-I signaling pathway induced by HCV infection and led to an increased viral replication (35).
Taking our results together, we propose that during acute infection of ZIKV, the ADAR1 promotes the viral replication through inhibiting the PKR activation and then enhancing the viral protein synthesis. The inhibitory effect of ADAR1 on the PKR activation could be both IFN-independent and IFN-dependent. The ADAR1-editing activity is not required for its interaction with PKR (30) and is not required for promoting the ZIKV replication. Overall, our study sheds the first light on the proviral role of ADAR1 in the ZIKV replication, suggesting that ADAR1 could be a potential target of antiviral drugs.
Experimental procedures
Cell culture
Human lung carcinoma epithelial cells (A549, ATCC CCL-185), human embryonic kidney cells (293T, ATCC CRL-3216), glioblastoma cells (SNB19, ATCC CRL-2219), and African green monkey kidney cells (Vero, ATCC CCL-81) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco) at 37 °C with 5% CO2. The media were added with 100 units/ml streptomycin and penicillin (Invitrogen).
Virus, virus infection, and titration
The ZIKV (H/PF/2013 strain) was provided by Guangzhou Centers for Disease Control and propagated in Vero cells. The supernatants were collected when the cytopathic effect appeared and the cellular debris was removed by centrifugation. Virus stocks were titered and stored at −80 °C.
In the single-step virus growth assay, A549 cells were infected with ZIKV at MOI 3. The cells were harvested at the indicated time points for Western blotting or real-time PCR. The supernatants were harvested at 24 h p.i. for virus titration. In the multistep virus growth assay, cells were infected with ZIKV at MOI 0.01. The supernatants were harvested at 24, 48, and 72 h p.i.
Virus titers of ZIKV were determined by a standard plaque assay on Vero cells. Serial 10-fold dilutions of each sample were prepared, and 100 μl/well of the diluted virus were added into the 12-well plates. The incubation medium was removed and cultured in the mixture of 2× Dulbecco's modified Eagle's medium (Invitrogen) and 2% methylcellulose (Sigma) (1:1). Visible plaques were counted at 3–4 days p.i.
Antibodies
Primary antibodies included anti-ADAR1, anti-PKR, anti-eIF2α (Santa Cruz Biotechnology, Inc.), anti-ZIKV envelope (E) (Biofront or Gene Tex), anti-ZIKV NS5 protein (Gene Tex), anti-phospho-PKR, anti-phospho-eIF2α (CST), anti-β-actin (Sigma), anti-tubulin (Beijing Ray Antibody Biotech), and anti-GAPDH (Proteintech). Secondary antibodies included IRDye 800 CW–conjugated anti-rabbit IgG, IRDye 680 CW–conjugated anti-mouse IgG (LI-COR), horseradish peroxidase–conjugated anti-mouse IgG (CST), anti-rabbit IgG (Bio-Rad), and anti-goat IgG (LI-COR).
Plasmid construction and transfection
The oligonucleotide sequences used for generation of sgRNA are listed in Table 1. A pair of forward and reverse oligonucleotides for generation of each sgRNA were annealed and then inserted into plasmid vectors LentiCRISPR v2 between BsmBI restriction sites. The resulting plasmids were designated as pLenti-sgADAR1-1, pLenti-sgADAR1-2 (targeting the ADAR1 gene), pLenti-sgIFNAR1 (targeting the IFNAR1 gene), pLenti-sgSTAT1 (targeting the STAT1 gene), and pLenti-sgRNase L (targeting the RNase L gene).
Table 1.
