

Abstract
There is a large unmet need for effective therapies for cholestatic disorders, including primary sclerosing cholangitis (PSC), a disease that commonly results in liver failure. Angiotensin (Ang) II of the renin Ang system (RAS) is a potent profibrotic peptide, and Ang converting enzyme 2 (ACE2) of the alternate RAS breaks down Ang II to antifibrotic peptide Ang‐(1‐7). In the present study, we investigated long‐term effects of ACE2 delivered by an adeno‐associated viral vector and short‐term effects of Ang‐(1‐7) peptide in multiple drug‐resistant gene 2‐knockout (Mdr2‐KO) mice. These mice develop progressive biliary fibrosis with pathologic features closely resembling those observed in PSC. A single intraperitoneal injection of ACE2 therapy markedly reduced liver injury (P < 0.05) and biliary fibrosis (P < 0.01) at both established (3‐6 months of age) and advanced (7‐9 months of age) disease compared to control vector‐injected Mdr2‐KO mice. This was accompanied by increased hepatic Ang‐(1‐7) levels (P < 0.05) with concomitant reduction in hepatic Ang II levels (P < 0.05) compared to controls. Moreover, Ang‐(1‐7) peptide infusion improved liver injury (P < 0.05) and biliary fibrosis (P < 0.0001) compared to saline‐infused disease controls. The therapeutic effects of both ACE2 therapy and Ang‐(1‐7) infusion were associated with significant (P < 0.01) reduction in hepatic stellate cell (HSC) activation and collagen expression. While ACE2 therapy prevented the loss of epithelial characteristics of hepatocytes and/or cholangiocytes in vivo, Ang‐(1‐7) prevented transdifferentiation of human cholangiocytes (H69 cells) into the collagen‐secreting myofibroblastic phenotype in vitro. We showed that an increased ratio of hepatic Ang‐(1‐7) to Ang II levels by ACE2 therapy results in the inhibition of HSC activation and biliary fibrosis. Conclusion: ACE2 therapy has the potential to treat patients with biliary diseases, such as PSC.

Abbreviations
- α‐SMA
alpha smooth muscle actin
- AAV
adeno‐associated viral
- ACE
angiotensin converting enzyme
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- Ang
angiotensin
- AST
aspartate aminotransferase
- AT1‐R
angiotensin II type 1 receptor
- BDL
bile duct ligation
- CK19
cytokeratin‐19
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- EMT
epithelial‐to‐mesenchymal transdifferentiation
- HSA
human serum albumin
- HSC
hepatic stellate cell
- IL‐6
interleukin‐6
- KO
knockout
- mACE2‐rAAV/8
adeno‐associated viral vector carrying murine angiotensin converting enzyme 2
- MasR
Mas receptor
- MCP‐1
monocyte chemoattractant protein 1
- Mdr2
multiple drug‐resistant gene 2
- PSC
primary sclerosing cholangitis
- RAS
renin angiotensin system
- TGF‐β1
transforming growth factor β1
The lack of effective medical therapies for primary sclerosing cholangitis (PSC) and other related cholestatic conditions has meant that these diseases remain a major indication for liver transplantation.1 Although, there are a number of antifibrotic compounds currently being evaluated in clinical studies in chronic cholestatic liver disease, the lack of liver specificity of these drugs has raised concerns about their off‐target effects and long‐term safety and tolerability.
A number of experimental studies have provided evidence that the renin angiotensin (Ang) system (RAS) plays a central role in the pathogenesis of liver fibrosis2 and in cholestatic liver disease, drugs that target Ang converting enzyme (ACE) or Ang II type 1 receptor (AT1‐R) of the RAS can inhibit biliary fibrosis. Mechanistic support for the therapeutic effects of RAS blockade includes the fact that in the cholestatic liver, ACE expression is up‐regulated, production of the profibrotic Ang peptide Ang II is increased, and activated hepatic stellate cells (HSCs) have high expression of AT1‐R.3, 4 However, drugs targeting the RAS are poorly tolerated in cirrhosis because they lower peripheral resistance, causing hypotension and renal dysfunction5; as a result, there is a lack of clinical studies that have investigated the possible therapeutic role of RAS blockers in established chronic cholestatic liver disease.
Several studies have suggested that the so‐called alternate axis of the RAS, which opposes many of the deleterious effects of Ang II, is a potential target for antifibrotic therapies.6 This alternate RAS axis is driven by ACE2 (a homolog of ACE), which breaks down profibrotic Ang II to antifibrotic heptapeptide Ang‐(1‐7).7, 8 We have previously demonstrated that in a short‐term model of biliary fibrosis induced by bile duct ligation (BDL) for 2 weeks, Ang‐(1‐7) infusion inhibited liver fibrosis.9 Furthermore, we recently showed that a liver‐specific adeno‐associated viral (AAV) vector carrying murine ACE2 (mACE2‐rAAV2/8) markedly reduced biliary fibrosis in a 2‐week mouse model and was accompanied by a decreased ratio of hepatic Ang II to Ang‐(1‐7) concentrations in the diseased liver.10
The aim of the current study was to determine the efficacy of this therapy in a long‐term model of liver disease that resembles chronic cholestatic liver disease in humans. We therefore determined the therapeutic effects of ACE2 overexpression in established (3‐6 months of age) as well as advanced (7‐9 months of age) biliary fibrosis, using the multiple drug‐resistant gene 2‐knockout (Mdr2‐KO) mouse model of progressive biliary disease, which has features similar to human PSC.1, 11 Mdr2‐KO mice, which lack the mdr2 gene, are unable to secrete phosphatidylcholine into bile; this leads to toxic bile acid stasis with subsequent development of progressive biliary disease characterized by portal inflammation, ductular proliferation, and fibrosis around 3 months of age.11, 12 AAV vectors have no known pathogenicity in humans and have been successfully used in multiple phase I‐III clinical trials.13 With rapidly evolving gene transfer technology, gene therapy applications have rapidly expanded to include a wide range of genetic diseases. A recent approval by the U.S. Food and Drug Administration to use AAV vector‐based gene therapy for a rare form of childhood blindness, a genetic disorder, has provided impetus for the development of gene therapy approaches for nongenetic diseases. In the present study, we demonstrate that a single injection of liver‐specific ACE2 therapy produced a major and long‐lasting antifibrotic effect in both established and advanced biliary fibrosis in the Mdr2‐KO model of human PSC.
Materials and Methods
Animal Model of Biliary Fibrosis
Mdr2‐KO male mice with the FVB/N mouse strain background were used in this study. All animals were housed with a 12‐hour light–dark cycle at room temperature (22°C‐24°C) with water and standard mouse chow ad libitum. Experimental procedures were approved by the Animal Ethics Committee of Austin Health and performed according to the National Health and Medical Research Council of Australia guidelines for animal experimentation. Different groups of Mdr2‐KO animals at established (3 months old) and advanced (7 months old) biliary fibrosis were treated with either ACE2 or control vector for 3 and 2 months, respectively. In another experiment, 3‐month‐old Mdr2‐KO animals were infused with either Ang‐(1‐7) peptide or saline for 4 weeks (see Supporting Materials for details).
Viral Vector Preparation and Cell Culture Experiments
Viral vector preparation for mACE2 (mACE2‐rAAV2/8) and control vector (human serum albumin [HSA]‐rAAV2/8) and in vitro experiments using H69 cells (immortalized human cholangiocytes) are described in detail in the Supporting Materials.
Biochemical and Histologic Analyses
Assessments of liver biochemistry, histology of liver inflammation and fibrosis, ACE2 activity, quantitative real‐time polymerase chain reaction, and liver Ang II and Ang‐(1‐7) levels were performed following the described protocols14 (see Supporting Materials for details of each procedure).
Immunohistochemistry
Immunohistochemistry for e‐cadherin, ACE2, Mas receptor (MasR), alpha smooth muscle actin (α‐SMA), cytokeratin‐19 (CK19), and mesenchymal marker S100A4 as well as western blot for B‐cell lymphoma‐extra large (Bcl‐XL) were performed (see Supporting Materials for details of each procedure).
Statistical Analysis
Means between groups were compared using either a two‐tailed unpaired Student t test or one‐way analysis of variance with Tukey post‐hoc test. All data are expressed as mean ± SEM. All statistical analyses were carried out using the computer package PRISM (GraphPad Prism 7.0). A value P < 0.05 was considered statistically significant.
