

Abstract
Estrogen receptor alpha (ERα) activity is associated with increased cancer cell proliferation. Studies aiming to understand the impact of ERα on cancer‐associated phenotypes have largely been limited to its transcriptional activity. Herein, we demonstrate that ERα coordinates its transcriptional output with selective modulation of mRNA translation. Importantly, translational perturbations caused by depletion of ERα largely manifest as “translational offsetting” of the transcriptome, whereby amounts of translated mRNAs and corresponding protein levels are maintained constant despite changes in mRNA abundance. Transcripts whose levels, but not polysome association, are reduced following ERα depletion lack features which limit translation efficiency including structured 5′UTRs and miRNA target sites. In contrast, mRNAs induced upon ERα depletion whose polysome association remains unaltered are enriched in codons requiring U34‐modified tRNAs for efficient decoding. Consistently, ERα regulates levels of U34‐modifying enzymes and thereby controls levels of U34‐modified tRNAs. These findings unravel a hitherto unprecedented mechanism of ERα‐dependent orchestration of transcriptional and translational programs that may be a pervasive mechanism of proteome maintenance in hormone‐dependent cancers.
Keywords: 5′ UTR, estrogen receptor, hormone‐dependent cancer, mRNA translation, U34 tRNA modification
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Protein Biosynthesis & Quality Control
In addition to its well‐established role in transcription, ERα also modulates levels of translation of selective transcripts that show alteration in their steady‐state levels following ERα perturbation, a phenomenon referred to as translational offsetting.

Introduction
Gene expression is regulated at multiple levels including transcription, nuclear mRNA export, storage, stability, and translation. These processes together with protein degradation govern proteome composition (Morris et al, 2010; Piccirillo et al, 2014; Bisogno & Keene, 2018). While mRNA levels are key determinants of the proteome under non‐stressed growth conditions, the contribution of mRNA translation remains contentious (Schwanhäusser et al, 2011; Li et al, 2014, 2017). In contrast, it is well established that altered translational efficiencies reshape the proteome during various dynamic responses including cellular differentiation and endoplasmic reticulum stress (Kristensen et al, 2013; Baird et al, 2014; Liu et al, 2016; Guan et al, 2017).
Translation is regulated globally, leading to altered translational efficiency of most cellular mRNAs, or selectively by modulating translation of limited subsets of mRNAs (Piccirillo et al, 2014). Most commonly, selective changes in translational efficiency are considered to allow modulation of protein levels in absence of corresponding changes in mRNA levels (Larsson et al, 2010). This is thought to be mediated by interactions between RNA elements in untranslated regions (UTRs), RNA‐binding proteins, and translation initiation factors (Koromilas et al, 1992; Hershey et al, 2012; Hinnebusch et al, 2016; Masvidal et al, 2017). Accordingly, 5′UTR length, structure, and presence of cis‐acting elements, such as upstream open reading frames (uORFs), and 3′UTR trans‐acting factors including microRNAs (miRNAs) play a pivotal role in translational control (Gebauer et al, 2012; Larsson et al, 2013; Gandin et al, 2016b; Hinnebusch et al, 2016). Although the vast majority of known RNA elements implicated in modulation of mRNA translation reside within UTRs, it has been reported that nucleotide sequence and/or elements in the open reading frame may also regulate translation (López et al, 2015; Thandapani et al, 2015). Indeed, selective alterations of transfer RNAs (tRNAs) were recently described to affect translation of mRNAs based on their codon usage (Goodarzi et al, 2016). Moreover, recent reports highlight alterations of tRNA modifications as an important mechanism underlying selective translation (Chan et al, 2015; Delaunay et al, 2016; Rapino et al, 2017, 2018) which are thought to act by maintaining protein homeostasis or driving an adaptive proteome (Nedialkova & Leidel, 2015).
Estrogen receptor alpha (ERα) is a key steroid receptor and transcription factor which drives tumorigenesis in hormone‐dependent cancers (Shanle & Xu, 2010). In prostate cancer, ERα expression is associated with increased cell proliferation. Moreover, ERα is overexpressed in genetically engineered mouse models of prostate cancer and high‐grade patient tumors (Chakravarty et al, 2014; Megas et al, 2015; Takizawa et al, 2015). In this context, the ERα transcriptional output is thought to direct a program distinct from the androgen receptor (AR) that may contribute to emergence of castrate‐resistant prostate cancer and aggressive tumor subtypes (Setlur et al, 2008). Intriguingly, in addition to its role in regulating transcription, ERα may directly or indirectly influence PI3K/AKT/mTOR and MAPK pathway signaling, as shown in several tissues (Levin, 2009) including the prostate (Takizawa et al, 2015). Studies in multiple cellular models have revealed that fluctuations in mTOR activity predominantly affect translation of mRNA subsets defined by long and highly structured 5′UTRs, extremely short 5′UTRs or 5′UTRs harboring a 5′‐terminal oligopyrimidine tract (TOP; Patursky‐Polischuk et al, 2009; Hsieh et al, 2012; Larsson et al, 2012; Meyuhas & Kahan, 2015; Gandin et al, 2016b; Masvidal et al, 2017). Moreover, MAPK signaling induces phosphorylation of eIF4E which also selectively modulates mRNA translation (Furic et al, 2010; Robichaud et al, 2015). Therefore, we investigated whether ERα, in addition to its well‐established role in transcription, also modulates translation.
Results
Changes in steady‐state mRNA levels upon depletion of ERα are largely offset at the level of translation
To study the impact of ERα on regulation of gene expression in prostate cancer, we used the BM67 cell line derived from the PTEN null mouse model of prostate cancer (Takizawa et al, 2015). BM67 cells express relatively high level of ERα. ERα was silenced using an shRNA to generate shERα BM67 cells (Fig 1A). To assess effects of ERα depletion on mRNA abundance and translation, we used polysome profiling quantified by DNA microarrays (Appendix Fig S1A and B). Polysome profiling generates parallel data on efficiently translated (i.e., those associated with > 3 ribosomes) and total mRNA (Fig 1A; Gandin et al, 2014). Changes in polysome‐associated mRNA can either be driven by congruent alterations in corresponding cytosolic mRNA levels or stem from changes in translational efficiencies which are not accompanied by fluctuations in mRNA levels. The anota2seq algorithm identifies changes in polysome‐associated and cytosolic mRNA and computes alterations in translational efficiency (Larsson et al, 2010; Oertlin et al, 2019). In line with the role of ERα as a transcription factor, widespread changes in total mRNA levels were observed between control and shERα BM67 cells as evidenced by a strong enrichment of transcripts with low P‐values (Fig 1B; Appendix Fig S1C). Substantially, fewer mRNAs displayed ERα‐associated changes in polysome association as indicated by a smaller enrichment of transcripts with low P‐values (Fig 1B; Appendix Fig S1C). Strikingly, when comparing fold changes for cytosolic and polysome‐associated mRNAs in shERα vs. control BM67 cells, we identified a population of genes showing changes in cytosolic mRNA without corresponding changes in their association with polysomes (Fig 1C). This suggests that shERα‐dependent alterations in mRNA levels may be buffered at the level of translation such that the amount of mRNA associated with polysomes is unaltered despite changes in corresponding mRNA levels. Translational buffering has been described in the context of transcript‐dosage compensation where it acts to maintain protein levels similar between different bacterial (Lalanne et al, 2018) and yeast species (Artieri & Fraser, 2014; McManus et al, 2014) and humans (Cenik et al, 2015). It has also been reported that gene dosage effects caused by aneuploidy may be compensated at the level of translation in a cell type‐specific context (Zhang & Presgraves, 2017) and that translational buffering may reduce transcriptional “noise” caused by acute stimulation of cells with growth factors (Tebaldi et al, 2012). In contrast to these modes of regulation, which appear to chiefly reduce “noise” in the proteome composition, transcriptional defects caused by ERα‐depletion appear to induce a form of translational buffering which can be activated to sustain an adaptive proteome and thus we refer to it as “translational offsetting”.
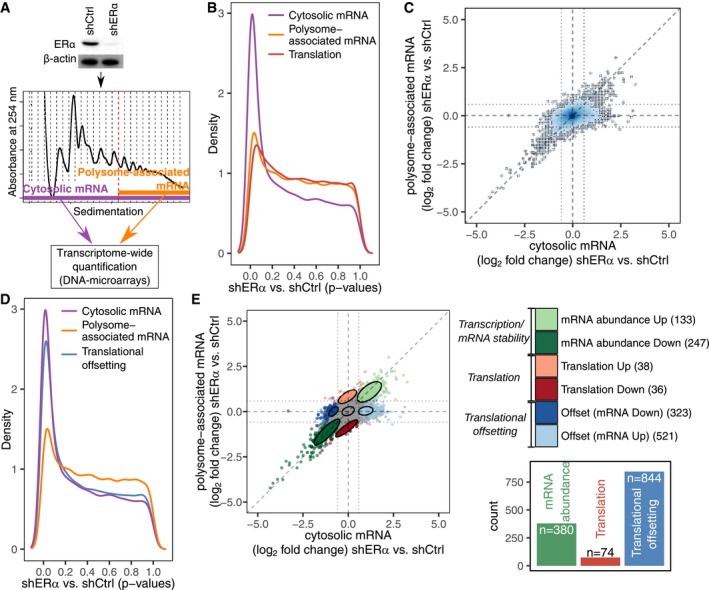
Figure 1. ERα‐dependent alterations in steady‐state mRNA levels are largely offset at the level of mRNA translation.
-
AExpression of ERα in control (shCtrl) and knockdown (shERα) BM67 cells was determined by Western blotting (β‐actin served as a loading control). Gene expression was determined using polysome profiling which quantifies both total mRNA and efficiently translated polysome‐associated mRNA (n = 3).
-
BDistributions (quantified by kernel density estimation) of P‐values for differential expression (shERα vs. shCtrl BM67 cells) using data from polysome‐associated mRNA (orange), cytosolic mRNA (purple) or from analysis of changes in translational efficiency (i.e., changes in polysome‐associated mRNA not paralleled by changes in cytosolic mRNA) leading to altered protein levels (red; a higher density of low P‐values indicates a higher frequency of changes in gene expression).
-
CScatter plot of polysome‐associated mRNA vs. cytosolic mRNA log2 fold changes (shERα vs. shCtrl). Areas of the plot are colored according to the density of data points [genes; dark blue corresponds to areas with many genes (see Appendix Supplementary Methods)].
-
DSame plot as in (B) but including a blue density of P‐values from the analysis of translational offsetting.
-
EA scatter plot similar to (C) but where genes are colored according to their mode of regulation derived from anota2seq analysis. A relaxed threshold (P < 0.05) was used to identify a set of transcripts regulated via translation, which did not pass thresholds used for identification of changes in mRNA abundance or translational offsetting (FDR < 0.1). Confidence ellipses (level 0.7) are overlaid for each mode of regulation. A bar graph indicates the number of mRNAs regulated via each mode.
Source data are available online for this figure.
We next implemented adjustments in anota2seq that allowed analysis of different forms of translational buffering including ERα‐dependent translational offsetting (Oertlin et al, 2019). This led to identification of a large number of mRNAs which changed in abundance in shERα BM67 vs. control cells but were translationally offset as illustrated by an abundance of low P‐values (Fig 1D; Appendix Fig S1D). Notably, translational offsetting appeared to be much more common than changes in translation which are expected to alter protein levels (i.e., changes in polysome‐associated mRNA not paralleled by changes in total mRNA as quantified by anota2seq; Fig 1D). Anota2seq also allows to categorize transcripts in three modes of regulation: (i) changes in mRNA abundance (congruent changes in total and polysome‐associated mRNA), (ii) changes in translation (changes in polysome‐associated mRNA without corresponding changes in total mRNA) and translational offsetting (changes in cytosolic mRNA without corresponding changes in their polysome association) (Fig 1E; Table EV1). Strikingly, as evidenced by the number of transcripts under each mode of regulation, translational offsetting was the predominant mode for regulation of gene expression following ERα depletion (Fig 1E). Importantly, comparable results were obtained when RNA sequencing (RNAseq) was used to quantify translatomes and transcriptomes instead of DNA microarrays (Fig EV1A–F). Collectively, these data suggest that the ERα‐dependent perturbations in mRNA levels are largely offset at the level of translation.