Sequences of primers used in CRISPR/Cas9 gene editing
| Gene | Primer | Sequence (5′–3′) | Targeting region |
|---|---|---|---|
| ADAR1 | sgADAR1–1 | Forward: CACCGCTCGGCCATTGATGACAACC | Exon 3 |
| Reverse: AAACGGTTGTCATCAATGGCCGAGC | |||
| sgADAR1–2 | Forward: CACCGAGCCATGACAATTCTGCTAG | Exon 3 | |
| Reverse: AAACCTAGCAGAATTGTCATGGCTC | |||
| IFNAR1 | sgIFNAR1 | Forward: CACCGGATCTAATGTTAAAGACTGG | Exon 8 |
| Reverse: AAACCCAGTCTTTAACATTAGATCC | |||
| STAT1 | sgSTAT1 | Forward: CACCGTTCCCTATAGGATGTCTCAG | Intron 2 |
| Reverse: AAACCTGAGACATCCTATAGGGAAC | |||
| RNase L | sgRNase L | Forward: CACCGGCAGATCACCCACAGTGTTC | Intron 7 |
| Reverse: AAACGAACACTGTGGGTGATCTGCC |
Plasmids pcDNA6-ADAR1-p150 and pcDNA6-ADAR1-p110 were mutated at synonymous sites in sgRNA2-targeted sequence present in the human ADAR1 ORF to resist the knockout by sgRNA. To obtain plasmid encoding the ADAR1 lacking adenosine deaminases activity, the His at aa 910 of ADAR1 was mutated to Gln, and the Glu at aa 912 of ADAR1 was mutated to Ala by site-directed mutagenesis (the sequences of primers are listed in Table 2). The mutations were verified by sequencing. To amplify the N-terminal part of ADAR1-p110 (aa 1–501, p110N), pcDNA6-ADAR1-p110 was used as a template. The PCR primer sequences are listed in Table 2. The p150, p150C, p110, p110C, or p110N fragments were amplified by PCR and inserted into the lentiviral vector CSII-EF-MCS-IRES2-Venus.
Table 2.
Sequences of oligonucleotides used in cloning
| Gene | Sequence (5′–3′) |
|---|---|
| ADAR1-p110 | 5F: ATTTGCGGCCGCATGGCCGAGATCAAGGAGAAAAT |
| 3R: CGCGGATCCCTATACTGGGCAGAGATAAAAG | |
| ADAR1-p150 | 5F: ATTTGCGGCCGCATGAATCCGCGGCAGGGGTATT |
| 3R: ATTTGCGGCCGCCTATACTGGGCAGAGATAAAA | |
| ADAR1 (sgRNA2) mutation | 5F: GACAATTCTGCTCGAAGAAGCCAAAGCC |
| 3R: GGCTTTGGCTTCTTCGAGCAGAATTGTC | |
| ADAR1 (H910Q/E912A) mutation | 5F: CAATGACTGCCAGGCAGCAATAATCTC |
| 3R: GGGAGATTATTGCTGCCTGGCAGTC | |
| ADAR1-p110N | 5F: ATTTGCGGCCGCATGGCCGAGATCAAGGAGAAAATC |
| 3R: CGCGGATCCTCACTTATCGTCGTCATCCTTGTAATCACCACCACCGTTCTCCCCAATCAAGACACGGAG | |
| shPKR90 | 5F: GATCCCCGCAGGGAGTAGTACTTAAATATTCAAGAGATATTTAAGTACTACTCCCTGCTTTTTA |
| 3R: AGCTTAAAAAGCAGGGAGTAGTACTTAAATATCTCTTGAATATTTAAGTACTACTCCCTGCGGG | |
| shPKR405 | 5F: GATCCCCGCATGGGCCAGAAGGATTTCATTCAAGAGATGAAATCCTTCTGGCCCATGCTTTTTA |
| 3R: AGCTTAAAAAGCATGGGCCAGAAGGATTTCATCTCTTGAATGAAATCCTTCTGGCCCATGCGGG |
Generation of knockout cells by CRISPR/Cas9 gene editing
293T cells were transfected with vectors containing sgRNA (pLenti-sgADAR1–1, pLenti-sgADAR1–2, and pLenti-sgIFNAR1, pLenti-sgSTAT1) and two packaging plasmids (psPAX2 and pMD2.G) using FuGENE® HD Transfection Reagent (Promega). Supernatants including lentivirus were collected at 2 days post-transfection and passed through a 0.45-μm filter. The lentivirus supernatants were transduced into A549 cells for 24 h. Then cells were transferred to a 10-cm dish and selected by 1 μg/ml puromycin for 8 days. Puromycin-resistant clones were sorted and confirmed by Western blotting and genome DNA sequencing. Genomic DNA was extracted using a Cell Genomic DNA extraction kit (Bioteke). Regions surrounding sgRNA target sequences were amplified by PCR. PCR products were cloned into pMD-18T (Takara) for sequencing as described previously (44).