Results
ACE2 Therapy and Ang‐(1‐7) Reduce Liver Enzymes in Mdr2‐KO Mice
Three‐month‐old Mdr2‐KO mice showed significantly (P < 0.05) elevated liver enzyme profiles compared to those in healthy controls (Fig. 1). Moreover, liver injury, as reflected by increased plasma levels of alkaline phosphatase (ALP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST), worsened during the course of disease progression at 6 months (Fig. 1A‐C) and 9 months (Fig. 1D‐F) of age. However, compared to the control vector‐injected animals, ACE2 therapy markedly improved liver injury in Mdr2‐KO mice, as reflected by a significant reduction in liver enzyme profiles at established (3‐6 months; P < 0.0001; Fig. 1A‐C) or advanced (7‐9 months; P < 0.05; Fig. 1D‐F) biliary fibrosis. Comparable results were obtained with Ang‐(1‐7) infusion, which caused a significant (P < 0.05) reduction in liver enzyme profiles in Mdr2‐KO mice compared to those in saline‐treated control Mrd2‐KO mice (Fig. 1G‐I).
Figure 1.
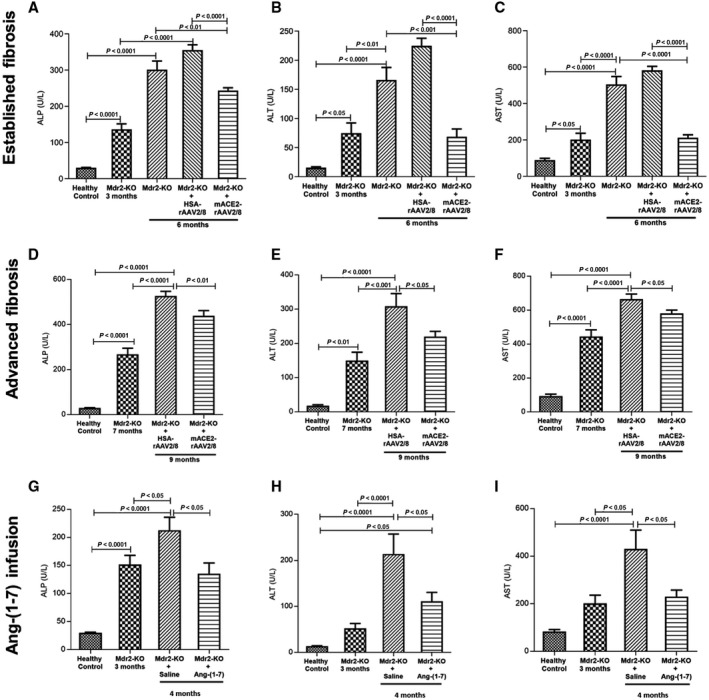
Plasma concentrations of ALP, ALT, and AST. A single intraperitoneal injection of ACE2 vector treatment at (A‐C) established and (D‐F) advanced stages of biliary disease caused significant reductions in plasma levels of ALP, ALT, and AST compared to control vector‐treated animals and age‐matched control Mdr2‐KO animals. In support of this, animals treated for 4 weeks with Ang‐(1‐7) showed significantly reduced levels of plasma (G) ALP, (H) ALT, and (I) AST levels compared to saline‐infused disease‐control Mdr2‐KO mice, indicating that the treatment improved liver injury. Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group and n = 5‐7 mice per Mdr2‐KO group without the vector.
ACE2 Therapy and Ang‐(1‐7) Reduce Liver Fibrosis in Mdr2‐KO Mice
We assessed the presence of activated HSCs by quantifying α‐SMA. Compared to healthy controls, α‐SMA gene expression was significantly (P < 0.0001) higher in Mdr2‐KO mice at 6 and 9 months of age (Fig. 2A,C). Importantly, compared to the control vector‐injected disease controls, a single dose of ACE2 therapy caused a significant (P < 0.01) reduction in α‐SMA expression in established biliary fibrosis (Fig. 2A). In the advanced biliary fibrosis group, the inhibitory effect of ACE2 therapy on α‐SMA was more than 95% (P < 0.0001), with the expression level almost reaching healthy control levels (Fig. 2C). Consistent with gene expression profiles, strong protein staining of α‐SMA in Mdr2‐KO livers was markedly reduced by ACE2 therapy (Fig. 3E).
Figure 2.
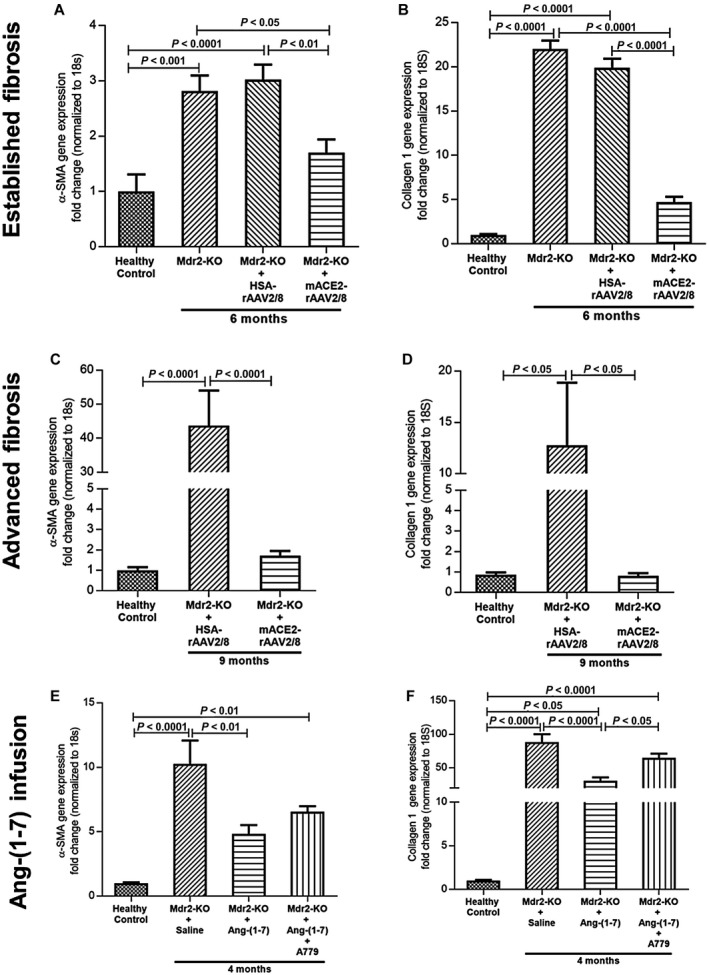
Gene expression of α‐SMA and collagen 1. (A‐D) α‐SMA and collagen 1 gene expression were significantly reduced by ACE2 therapy in both (A,B) established and (C,D) advanced biliary fibrosis compared to control vector‐injected Mdr2‐KO mice. (E,F) Similar to ACE2 treatment, Ang‐(1‐7) infusion significantly reduced their expressions in the liver of Mdr2‐KO mice whereas simultaneous infusion with MasR blocker A779 abrogated the effect of Ang‐(1‐7). Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group and n = 5‐7 mice per Mdr2‐KO group without the vector.
Figure 3.