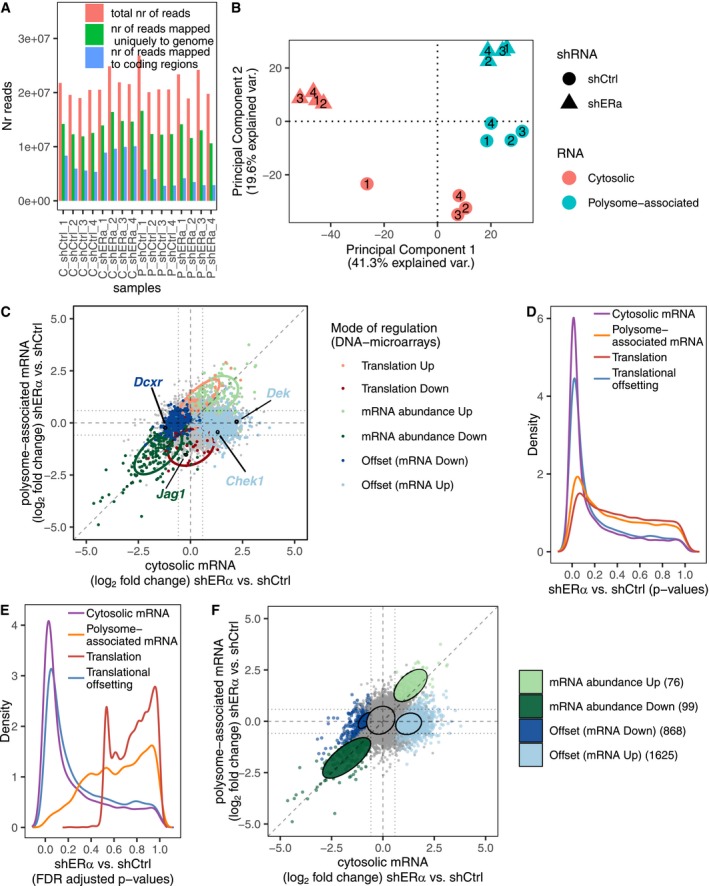
Figure EV1. Validation of translational offsetting by RNA sequencing.
-
ANumber of RNAseq reads sequenced and after each preprocessing step.
-
BA principal component analysis, using per gene‐centered expression data, was performed on samples from the transcriptome‐wide polysome profiling of ERα depletion (normalized RNAseq data, N = 4). Scores were plotted for the first two principal components.
-
CShown is a scatter plot of RNAseq quantification of polysome‐associated mRNA vs. cytosolic mRNA log2 fold change (shERα vs. shCtrl). Genes are colored according to their identified mode of regulation (i.e., from Fig 1E). Confidence ellipses (based on RNAseq data, level 0.7) are overlaid for each mode of regulation. Selected targets (also quantified using Nanostring, Western blotting) are indicated.
-
D–FA similar analysis as Fig 1B–E was performed on RNAseq data instead of DNA microarrays data. Distributions (quantified by kernel density estimation) of P‐values (D) and FDR‐adjusted P‐values (E) for differential expression between shERα and shCtrl BM67 cells using data from polysome‐associated mRNA (orange) or cytosolic mRNA (purple), for analysis of differences in translational efficiencies leading to altered protein levels (red) and analysis of translational offsetting (blue). (F) A scatter plot similar to (Fig 1E) where genes are colored according to their mode of regulation derived from anota2seq analysis on RNAseq data.
ERα‐dependent translational offsetting opposes changes in protein levels despite alterations in corresponding mRNA abundance
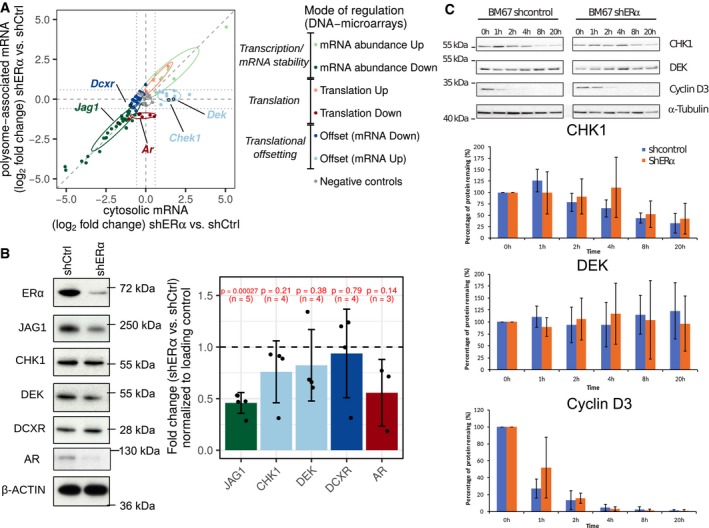
To validate observed ERα‐dependent changes in gene expression, we selected 86 candidate genes from the three modes of regulation (i.e., mRNA abundance, translation, and translational offsetting), together with negative controls. Using this set of genes, we employed Nanostring technology (Geiss et al, 2008) and validated all modes of ERα‐dependent regulation of gene expression observed using DNA microarrays/RNAseq (Fig 2A; Appendix Fig S2; Table EV2). We next selected genes regulated at the level of translation (AR), mRNA abundance (JAG1) or translational offsetting (CHEK1, DEK, and DCXR) and assessed corresponding protein levels using Western blotting. AR and JAG1 were downregulated in shERα as compared to control BM67 cells, which corresponded to the observed decrease in their polysome association (Fig 2B). In turn, levels of proteins encoded by translationally offset mRNAs remained comparable between ERα depleted and control BM67 cells (Fig 2B). To exclude the possibility that observed increase in DEK and CHK1 mRNA but not protein levels in ERα depleted vs. control BM67 cells stems from reduces protein stability, we performed cycloheximide chase assay. These experiments revealed that depletion of ERα does not decrease stability of DEK and CHK1 protein (Fig 2C). Altogether, these data suggest that ERα depletion leads to translational offsetting which opposes changes in protein levels despite alterations in mRNA abundance.
Figure 2. ERα‐dependent translational offsetting opposes changes in protein levels despite alterations in corresponding mRNA abundance.
-
ATargets from each mode of regulation (cf. Fig 1E) were selected for validation by Nanostring. Shown is a scatter plot of Nanostring quantification of polysome‐associated mRNA vs. cytosolic mRNA log2 fold change (shERα vs. shCtrl; n = 3). Genes are colored according to their identified mode of regulation (i.e., from Fig 1E). Confidence ellipses (based on Nanostring data, level 0.7) are overlaid for each mode of regulation. Selected genes from each mode of regulation are indicated.
-
BLevels of indicated proteins from shERα and control BM67 cells were determined by Western blotting with their quantifications. β‐Actin served as loading control. Graphs represent mean ± standard deviation (n = 3–5 as indicated). Protein expression between groups was compared using two‐sided paired Student's t‐tests.
-
CImmunoblotting of BM67 cell lines extract and its quantitation following incubation with 100 μg/ml cycloheximide as indicated. Graphs represent mean ± standard deviation (n = 5).
Source data are available online for this figure.
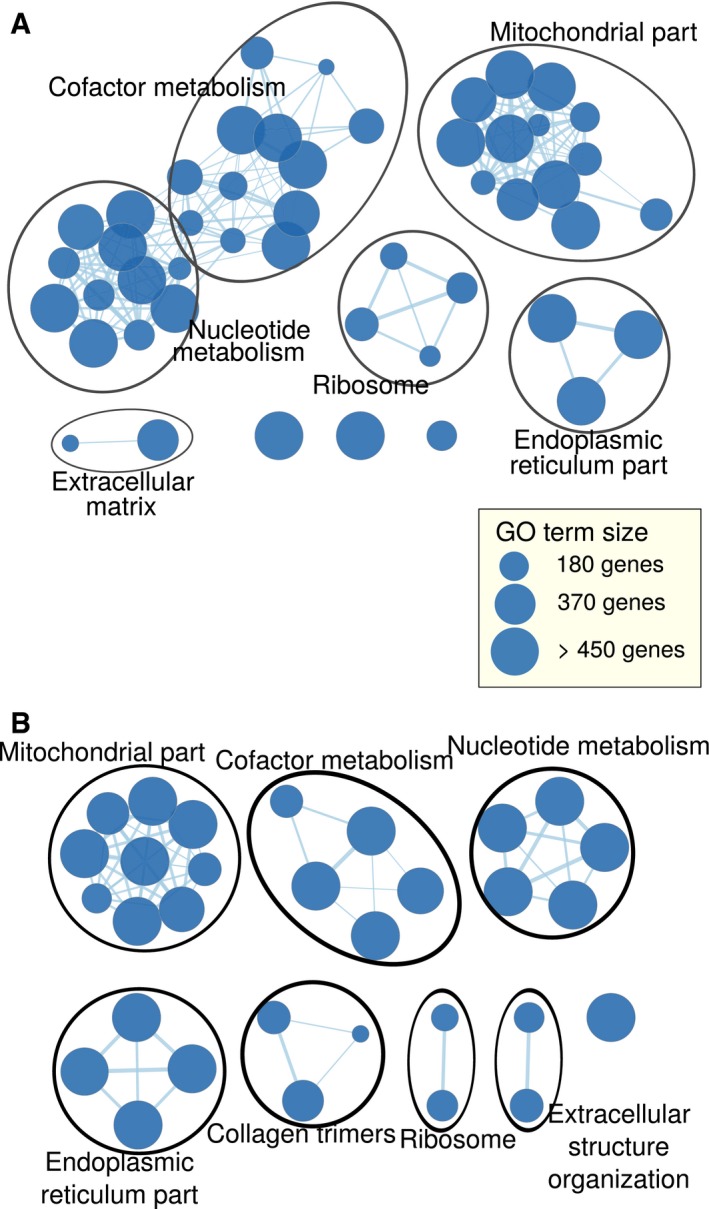
We next sought to establish functional relationships between the ERα‐dependent genes which underwent translational offsetting by performing gene set enrichment analyses using Gene Ontology (GO) annotations (Gene Ontology Consortium, 2015). No functions passed selected thresholds for enrichment among proteins encoded by mRNAs induced in shERα vs. control BM67 cells but translationally offset. In contrast, metabolism and mitochondria‐related functions were enriched (false discovery rate (FDR)‐adjusted P‐value = 0.008 and 0.001, respectively) among proteins encoded by mRNAs whose levels were suppressed but translationally offset upon ERα depletion (Fig EV2A; Table EV3). As expected, this largely overlapped with the enrichment of cellular functions among proteins encoded by mRNAs whose total mRNA was reduced upon ERα depletion (irrespective of whether this was paralleled by changes in their polysome association or not; Fig EV2A and B). In contrast, there was no significant enrichment in cellular functions among proteins encoded by mRNAs whose polysome association was reduced upon ERα depletion. Therefore, subsets of mRNAs which are offset at the level of translation upon ERα depletion are functionally related and are implicated in essential cellular functions.
Figure EV2. ERα depletion‐associated translational offsetting modulates expression of genes belonging to distinct cellular functions.
-
A, BGO terms enriched among proteins encoded by mRNAs suppressed but offset upon ERα depletion with FDR < 0.15 (A) and enriched among proteins encoded by cytosolic mRNAs downregulated (FDR < 0.15) (B) are visualized as a network. Each node is a GO term connected to other GO terms whose enriched genes overlap. The size of each node reflects the number of genes associated with its corresponding term and the width of the edge illustrates the size of the overlap between two connected nodes.