RNA interference
The sequences of two siRNAs targeting human ADAR1 RNA were CGCAGAGUUCCUCACCUGUA and GCUUCAACACUCUGACUAA (Invitrogen). A control siRNA with scrambled sequence was used as negative control (siNC). Transfection was carried out with 40 nm siRNA using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's instructions. At 48 h post-transfection, cells were harvested or used for further analysis.
Generation of PKR knockdown cells
A549 cells were transfected with plasmid pSUPER.retro.puro expressing shPKR90 or shPKR405 using Lipofectamine 2000 reagent (Invitrogen) (32). After 1 day, the cells were diluted in 10-cm plates and selected by 1 μg/ml puromycin for 8 days. Puromycin-resistant clones were sorted and confirmed by Western blotting.
qRT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Total RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (Promega). The qRT-PCR analyses were performed with SYBR® Select Master Mix for CFX (Applied Biosystems) in a Bio-Rad CFX96 machine (primer sequences were listed in Table 3). Data analyses for differences in gene expression by qRT-PCR were done by using ΔCT values as described previously (45).
Table 3.
Sequences of primers used in qRT-PCR
| Gene | Sequence (5′–3′) |
|---|---|
| ADAR1 | 5F: ACTACGAGACGGCCAAGAAC |
| 3R: TGTGATGAGGAATGCTACGAC | |
| ZIKV NS1 | 5F: GTCAGAGCAGCAAAGACAA |
| 3R: CAGCCTCCTTTCCCTTAACA | |
| β-Actin | 5F: GCTCCTCCTGAGCGCAAG |
| 3R: CATCTGCTGGAAGGTGGACA | |
| IFN-β | 5F: AAACTCATGAGCAGTCTGCA |
| 3R: AGGAGATCTTCAGTTTCGGAGG |
Western blotting
Cells were lysed in radioimmune precipitation assay lysis buffer (pH 7.4) (50 mm Tris-HCl, 0.5% (v/v) Nonidet P-40, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA, 1 mm phenylmethanesulfonyl fluoride (PMSF), 1% protease inhibitor mixtures, 1 mm sodium orthovanadate (Na3VO4), and 1 mm sodium fluoride (NaF)). Proteins were separated on SDS-PAGE and transferred onto nitrocellulose membranes, followed by blocking in 0.1% PBST with 5% BSA (New England Biolabs) and incubating with primary antibodies at 4 °C overnight. Detection was performed with IRDye 800 CW–conjugated anti-rabbit IgG and IRDye 680 CW–conjugated anti-mouse IgG secondary antibody (LI-COR) according to the manufacturer's protocols or horseradish peroxidase–conjugated secondary antibodies (Bio-Rad). Immunoreactive bands were visualized using an Odyssey IR imaging system (LI-COR) as described previously (39). The Western blotting bands were quantified by Quantity One (Bio-Rad).
Flow cytometry analysis
The control cells and ADAR1 knockout cells were fixed by eBioscienceTM IC Fixation Buffer (Invitrogen) and incubated with anti-WNV envelope protein antibody (E53) (46), followed by incubation with Alexa FluorTM 647 goat anti-mouse IgG antibody (Invitrogen). Labeled cells were analyzed by flow cytometry (CytoFLEX).