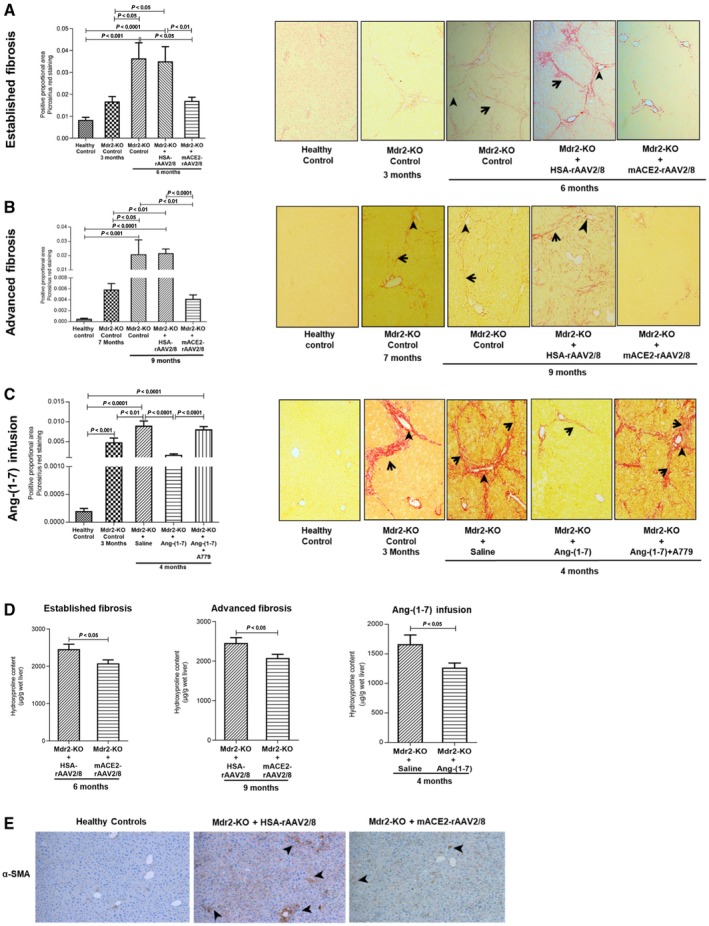
Quantification of liver collagen by picrosirius red staining and hydroxyproline content and HSC activation using α‐SMA immunohistochemistry in Mdr2‐KO mice with established and advanced biliary fibrosis following ACE2 therapy and Ang‐(1‐7) infusion. (A) ACE2 gene therapy significantly reduced picrosirius‐stained area, reflecting reduced liver collagen content at 3 months post‐treatment in 6‐month‐old Mdr2‐KO mice compared to control vector‐injected mice. (B) Liver collagen content, which was significantly increased from 7 to 9 months of age in Mdr2‐KO mice, was significantly reduced by ACE2 gene therapy at 2 months post‐treatment compared to 9‐month‐old control vector‐injected Mdr2‐KO mice. (C) Ang‐(1‐7) infusion for 4 weeks into 3‐month‐old Mdr2‐KO mice significantly reduced liver collagen content compared to saline‐infused Mdr2‐KO mice. However, simultaneous infusion of MasR blocker A779 completely abolished the effect of Ang‐(1‐7). (A‐C) Representative images of picrosirius red‐stained liver sections from different groups. Arrow shows bridging fibrosis; arrowhead shows periportal fibrosis (magnification ×200). (D) Similarly, liver hydroxyproline content was significantly reduced by ACE2 therapy and Ang‐(1‐7) treatment in Mdr2‐KO animals compared to controls. (A‐D) Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group and n = 5‐7 mice per Mdr2‐KO group without the vector. (E) Analysis of α‐SMA immunohistochemistry showed strong staining in liver sinusoids consistent with localization of activated HSCs in control vector‐injected Mdr2‐KO mice (arrowheads) compared to ACE2‐treated mice (magnification ×100).
We then assessed gene expression of a major extracellular matrix (ECM) protein collagen 1 (type 1α1). As expected, there was a significant (P < 0.05) increase in collagen 1 expression in Mdr2‐KO mice at both stages compared to those in the healthy controls (Fig. 2B,D). However, compared to control vector‐injected disease controls, treatment with ACE2 inhibited collagen 1 expression by more than 50% (P < 0.0001) in established biliary fibrosis (Fig. 2B). This inhibitory effect of ACE2 was even more profound in the advanced stage of biliary fibrosis, with more than 90% reduction and reaching healthy control levels (Fig. 2D).
We have previously shown ACE2 therapy increases production of Ang‐(1‐7)10 and this contributes to its antifibrotic effect. Here, we tested the effect of direct infusion of Ang‐(1‐7) on HSC activation and collagen expression in early biliary fibrosis. We found that Ang‐(1‐7) infusion for 4 weeks inhibited HSC activation, as reflected by more than 50% reduction (P < 0.01) in α‐SMA gene expression (Fig. 2E), and reduced collagen 1 gene expression by more than 60% (Fig. 2F). To further explore the mechanism by which Ang‐(1‐7) mediated this inhibitory effect, we co‐infused mice with A779, a putative Ang‐(1‐7) receptor Mas blocker, and found that A779 reversed the effects of Ang‐(1‐7) on the expression of α‐SMA and collagen 1 (Fig. 2E,F).
We further assessed the antifibrotic effects of ACE2 therapy in Mdr2‐KO mice by quantifying hepatic collagen protein deposition by picrosirius red staining. As expected, there was a rapid progression (P < 0.001) of fibrosis from 3 to 6 months (established biliary fibrosis) and 7 to 9 months (advanced biliary fibrosis) of age in Mdr2‐KO mice compared to those in healthy controls (Fig. 3). However, in keeping with the profound reduction in collagen 1 gene expression, ACE2 therapy markedly reduced collagen deposition in established biliary fibrosis (>50%; Fig. 3A,B) and by more than 80% in advanced biliary fibrosis compared with control vector‐injected disease controls (Fig. 3C).
We found that consistent with collagen 1 gene expression (Fig. 2F), hepatic collagen protein deposition at 3 months and at approximately 4 months (i.e., 4 weeks after saline infusion) of age was significantly (P < 0.001) higher in Mdr2‐KO mice than in healthy controls (Fig. 3C). However, confirming its effect on collagen 1 expression, Ang‐(1‐7) infusion inhibited collagen protein deposition by more than 75% (P < 0.0001) compared with saline‐infused disease controls. Mechanistically, this inhibition on collagen protein secretion by Ang‐(1‐7) was mediated by MasR because MasR blocker A779 completely abolished this response. These results were further supported by liver hydroxyproline content, which was significantly (P < 0.05) reduced by both ACE2 therapy and Ang‐(1‐7) infusion (Fig. 3D). Moreover, the histopathological assessment clearly indicated that, compared to controls, ACE2‐treated as well as Ang‐(1‐7) infused Mdr2‐KO mice had regression of fibrosis (Table 1).
Table 1.
Hepatic Inflammation and Fibrosis in Mdr2‐KO Mice Treated With ACE2 and Ang‐(1‐7)
| Area of Inflammation/Fibrosis | Animal Groups | ||||
|---|---|---|---|---|---|
| Healthy Control | Mdr2‐KO + HSA‐rAAV2/8 | Mdr2‐KO + mACE2‐rAAV2/8 | Mdr2‐KO + Saline | Mdr2‐KO + Ang‐(1‐7) | |
| Portal Inflammation | NI | 1 ± 0.45 | 0.75 ± 0.37 | 1.8 ± 0.8 | 1.2 ± 0.54 |
| Lobular Inflammation | NI | NI | NI | 1.6 ± 0.7 | 0.4 ± 0.18* |
| Fibrosis | NF | 3.4 ± 0.15 | 2.4 ± 1.07† | 3.5 ± 1.56 | 1.4 ± 0.62‡ |
P < 0.05 versus saline‐infusion;
P < 0.05 versus control HSA vector injection;
P < 0.05 versus saline‐infusion. Histopathological assessment of fibrosis was made by using Metavir scoring.
Abbreviations: NF, no fibrosis; NI, no inflammation.
ACE2 Therapy Ameliorates the Expression of Proinflammatory and Profibrotic Cytokines
In biliary fibrosis, activation of HSCs into myofibroblasts and the resulting increase in ECM secretion is triggered by proinflammatory and profibrogenic mediators produced by activated resident macrophages, Kupffer cells, and dying hepatocytes. Therefore, we investigated the effects of ACE2 therapy on gene expression of proinflammatory cytokines, such as monocyte chemoattractant protein 1 (MCP‐1) and interleukin‐6 (IL‐6), and profibrotic cytokines, such as connective tissue growth factor (CTGF) and transforming growth factor β1 (TGF‐β1). The expression of MCP‐1 (Fig. 4A,B) and CTGF (Fig. 4C,D) was markedly (P < 0.001) up‐regulated at both established and advanced fibrosis in Mdr2‐KO mice compared with healthy controls. Consistent with our published data,10 ACE2 therapy significantly (P < 0.05) reduced the expression of these cytokines (Fig. 4A‐C), providing evidence that ACE2 therapy caused a reduction in liver inflammation, leading to a reduction in liver fibrosis, although it did not affect CTGF expression at advanced biliary fibrosis (Fig. 4D). On the other hand, IL‐6 expression was down‐regulated at both established and advanced biliary fibrosis in Mdr2‐KO mice compared to healthy controls and ACE2 therapy had no effect on its expression (Fig. 4E,F). Contrary to this, TGF‐β1, the expression of which was increased at established biliary fibrosis in Mdr2‐KO mice compared to healthy controls, was not affected by ACE2 therapy (Fig. 4G). In marked contrast to the expression pattern of these cytokines and findings of our previous study using a short‐term biliary fibrotic model,10 what is intriguing to note in this long‐term biliary fibrotic model is that the expression of TGF‐β1 in advanced disease was indeed down‐regulated by approximately 99% (P < 0.0001) compared with healthy controls (Fig. 4H). Making this more complicated, ACE2 therapy in fact rescued the expression of this potent profibrotic cytokine by increasing its expression from 1% to approximately 23% compared with healthy controls (Fig. 4H).