Short, unstructured 5′UTRs characterize mRNAs which are downregulated but translationally offset upon ERα depletion
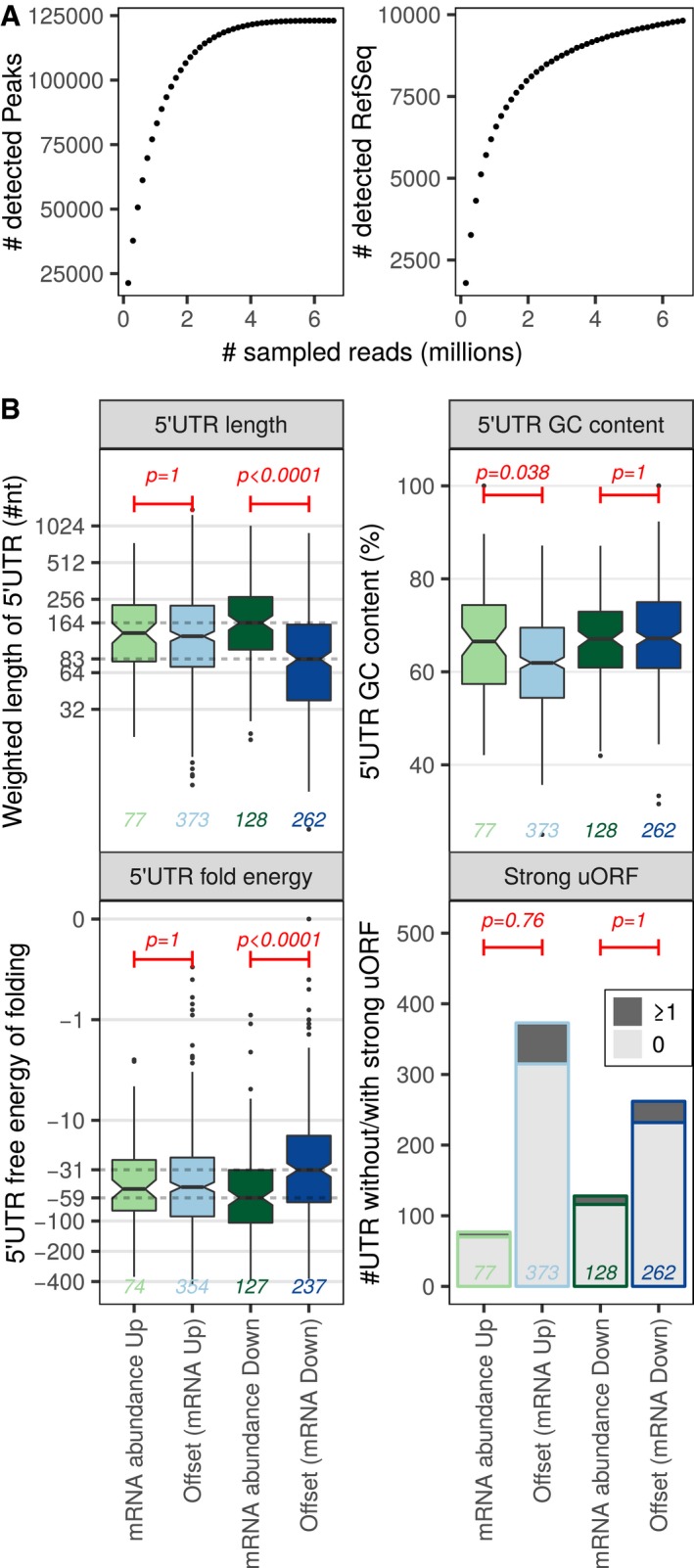
We next sought to identify mRNA features which underpin ERα‐dependent translational offsetting. To this end, we compared mRNAs which changed their level and polysome association in ERα‐depleted vs. control cells and contrasted them to those that maintained their polysome occupancy despite changes in their abundance. We initially focused on 5′UTR features as they play key roles in determining translation efficiency (Hinnebusch et al, 2016). To achieve this, we performed transcription start site profiling in shERα BM67 cells using nano‐cap analysis of gene expression (nanoCAGE). At a sequencing depth close to saturation, approximately 10,000 5′UTRs were detected (Fig 3A). Using these data, we contrasted genes whose mRNA abundance and polysome association changed in parallel to those that were translationally offset for: 5′UTR length, GC content, free energy of folding, and presence of uORFs in a strong Kozak context. Strikingly, transcripts whose levels were reduced upon ERα depletion but were translationally offset had a median 5′ UTR lengths ~ 50% shorter and were less structured relative to downregulated but non‐offset mRNAs (Fig 3B). In contrast, transcripts induced upon ERα depletion but translationally offset exhibited comparable 5′UTR length and folding free energy but slightly lower GC content as compared to non‐offset mRNAs (Fig 3B). Furthermore, there were no differences in proportion of mRNA harboring uORFs between offset and non‐offset transcripts (Fig 3B). Collectively, these findings suggest that mRNAs that decrease in abundance but are offset at the level of translation in ERα‐depleted cells contain shorter and less structured 5′UTRs.
Figure 3. Genes repressed upon ERα depletion but translationally offset have short and less stable 5′ UTRs.
-
AnanoCAGE sequencing was applied to determine transcription start sites in shERα BM67 cells. The number of detected transcription start sites (peaks) and RefSeq transcripts when sampling increasing number of RNAseq reads are indicated to evaluate the complexity of nanoCAGE RNAseq libraries.
-
BBoxplots for offset and non‐offset mRNAs comparing 5′ UTR weighted lengths (log2 scale), GC content (%), free energy (log10 scale, kcal/mole). In boxplots, the solid horizontal middle line is the median; the lower and upper box limits correspond to the first and third quartiles; the whiskers extend from the box limits to the most extreme value but not further than ± 1.5 × the inter‐quartile range (IQR) from the box limits. The notches extend to 1.58 × the IQR/√n where n is the number of data points. Data beyond the end of the whiskers are plotted as points. Wilcoxon–Mann–Whitney tests (two‐sided) were used to assess differences between translationally offset and non‐offset mRNAs. Number of transcripts harboring at least one (dark gray) or no (light gray) uORF in a strong Kozak context were visualized as a bar chart and differences were tested using one‐sided Fisher's exact tests. A global Bonferroni adjustment was applied on P‐values. Analysis was performed on nanoCAGE data from three replicates of shERα BM67 cells; the numbers of transcripts in each group are indicated in the figure.
Transcripts that are downregulated but translationally offset upon ERα depletion are largely devoid of miRNAs target sites
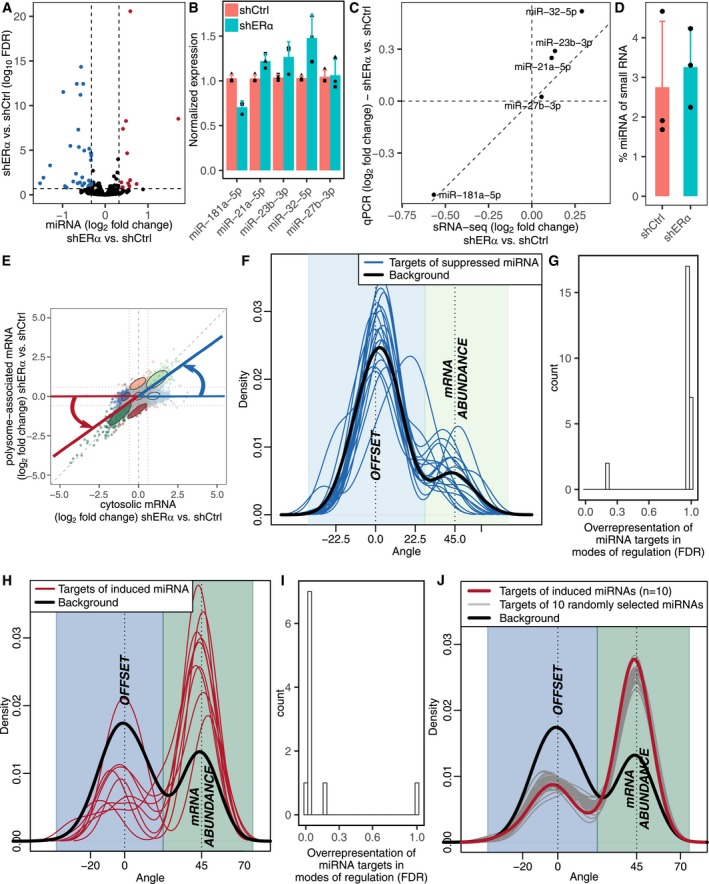
ERα modulates expression of multiple miRNAs (Castellano et al, 2009; Maillot et al, 2009; Klinge, 2012; Bailey et al, 2015), which led us to investigate the role of miRNAs in translational offsetting. To address this, we performed small RNAseq in shERα and control BM67 cells (Appendix Fig S3). ERα depletion led to alterations in levels of a subset of miRNAs (Fig 4A). Among these, five miRNAs (miR‐181a‐5p, miR‐21a‐5p, miR‐23b‐3p, miR‐32‐5p, and miR‐27b‐3p) were selected and their expression was validated using qPCR (Fig 4B and C). As RNAseq involves relative quantification of miRNAs, we also performed bioanalyzer‐based quantification of small RNAs to assess global changes in miRNA expression. These experiments unraveled that there were no major differences in total miRNA expression between shERα and control BM67 cells (Fig 4D). Next, we assessed whether targets of miRNAs with ERα‐dependent expression were selectively offset. Transcripts upregulated but translationally offset in shERα vs. control BM67 cells showed no enrichment of miRNA target sites for downregulated miRNAs (Fig 4E–G). In contrast, downregulated mRNAs that were translationally offset were largely devoid of the target sites for miRNAs which were upregulated in ERα‐depleted cells (Fig 4H and I). Importantly, such a strong underrepresentation of miRNA target sites among transcripts whose abundance was reduced but translationally offset upon ERα depletion was also observed when selecting random sets of miRNAs (Fig 4J). This suggests that there is no link between ERα‐regulated miRNAs and translational offsetting, but rather that there is a general lack of miRNA target sites within this subset of transcripts. In summary, mRNAs whose levels are reduced but offset upon ERα depletion have short and less structured 5′UTRs and harbor less miRNA target sites in their 3′ UTRs as compared to non‐offset transcripts.
Figure 4. Upon ERα depletion, target sites for miRNAs are under‐represented among suppressed but offset mRNAs.
-
AVolcano plot of ERα‐dependent miRNA expression in BM67 cells quantified by small RNAseq. Down‐ and up‐regulated miRNAs (FDR < 0.2 and fold change > 1.25) are colored in blue and red, respectively.
-
B, CValidation of miRNA expression using qPCR. Normalized expression (mean ± standard deviation; n = 3) (B) and expression fold change (shERα vs. shCtrl) obtained from small RNAseq vs. qPCR (C).
-
DPercentage of miRNA among small RNAs in shERα and shCtrl BM67 cells (mean ± standard deviation; n = 3).
-
E–JAssessment of the relationship between presence of miRNA target sites and translational offsetting. (E) For visualization, mRNAs were assigned angles describing their mode of regulation such that mRNAs with congruently modulated total and polysome‐associated mRNA obtain an angle of ˜45°, whereas offset mRNAs are assigned an angle of ˜0°. (F) The distributions (quantified by kernel density estimation) of such angles were then compared between mRNAs whose levels were induced upon ERα depletion, which were also targets of each suppressed miRNAs separately (blue lines); and the set of background transcripts (mRNAs induced upon ERα depletion but not targets of suppressed miRNAs; black). (G) For such subsets of miRNA targets (i.e., from F), the relative number of offset and non‐offset mRNAs (as determined by anota2seq analysis) was also compared to the background (same as in F) using Fisher's exact test, and the resulting adjusted P‐values (FDRs; across all assessed miRNAs) were visualized as a histogram. (H, I) A similar assessment as in (F, G) but for suppressed mRNAs and induced miRNAs. (J) A similar assessment as in (H) but for randomly selected groups of 10 miRNAs (gray lines) compared to background (black) and targets of all induced miRNA (red; i.e., from H).
Transcripts whose levels are induced by ERα depletion, but translationally offset are decoded by a distinct set of tRNAs
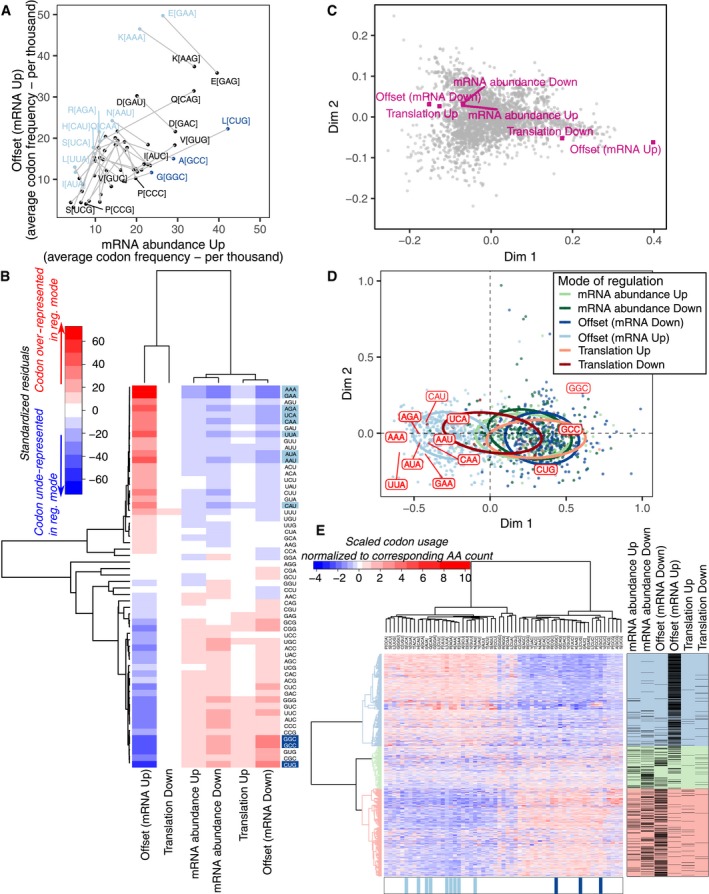
Because no distinct 5′ or 3′UTR features were observed among mRNAs which are upregulated but translationally offset in ERα‐depleted vs. control cells, we next investigated their codon usage. Transcripts expressed during proliferation vs. differentiation exhibit distinct codon usage and thus appear to require different subsets of tRNAs for their translation (Gingold et al, 2014). We therefore considered that a mismatch between tRNA demand (codon usage from expressed mRNAs) and tRNA expression could lead to translational offsetting. Indeed, there was a strong association between the mode of regulation and codon composition (P < 0.001, Pearson's chi‐squared test) as mRNAs whose upregulation was translationally offset following ERα depletion showed a striking enrichment for a distinct subset of codons (Fig 5A and B). This was confirmed by analyses of codon bias using a set of highly expressed genes as reference (top panels) and measures of adaptation to the tRNA pool by the tRNA adaptation index (tAI) whereby relative tRNA levels are assumed to be mirrored by their genomic copy numbers (Fig EV3A). Moreover, reduced tAI was observed at all sextiles along the coding sequences of upregulated mRNAs which were translationally offset (Fig EV3B). We next assessed how codon usage in herein identified modes of regulation of gene expression compared to codon usage in transcripts encoding proteins with distinct cellular function [which was used to detect codon bias between proliferation and differentiation‐associated transcripts previously (Gingold et al, 2014)]. To this end, we first visualized differences in codon usage between mRNAs grouped based on cellular functions (i.e., within GO terms) using correspondence analysis. Codon usage of different modes of regulation of gene expression was then projected in the same dimensions (Fig 5C, Table EV4). Proliferation‐ and differentiation‐related functions showed extreme positive and negative values, respectively, in the first dimension (Table EV4). Strikingly, mRNAs which were induced at the total transcript level but translationally offset following ERα depletion showed a more positive value in dimension one than any GO term (Fig 5C). Notably, in the gene set enrichment analysis (Fig EV2A), while the most enriched GO terms found among proteins encoded by mRNAs whose upregulation was offset at the level of translation were related to cellular proliferation, this enrichment did not pass the thresholds for statistical significance (Table EV2). To further characterize the nature of the differences in codon usage, we selected the first quintile of codons (12 codons; Fig 5B; Appendix Table S1) with highest positive residuals from independence between codon composition and mode of regulation. Out of these, 9 and 3 codons were over‐represented among mRNAs whose upregulation and downregulation were, respectively, translationally offset, following ERα depletion (Fig 5B and D). Notably, these three codons showed a stronger depletion among mRNAs that were upregulated but translationally offset as compared to their enrichment among mRNAs which were downregulated but offset. Yet, the codon adaptation indexes of mRNAs whose downregulation was offset at the level of translation were all consistently higher compared to those of non‐offset mRNAs (Figs 5B and EV3A–C; Appendix Table S1). These analyses were based on codon frequency (which is affected by amino acid frequency) but comparable results were obtained following normalization to amino acid counts (Figs 5E and EV3D–F). Notably, codons enriched among induced and offset (nine codons) or repressed and offset (three codons) mRNAs show strong co‐variation across expressed transcripts (Fig EV3G). In summary, mRNAs whose levels are induced but offset at the level of translation show distinct codon usage, which suggests that their translational offset could stem from a mismatch between tRNA abundance and demand.