Subcellular fractionation
Cells were washed twice with PBS and lysed in cytoplasmic extract buffer (10 mm HEPES, 60 mm KCl, 1 mm EDTA, 0.075% (v/v) Nonidet P-40, 1 mm DTT, and 1 mm PMSF). The mixture was spun at 14,000 rpm for 5 min to separate the cytoplasmic extract (supernatant). The precipitate was washed with cytoplasmic extract buffer and then lysed in nuclear extract buffer (20 mm HEPES, 420 mm NaCl, 10 mm KCl, 1 mm EDTA, 1 mm PMSF, and 20% (v/v) glycerol). The mixture was spun at 14,000 rpm for 5 min to separate the nuclear extract (supernatant).
Immunofluorescence microscopy
Cells were washed twice with PBS and fixed in 4% (v/v) paraformaldehyde for 45 min. Cells were permeabilized in 0.02% Triton X-100 for 15 min and blocked in blocking buffer (5% BSA in PBS) for 1 h, followed by incubation with primary antibodies at 4 °C overnight. Cells were incubated with Alexa Fluor 488–conjugated anti-mouse-IgG (Invitrogen) and Cy3-conjugated goat anti-rabbit-IgG (Millipore) for 1 h. Cells were then stained with 0.2 mg/ml DAPI (Invitrogen) and visualized using a Zeiss fluorescence microscope. The co-localization analysis was performed by ImageJ (Bio-Rad).
Replicon assay
The replicon plasmids pFK-SGR and pFK-SGR-GDD (29) were linearized with MluI (New England Biolabs). Plasmid DNA was phenol-chloroform–extracted and precipitated with sodium acetate. The linearized DNA template was transcribed using a mMESSAGE mMACHINE® T7 kit (Ambion) according to the manufacturer's protocol. The product of transcription was purified by lithium chloride (LiCl) precipitation. The replicon RNAs (0.4 μg/well) were transfected into each well of Vero or A549 cells in a 24-well plate using Lipofectamine® 2000 reagent (Invitrogen). At the indicated time points, the cells were washed with PBS and lysed using passive lysis buffer (Promega). The cells were scraped from plates and stored at −80 °C. Once samples for all time points had been collected, luciferase activities were measured using a GLOMAXTM 96 microplate luminometer (Promega).
Ribosomal RNA (rRNA) cleavage assay
Control cells and ADAR1 KO cells were transfected with mock, poly(I:C), or WT ZIKV replicon RNAs. At 6 h post-transfection, cells were harvested in TRIzol (Invitrogen) for total RNA extraction. The integrity of 28S and 18S rRNA was examined by RNA electrophoresis or measured by an Agilent 2100 BioAnalyzer.
Statistical analysis
All statistical analysis of viral RNA levels or titers was performed with an unpaired, two-tailed Student's t test. Data are presented as mean ± S.D. from at least three independent experiments. The differences were considered statistically significant when p was <0.05, as described previously (39).
Author contributions
S. Z. and P. Z. conceptualization; S. Z., C. Y., F. Z., Y. H., Y. L., C. H., X. M., J. D., and Y. W. data curation; S. Z., F. Z., X. M., and J. D. formal analysis; S. Z., C. Y., Y. H., Y. L., C. H., X. M., Y. W., G. L., J. H., and P. Z. methodology; S. Z. and J. H. writing-original draft; C. Y., Y. L., C. L., and P. Z. investigation; C. Y., Y. H., Y. W., G. L., J. H., C. L., and P. Z. project administration; F. Z. and G. L. resources; Y. L., J. D., and P. Z. validation; G. L., C. L., and P. Z. supervision; J. H., C. L., and P. Z. funding acquisition; J. H., C. L., and P. Z. writing-review and editing.
Acknowledgment
We thank Dr. Charles Samuel (University of California, Santa Barbara) for generously providing pcDNA6-ADAR1 plasmids.