Figure 4.
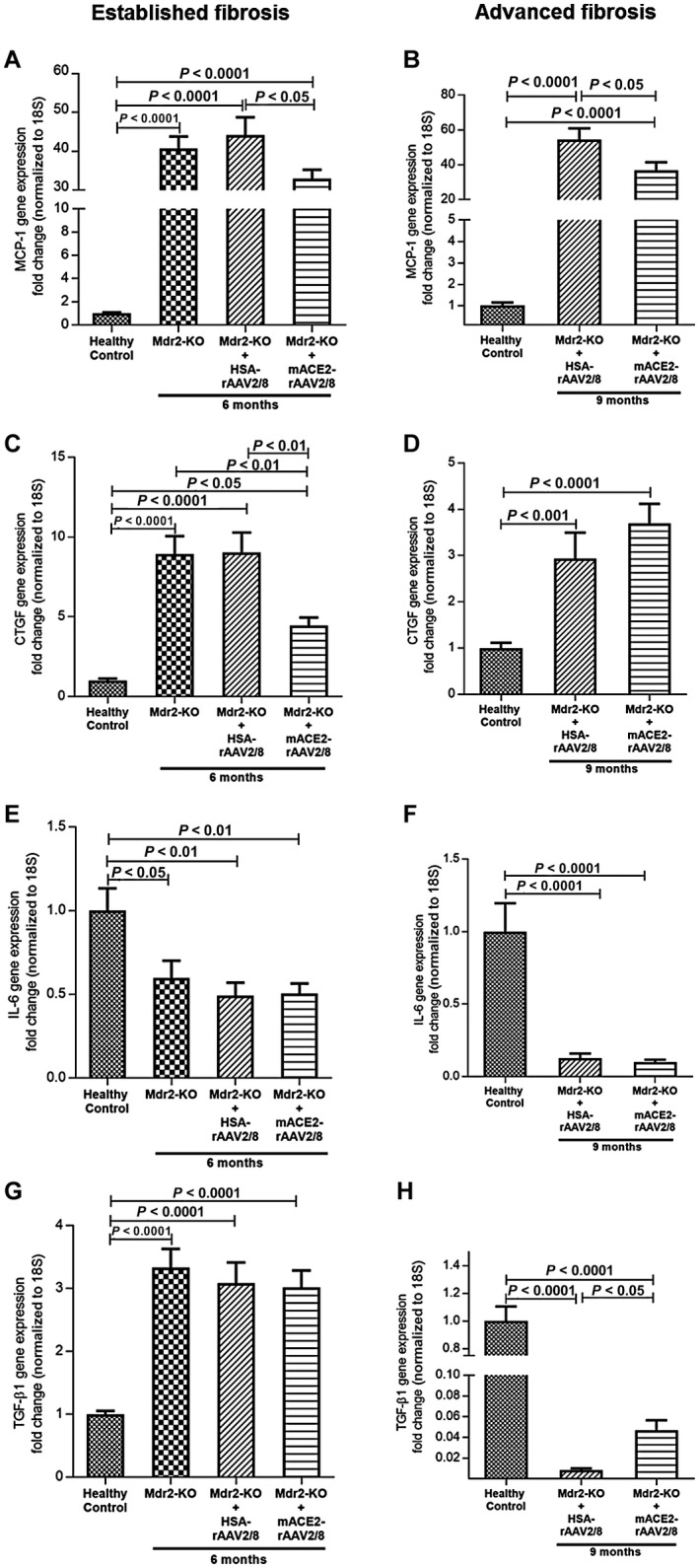
Proinflammatory and profibrotic cytokine gene expression in established and advanced biliary fibrosis. Gene expression of (A,B) MCP‐1 at both established and advanced fibrosis and (C,D) CTGF at established fibrosis was significantly reduced by ACE2 gene therapy compared to control vector‐injected mice. (E,F) In contrast, IL‐6 expression, which was down‐regulated at both stages of fibrosis, was unaffected by ACE2 therapy. (G) Moreover, TGF‐β1, which was up‐regulated at established fibrosis, was also unaffected by ACE2 therapy. (H) In marked contrast, TGF‐β1 expression, which was significantly down‐regulated at advanced fibrosis, was restored to a greater extent by ACE2 therapy. Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group and n = 5‐7 mice per Mdr2‐KO group without the vector.
Similar to their expression at 6 months of age, the expression of IL‐6, MCP‐1, CTGF, and TGF‐β1 at 4 months of age in Mdr2‐KO mice was significantly (P < 0.05) elevated compared with the healthy controls (Fig. 5). However, with the exception of MCP‐1, Ang‐(1‐7) infusion caused a profound (P < 0.0001) reduction in the expression of other cytokines compared with control vector‐injected disease controls (Fig. 5C,D). In contrast to reduced expression at 6 and 9 months of age, liver IL‐6 expression, which was significantly (P < 0.001) increased at 4 months of age (early biliary fibrosis) in Mdr2‐KO mice compared to healthy controls, was significantly (P < 0.0001) inhibited by Ang‐(1‐7) infusion (Fig. 5A). Importantly, the inhibitory effect of Ang‐(1‐7) on CTGF and TGF‐β1 was completely abolished when the animals were co‐infused with MasR blocker A779 (Fig. 5C,D), suggesting that Ang‐(1‐7) derived from Ang II by ACE2 action plays a prominent role in the ACE2‐driven therapeutic effect. Apart from the therapeutic effect of ACE2 therapy and Ang‐(1‐7), we tested whether cholangiocyte senescence was attributable to the reduced inflammation in ACE2‐treated Mdr2‐KO mice. We found that protein levels of Bcl‐XL, a marker of senescence, were not altered in ACE2‐treated Mdr2‐KO mice, suggesting that cholangiocyte senescence was highly unlikely to be associated with the reduced inflammation (Supporting Fig. S1B).
Figure 5.
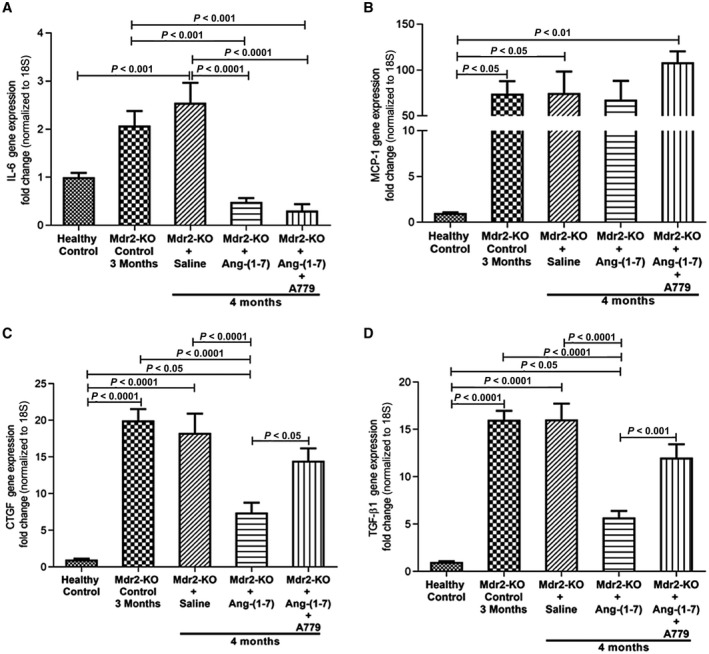
Changes in proinflammatory and profibrogenic gene expression in Ang‐(1‐7)‐infused mice. Expression of hepatic proinflammatory cytokines (A) IL‐6 and (B) MCP‐1 and profibrotic cytokines (C) CTGF and (D) TGF‐β1 in Mdr2‐KO mice at 4 weeks post‐treatment with Ang‐(1‐7) and combined treatment with Ang‐(1‐7) and A779 is shown. Ang‐(1‐7) infusion inhibited gene expression of (A) IL‐6, (C) CTGF, and (D) TGF‐β1 but not (B) MCP‐1 in 4‐month‐old Mdr2‐KO mice. While the effect of Ang‐(1‐7) on IL‐6 expression was unaffected by simultaneous infusion with MasR blocker A779, Ang‐(1‐7)‐induced down‐regulation of (C) CTGF and (D) TGF‐β1 was completely prevented by MasR antagonism with A779. Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group.