Figure 5. Transcripts induced but translationally offset upon ERα depletion are characterized by distinct codon usage.
-
AFor each codon, the average frequency (per thousand) is compared between transcripts induced but offset vs. non‐offset (i.e., abundance mode of regulation) upon ERα depletion in BM67 cells. Codons for the same amino acid are connected by a gray line.
-
BHeatmap of standardized residuals from a chi‐squared contingency table test. Shown in red and blue are cells with counts significantly higher and lower, respectively, than expected counts under the null hypothesis (i.e., independence between mode for regulation of gene expression and codon composition).
-
CA correspondence analysis for average codon frequency among transcripts annotated to each GO term. Each gray dot corresponds to one GO term. The gene expression modes observed upon ERα depletion (Fig 1E) were then projected into the same dimensions based on the average codon frequencies of included transcripts.
-
DA correspondence analysis for codon frequency in each regulated mRNA (from Fig 1E). Each dot represents one mRNA and is colored according to its mode of regulation. Codons identified as over‐represented among translationally offset mRNAs are also indicated.
-
EUnsupervised clustering of gene level codon usage normalized by amino acid counts. All regulated mRNAs (from Fig 1E) are shown in rows and all codons in columns. Codons identified as over‐represented among mRNAs whose levels were induced but offset or suppressed but offset are indicated in light and dark blue, respectively.
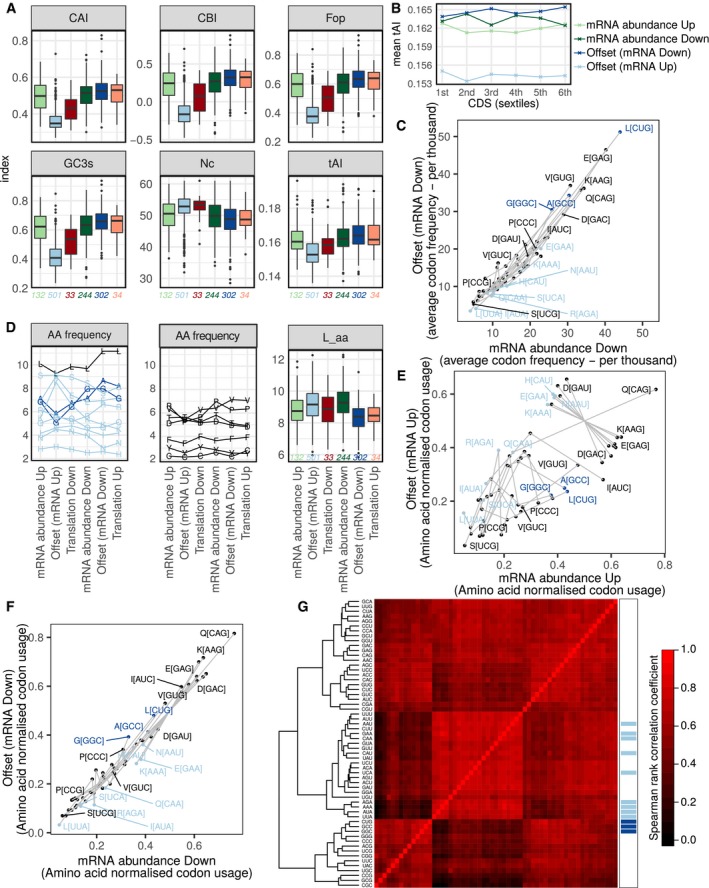
Figure EV3. Analyzes of codon usage.
-
ABoxplots (plotted as in Fig 3B) of mRNAs under all modes for regulation of gene expression comparing six measurements of codon usage: Codon Adaptation Index (CAI), Codon Bias Index (CBI), Frequency of optimal codons (Fop), GC content at the codon third position (GC3s), Effective number of codons (Nc), tRNA adaptation index (tAI). The numbers of mRNAs in each group are indicated in the figure.
-
BCDS are divided into sextiles, and the mean tRNA adaptation index is plotted for each mode of regulation at these CDS positions.
-
CFor each codon, the average frequency (per thousand) is compared between transcripts suppressed but offset vs. non‐offset upon ERα depletion. Codons coding for the same amino acid are connected by a gray line.
-
DLeft panel shows average frequency (percent) of amino acids encoded by codons previously identified as over‐represented among induced or suppressed but offset mRNAs (colored in light or dark blue, respectively; leucine (L, black) is encoded by codons in both groups). Middle panel shows average frequency (percent) of all other amino acids. Right panel shows boxplots (plotted as in Fig 3B) for all mRNAs within each mode for regulation of gene expression comparing number of amino acids (log2, L_aa, right panel). The numbers of mRNAs in each group are indicated in the figure.
-
E, FFor each codon, the codon usage normalized by amino acid count is compared between transcripts induced (E) or suppressed (F) but offset vs. non‐offset (i.e., abundance mode of regulation) upon ERα depletion. Codons coding for the same amino acid are connected by a gray line.
-
GUnsupervised clustering of a Spearman correlation matrix of codon frequency of regulated mRNAs. Codons identified as over‐represented among induced but offset and suppressed but offset mRNAs are indicated in light and dark blue, respectively.
Transcripts whose upregulation is translationally offset are enriched in codons decoded by U34‐modified tRNAs
To explore whether ERα‐dependent alterations in tRNA expression may underpin translational offsetting, we employed RNAseq of small RNAs. Notably, tRNA modifications result in short RNA sequencing reads, which do not consistently allow locus‐specific expression data (Cozen et al, 2015). Nevertheless, we obtained expression data on tRNAs irrespective of loci which allowed quantification of tRNA expression (Appendix Fig S4). When comparing shERα to control BM67 cells, however, no significant change (FDR < 0.05) in tRNA levels was observed (Fig EV4A). We next grouped tRNAs corresponding to codons identified as over‐represented in mRNAs whose alterations were translationally offset. No differences in expression of any of the tRNA groups were, however, observed between shERα and control BM67 cells (Fig EV4B). In addition to alteration of their expression, tRNA function is regulated by post‐transcriptional modifications (El Yacoubi et al, 2012). Modifications present at the anticodon loop affect translational rates, sometimes in a codon‐specific manner, by modulating the stability of codon–anticodon pairing, and thus limiting decoding to specific nucleotides in the wobble position (Rezgui et al, 2013; Deng et al, 2015). Among the nine codons which were over‐represented in mRNAs whose upregulation was translationally offset, seven had an adenosine in the 3′ position. These codons are decoded by tRNAs harboring modified uridine at position 34 (U34; Appendix Table S1). Moreover, Dek mRNA, which encodes a tumor‐promoting protein and whose upregulation is offset at the level of translation upon ERα depletion (Fig 2A and B), is dependent on 5‐methoxycarbonyl‐methyl‐2‐thiouridine (mcm5s2U) modification at U34 of corresponding tRNAs (Delaunay et al, 2016). In yeast, this modification is present on tRNAUUC Glu, tRNAUUU Lys, and tRNAUUG Gln, wherein it facilitates decoding of GAA, AAA, and CAA codons (Johansson et al, 2008). Strikingly, these codons are among those that were over‐represented in mRNAs whose induction is translationally offset in ERα‐depleted cells (Fig 5B). Thus, we speculated that although tRNA expression did not change following ERα depletion, alterations in mcm5s2U modifications may underlie translational offsetting of mRNAs whose levels are induced.
Figure EV4. ERα depletion‐associated translational offsetting cannot be explained by modulations of tRNA levels.
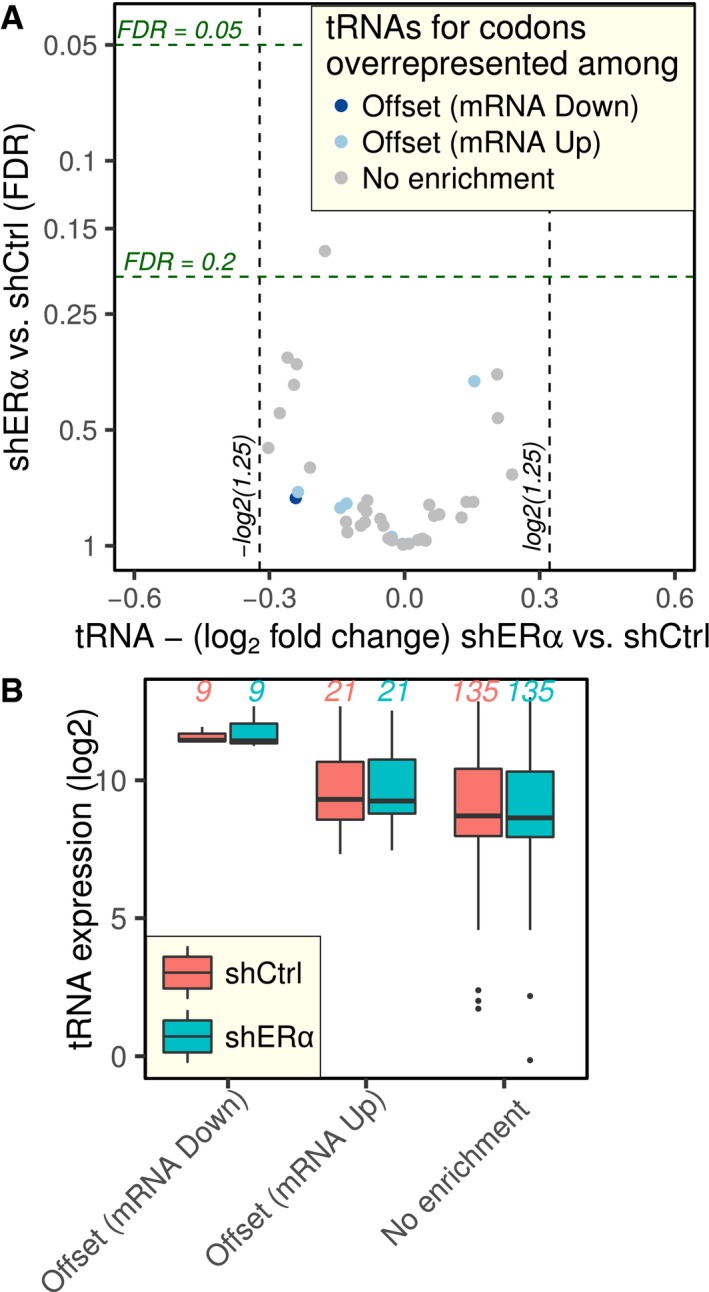
-
AVolcano plot of changes in tRNA level (shERα vs. shCtrl). Each tRNA is colored according to the mode for regulation of gene expression it is enriched in.