This work was supported by National Natural Science Foundation of China Grant 31970887, Guangdong Science and Technology Program Grants 2017A050501012 and 2018A050506029, and Natural Science Foundation of Guangdong Province Grant 2017A030313504. The authors declare that they have no conflicts of interest with the contents of this article.
- ZIKV
- Zika virus
- E
- ZIKV envelope protein
- ADAR
- adenosine deaminase acting on dsRNA
- dsRBD
- dsRNA-binding domain
- IFN
- interferon
- VSV
- vesicular stomatitis virus
- PKR
- dsRNA-regulated protein kinase
- MOI
- multiplicity of infection
- qRT-PCR
- quantitative reverse transcription PCR
- p.i.
- postinfection
- siNC
- negative control siRNA
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- sgRNA
- single guide RNA
- aa
- amino acid(s)
- PMSF
- phenylmethanesulfonyl fluoride
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Musso D., and Gubler D. J. (2016) Zika virus. Clin. Microbiol. Rev. 29, 487–524 10.1128/CMR.00072-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffy M. R., Chen T. H., Hancock W. T., Powers A. M., Kool J. L., Lanciotti R. S., Pretrick M., Marfel M., Holzbauer S., Dubray C., Guillaumot L., Griggs A., Bel M., Lambert A. J., Laven J., et al. (2009) Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 360, 2536–2543 10.1056/NEJMoa0805715 [DOI] [PubMed] [Google Scholar]
- 3. Kumar A., Hou S., Airo A. M., Limonta D., Mancinelli V., Branton W., Power C., and Hobman T. C. (2016) Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 17, 1766–1775 10.15252/embr.201642627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chaudhary V., Yuen K. S., Chan J. F., Chan C. P., Wang P. H., Cai J. P., Zhang S., Liang M., Kok K. H., Chan C. P., Yuen K. Y., and Jin D. Y. (2017) Selective activation of type II interferon signaling by Zika virus NS5 protein. J. Virol. 91, e00163–17 10.1128/JVI.00163-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant A., Ponia S. S., Tripathi S., Balasubramaniam V., Miorin L., Sourisseau M., Schwarz M. C., Sánchez-Seco M. P., Evans M. J., Best S. M., and García-Sastre A. (2016) Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 10.1016/j.chom.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lazear H. M., and Diamond M. S. (2016) Zika virus: new clinical syndromes and its emergence in the Western hemisphere. J. Virol. 90, 4864–4875 10.1128/JVI.00252-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pierson T. C., and Diamond M. S. (2018) The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581 10.1038/s41586-018-0446-y [DOI] [PubMed] [Google Scholar]
- 8. Wang B., Thurmond S., Hai R., and Song J. (2018) Structure and function of Zika virus NS5 protein: perspectives for drug design. Cell Mol. Life Sci. 75, 1723–1736 10.1007/s00018-018-2751-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsetsarkin K. A., Kenney H., Chen R., Liu G., Manukyan H., Whitehead S. S., Laassri M., Chumakov K., and Pletnev A. G. (2016) A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. MBio 7, pii: e01114–16 10.1128/mBio.01114-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pierson T. C., and Graham B. S. (2016) Zika virus: immunity and vaccine development. Cell 167, 625–631 10.1016/j.cell.2016.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shao Q., Herrlinger S., Zhu Y. N., Yang M., Goodfellow F., Stice S. L., Qi X. P., Brindley M. A., and Chen J. F. (2017) The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 144, 4114–4124 10.1242/dev.156752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haddow A. D., Nasar F., Guzman H., Ponlawat A., Jarman R. G., Tesh R. B., and Weaver S. C. (2016) Genetic Characterization of Spondweni and Zika viruses and susceptibility of geographically distinct strains of Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus (Diptera: Culicidae) to Spondweni virus. PLoS Negl. Trop. Dis. 10, e0005083 10.1371/journal.pntd.0005083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piontkivska H., Frederick M., Miyamoto M. M., and Wayne M. L. (2017) RNA editing by the host ADAR system affects the molecular evolution of the Zika virus. Ecol. Evol. 7, 4475–4485 10.1002/ece3.3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khrustalev V. V., Khrustaleva T. A., Sharma N., and Giri R. (2017) Mutational pressure in Zika virus: local ADAR-editing areas associated with pauses in translation and replication. Front. Cell Infect. Microbiol. 