It is interesting to note that the expression levels of proinflammatory and profibrotic cytokines early in disease progression in Mdr2‐KO mice differed markedly from those at an advanced stage of disease progression. For example, the expression levels of these cytokines at 3‐4 months of age, at which time they were treated with Ang‐(1‐7) infusion, were highest and gradually declined toward 9 months of age compared to healthy controls. In addition to gene expression, histopathological assessments of liver sections showed an increased (P < 0.05) lobular inflammation in saline‐infused 4‐month‐old Mdr2‐KO mice that was significantly (P < 0.05) reduced by Ang‐(1‐7) infusion, although the peptide infusion did not affect portal inflammation in these animals (Table 1). In marked contrast, 6‐month‐old control vector‐injected Mdr2‐KO mice showed reduced portal inflammation compared to 4‐month‐old saline‐infused controls and ACE2 therapy did not affect portal inflammation. Moreover, lobular inflammation could only be seen in 4‐month‐old but not in 6‐month‐old Mdr2‐KO mice. This is likely attributable to the fact that Mdr2‐KO mice are known to display varying degrees of inflammation during their lifetime.12
ACE2 Therapy Reduces Profibrotic Peptide Ang II and Increases Antifibrotic Ang‐(1‐7) Levels in the Liver
We have previously reported that the ACE2 vector is liver specific10; this was further confirmed in Mdr2‐KO mice (Supporting Fig. S2A). A single injection of ACE2 vector significantly increased ACE2 gene expression (P < 0.0001), protein levels (P < 0.01), and protein activity (P < 0.0001) in Mdr2‐KO livers at 6 months of age compared to control vector‐injected animals (Fig. 6A,B,E). Moreover, Mdr2‐KO mice treated with ACE2 at advanced biliary fibrosis (7 months) and killed at 9 months of age showed a 150‐fold increase (P < 0.0001) in ACE2 gene expression in the liver compared to control vector‐injected diseased mice and healthy controls (Fig. 6B). In line with this, we found increased ACE2 protein expression in liver specimens from patients with PSC (Supporting Fig. S1A) compared to healthy human livers. Moreover, MasR gene and protein expression also showed a marked up‐regulation in response to ACE2 therapy in Mdr2‐KO mice compared to those of control vector‐injected mice (Fig. 6D,E).
Figure 6.
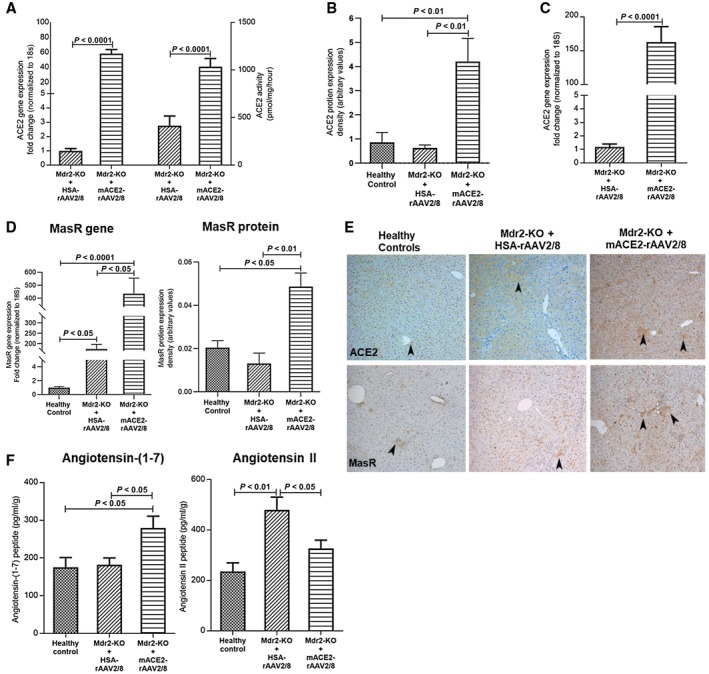
Hepatic ACE2 gene and protein expression, protein activity, MasR gene and protein expression, and hepatic angiotensin peptide levels in Mdr2‐KO mice treated with ACE2. ACE2 treatment significantly increased (A,B) ACE2 gene and protein expression and protein activity in 6‐month‐old Mdr2‐KO mice with 3 months post‐treatment and (C) ACE2 gene expression in 9‐month‐old Mdr2‐KO mice with 2 months post‐treatment. (D) Also, ACE2 therapy markedly increased hepatic MasR gene and protein expression. (E) Representative images of ACE2 and MasR protein staining in Mdr2‐KO liver sections. Arrowhead shows positive staining (magnification ×100). (F) As expected, ACE2 therapy significantly increased hepatic levels of antifibrotic peptide Ang‐(1‐7) by breaking down hepatic Ang II peptide, resulting in a reduction in hepatic levels of profibrotic peptide Ang II in 6‐month‐old Mdr2‐KO mice with 3 months post‐treatment compared to control vector HSA‐rAAV2/8‐injected mice. Each bar represents the mean ± SEM profile from n = 12‐15 mice per treatment group.
Mechanistically, hepatic ACE2 overexpression would be expected to produce dual benefits: degradation of the potent profibrotic peptide Ang II and generation of the antifibrotic peptide Ang‐(1‐7) within the liver.9 We therefore measured hepatic Ang peptide levels in healthy mice and in mice with liver disease that received ACE2 or control HSA vector at 3 months of age and were killed 3 months after treatment. As expected, ACE2 therapy produced significantly (P < 0.05) increased Ang‐(1‐7) peptide levels (Fig. 6F) in Mdr2‐KO mouse livers compared to the disease control group. These elevated Ang‐(1‐7) levels were accompanied by a significant (P < 0.05) reduction in hepatic Ang II peptide levels in ACE2‐treated mouse livers compared to levels in the control vector‐injected animals (Fig. 6F), reflecting that ACE2 shifted the balance between hepatic Ang peptide levels, leading to an increased ratio of hepatic Ang‐(1‐7) to Ang II (mean ± SEM, 0.92 ± 0.18 and 0.49 ± 0.03 in ACE2 and control HSA vector‐treated groups, respectively; P < 0.05).
ACE2 Therapy and Ang‐(1‐7) Peptide Affect Epithelial‐To‐Mesenchymal Transdifferentiation in Cholangiocytes
It has been suggested that epithelial‐to‐mesenchymal transdifferentiation (EMT) is an alternate source of the periportal myofibroblastic cell population that may contribute to biliary fibrosis.15 Cholangiocytes, the biliary epithelial cells, undergo EMT and are thought to be actively involved in biliary fibrosis.16 Indeed, we found a faint brown staining for CK19, an epithelial marker, in cholangiocytes of control vector‐injected Mdr2‐KO mouse livers compared to a high level of staining in healthy controls, suggesting that cholangiocytes in the diseased liver lose their epithelial characteristics. Most importantly, ACE2 therapy restored the epithelial phenotype of cholangiocytes in the Mdr2‐KO liver, as reflected by strong staining for CK19 (Fig. 7A). On the other hand, we found that the mesenchymal marker S100A4 stained positive in cholangiocytes of Mdr2‐KO mouse livers compared to the absence of its staining in healthy control livers. As expected, S100A4 staining of ACE2‐treated mouse livers was absent, suggesting that ACE2 therapy prevented EMT of cholangiocytes in this model.
Figure 7.