-
BtRNAs were grouped according to the mode for regulation of gene expression they are enriched in, and their expression is compared using boxplots (plotted as in Fig 3B). The numbers of tRNAs in each group are indicated in the figure.
ERα regulates expression of tRNA U34 modification enzymes leading to selective translational offsetting
The mcm5s2U modification is catalyzed by a cascade of enzymes including ELP3, ALKBH8, and CTU1/2 which sequentially modify U34 to generate cm5U, mcm5U, and mcm5s2U, respectively (Rapino et al, 2017). Upon ERα depletion in BM67 cells, Elp3 (P = 0.03) showed a reduction in the amount of polysome‐associated mRNA (data from polysome profiling in Figs 1 and EV1). Consistently, ELP3 protein level was decreased in shERα BM67 as compared to control cells (Fig 6A). To further explore the relationship between ERα, ELP3, and translational offsetting of DEK, we generated 5 Esr1 (encoding for ERα) knockout BM67 cell lines using CRISPR/Cas9 and two different guide RNAs (gRNAs). As expected, ELP3 protein levels were reduced across all cell lines with abrogated Esr1 as compared to the controls (Fig 6B upper panels). Moreover, although DEK protein levels were on average unchanged across the 5 Esr1 KO cell lines as compared to control, mRNA levels were on average increased (Fig 6B lower left; 4 out of 5 of the individual cell lines showed offsetting between protein and mRNA levels). To further establish the relationship between ELP3 expression and translational offsetting, we abrogated ELP3 expression in BM67 cells using CRISPR/Cas9. We also rescued ELP3 expression in ELP3 KO cells using a construct which is not targeted by ELP3 gRNAs. Similarly to Esr1 abrogation, ELP3 depletion resulted in translational offsetting of DEK whereby DEK protein levels remained unchanged notwithstanding increase in its mRNA levels as compared to the control (Fig 6C). Accordingly, offsetting was reversed by re‐expression of ELP3 in ELP3 KO cells (Fig 6D). These data suggest that ELP3 mediates the effects of ERα on translational offsetting.
Figure 6. Translational offsetting of transcripts induced following ERα depletion is mediated by U34 tRNA modification enzymes.

-
AImmunoblotting of BM67 cell extract for ELP3 after stable shRNA‐mediated ERα knockdown in BM67 cells. Graphs represent mean ± standard deviation of n = 3.
-
BqPCR analysis and immunoblotting of BM67 cell extract for indicated proteins with its quantitation following deletion of Esr1 gene by CRISPR‐Cas9 technology. Graphs represent mean ± standard deviation of n = 5.
-
CqPCR analysis and immunoblotting of BM67 cell extract for ELP3 and DEK with its quantitation following deletion of Elp3 gene by CRISPR‐Cas9 technology. Graphs represent mean ± standard deviation of n = 4.
-
DImmunoblotting and qPCR analysis comparing cell extracts obtained from BM67 cells transduced with pLentiCRISPRV2 empty vector, Elp3‐null BM67 cells, and Elp3‐null BM67 cells overexpressing ELP3 protein containing silent mutations in the sgRNA binding site. Graphs represent mean ± standard deviation of n = 3–4.
To assess the functional consequences of the observed phenomena, we monitored effects of fulvestrant and E2 on proliferation of ELP3 KO BM67 cells. At the baseline, proliferation of BM67 cells was significantly reduced by abrogation of ELP3 expression (Fig EV5). Moreover, whereas fulvestrant reduced and E2 induced proliferation of control cells, these effects were attenuated in ELP3 KO cells. This suggests that ELP3 is implicated in regulating proliferation downstream of ERα.
Figure EV5. Selective estrogen receptor modulators (SERMs) treatment in parental and Elp3‐null BM67 cells.
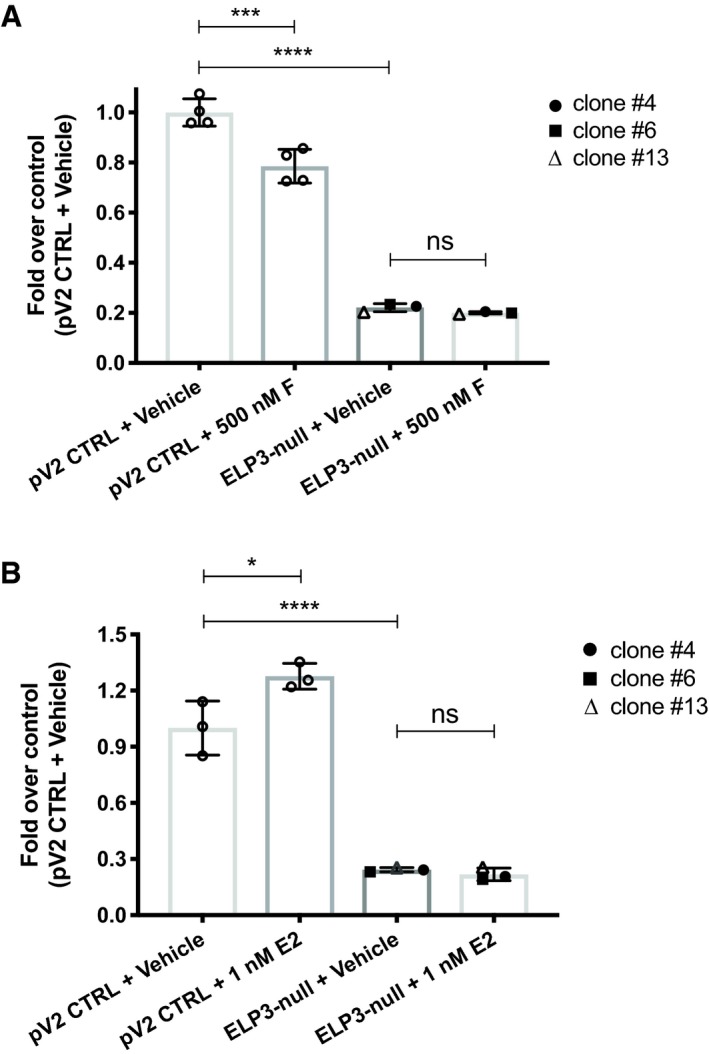
-
A, BTotal cell number analysis using the sulforhodamine B assay in BM67 cells transduced with pLentiCRISPRV2 empty vector vs. Elp3‐null BM67 cells in response to (A) ICI‐182780 (500 nM; 72 h) and (B) E2 (1 nM; 72 h). Graphs represent mean ± SD of n = 3 experiments. Data were analyzed by one‐way ANOVA. ns P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
ERα regulates mcm5s2‐U tRNA modification
There are two approaches to studying estrogen signaling, depleting the receptor, or modulating its activity with ligands. Using BM67 cells with stable knockdown of ERα allowed us to exclude potential confounding effects of ERβ and established the translational offsetting as a sustained response to repression of ERα signaling. Yet, this experimental model does not distinguish between direct effects of ERα vs. indirect effects which are mediated by its transcriptional targets (Katchy et al, 2012) and/or secondary adaptive mechanisms which are activated in response to the chronic reduction in ERα signaling. Therefore, to confirm that ERα modulates activity of the ELP3/ALKBH8/CTU1/2 pathway and to exclude cell line‐dependent biases, we employed MCF7 human breast cancer cells, which is a commonly used experimental model of ligand‐induced ERα activity (Hewitt & Korach, 2018). Strikingly, a previously published dataset wherein MCF7 cells were starved and then treated with estradiol (E2) or vehicle for 24 h in the presence or absence of selective estrogen receptor modulators (SERMs; Wardell et al, 2012; Data Ref: McDonnell et al, 2012) revealed that ERα activity is directly proportional to ELP3, ALKBH8, and CTU2 mRNA levels (Fig 7A; CTU1 was not quantified in Data Ref: McDonnell et al, 2012). To corroborate these findings, we employed the same experimental setup using ER antagonist fulvestrant (ICI 182780) and monitored levels of enzymes which catalyze mcm5s2‐U modifications. E2‐dependent induction of transcriptional activity of ERα was confirmed by upregulation of its well‐established target c‐MYC (Wang et al, 2011). This was paralleled by an increase in levels of ELP3 and CTU1 but not ALKBH8 proteins (Fig 7B), which was abrogated by ICI‐182780 (Fig 7B). Consistent with translational offsetting, DEK protein levels were unaltered by ligand‐dependent modulation of ERα activity (Fig 7B). To evaluate whether ER‐mediated regulation of ELP3 and CTU1 expression was direct, we examined ChIP‐seq data for potential ER binding sites proximal to these genes. In multiple ER cistromes derived from distinct cell line models of breast cancer and primary breast tumors, we found evidence for E2‐dependent ERα binding at ELP3 and CTU1 (Fig 7C, Appendix Fig S5). We validated one of the ELP3 binding sites by ChIP‐qPCR (Appendix Fig S5B). Thus, ERα appears to directly regulate the levels of enzymes implicated in mcm5s2‐U34 tRNA modification.
Figure 7. ERα regulates mcm5s2‐U tRNA modifications by controlling the expression of mcm5s2‐U‐modifying enzymes in MCF7 cells.
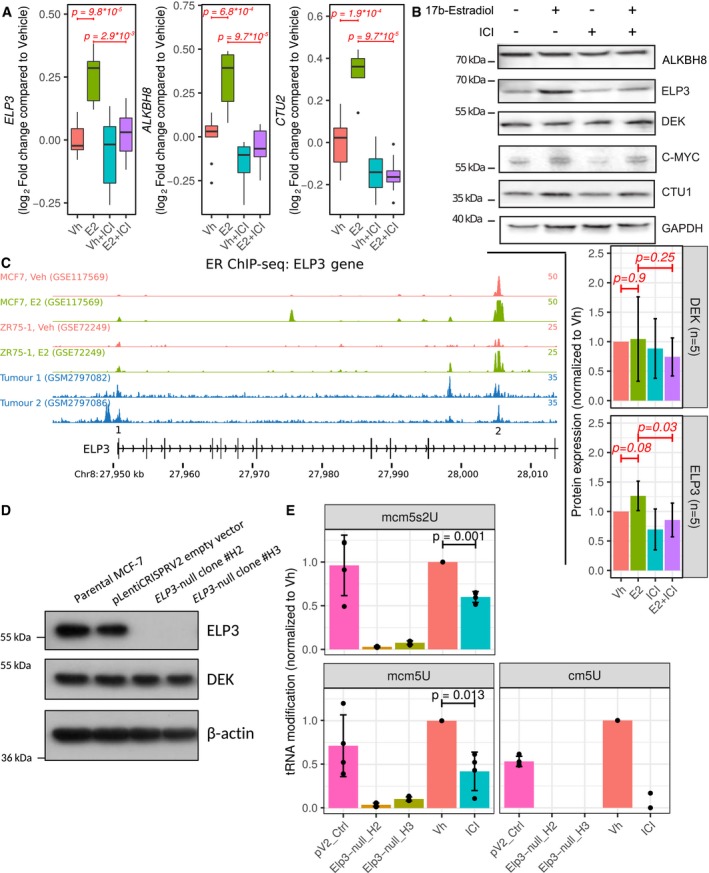
-
ABoxplots (plotted as in Fig 3B) of gene expression of ELP3, ALKBH8, and CTU2 upon treatment with 17β‐E2 and/or ICI‐182780 in MCF7 cells (extracted from Wardell et al, 2012; Data Ref: McDonnell et al, 2012). Wilcoxon–Mann–Whitney tests (two‐sided) were used to assess differences between conditions (n = 10) and a global Bonferroni adjustment was applied on P‐values.
-
BImmunoblotting of MCF7 cell extracts. MCF‐7 cells were grown in phenol red‐free RPMI media supplemented with 5% charcoal‐stripped serum for 24 h and then treated with 1 nM E2 and/or 100 nM ICI‐182780 for 24 h. Graphs represent mean ± SD of n = 5 experiments. Data were analyzed using two‐sided paired Student's t‐tests.
-
CChIP‐seq reveals ER binding sites proximal to the ELP3 gene. Each track represents a distinct ChIP‐seq dataset.
-
DImmunoblotting of MCF‐7 cell extract for indicated proteins following deletion of ELP3 gene by CRISPR‐Cas9 technology.
-
EQuantification of mcm5S2‐U, mcm5U, and cm5U levels in ELP3‐null MCF‐7 cells and in MCF‐7 cells treated with 100 nM ICI‐182780 for 72 h vs. their respective controls by liquid chromatography–mass spectrometry (LC‐MS) analysis. The MCF‐7 cells were grown in phenol red‐free RPMI media supplemented with 5% charcoal‐stripped serum for 24 h prior to ICI‐182780 treatment. Graphs represent mean ± SD of n = 4. Data were analyzed using two‐sided paired Student's t‐tests. Vh, Vehicle; ICI, ICI‐182780.
Source data are available online for this figure.