7, 44 10.3389/fcimb.2017.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomaselli S., Locatelli F., and Gallo A. (2014) The RNA editing enzymes ADARs: mechanism of action and human disease. Cell Tissue Res. 356, 527–532 10.1007/s00441-014-1863-3 [DOI] [PubMed] [Google Scholar]
- 16. Samuel C. E. (2011) Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411, 180–193 10.1016/j.virol.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Z., Wolff K. C., and Samuel C. E. (2010) RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 396, 316–322 10.1016/j.virol.2009.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. George C. X., Gan Z., Liu Y., and Samuel C. E. (2011) Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interferon Cytokine Res. 31, 99–117 10.1089/jir.2010.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rice G. I., Kasher P. R., Forte G. M., Mannion N. M., Greenwood S. M., Szynkiewicz M., Dickerson J. E., Bhaskar S. S., Zampini M., Briggs T. A., Jenkinson E. M., Bacino C. A., Battini R., Bertini E., Brogan P. A., et al. (2012) Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 10.1038/ng.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiden M. D., Hoshino S., Levy D. N., Li Y., Kumar R., Burke S. A., Dawson R., Hioe C. E., Borkowsky W., Rom W. N., and Hoshino Y. (2014) Adenosine deaminase acting on RNA-1 (ADAR1) inhibits HIV-1 replication in human alveolar macrophages. PLoS One 9, e108476 10.1371/journal.pone.0108476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ward S. V., George C. X., Welch M. J., Liou L. Y., Hahm B., Lewicki H., de la Torre J. C., Samuel C. E., and Oldstone M. B. (2011) RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 331–336 10.1073/pnas.1017241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Casey J. L. (2012) Control of ADAR1 editing of hepatitis δ virus RNAs. Curr. Top. Microbiol. Immunol. 353, 123–143 10.1007/82_2011_146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gélinas J. F., Clerzius G., Shaw E., and Gatignol A. (2011) Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J. Virol. 85, 8460–8466 10.1128/JVI.00240-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomaselli S., Galeano F., Locatelli F., and Gallo A. (2015) ADARs and the balance game between virus infection and innate immune cell response. Curr. Issues Mol. Biol. 17, 37–51 [PubMed] [Google Scholar]
- 25. Yang S., Deng P., Zhu Z., Zhu J., Wang G., Zhang L., Chen A. F., Wang T., Sarkar S. N., Billiar T. R., and Wang Q. (2014) Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 193, 3436–3445 10.4049/jimmunol.1401136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie Y., Hammond G. L., and Yang J. H. (2007) Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 81, 917–923 10.1128/JVI.01527-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. George C. X., Li Z., Okonski K. M., Toth A. M., Wang Y., and Samuel C. E. (2009) Tipping the balance: antagonism of PKR kinase and ADAR1 deaminase functions by virus gene products. J. Interferon Cytokine Res. 29, 477–487 10.1089/jir.2009.0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okonski K. M., and Samuel C. E. (2013) Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 87, 756–766 10.1128/JVI.02270-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao F., Xu Y., Lavillette D., Zhong J., Zou G., and Long G. (2018) Negligible contribution of M2634V substitution to ZIKV pathogenesis in AG6 mice revealed by a bacterial promoter activity reduced infectious clone. Sci. Rep. 8, 10491 10.1038/s41598-018-28890-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nie Y., Ding L., Kao P. N., Braun R., and Yang J. H. (2005) ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol. Cell. Biol. 25, 6956–6963 10.1128/MCB.25.16.6956-6963.