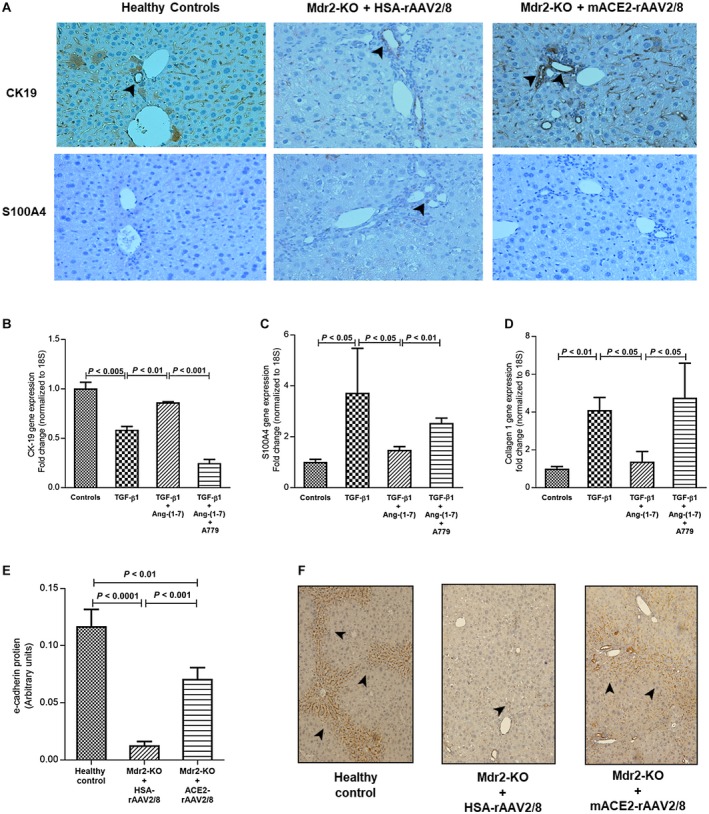
Effect of ACE2 treatment on protein expression of epithelial (CK‐19) and mesenchymal (S100A4) markers; effect of Ang‐(1‐7) on gene expression of CK‐19, S100A4, and collagen 1 in TGF‐β1‐activated cholangiocytes (H69 cells); and effect of ACE2 treatment on e‐cadherin protein in Mdr2‐KO mice. (A) ACE2 therapy restored epithelial characteristics of cholangiocytes, as reflected by increased CK‐19 expression, and prevented transdifferentiation into a mesenchymal phenotype, as reflected by reduced S100A4 expression in Mdr2‐KO mice. In support of the inhibition of EMT of cholangiocytes by ACE2‐derived Ang‐(1‐7) in the animal model, Ang‐(1‐7) prevented TGF‐β1‐induced (B) down‐regulation of CK‐19 expression and (C) up‐regulation of S100A4 expression, leading to (D) inhibition of collagen 1 expression. Thus, Ang‐(1‐7) inhibited EMT of cholangiocytes, which was completely blocked by MasR blocker A779. (E,F) Moreover, ACE2 therapy restored protein expression of e‐cadherin (another epithelial marker known to protect from cholangiopathies) in Mdr2‐KO mice. (A,F) Arrowhead shows positive staining (magnification ×200). Each bar in (B‐D) represents the mean ± SEM profile from three independent experiments. Each bar in (E) represents the mean ± SEM profile from n = 12‐15 mice per treatment group.
To further support the role of alternate RAS on EMT, we investigated the effect of Ang‐(1‐7) and MasR blocker A779 on gene expression of CK‐19, S100A4, and the fibrosis marker collagen 1 in TGF‐β1‐activated H69 cells because TGF‐β1 is considered to be a potent inducer of EMT.16, 17 TGF‐β1‐activated H69 cells showed a significant (P < 0.005) reduction in CK‐19 expression compared to untreated cells (Fig. 7B). This reduction in CK‐19 was completely abolished (P < 0.01) by Ang‐(1‐7) treatment (Fig. 7B). The reduced epithelial characteristics of activated H69 cells were accompanied by significantly (P < 0.05) increased expression of mesenchymal marker S100A4 (Fig. 7C). Consistent with the up‐regulation of S100A4 expression, the fibrosis marker gene collagen 1 expression was significantly (P < 0.01) increased in activated cholangiocytes (Fig. 7D). Strikingly, the phenotypic changes resulting from TGF‐β1‐induced activation of H69 cells, which included the loss of epithelial characteristics and acquiring a mesenchymal collagen‐secreting phenotype, were completely abolished (P < 0.05) by Ang‐(1‐7) treatment (Fig. 7B‐D). Moreover, we found that the inhibitory effects of Ang‐(1‐7) on EMT were mediated by MasR because specific MasR antagonist A779 completely blocked the inhibitory effects of the peptide (Fig. 7B‐D). Further supporting this phenomenon, we found that Ang‐(1‐7) by binding to MasR triggers MasR gene up‐regulation in these cells (Supporting Fig. S2B).
The in vitro findings with H69 cells were corroborated by in vivo findings that the level of e‐cadherin protein, an epithelial cell marker known to prevent the development of sclerosing cholangitis, peribiliary fibrosis, and EMT,18 was dramatically reduced (P < 0.0001) in Mdr2‐KO mice at 6 months of age (Fig. 7E,F). However, in support of the protective effect of Ang‐(1‐7) on the epithelial characteristics of cholangiocytes (Fig. 7B‐D), ACE2 therapy markedly (P < 0.001) inhibited the loss of e‐cadherin protein expression in Mdr2‐KO mice (Fig. 7E,F).
Discussion
We have recently reported for the first time that a single dose of mACE2‐rAAV2/8 vector produces a sustained overexpression of ACE2 in the fibrotic liver of a short‐term (2‐week) mouse model of biliary fibrosis.10 We now report that compared to this 2‐week biliary fibrosis model, a single dose of mACE2‐rAAV2/8 leads to high levels of ACE2 expression and activity in a long‐term genetic model of biliary fibrosis. The increased hepatic ACE2 protein expression and activity produced a large reduction (>50%) in ECM deposition at 6 months of age (established biliary fibrosis) and, most importantly, at 9 months of age (>80%) (advanced biliary fibrosis) in Mdr2‐KO mice compared to vector controls. This was accompanied by a marked increase in the ratio of hepatic Ang‐(1‐7) to Ang II and a reduction in HSC activation and release of proinflammatory and profibrotic mediators. The effect of ACE2 at advanced biliary fibrosis is particularly impressive because it is generally thought that vector expression and its ability to alter the course of disease may be limited in animals with more advanced liver disease.19 Thus, we confirm that a single dose of this ACE2 viral vector provides much longer term inhibition of biliary fibrosis.
The Mdr2‐KO mouse model, which has pathological features closely resembling those of human PSC and results in advanced biliary fibrosis and cirrhosis over 7‐10 months due to the lack of biliary phospholipid secretion,20 is a well‐characterized long‐term model widely used in studies.21 Therapies targeting to inhibit or reverse biliary fibrosis in short‐term models, such as those reported by us,10 are necessary as proof of concept; however, physiologically relevant longer term models are required to elucidate the therapeutic efficacy of a novel strategy that can then be translated to therapeutic applications for human cholangiopathies.
We demonstrate that ACE2 therapy in established and advanced biliary disease as well as Ang‐(1‐7) infusion in early biliary fibrosis improved liver injury as assessed by plasma ALT, ALP, and AST levels. Inflammation is a hallmark of tissue injury and drives fibrogenesis when it is persistent. In the liver, it is characterized by increased release of proinflammatory cytokines, which precede liver fibrosis. One of the best characterized proinflammatory cytokines in liver fibrosis is MCP‐1, which is released not only by activated resident macrophages (Kupffer cells) but also by activated HSCs.22 Although the expression of MCP‐1 was unaffected by Ang‐(1‐7) infusion at 4 months of age, it was attenuated by ACE2 treatment at 6 and 9 months of age compared to control vector‐injected Mdr2‐KO mice; this is in line with improved liver injury. Despite this, profibrotic cytokine CTGF was inhibited by Ang‐(1‐7) at 4 months of age and by ACE2 at 6 months but not at 9 months of age. Ang‐(1‐7) infusion inhibited the expression of the potent profibrotic cytokine TGF‐β1 in the early development of fibrosis leading up to 4 months. However, ACE2 treatment failed to suppress increased expression of this cytokine during established fibrosis leading up to 6 months of age. In fact, TGF‐β1 expression was massively down‐regulated during advanced fibrosis leading up to 9 months of age. Thus, these findings suggest that this cytokine is a key driver during early development of biliary fibrosis in the Mdr2‐KO model. This is consistent with the report that the expression of these cytokines in Mdr2‐KO mice is variable and has distinct roles, depending on the stage of disease progression.20 This agrees with current findings that Ang‐(1‐7) at 4 months but not ACE2 therapy at 6 months improved infiltrating immune cells in Mdr2‐KO mice.
It is widely accepted that Ang II plays a pivotal role in hepatic fibrogenesis by activating HSCs and other myofibroblastic cell populations during tissue injury.23 Unlike Ang II, which has a well‐characterized profibrotic role in liver fibrosis, the counter‐regulatory role of Ang‐(1‐7) in biliary fibrosis has received less attention. Ferrario24 provided evidence that Ang‐(1‐7) is an antifibrotic peptide that counteracts the deleterious effects of Ang II in the heart and kidneys. Supporting these observations, studies from our laboratory have shown that intraperitoneal infusion of Ang‐(1‐7) improved biliary fibrosis in a short‐term 2‐week rat BDL.9 The present findings that direct infusion of Ang‐(1‐7) peptide markedly reduces liver fibrosis in Mdr2‐KO mice provide strong evidence to suggest that the antifibrotic effects of ACE2 therapy on biliary fibrosis were in large part due to increased generation of hepatic Ang‐(1‐7) levels from hepatic Ang II (Fig. 6).