Thus far, our data point out that regulation of mcm5s2‐U34 tRNA‐modifying enzymes plays a major role in translational offsetting as a function of alterations in ERα signaling. Intriguingly however, ERα‐dependent suppression or decrease in the expression of U34‐modifying enzymes did not correlate with global changes in mcm5s2‐U‐modified tRNAs as quantified by the [(N‐acryloylamino)phenyl]mercuric chloride (APM) method [Igloi, 1988; as reported in a recent study (Rapino et al, 2018)] or mass spectrometry (Appendix Fig S6A and B). We reasoned that this may be a consequence of accumulation of modified tRNAs during chronic ERα depletion (e.g., due to reduction in global protein synthesis). We therefore performed experiments under conditions wherein ERα was inhibited by fulvestrant in MCF7 cells for a shorter period of time (72 h), followed by cm5U, mcm5U, and mcm5s2U quantification by mass spectrometry. As a positive control, we used cells wherein ELP3 expression was abolished using CRISPR/Cas9 (Fig 7D). Indeed, similarly to ELP3 loss, fulvestrant dramatically decreased cm5U, mcm5U, and mcm5s2U modifications by ~ 95, ~ 65, and ~ 40%, respectively, as compared to vehicle‐treated control cells (Fig 7E). These data suggest that ERα stimulates mcm5s2‐U tRNA modifications likely by upregulation of ELP3 and other mcm5s2U‐modifying enzymes. Thus, downregulation of U34‐modifying enzymes following ERα‐depletion leads to translational offsetting of its upregulated transcriptional targets.
Discussion
Improved experimental and analytical methods for transcriptome‐wide analysis of translation have been essential for identifying hitherto unprecedented mechanisms of translation regulation (Truitt & Ruggero, 2016; Ingolia et al, 2018; Yordanova et al, 2018). Using such methods, upon depletion of ERα in prostate cancer, we observed that translational offsetting appears to be a pervasive mechanism which maintains proteome composition. Transcription start site profiling and sequencing of small RNAs revealed that translational offsetting of mRNAs whose levels are decreased is linked to distinct 5′ and 3′ UTR features. The length of the 5′UTR is thought to strongly impact on translation whereby short (e.g., < 30 bases) and very long (e.g., > 150 bases) 5′UTRs are associated with reduced translational efficiencies (Pelletier & Sonenberg, 1987; Koromilas et al, 1992; Arribere & Gilbert, 2013; Sinvani et al, 2015; Gandin et al, 2016b). In contrast, the median 5′UTR length (85 nt) of mRNAs whose downregulation is offset at the level of translation corresponds to what has been described as the “optimal” 5′UTR length for translation in mammalian cells (Kozak, 1987). Moreover, target sites for miRNAs, which mediate translational suppression (Filipowicz et al, 2008), are largely absent in mRNAs whose suppression is translationally offset. Finally, tRNAs that decode mRNAs whose downregulation is translationally offset appear to be expressed at higher levels as compared to other identified tRNA groups (Fig EV4B) and such transcripts also appear to exert more optimal codon usage as compared to non‐offset mRNAs (Fig EV3A and B). Therefore, this subset of mRNAs exhibits multiple features which would be expected to facilitate translation and thus offset reduced mRNA levels.
We also observed widespread translational offsetting for mRNAs whose levels were induced in ERα‐depleted vs. control cells. In this case, translational offsetting may be attributed to codon usage of these transcripts. Indeed, the frequency of codons which are decoded more efficiently by the U34‐modified tRNAs was substantially higher in transcripts whose induction after ERα depletion was translationally offset as compared to non‐offset mRNAs. Consistently, ERα depletion reduced expression of ELP3 and other U34 modification enzymes, which appear to play a major role in tumorigenesis (Ladang et al, 2015; Delaunay et al, 2016; Rapino et al, 2018). In this context, genes such as DEK (Delaunay et al, 2016) and HIF1α (Rapino et al, 2018) were characterized as key downstream effectors, which mediate the pro‐neoplastic effects of U34 modification enzymes. Collectively, these findings suggest that ERα‐dependent modulation of U34‐modification enzymes expression results in translational offset of transcripts whose levels are induced upon ERα depletion.
In addition to translation initiation, it has recently been revealed that translation elongation is also dysregulated in cancer, which in part appears to be determined by codon composition of mRNAs (Leprivier et al, 2013; Faller et al, 2015). Herein, we have identified regulation of U34‐modification enzymes as a process by which ERα modulates translation of a subset of mRNAs with distinct codon usage in prostate cancer cells. This regulation is mediated at the level of mRNA translation by offset of transcriptional targets requiring U34‐modified tRNAs. At the same time, a second set of transcripts which harbor optimal 5′UTRs and codon composition but lack target sites for miRNAs are translationally offset when mRNA levels decrease. We speculate that translational offsetting plays a role in mediating biological effects of ERα in neoplastic tissues. Indeed, using a polysome‐profiling data set comparing tamoxifen‐sensitive vs. resistant cells (Geter et al, 2017a; Data ref: Geter et al, 2017b), we observed translational activation of mRNAs with higher requirements for U34‐modified tRNAs and increased expression of ALKBH8 in tamoxifen‐resistant cells (Appendix Fig S7). Thus, modulation of translation via ERα‐dependent changes in U34‐modifications may be associated with drug resistance and our findings therefore may have important implications in understanding alterations in gene expression programs following treatment with ERα antagonists.
In conclusion, this study establishes translational offsetting as a distinct subtype of a widespread buffering mechanism which allows adaptation to short‐term (E2 treatment) and long‐term (ERα depletion) alterations in ERα‐dependent transcriptomes. Moreover, these findings unravel a previously unappreciated cooperation between transcriptional and translational programs which suggest a hitherto unappreciated plasticity of gene expression machinery in shaping adaptive proteomes.
Materials and Methods
Cell culture, antibodies, Western blot
MCF7 cells were purchased from America Type Tissue Culture Collection and used at low passage for less than 2 months before thawing a new vial. The PTEN‐deficient prostate cancer cell line (BM67) derived from the PB‐Cre;PtenFlox/Flox mouse model of prostate cancer (Wang et al, 2003) has been described previously (Takizawa et al, 2015) and used at low passage. Cells were maintained on 4 μg/ml of puromycin (Sigma). Unless stated otherwise, cells were grown in RPMI‐1640 (Gibco) and supplemented to a final concentration of 5% fetal bovine serum (FBS, Sigma) and 100 IU/ml penicillin and 10 μg/ml streptomycin (P/S, Invitrogen) and kept in a humidified incubator at 37°C supplemented with 5% CO2. All cell lines were routinely tested for mycoplasma (in house service, Peter MacCallum Cancer Centre). Western blot information, CRISPR/Cas9 method, as well as a description of the cycloheximide chase assay are provided in Appendix Supplementary Methods.
RNA extraction
Polysome profiling was performed on four replicates of each condition as previously described (Gandin et al, 2014). Briefly, cytosolic lysates were loaded on a 5–50% sucrose gradient allowing for isolation of mRNAs associated with more than three ribosomes (hereafter referred to as polysome‐associated mRNA) after ultracentrifugation. Total cytosolic RNA was isolated in parallel (Gandin et al, 2014).
DNA microarray assays and data processing
Cytosolic and polysome‐associated RNA were quantified using the Affymetrix Mouse Gene 1.1 ST array as described previously (Gandin et al, 2016a) by the Bioinformatics and Expression analysis core facility at Karolinska Institutet. Poor quality arrays were obtained for one replicate of each condition leading to exclusion of the whole replicate (cytosolic and polysome‐associated samples from both conditions) from all analyses. Gene expression was normalized using Robust Multiarray Averaging and annotated with a custom probeset definition (mogene11st_Mm_ENTREZG; Dai et al, 2005; Sandberg & Larsson, 2007).
Analysis of polysome‐profiling data
Changes in translational efficiency leading to altered protein levels were quantified using analysis of partial variance (Larsson et al, 2010, 2011) as implemented in the anota2seq R/Bioconductor package version 1.2.0 (Oertlin et al, 2019). Differential expression of cytosolic and polysome‐associated RNA was also assessed using the anota2seq package. For such analyses, Benjamini–Hochberg correction was used to account for multiple testing and a random variance model was used to increase statistical power (Wright & Simon, 2003; Larsson et al, 2010). Translational offsetting was defined by mRNAs showing changes in cytosolic RNA which are not reflected in polysome loading as implemented in anota2seq. Details of the analysis are provided in Appendix Supplementary Methods.
Preparation of RNAseq libraries, sequencing, and analysis
RNAseq libraries were prepared using the Smart‐seq2 method as described previously (Picelli et al, 2014), using 10 ng RNA as input material. After pre‐amplification PCR, 80 pg of cDNA was used for tagmentation using the Nextera XT Kit (Illumina) in a total volume of 25 μl. Sequencing was performed using the HiSeq2500 platform (HiSeq Control Software 2.2.58/RTA 1.18.64) with a 1 × 51 setup using “HiSeq Rapid SBS Kit v2” chemistry. FastQ conversion was performed using bcl2fastq_v2.19.1.403 from the CASAVA software suite. The quality scale used is Sanger/phred33/Illumina 1.8+. Details of the analysis are provided in Appendix Supplementary Methods.
Nanostring gene expression quantification and analysis
145 genes were selected for quantification by Nanostring (Geiss et al, 2008). Within each mode of regulation (as defined in Fig 1E), genes among the top smallest P‐values were randomly selected. Additional genes within each mode of regulation (not belonging to the most significant sets) as well as 11 negative controls (non‐regulated genes) were included as well. Details about methodology and analysis are given in Appendix Supplementary Methods.
GO enrichment analysis
A generally applicable gene set enrichment for pathway analysis (Luo et al, 2009) was performed to identify enrichment of key cellular functions represented by GO terms (Gene Ontology Consortium, 2015). Additional information is provided in Appendix Supplementary Methods.
nanoCAGE library preparation, sequencing, and analysis
nanoCAGE libraries of cytosolic mRNA from shERα BM67 cells were prepared as described previously (Gandin et al, 2016b) with several modifications detailed in Appendix Supplementary Methods where description of preprocessing and analysis is also provided.
RNAseq of small RNAs
RNA was extracted in triplicates from ERα shRNA and control BM67 cells using the RNeasy Plus Mini Kit (Qiagen). RNAseq libraries were prepared according to the Illumina TruSeq Small RNA Library Preparation protocol with small RNA enrichment on the Agilent Bravo Liquid Handling Platform and sequenced on HiSeq2500 (HiSeq Control Software 2.2.58/RTA 1.18.64) with a 1 × 51 setup. Library preparation and sequencing were performed at Science for Life Laboratory National Genomics Infrastructure. Preprocessing and analysis of RNAseq of small RNAs data are described in Appendix Supplementary Methods.
Validation of miRNA expression using qPCR
cDNA was synthesized using miSCRIPT II RT kit (Qiagen; three replicates) followed by specific miRNA amplification using miSCRIPT SYBR Green PCR kit (Qiagen) using the following miScript specific primers mmu‐miR‐21a‐5p (5′‐UAGCUUAUCAGACUGAUGUUGA‐3′), mmu‐miR‐181a‐5p (5′‐AACAUUCAACGCUGUCGGUGAGU‐3′), mmu‐miR‐32‐5p (5′‐UAUUGCACAUUACUAAGUUGCA‐3′), mmu‐miR‐23b‐3p (5′‐AUCACAUUGCCAGGGAUUACC‐3′) and mmu‐miR‐27b‐3p (5′‐UUCACAGUGGCUAAGUUCUGC‐3′).
Analysis of codon usage
A detailed explanation is given in Appendix Supplementary Methods. Briefly, the longest coding sequences of all regulated mRNAs were extracted from the consensus coding sequence database (Pruitt et al, 2009) to retrieve their codon composition. The codon usage indexes were computed using the codonW (available at: https://sourceforge.net/projects/codonw/files/codonw/SourceCode-1.4.4%28gz%29/ [Accessed September 18, 2019]) and tAI (dos Reis et al, 2003, 2004; dos Reis) packages.
Quantitative real‐time polymerase chain reaction (qRT–PCR)
RNA was isolated using Qiagen RNeasy Mini Kit (Qiagen 74106) as per manufacturers’ protocols. Eluted RNA concentration was quantified with a Nanodrop ND1000 spectrophotometer (Thermo Fisher Scientific). Total RNA equivalent to equal amount of total RNA (500 ng) was treated with RNase‐free DNAse (Promega) at 37°C for 15 min followed by 15 min incubation at 70°C. cDNA was synthesized with 60 U Superscript III (Life Technologies 18080‐044) and 12.25 ng random hexamer primers (Promega C1181) at room temperature for 5 min, 37°C for 5 min, 47°C for 120 min and 70°C for 15 min according to the manufacturer's instructions. Synthesized cDNA was diluted with sterile MilliQ water and stored at −20°C.
qRT–PCR samples consisting of 8 μl cDNA sample and 12 μl of 0.1 μM oligo‐primers (forward–reverse mix) and Fast SYBR Green master mix (Applied Biosystem 4385612) were plated in triplicate in MicroAMP Optical 96‐well reaction plates (Applied Biosystems N8010560). The qRT–PCRs were performed by the StepOne Plus Real‐Time PCR system (Applied Biosystem 4376600), and fold change was determined quantitatively by 2−(ΔΔCt).