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dauber B., and Wolff T. (2009) Activation of the antiviral kinase PKR and viral countermeasures. Viruses 1, 523–544 10.3390/v1030523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang P., and Samuel C. E. (2007) Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 81, 8192–8200 10.1128/JVI.00426-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y., Banerjee S., Goldstein S. A., Dong B., Gaughan C., Rath S., Donovan J., Korennykh A., Silverman R. H., and Weiss S. R. (2017) Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6, e25687 10.7554/eLife.25687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Whelan J. N., Li Y., Silverman R. H., and Weiss S. R. (2019) Zika virus production is resistant to RNase L antiviral activity. J. Virol. 93, e00313–19 10.1128/JVI.00313-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pujantell M., Franco S., Galván-Femenia I., Badia R., Castellví M., Garcia-Vidal E., Clotet B., de Cid R., Tural C., Martínez M. A., Riveira-Muñoz E., Esté J. A., and Ballana E. (2018) ADAR1 affects HCV infection by modulating innate immune response. Antiviral Res. 156, 116–127 10.1016/j.antiviral.2018.05.012 [DOI] [PubMed] [Google Scholar]
- 36. Wang Q., Li X., Qi R., and Billiar T. (2017) RNA editing, ADAR1, and the innate immune response. Genes (Basel) 8, E41 10.3390/genes8010041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. George C. X., Ramaswami G., Li J. B., and Samuel C. E. (2016) Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J. Biol. Chem. 291, 6158–6168 10.1074/jbc.M115.709014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bayer A., Lennemann N. J., Ouyang Y., Bramley J. C., Morosky S., Marques E. T. Jr., Cherry S., Sadovsky Y., and Coyne C. B. (2016) Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19, 705–712 10.1016/j.chom.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y., Chen X., Xie J., Zhou S., Huang Y., Li Y. P., Li X., Liu C., He J., and Zhang P. (2019) RNA helicase A is an important host factor involved in Dengue virus replication. J. Virol. 93, e01306–18 10.1128/JVI.01306-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung H., Calis J. J. A., Wu X., Sun T., Yu Y., Sarbanes S. L., Dao Thi V. L., Shilvock A. R., Hoffmann H. H., Rosenberg B. R., and Rice C. M. (2018) Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824 e814.e14 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gannon H. S., Zou T., Kiessling M. K., Gao G. F., Cai D., Choi P. S., Ivan A. P., Buchumenski I., Berger A. C., Goldstein J. T., Cherniack A. D., Vazquez F., Tsherniak A., Levanon E. Y., Hahn W. C., and Meyerson M. (2018) Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 9, 5450 10.1038/s41467-018-07824-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dabo S., and Meurs E. F. (2012) dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4, 2598–2635 10.3390/v4112598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Toth A. M., Li Z., Cattaneo R., and Samuel C. E. (2009) RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 284, 29350–29356 10.1074/jbc.M109.045146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao H., Lin Y., He J., Zhou S., Liang M., Huang C., Li X., Liu C., and Zhang P. (2019) Role of heparan sulfate in the Zika virus entry, replication, and cell death. Virology 529, 91–100 10.1016/j.virol.2019.01.019 [DOI] [PubMed] [Google Scholar]
- 45. Zhang P., Li Y., Xia J., He J., Pu J., Xie J., Wu S., Feng L., Huang X., and Zhang P. (2014) IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J. Cell Sci. 127, 2471–2482 10.1242/jcs.139626 [DOI] [PubMed] [Google Scholar]
- 46. Cherrier M. V., Kaufmann B., Nybakken G. E., Lok S. M., Warren J. T., Chen B. R., Nelson C. A., Kostyuchenko V. A., Holdaway H. A., Chipman P. R., Kuhn R. J., Diamond M. S., Rossmann M. G., and Fremont D. H. (2009) Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 28, 3269–3276 10.1038/emboj.2009.245 [DOI] [PMC free article] [PubMed] [Google Scholar]