Perpetuation of inflammation, which leads to activation of profibrogenic stimuli, is often associated with the activation of quiescent HSCs, leading to secretion of excess ECM.3 The present findings support published data10 showing that ACE2 therapy as well as Ang‐(1‐7) peptide infusion ameliorated increased HSC activity, as reflected by approximately 50%‐80% reductions in liver α‐SMA gene and protein expression (Figs. 2 and 3). Consistent with a markedly attenuated activity of the myofibroblastic cell population, we found that in the livers of ACE2‐ and Ang‐(1‐7)‐treated mice, the expression of collagen 1, a major ECM protein secreted by activated HSCs, was dramatically reduced by more than 50% compared to control vector or saline‐infused controls. These changes led to more than 50% reduction in fibrosis early in disease progression and reached more than 80% in advanced biliary disease (Fig. 3; Table 1). Consistent with our published study,25 the present study confirmed that the beneficial effects of Ang‐(1‐7) peptide in Mdr2‐KO mice are mediated by MasR, as reflected by the inhibition of Ang‐(1‐7) effects by MasR antagonist A779. Moreover, apart from signal transduction, the positive feedforward effect of Ang‐(1‐7) peptide on MasR is reflected by up‐regulation of MasR by Ang‐(1‐7) in H69 cells and ACE2‐derived Ang‐(1‐7) in Mdr2‐KO mice26 (Fig. 6D; Supporting Fig. S2B). Further supporting the role of MasR in mediating Ang‐(1‐7) action in biliary fibrosis, the inhibitory effect of Ang‐(1‐7) on profibrotic mediators, such as CTGF, TGF‐β1, and collagen 1, was completely abrogated by MasR blockade. This is in agreement with the finding that infusion of MasR antagonist A779 to BDL mice worsened liver fibrosis.27
While HSCs are the main cell type responsible for liver fibrosis,28, 29 there is evidence that other cell types are also involved in ECM deposition in the injured liver. For example, portal fibroblasts that originate from hematopoietic stem cells and migrate to injury sites are capable of differentiating into myofibroblasts that secrete ECM.28 It has also been reported that following liver injury, biliary epithelial cells (cholangiocytes), which contribute to the ductular reaction in biliary fibrosis, proliferate and transform from a quiescent state to a reactive state. Once activated, they begin to proliferate, lose their epithelial phenotype, and express inflammatory and profibrogenic cytokines and growth factors, leading to increased deposition of ECM in the periportal region.15, 30 This is supported by observations from in vivo and in vitro studies that cholangiocytes can undergo EMT and collagen production during biliary injury.15, 16, 31 Consistent with these studies, we demonstrated that following activation by TGF‐β1, immortalized human cholangiocytes underwent transdifferentiation from an epithelial to a mesenchymal phenotype, as reflected by decreased expression of epithelial marker CK‐19 and increased expression of mesenchymal marker S100A4. Importantly, this phenotypic transdifferentiation of cholangiocytes was accompanied by increased expression of collagen 1, providing evidence that EMT may represent an alternative source of the myofibroblastic cell population in the periportal region that may have also contributed to biliary fibrosis in Mdr2‐KO mice.32 Because TGF‐β1 has been implicated in the activation of cholangiocytes,33 it can be postulated that the increased expression of this cytokine in early and established biliary fibrosis of Mdr2‐KO mice may drive in vivo activation of cholangiocytes, which in turn may have a direct role in biliary fibrosis. On the other hand, it is likely that TGF‐β1 may not be the driving force in advanced biliary fibrosis of this model and perhaps CTGF plays a role as profibrotic mediator at this stage.19 Most importantly, the present findings that Ang‐(1‐7) treatment restored the expression of epithelial marker CK‐19 and prevented up‐regulation of mesenchymal marker S100A4 and collagen 1 expression in TGF‐β1 activated human cholangiocytes support the notion that ACE2 therapy, which increases local Ang‐(1‐7) levels in the injured liver, has the potential to ameliorate EMT of cholangiocytes. Indeed, liver CK‐19 protein, which was down‐regulated in Mdr2‐KO mice, was restored by ACE2 therapy and prevented the transdifferentiation into a mesenchymal phenotype. Moreover, intervention with the MasR antagonist A779 in these cells provides direct evidence that the inhibitory effect of Ang‐(1‐7) on cholangiocyte activation is mediated via the MasR.
In marked contrast, liver TGF‐β1 expression in Mdr2‐KO mice with advanced biliary fibrosis at 9 months of age was down‐regulated by approximately 99% compared to healthy control mice of the same background. Unexpectedly, ACE2 treatment, which is expected to inhibit the expression of this profibrotic cytokine, in fact stimulated its expression. This is not surprising because it has been shown that TGF‐β1 is a multifaceted molecule that both suppresses tumor growth as well as promotes tumor progression and invasion.19 Depending on the stage of tumor development and its microenvironment, an array of intracellular signaling molecules either activate or suppress the cytokine action.19 Indeed, Mdr2‐KO mice with advanced biliary disease have been shown to develop hepatocellular cancer,34 and TGF‐β1 may suppress cancer growth.19 Thus, it is likely that down‐regulation of TGF‐β1 in 9‐month‐old Mdr2‐KO mice might favor cancer growth and that up‐regulation of TGF‐β1 expression by ACE2 therapy could oppose this. Indeed, we found that a majority of Mdr2‐KO mice at 9 months of age had developed liver tumor nodules that were absent in ACE2‐treated mice (data not shown), supporting the work showing inhibition of lung cancer metastasis by ACE2.35 In further support of this argument, ACE2 therapy restored the loss of e‐cadherin in cholangiocytes and/or hepatocytes, and the loss of e‐cadherin is strongly linked to the development of sclerosing cholangitis, peribiliary fibrosis, EMT, and hepatocellular carcinoma.18, 35 However, inhibitory effects of ACE2 on tumor growth are not discussed in this report as it is beyond the scope of the present study. Nevertheless, it appears that the ACE2‐induced increased expression of TGF‐β1 compared to control vector‐injected mice had minimal impact on biliary fibrosis because collagen deposition was dramatically reduced in ACE2‐treated Mdr2‐KO mice, suggesting that the progression of biliary fibrosis at an advanced stage is TGF‐β1 independent (Figs. 2 and 3). On the other hand, TGF‐β1‐dependent biliary fibrosis was evident at an early stage of biliary fibrosis in Mdr2‐KO mice in which Ang‐(1‐7), a product of ACE2 action, inhibited the cytokine expression, leading to a reduction in biliary fibrosis (Fig. 3).
In summary, this study demonstrates a therapeutic approach for biliary diseases using liver‐specific overexpression of ACE2 in a murine model that spontaneously develops chronic biliary fibrosis with pathological features closely resembling those of human PSC. A remarkable feature of this model is that the pathogenesis of biliary fibrosis is both TGF‐β1 dependent, as seen in early fibrosis development, as well as TGF‐β1 independent, as seen in advanced fibrosis development. Irrespective of its role, ACE2 therapy and its peptide product Ang‐(1‐7) show strong inhibitory properties on inflammatory and profibrotic cytokine secretion and myofibroblast activation, leading to a marked reduction in chronic biliary fibrosis in Mdr2‐KO mice. It is thus possible that increased ACE2 protein expression in PSC livers may imply a counter‐regulatory role of the enzyme in human biliary fibrosis (Supporting Fig. S1A). Mechanistically, the beneficial effects of ACE2 likely resulted from a marked reduction in profibrotic Ang II peptide levels with a concomitant increase in antifibrotic Ang‐(1‐7) peptide levels by the catalytic activity of ACE2. In conjunction with reduced activity of the profibrotic hepatic Ang II/AT1‐R axis, increased local production of Ang‐(1‐7), which acts through MasR, inhibits downstream signaling, including NADPH oxidase‐mediated reactive oxygen species production,4, 36, 37 resulting in a marked reduction in biliary fibrosis. This phenomenon has been illustrated in Fig. 8. Thus, the findings from this study provide strong evidence that ACE2 gene therapy has potential for the treatment of patients with biliary diseases, such as PSC.
Figure 8.