Gene deletion by CRISPR/Cas9 technology and target rescue
Elp3 and Esr1 genes were deleted from BM67 cells by CRISPR/Cas9 technology using plentiCRISPRv2. HEK293T cells were transfected with gRNA constructs and viral packaging plasmid (ViraPower™ Lentiviral Packaging Mix, Invitrogen K497500) using Lipofectamine 3000 system (Invitrogen). BM67 and MCF‐7 cells were infected with viral particles and selected with puromycin. Guide sequences for BM67 are as follows:
Esr1 (GTAACACTTGCGCAGCCGAC; TCTGACAATCGACGCCAGAA),
Elp3 (TGTCCACACATCAGTTTCAC; CTGCAGCGATGATATCCACC),
Guide sequences for MCF7 are as follows:
ELP3 (GATGCCTGACCTGCCAAACG; GAGTTACTCTCCTAGTGACC)
Single colonies were obtained using a BD FACSAria™ Fusion5 (BD Biosciences). The colonies were expanded into 24‐well plates and screened for depletion of the targeted gene product by immunoblotting.
For target gene rescue, Elp3‐null BM67 cells were incubated with a mixture containing 7.5 μl of Lipofectamine 3000 (Invitrogen) complexed with 2.5 μg of empty vector plasmid or plasmid harboring ELP3 (pcDNA3.1/Hygro(+)‐Elp3, Genscript Clone ID OMu06165C with silent mutations on the gRNA binding site: TGTCCACACATCAGTTTCAC > TGCCCTCATATAAGCTTTAC) for 24 h followed by hygromycin selection. Samples were then processed for immunoblotting analysis.
Quantification and analysis of tRNA levels
RNAseq of small RNAs data described above was used for tRNA quantification using the ARM‐seq bioinformatics pipeline from Cozen et al (2015) with a few modifications detailed in Appendix Supplementary Methods.
mcm5s2‐U tE(UUC) detection and quantification by APM
For quantification of mcm5s2‐U‐modified tE(UUC), tRNA was purified using the miRvana kit (Roche; four replicates). 0.5 μg RNA was resolved on an 8% acrylamide gels containing 0.5× TBE, 7 M urea, and 50 mg/ml [(N‐acryloylamino)phenyl]mercuric chloride (APM; Igloi, 1988). Northern blot analysis was performed essentially as described in Leidel et al (2009), using 5′‐TTCCCATACCGGGAGTCGAACCCG‐3′ as probe to detect tE(UUC).
cm5U, mcm5U and mcm5s2U detection and quantification by LC‐MS/MS
mcm5U and mcm5s2U were synthesized by Dr. Malkiewicz and colleagues, lyophilized and stored at −20°C. cm5U was purchased from the AA BLOCKS LLC. (San Diego, CA, USA). [13C][15N]‐G was purchased from the Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA). All the nucleoside standards were characterized individually by liquid chromatography coupled with both UV and mass spectrometric detection. If degradation or impurities were present, the samples were discarded from further analysis. Acetonitrile (LC‐MS Optima) was purchased from Fisher.
A volume of 4 μl (96 to 487 ng/μl) total RNA was used to evaluate levels of cm5U, mcm5U, and mcm5s2U, by LC‐MS/MS using a similar method as described (Basanta‐Sanchez et al, 2016; Tardu et al, 2019). Briefly, prior to UHPLC‐MS analysis, each sample was mixed with 0.4 pg/μl of internal standard (IS), isotopically labeled guanosine, [13C][15N]‐G. The enzymatic digestion was carried out using Nucleoside Digestion Mix (New England BioLabs) according to the manufacturer's instructions. Finally, the digested samples were lyophilized and reconstituted in 100 μl of RNAse‐free water, 0.01% formic acid prior to UHPLC‐MS/MS analysis. The UHPLC‐MS analysis was accomplished on a Waters XEVO TQ‐S™ (Waters Corporation, USA) triple quadruple mass spectrometer equipped with an electrospray source (ESI) source maintained at 150°C and a capillary voltage of 1 kV. Nitrogen was used as the nebulizer gas which was maintained at 7 bars pressure, flow rate of 500 l/h and at a temperature of 500°C. UHPLC‐MS/MS analysis was performed in ESI positive‐ion mode using multiple reaction monitoring (MRM) from ion transitions previously determined for mcm5s2U. A Waters ACQUITY UPLC™ HSS T3 guard column, 2.1 × 5 mm, 1.8 μm, attached to an HSS T3 column, 2.1 × 50 mm, 1.7 μm were used for the separation. Mobile phases included RNAse‐free water (18 MΩ cm−1) containing 0.01% formic acid (Buffer A) and 50:50 acetonitrile in Buffer A (Buffer B). The digested nucleotides were eluted at a flow rate of 0.5 ml/min with a gradient as follows: 0–2 min, 0–10% B; 2–3 min, 10–15% B; 3–4 min, 15–100% B; 4–4.5 min, 100% B. The total run time was 7 min. The column oven temperature was kept at 35°C, and sample injection volume was 10 μl. Three injections were performed for each sample. Data acquisition and analysis were performed with MassLynx V4.1 and TargetLynx. Quantification was performed based on nucleoside‐to‐base ion transitions using standard curves of pure nucleosides and stable isotope‐labeled guanosine internal standards. Calibration curves were plotted using linear regression.
Analysis of public dataset for E2‐dependent expression of ELP3, ALKBH8, and CTU2
Expression of enzymes catalyzing the mcm5s2‐U34 tRNA modification upon treatment with E2 and/or ICI‐182780 was analyzed using a publicly available data (Wardell et al, 2012; Data Ref: McDonnell et al, 2012) as detailed in Appendix Supplementary Methods.
Selective estrogen receptor modulator treatment
Cells were seeded in standard cell culture conditions to achieve 25–40% confluency. Twenty‐four hours later, the culture medium was replaced with fresh standard culture medium (FBS). 24 h later, SERM was administered at a concentration of 1 nM 17β‐estradiol (E2) or 100 nM of Fulvestrant (ICI‐182780).
ER DNA binding analysis by chromatin immunoprecipitation (ChIP)
ER binding proximal to the ELP3, CTU1, and ALKBH8 genes was assessed using published ChIP‐sequencing data (Helzer et al, 2018a; Swinstead et al, 2016a; Severson et al, 2018; Data Ref: Helzer et al, 2018b; Data Ref: Swinstead et al, 2016b; Data Ref: Schuurman et al, 2018). ChIP‐qPCR of ER from ZR‐75‐1 cells was done essentially as described previously (Mohammed et al, 2015) using ER sc‐543x antibody (Santa Cruz). Briefly, cells were cultured for 3 days in phenol red‐free RPMI‐1640 media supplemented with 5% steroid‐stripped FBS then treated with E2 (10 nM) for 4 h prior to harvest. Four independent experiments were carried out.
Drug dose–response sulforhodamine B proliferation assays
The cell lines were seeded into 96‐well plates for 24 h before the addition of drug as indicated in figure legends. Cold trichloroacetic acid (Merck) (final concentration 10%) was added to the plates to fix the cells in situ at 4°C for 1 h. The plates were then washed with cold water and left to dry overnight. Cells were stained using 0.4% (w/v) sulforhodamine B (SRB) (Sigma) in 1% (v/v) acetic acid (Sigma) at room temperature for 1 h and washed with cold water and then 1% (v/v) acetic acid. The bound dye was solubilized in 10 mM Tris, and the absorbance was measured at 550 nm using an iMark™ Microplate Absorbance Reader (Bio‐Rad).
Statistics
Unless stated otherwise, all statistical tests are two‐sided.
Author contributions
ITo, OL, and LF conceived and designed the study; EPK, VH, RJR, ML, JR, SC, BS, PBa, FR, PBu, KS, ITa, and QL performed experiments; JL, EPK, VH, RJR, ML, JR, MGL, KJS, FR, PC, KS, LAS, ITo, OL, and LF performed analysis and interpreted the data; PC, GPR, LAS, SAL, QL, ITo, OL, and LF supervised the research; and JL, EPK, ITo, OL, and LF wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The authors acknowledge A. Hanson and G. Laven‐Law for expert technical assistance with ChIP‐qPCR, A. Puri for technical assistance with Western blots, and Dr. T. Begley for advice with tRNA modification analysis. The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure (NGI) and Bioinformatics and Expression Analysis (BEA, Karolinska Institutet) core facilities and Uppmax for providing assistance in massive parallel sequencing, DNA microarray processing and computational infrastructure. The research was supported by the Swedish Research Council, the Wallenberg Academy Fellow Program, the Swedish Cancer Society, the Cancer Society in Stockholm and STRATCAN (to OL); National Health and Medical Research Council (NHMRC) grants APP1141339 to ITo, OL, LF; APP 1102752 to GPR and APP 1145777 to LAS; the Department of Health and Human Services acting through the Victorian Cancer Agency (MCRF16007) to LF and (MCRF18017) to MGL; EJ Whitten Foundation to LF; DFG grant [LE 3260/3‐1] to SAL; Walloon Excellence in Life Sciences and Biotechnology (WELBIO) to PC; Canadian Institutes for Health Research (MOP‐363027) and National Institutes of Health grants (R01 CA 202021‐01‐A1) (to ITo); Joint Canada‐Israel Health Research Program (JCIHRP) (108589‐001) (to ITo and OL). ITo is supported by Junior 2 award from Fonds de Recherche du Québec—Santé (34872). This project was further facilitated by funding from the Swedish foundation for international cooperation in research and higher education (STINT) to ITo, OL, and LF.
The EMBO Journal (2019) 38: e101323
See also: I Kozlovski & R Agami (December 2019)
Contributor Information
Ivan Topisirovic, Email: ivan.topisirovic@mcgill.ca.
Ola Larsson, Email: ola.larsson@ki.se.
Luc Furic, Email: luc.furic@monash.edu.
Data availability
The DNA microarrays, RNAseq of full length or small RNAs, and nanoCAGE data generated and analyzed during the current study are available in the NCBI Gene Expression Omnibus repository under accession number GSE120917 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120917).
References
- Arribere JA, Gilbert WV (2013) Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res 23: 977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Fraser HB (2014) Evolution at two levels of gene expression in yeast. Genome Res 24: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey ST, Westerling T, Brown M (2015) Loss of estrogen‐regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer Res 75: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird TD, Palam LR, Fusakio ME, Willy JA, Davis CM, McClintick JN, Anthony TG, Wek RC (2014) Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKα . Mol Biol Cell 25: 1686–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanta‐Sanchez M, Temple S, Ansari SA, D'Amico A, Agris PF (2016) Attomole quantification and global profile of RNA modifications: epitranscriptome of human neural stem cells. Nucleic Acids Res 44: e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno LS, Keene JD (2018) RNA regulons in cancer and inflammation. Curr Opin Genet Dev 48: 97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano L, Giamas G, Jacob J, Coombes RC, Lucchesi W, Thiruchelvam P, Barton G, Jiao LR, Wait R, Waxman J et al (2009) The estrogen receptor‐alpha‐induced microRNA signature regulates itself and its transcriptional response. Proc Natl Acad Sci USA 106: 15732–15737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik C, Cenik ES, Byeon GW, Grubert F, Candille SI, Spacek D, Alsallakh B, Tilgner H, Araya CL, Tang H et al (2015) Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res 25: 1610–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M et al (2014) The oestrogen receptor alpha‐regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun 5: 5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CTY, Deng W, Li F, DeMott MS, Babu IR, Begley TJ, Dedon PC (2015) Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon‐biased genes. Chem Res Toxicol 28: 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Hrabeta‐Robinson E, Phizicky EM, Lowe TM (2015) ARM‐seq: AlkB‐facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods 12: 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H et al (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33: e175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay S, Rapino F, Tharun L, Zhou Z, Heukamp L, Termathe M, Shostak K, Klevernic I, Florin A, Desmecht H et al (2016) Elp3 links tRNA modification to IRES‐dependent translation of LEF1 to sustain metastasis in breast cancer. J Exp Med 213: 2503–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Babu IR, Su D, Yin S, Begley TJ, Dedon PC (2015) Trm9‐catalyzed tRNA modifications regulate global protein expression by codon‐biased translation. PLoS Genet 11: e1005706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Bailly M, de Crécy‐Lagard V (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46: 69–95 [DOI] [PubMed] [Google Scholar]
- Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB et al (2015) mTORC1‐mediated translational elongation limits intestinal tumour initiation and growth. Nature 517: 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post‐transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114 [DOI] [PubMed] [Google Scholar]
- Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA et al (2010) eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci USA 107: 14134–14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Sikström K, Alain T, Morita M, McLaughlan S, Larsson O, Topisirovic I (2014) Polysome fractionation and analysis of mammalian translatomes on a genome‐wide scale. J Vis Exp 87: e51455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Masvidal L, Cargnello M, Gyenis L, McLaughlan S, Cai Y, Tenkerian C, Morita M, Balanathan P, Jean‐Jean O et al (2016a) mTORC1 and CK2 coordinate ternary and eIF4F complex assembly. Nat Commun 7: 11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Masvidal L, Hulea L, Gravel S‐P, Cargnello M, McLaughlan S, Cai Y, Balanathan P, Morita M, Rajakumar A et al (2016b) nanoCAGE reveals 5′ UTR features that define specific modes of translation of functionally related MTOR‐sensitive mRNAs. Genome Res 26: 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Preiss T, Hentze MW (2012) From cis‐regulatory elements to complex RNPs and back. Cold Spring Harb Perspect Biol 4: a012245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T et al (2008) Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol 26: 317–325 [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2015) Gene Ontology Consortium: going forward. Nucleic Acids Res 43: D1049–D1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geter PA, Ernlund AW, Bakogianni S, Alard A, Arju R, Giashuddin S, Gadi A, Bromberg J, Schneider RJ (2017a) Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Genes Dev 31: 2235–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geter P, Schneider RJ, Ernlund A (2017b) Gene Expression Omnibus GSE107590 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107590) [DATASET]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD et al (2014) A dual program for translation regulation in cellular proliferation and differentiation. Cell 158: 1281–1292 [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF (2016) Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165: 1416–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B‐J, van Hoef V, Jobava R, Elroy‐Stein O, Valasek LS, Cargnello M, Gao X‐H, Krokowski D, Merrick WC, Kimball SR et al (2017) A unique ISR program determines cellular responses to chronic stress. Mol Cell 68: 885–900.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helzer KT, Szatkowski Ozers M, Meyer MB, Benkusky NA, Solodin N, Reese RM, Warren CL, Pike JW, Alarid ET (2018a) The phosphorylated estrogen receptor α (ER) cistrome identifies a subset of active enhancers enriched for direct ER‐DNA binding and the transcription factor GRHL2. Mol Cell Biol 39: e00417‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helzer KT, Ozers MS, Meyer MB, Benkusky NA, Solodin N, Reese RM, Warren CL, Pike JW, Alarid ET (2018b) Gene Expression Omnibus GSE117569 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117569) [DATASET]
- Hershey JWB, Sonenberg N, Mathews MB (2012) Principles of translational control: an overview. Cold Spring Harb Perspect Biol 4: a011528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Korach KS (2018) Estrogen receptors: new directions in the new millennium. Endocr Rev 39: 664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5′‐untranslated regions of eukaryotic mRNAs. Science 352: 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ et al (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igloi GL (1988) Interaction of tRNAs and of phosphorothioate‐substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry 27: 3842–3849 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Hussmann JA, Weissman JS (2018) Ribosome profiling: global views of translation. Cold Spring Harb Perspect Biol 11: a032698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MJO, Esberg A, Huang B, Björk GR, Byström AS (2008) Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol 28: 3301–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchy A, Edvardsson K, Aydogdu E, Williams C (2012) Estradiol‐activated estrogen receptor α does not regulate mature microRNAs in T47D breast cancer cells. J Steroid Biochem Mol Biol 128: 145–153 [DOI] [PubMed] [Google Scholar]
- Klinge CM (2012) miRNAs and estrogen action. Trends Endocrinol Metab 23: 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koromilas AE, Lazaris‐Karatzas A, Sonenberg N (1992) mRNAs containing extensive secondary structure in their 5′ non‐coding region translate efficiently in cells overexpressing initiation factor eIF‐4E. EMBO J 11: 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1987) An analysis of 5′‐noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15: 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, Foster LJ (2013) Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol Syst Biol 9: 689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladang A, Rapino F, Heukamp LC, Tharun L, Shostak K, Hermand D, Delaunay S, Klevernic I, Jiang Z, Jacques N et al (2015) Elp3 drives Wnt‐dependent tumor initiation and regeneration in the intestine. J Exp Med 212: 2057–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalanne J‐B, Taggart JC, Guo MS, Herzel L, Schieler A, Li G‐W (2018) Evolutionary convergence of pathway‐specific enzyme expression stoichiometry. Cell 173: 749–761.e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Sonenberg N, Nadon R (2010) Identification of differential translation in genome wide studies. Proc Natl Acad Sci USA 107: 21487–21492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Sonenberg N, Nadon R (2011) anota: Analysis of differential translation in genome‐wide studies. Bioinformatics 27: 1440–1441 [DOI] [PubMed] [Google Scholar]
- Larsson O, Morita M, Topisirovic I, Alain T, Blouin M‐J, Pollak M, Sonenberg N (2012) Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc Natl Acad Sci USA 109: 8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Tian B, Sonenberg N (2013) Toward a genome‐wide landscape of translational control. Cold Spring Harb Perspect Biol 5: a012302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Pedrioli PGA, Bucher T, Brost R, Costanzo M, Schmidt A, Aebersold R, Boone C, Hofmann K, Peter M (2009) Ubiquitin‐related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458: 228–232 [DOI] [PubMed] [Google Scholar]
- Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo A‐RF, Kool M, Agnihotri S, El‐Naggar A, Yu B, Prakash Somasekharan S et al (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153: 1064–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ER (2009) Membrane oestrogen receptor alpha signalling to cell functions. J Physiol 587: 5019–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Bickel PJ, Biggin MD (2014) System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2: e270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Chew G‐L, Biggin MD (2017) Quantitating translational control: mRNA abundance‐dependent and independent contributions and the mRNA sequences that specify them. Nucleic Acids Res 45: 11821–11836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, Aebersold R (2016) On the dependency of cellular protein levels on mRNA abundance. Cell 165: 535–550 [DOI] [PubMed] [Google Scholar]
- López I, Tournillon A‐S, Nylander K, Fåhraeus R (2015) p53‐mediated control of gene expression via mRNA translation during endoplasmic reticulum stress. Cell Cycle 14: 3373–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10: 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillot G, Lacroix‐Triki M, Pierredon S, Gratadou L, Schmidt S, Bénès V, Roché H, Dalenc F, Auboeuf D, Millevoi S et al (2009) Widespread estrogen‐dependent repression of micrornas involved in breast tumor cell growth. Cancer Res 69: 8332–8340 [DOI] [PubMed] [Google Scholar]
- Masvidal L, Hulea L, Furic L, Topisirovic I, Larsson O (2017) mTOR‐sensitive translation: cleared fog reveals more trees. RNA Biol 14: 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, Wardell SE, Kazmin DA (2012) Gene Expression Omnibus GSE35428 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35428) [DATASET]
- McManus CJ, May GE, Spealman P, Shteyman A (2014) Ribosome profiling reveals post‐transcriptional buffering of divergent gene expression in yeast. Genome Res 24: 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megas G, Chrisofos M, Anastasiou I, Tsitlidou A, Choreftaki T, Deliveliotis C (2015) Estrogen receptor (α and β) but not androgen receptor expression is correlated with recurrence, progression and survival in post prostatectomy T3N0M0 locally advanced prostate cancer in an urban Greek population. Asian J Androl 17: 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyuhas O, Kahan T (2015) The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta Gene Regul Mech 1849: 801–811 [DOI] [PubMed] [Google Scholar]
- Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A et al (2015) Progesterone receptor modulates ERα action in breast cancer. Nature 523: 313–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, Keene JD (2010) Systematic analysis of posttranscriptional gene expression. Wiley Interdiscip Rev Syst Biol Med 2: 162–180 [DOI] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertlin C, Lorent J, Murie C, Furic L, Topisirovic I, Larsson O (2019) Generally applicable transcriptome‐wide analysis of translation using anota2seq. Nucleic Acids Res 47: e70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patursky‐Polischuk I, Stolovich‐Rain M, Hausner‐Hanochi M, Kasir J, Cybulski N, Avruch J, Ruegg MA, Hall MN, Meyuhas O (2009) The TSC‐mTOR pathway mediates translational activation of TOP mRNAs by insulin largely in a raptor‐ or rictor‐independent manner. Mol Cell Biol 29: 640–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N (1987) The involvement of mRNA secondary structure in protein synthesis. Biochem Cell Biol 65: 576–581 [DOI] [PubMed] [Google Scholar]
- Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O (2014) Translational control of immune responses: from transcripts to translatomes. Nat Immunol 15: 503–511 [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R (2014) Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc 9: 171–181 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ et al (2009) The consensus coding sequence (CCDS) project: identifying a common protein‐coding gene set for the human and mouse genomes. Genome Res 19: 1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Zhou Z, Chariot A, Close P (2017) tRNA modification: is cancer having a wobble? Trends Cancer 3: 249–252 [DOI] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J et al (2018) Codon‐specific translation reprogramming promotes resistance to targeted therapy. Nature 558: 605–609 [DOI] [PubMed] [Google Scholar]
- dos Reis M GitHub—mariodosreis/tai: the tRNA adaptation index. https://github.com/mariodosreis/tai [Accessed June 8, 2018]
- dos Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Wernisch L, Savva R (2003) Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K‐12 genome. Nucleic Acids Res 31: 6976–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezgui VAN, Tyagi K, Ranjan N, Konevega AL, Mittelstaet J, Rodnina MV, Peter M, Pedrioli PGA (2013) tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine‐tune protein translation by promoting ribosome A‐site binding. Proc Natl Acad Sci USA 110: 12289–12294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L et al (2015) Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP‐3. Oncogene 34: 2032–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Larsson O (2007) Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics 8: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurman K, Joosten S, Kim Y, Severson TM, van der Groep P, van Diest P, Zwart W (2018) Gene Expression Omnibus GSE104399 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104399) [DATASET]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, Perner S, Sboner A, Pawitan Y, Andrén O, Johnson LA et al (2008) Estrogen‐dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst 100: 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson TM, Kim Y, Joosten SEP, Schuurman K, van der Groep P, Moelans CB, ter Hoeve ND, Manson QF, Martens JW, van Deurzen CHM et al (2018) Characterizing steroid hormone receptor chromatin binding landscapes in male and female breast cancer. Nat Commun 9: 482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanle EK, Xu W (2010) Selectively targeting estrogen receptors for cancer treatment. Adv Drug Deliv Rev 62: 1265–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Tamarkin‐Ben‐Harush A, Viollet B, Dikstein R (2015) Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1‐eIF4GI cooperation in start codon selection. Cell Metab 21: 479–492 [DOI] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD et al (2016a) Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell 165: 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinstead EE, Baek S, Hager GL (2016b) Gene Expression Omnibus GSE72249 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72249) [DATASET]
- Takizawa I, Lawrence MG, Balanathan P, Rebello R, Pearson HB, Garg E, Pedersen J, Pouliot N, Nadon R, Watt MJ et al (2015) Estrogen receptor alpha drives proliferation in PTEN‐deficient prostate carcinoma by stimulating survival signaling, MYC expression and altering glucose sensitivity. Oncotarget 6: 604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardu M, Jones JD, Kennedy RT, Lin Q, Koutmou KS (2019) Identification and quantification of modified nucleosides in Saccharomyces cerevisiae mRNAs. ACS Chem Biol 14: 1403–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebaldi T, Re A, Viero G, Pegoretti I, Passerini A, Blanzieri E, Quattrone A (2012) Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC Genom 13: 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P, Song J, Gandin V, Cai Y, Rouleau SG, Garant J‐M, Boisvert F‐M, Yu Z, Perreault J‐P, Topisirovic I et al (2015) Aven recognition of RNA G‐quadruplexes regulates translation of the mixed lineage leukemia protooncogenes. Elife 4: e06234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, Ruggero D (2016) New frontiers in translational control of the cancer genome. Nat Rev Cancer 16: 288–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy‐Burman P, Nelson PS et al (2003) Prostate‐specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4: 209–221 [DOI] [PubMed] [Google Scholar]
- Wang C, Mayer JA, Mazumdar A, Fertuck K, Kim H, Brown M, Brown PH (2011) Estrogen induces c‐myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP‐1 transcription factor. Mol Endocrinol 25: 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell SE, Kazmin D, McDonnell DP (2012) Research resource: transcriptional profiling in a cellular model of breast cancer reveals functional and mechanistic differences between clinically relevant SERM and between SERM/estrogen complexes. Mol Endocrinol 26: 1235–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GW, Simon RM (2003) A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics 19: 2448–2455 [DOI] [PubMed] [Google Scholar]
- Yordanova MM, Loughran G, Zhdanov AV, Mariotti M, Kiniry SJ, O'Connor PBF, Andreev DE, Tzani I, Saffert P, Michel AM et al (2018) AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nature 553: 356–360 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Presgraves DC (2017) Translational compensation of gene copy number alterations by aneuploidy in Drosophila melanogaster . Nucleic Acids Res 45: 2986–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The DNA microarrays, RNAseq of full length or small RNAs, and nanoCAGE data generated and analyzed during the current study are available in the NCBI Gene Expression Omnibus repository under accession number GSE120917 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120917).