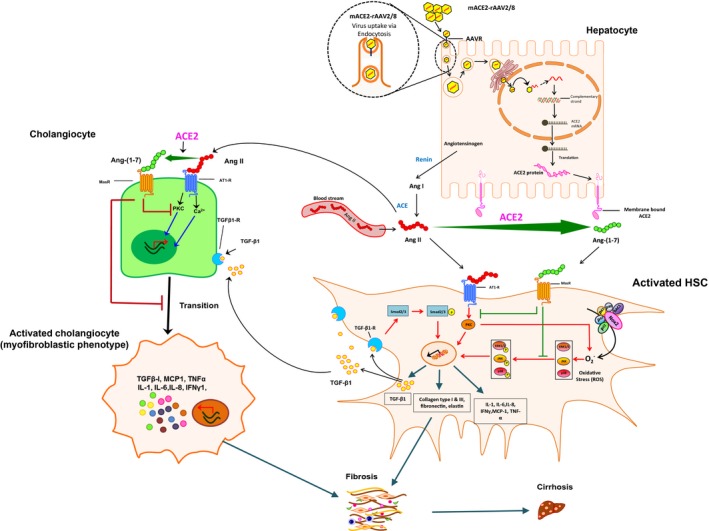
Depicted mechanisms by which Ang‐(1‐7) (generated by ACE2) inhibits activation of HSCs and cholangiocytes, resulting in improved biliary fibrosis in Mdr2‐KO mice. Abbreviations: IFN, interferon; mRNA, messenger RNA; ROS, reactive oxygen species; Smad2/3, SMAD family member 2/3.
Supporting information
Acknowledgment
We acknowledge the technical assistance on immunohistochemistry provided by the University of Melbourne histology platform and Australian Phenomics Network histopathology service.
Supported by the Australian National Health and Medical Research Council (grants APP1062372 and APP1124125 to P.W.A. and C.B.H.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Fickert P, Pollheimer MJ, Beuers U, Lackner C, Hirschfield G, Housset C, et al. International PSC Study Group (IPSCSG). Characterization of animal models for primary sclerosing cholangitis (PSC). J Hepatol 2014;60:1290‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paizis G, Cooper ME, Schembri JM, Tikellis C, Burrell LM, Angus PW. Up‐regulation of components of the renin‐angiotensin system in the bile duct‐ligated rat liver. Gastroenterology 2002;123:1667‐1676. Erratum. In: Gastroenterology 2004;126:634. [DOI] [PubMed] [Google Scholar]
- 3. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grace JA, Herath CB, Mak KY, Burrell LM, Angus PW. Update on new aspects of the renin‐angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond) 2012;123:225‐239. [DOI] [PubMed] [Google Scholar]
- 5. Tandon P, Abraldes JG, Berzigotti A, Garcia‐Pagan JC, Bosch J. Renin‐angiotensin‐aldosterone inhibitors in the reduction of portal pressure: a systematic review and meta‐analysis. J Hepatol 2010;53:273‐282. [DOI] [PubMed] [Google Scholar]
- 6. Herath CB, Mak KY, Angus PW. Role of the alternate RAS in liver disease and the GI tract In: Unger T, Steckelings UM, dos Santos RAS. eds. The Protective Arm of the Renin Angiotensin System: Functional Aspects and Therapeutic Implications, 1st ed London, United Kingdom: Academic Press; 2015:239‐247. [Google Scholar]
- 7. Herath CB, Lubel JS, Jia Z, Velkoska E, Casley D, Brown L, et al. Portal pressure responses and angiotensin peptide production in rat liver are determined by relative activity of ACE and ACE2. Am J Physiol Gastrointest Liver Physiol 2009;297:G98‐G106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem 2000;275:33238‐33243. [DOI] [PubMed] [Google Scholar]
- 9. Lubel JS, Herath CB, Tchongue J, Grace J, Jia Z, Spencer K, et al. Angiotensin‐(1‐7), an alternative metabolite of the renin‐angiotensin system, is up‐regulated in human liver disease and has antifibrotic activity in the bile‐duct‐ligated rat. Clin Sci (Lond) 2009;117:375‐386. [DOI] [PubMed] [Google Scholar]
- 10. Mak KY, Chin R, Cunningham SC, Habib MR, Torresi J, Sharland AF, et al. ACE2 therapy using adeno‐associated viral vector inhibits liver fibrosis in mice. Mol Ther 2015;23:1434‐1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, et al. Mice with homozygous disruption of the mdr2 P‐glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol 1994;145:1237‐1245. [PMC free article] [PubMed] [Google Scholar]
- 12. Katzenellenbogen M, Pappo O, Barash H, Klopstock N, Mizrahi L, Olam D, et al. Multiple adaptive mechanisms to chronic liver disease revealed at early stages of liver carcinogenesis in the Mdr2‐knockout mice. Cancer Res 2006;66:4001‐4010. [DOI] [PubMed] [Google Scholar]
- 13. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 2014;371:1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rajapaksha IG, Mak KY, Huang P, Burrell LM, Angus PW, Herath CB. The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci Rep 2018;8:10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao YL, Zhu RT, Sun YL. Epithelial‐mesenchymal transition in liver fibrosis. Biomed Rep 2016;4:269‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, et al. Hedgehog signaling regulates epithelial‐mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest 2008;118:3331‐3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalluri R, Neilson EG. Epithelial‐mesenchymal transition and its implications for fibrosis. J Clin Invest 2003;112:1776‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakagawa H, Hikiba Y, Hirata Y, Font‐Burgada J, Sakamoto K, Hayakawa Y, et al. Loss of liver E‐cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc Natl Acad Sci U S A 2014;111:1090‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Derynck R, Akhurst RJ, Balmain A. TGF‐[beta] signaling in tumor suppression and cancer progression. Nat Genet 2001;29:117‐129. Erratum. In: Nat Genet 2001;29:351. [DOI] [PubMed] [Google Scholar]
- 20. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2(Abcb4) knockout mice. Gastroenterology 2004;127:261‐274. [DOI] [PubMed] [Google Scholar]
- 21. Tabibian JH, O'Hara SP, Trussoni CE, Tietz PS, Splinter PL, Mounajjed T, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016;63:185‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu C, Tao Q, Sun M, Wu JZ, Yang W, Jian P, et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Invest 2010;90:1805‐1816. [DOI] [PubMed] [Google Scholar]
- 23. Bataller R, Ginès P, Nicolás JM, Görbig MN, Garcia‐Ramallo E, Gasull X, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000;118:1149‐1156. [DOI] [PubMed] [Google Scholar]
- 24. Ferrario CM. ACE2: more of Ang‐(1‐7) or less Ang II? Curr Opin Nephrol Hypertens 2011;20:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lubel John S, Herath Chandana B, Tchongue J, Grace J, Jia Z, Spencer K, et al. Angiotensin‐(1‐7), an alternative metabolite of the renin–angiotensin system, is up‐regulated in human liver disease and has antifibrotic activity in the bile‐duct‐ligated rat. Clin Sci (Lond) 2009;117:375‐386. [DOI] [PubMed] [Google Scholar]
- 26. Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin‐converting enzyme 2 and angiotensin‐(1‐7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol 2005;289:H2281‐H2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pereira RM, dos Santos RAS, Teixeira MM, Leite VHR, Costa LP, da Costa Dias FL, et al. The renin angiotensin system in a rat model of hepatic fibrosis: evidence for a protective role of angiotensin‐(1‐7). J Hepatol 2007;46:674‐681. [DOI] [PubMed] [Google Scholar]
- 28. Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology 2010;51:1438‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, et al. Expression of platelet‐derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol 1996;148:785‐800. [PMC free article] [PubMed] [Google Scholar]
- 31. Xia J‐L, Dai C, Michalopoulos GK, Liu Y. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am J Pathol 2006;168:1500‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenbaum LE, Wells RG. The Role of Stem Cells in Liver Repair and Fibrosis. Int J Biochem Cell Biol 2011;43:222‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor‐beta activation and retards biliary fibrosis progression. Gastroenterology 2008;135:660‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katzenellenbogen M, Mizrahi L, Pappo O, Klopstock N, Olam D, Jacob‐Hirsch J, et al. Molecular mechanisms of liver carcinogenesis in the Mdr2‐knockout mice. Mol Cancer Res 2007;5:1159‐1170. [DOI] [PubMed] [Google Scholar]
- 35. Qian Y, Guo Y, Wan H, Fan L, Feng Y, Ni L, et al. Angiotensin‐converting enzyme 2 attenuates the metastasis of non‐small cell lung cancer through inhibition of epithelial‐mesenchymal transition. Oncol Rep 2013;29:2408‐2414. [DOI] [PubMed] [Google Scholar]
- 36. Mak KY, Rajapaksha IG, Angus PW, Herath CB. The adeno‐associated virus ‐ a safe and promising vehicle for liverspecific gene therapy of inherited and non‐inherited disorders. Curr Gene Ther 2017;17:4‐16. [DOI] [PubMed] [Google Scholar]
- 37. Bataller R, Gäbele E, Parsons CJ, Morris T, Yang L, Schoonhoven R, et al. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct‐ligated rats. Hepatology 2005;41:1046‐1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials