

Abstract
Risks of pathogenic bacteria to the health of both human beings and water ecosystems have been widely acknowledged. However, traditional risk assessment methods based on fecal indicator bacteria and/or pure culture are not comprehensive at the community level, mainly owing to the limited taxonomic coverage. Here, we combined the technique of high-throughput sequencing and the concept of metacommunity to assess the potential pathogenic bacterial communities in an economically and ecologically crucial but highly polluted river—the North Canal River (NCR) in Haihe River Basin located in North China. NCR presented a significant environmental gradient, with the highest, moderate, and lowest levels of pollution in the up-, middle, and downstream. After multiple analyses, we successfully identified 48 genera, covering nine categories of potential pathogens (mainly human pathogens). The most abundant genus was Acinetobacter, which was rarely identified as a pathogen bacterium in previous studies of NCR. At the community level, we observed significant geographical variation of community composition and structure. Such a high level of geographical variation was mainly derived from differed abundance of species among sections along the river, especially the top seven Operational Taxonomic Units (OTUs). For example, relative abundance of OTU1 (Gammaproteobacteria/Acinetobacter) increased significantly from upstream towards downstream. Regarding the underlying mechanisms driving community geographical variation, environmental filtering was identified as the dominant ecological process and total nitrogen as the most influential environmental variable. Altogether, this study provided a comprehensive profile of potential pathogenic bacteria in NCR and revealed the underlying mechanisms of community succession. Owing to their high abundance and wide geographical distribution, we suggest that potential pathogens identified in this study should be incorporated into future monitoring and management programs in NCR. By revealing the correlation between environmental factors and community composition, the results obtained in this study have significant implications for early warning and risk assessment of potential pathogen bacteria, as well as management practices in highly polluted river ecosystems.
Electronic supplementary material
The online version of this article (10.1007/s13280-019-01184-z) contains supplementary material, which is available to authorized users.
Keywords: Community structure, Early warning, Environmental filtering, Potential pathogen bacteria, Risk assessment, Water pollution
Introduction
Pathogen pollution has caught significant public attentions due to its widespread distributions, frequent occurrences, and severe risks in the health of both ecosystems and human beings (Wilkes et al. 2013; Pandey et al. 2014). For example, previous studies have reported frequent outbreaks of water-borne diseases (i.e., cholera, typhoid fever, and bacillary dysentery) caused by bacteria, viruses, and protozoa (Cabral 2010; Wilkes et al. 2013; Pandey et al. 2014). Thus, microbial pathogens in aquatic ecosystems could directly jeopardize the health of human beings and other wildlife, especially those with direct contact and frequent exposure (Yang et al. 2012). In addition, risks of microbial pathogens could also be functional through indirect pathways. For example, the explosion of harmful bacteria or viruses could lead to deterioration or toxicity of water quality (Cabral 2010; James and Joyce 2004). With the aggravation of water pollution and associated emergence of abundant aquatic microbes, the potential risk of pathogenic bacteria or viruses can be high. Therefore, great efforts should be made to monitor and manage potential pathogenic microbes in river ecosystems, particularly those with a high level of pollution in developing countries (Dudgeon et al. 2006; Abraham 2010).
Currently, there are two principal ways to monitor potential pathogenic bacteria in aquatic ecosystems. The first and also the most commonly used method is to use fecal indicator bacteria (FIB) on behalf of pathogenic bacteria, coupled by quantitative microbial risk assessment (QMRA) (Edberg et al. 2000; Cabral 2010; Wilkes et al. 2013). The underlying hypothesis is that the abundance of pathogenic bacteria scales to the concentration or abundance of FIB. Hitherto, Escherichia coli is considered as the best and the most widely used biological indicator of drinking water for public health protection (Edberg et al. 2000; Chen et al. 2019). For example, in China, fecal coliform bacteria are listed as an indicator to assess surface water quality (Chinese national standard GB3838-2002), and six bacteria (e.g., Escherichia coli, fecal coliform bacteria) are listed as indicators to assess drinking water quality (Chinese national standard GB5749-2006). Unfortunately, many studies revealed the lack of correlation between host-specific markers and pathogens, as reviewed by Field and Samadpour (2007) and Santo Domingo et al. (2007). Therefore, relying exclusively on this method might cloud the understanding of true risks, leading to unreasonable judgments and untimely regulations. The second commonly used method is to directly assess the risks of pathogens using pure cultures or PCR-based methods (Vital et al. 2010; Ibekwe et al. 2013). This approach has proved to be useful in guiding early warning systems via detecting various pathogens, such as Listeria, Vibrio, Salmonella, and many others (Maugeri et al. 2004; Vital et al. 2010). However, previous studies mainly focus on traditional pathogens and could only identify a limited number of known taxonomic groups (Abraham 2010; Wilkes et al. 2011). The aggravation of water pollution might broaden the spectrum of potential pathogens, especially newly emerging or uncultivable ones. Traditional methods concentrating on one or several known categories might fail to picture the full profile of potential pathogenic bacteria communities. Thus, it is urgently needed to comprehensively understand geographical distribution and risks of pathogenic bacteria communities in river ecosystems using the state-of-the-art techniques and conceptual frameworks.
The proposal and application of metacommunity has recently spurred the exploration of community ecology, which also can provide insightful perspectives for research of pathogen bacteria. By integrating a set of local communities that are connected by potential dispersal of interacting species, metacommunity links up local communities to regional communities (Leibold et al. 2004). The application of metacommunity framework has revolutionized current research of temporal–spatial dynamics of aquatic communities and associated mechanisms (Heino et al. 2015; Soininen et al. 2018; Yang et al. 2019a, b). In addition, pioneering studies have confirmed that environmental filtering is the dominant ecological process in driving geographical distribution patterns of aquatic communities (i.e., ‘fine-scale species sorting hypothesis’ proposed by Xiong et al. (2017)). For this hypothesis, local communities are selected by environmental gradients, and species only occur at environmentally suitable habitats given that dispersal is sufficient (Leibold et al. 2004; Heino et al. 2015; Xiong et al. 2017; Tolonen et al. 2018; Yang et al. 2018). Environmental gradients along rivers mainly resulted from differences of local environmental factors, which were due to varied anthropogenic impacts along rivers (Dudgeon et al. 2006; Xiong et al. 2016, 2017; Peng et al. 2018; Yang et al. 2018). Consequently, the identification of local environmental factors responsible for geographical distribution of biodiversity could benefit early warning systems by predicting potential spread routes and explosion ranges of pathogens. Identification of influential factors is also crucial in guiding control strategies and management practices. Regarding potential pathogenic bacteria, they are by themselves an effective unit of a community. Therefore, the application of metacommunity concept would benefit the understanding of geographical distribution of pathogenic bacteria communities, reveal crucial influential environmental factors for observed distribution patterns, and improve the effectiveness of early warning and management programs. These knowledge and programs are particularly important for rivers that support cities with large populations and various industries.
North Canal River (NCR), located in Haihe River Basin in North China, runs through two megacities—Beijing (population in 2017: 21.7 million) and Tianjin (population in 2017: 15.56 million). In the past half century, the Beijing–Tianjin region shows the highest growth rates in both economy and population in China (Pernet-Coudrier et al. 2012). Due to high population density and severe anthropogenic disturbance, NCR suffers from serious pollution and deserves particular attentions, just similar to many other urban rivers globally (Abraham 2010). Dozens of wastewater treatment plants (WWTPs) are scattered along the river, and effluent from WWTPs is the major water resource of NCR, especially in the arid season (Heeb et al. 2012; Pernet-Coudrier et al. 2012). As a typical wastewater-receiving river, NCR inevitably faces the spread of microbiological pollution and risk of pathogenic bacteria. For example, previous studies have reported the occurrence of fecal bacteria and pathogenic bacteria in NCR (Yang et al. 2012; Chen et al. 2017; Wang et al. 2017). However, these research mainly focused on one section of NCR (especially the upstream and downstream), while ignored the connectivity of the whole river (Chen et al. 2017; Wang et al. 2017). Due to the continuous flow, a comprehensive survey from the perspective of a whole river is crucial for future monitoring and management (Vannote et al. 1980). In addition, those studies only focused on traditionally known microbial groups (Yang et al. 2012; Chen et al. 2017; Wang et al. 2017). Therefore, to develop future monitoring and management strategies, it is urgently necessary to perform a comprehensive survey at the community level of the whole river, as well as to investigate the underlying mechanisms driving the geographical distribution patterns of pathogenic bacteria.
Despite the acknowledgement of risks of aquatic pathogens and socio-economic values of rivers, there is a lack of comprehensive research related to pathogen risks in NCR. Thus, this study aims to (i) characterize the composition and structure of potential pathogen communities; (ii) investigate the dominant processes and factors leading to the geographical distribution patterns of potential pathogens; and (iii) extract promising perspectives for future monitoring and management practices based on patterns obtained from this study.
Materials and methods
Sample collection and measurement of physicochemical parameters
Based on our previous surveys (Yang et al. 2019a), NCR showed a significant pollution gradient, with the highest level of pollution in the upstream and the lowest level of pollution in the downstream. Accordingly, the whole river was divided into Sections I to III, corresponding to up-, middle, and downstream, respectively (Fig. 1). In total, 39 sites were sampled, with 18, 12, and 9 sites from Sections I to III, respectively. Water samples (0.5 L) were filtered through 0.22 μm mixed cellulose ester membranes (Millipore, Bedford, MA) to collect microbial cells. After sampling, membranes were stored at − 80 °C until further analyses. We recorded the geographical locations of each sampling site using Garmin Handheld GPS (Garmin Ltd., Kansas, USA) navigator.
Fig. 1.

Study area and sampling sites in North Canal River (NCR) revised from Yang et al. (2019a). The small map in the left bottom showed the Haihe River Basin of China, where NCR is located
In the field, we measured the water temperature (T), electric conductivity (EC), pH, oxidation–reduction potential (ORP), total dissolved solid (TDS), dissolved oxygen (DO), and concentration of chlorophyll a (Chl_a). In the laboratory, we measured the concentration of total nitrogen (TN), nitrate (NO3-N), ammonia (NH4-N), total phosphorous (TP), soluble reactive phosphorous (SRP), chemical oxygen demand (COD), and total organic carbon (TOC). We also measured the concentration of common metals and heavy metals, including potassium (K), calcium (Ca), sodium (Na), magnesium (Mg), chromium (Cr), iron (Fe), manganese (Mn), nickel (Ni), copper (Cu), zinc (Zn), arsenic (As), and lead (Pb). The measurement of these parameters followed the methods described by Xiong et al. (2016) and Yang et al. (2019b).
DNA extraction, PCR amplification, and high-throughput sequencing
Total genomic DNA of each sample was extracted from the membranes using a modified CTAB protocol (Yang et al. 2016). The primer pair of 515F/806R was used to amplify the hypervariable V4 region of 16S rRNA (Caporaso et al. 2011). To distinguish each sample, a twelve-nucleotide tag was added to the 5′-end of each primer pair. Three replicates were performed for each sample to recover species in the whole community (Zhan et al. 2014; Yang et al. 2016). Three replicates of each sample were then pooled together and purified using the Sangon column PCR product purification kit (Sangon Biotech, Shanghai, China). Finally, sequencing was performed using the Illumina Miseq PE250 platform.
Bioinformatics analysis and functional annotations
Raw data was de-multiplexed according to unique tags. Paired-end reads were then merged before trimming primers. We discarded low-quality sequences (i) containing ‘N’ (undetermined nucleotide); (ii) with quality score less than 20; (iii) with length shorter than 200 bp (Yang et al. 2016). The remained sequences were clustered into operational taxonomic units (OTUs) with the threshold of 97% similarity using UPARSE pipeline (Edgar 2013). Taxonomic information of representative sequences was determined by searching against the SILVA_128 database using the Ribosomal Database Project (RDP) classifier (Wang et al. 2007). OTU table was normalized by rarefying down to the smallest number of total sequences for each sample. The number of sequences assigned to each OTU was taken as a proxy for the abundance of OTUs for subsequent analyses (Zhan et al. 2014).
FAPROTAX (Functional Annotation of Prokaryotic Taxa) was used to investigate the potential functions of bacterial communities (Louca et al. 2016). This software functions with inputs of OTU table and classifier information of each OTU. Based on previous records of the lowest taxon, each OTU was categorized into a specific functional group. A total of 69 functional groups were identified (Yang et al. 2019a), and we further extracted OTUs belonging to at least one pathogenic group to create community of potential pathogenic bacteria. This new OTU table (a sub-table of the original one) was used for downstream analyses. As FAPROTAX relies on records of cultivable species (Louca et al. 2016), this research provided relatively conservative results. FAPROTAX used in this study was embedded in the in-house pipeline Galaxy (http://mem.rcees.ac.cn:8080/root).
Ecological and statistical analyses
Environmental variables (except pH) were first log10 (x + 1) transformed to improve normality. The clustering result of environmental variables was illustrated using principal component analysis (PCA). The significance of dissimilarity of environmental variables among three sections was tested using multi-response permutation procedure (MRPP), analysis of similarity (ANOSIM), and analysis of distance matrices (ADONIS). All these analyses were performed using R ‘vegan’ package (Oksanen et al. 2013).
In order to investigate the influence of dispersal limitation on community distribution patterns, a spatial eigenvector matrix was built to represent dispersal limitation. This matrix was created using the Principal Coordinates of Neighbor Matrices (PCNM) analysis (Borcard and Legendre 2002; Legendre 2008). Through eigenvector decomposition, PCNM created a truncated matrix of geographic distance with longitude and latitude data. Spatial explanatory variables were then selected when they corresponded to positive eigenvectors and showed positive spatial correlation. PCNM analysis was performed using the ‘pcnm’ function in R (R core Team 2015).
The clustering of potential pathogen communities was illustrated using non-metric multidimensional scaling (NMDS). The significance of dissimilarity of community composition among three sections was tested using MRPP, ANOSIM, and ADONIS. To investigate the influences of environmental and spatial variables, Mantel test was performed to select variables which were significantly correlated with community composition. Forward selection was further performed to select relatively more important ones from these selected variables (Blanchet et al. 2008). Two parsimonious redundancy models (RDA) were then constructed for environmental and spatial variables. All these analyses were performed in R ‘vegan’ package (Oksanen et al. 2013).
Finally, in order to dissect the relative importance of environmental filtering and dispersal limitation in affecting the geographical distribution patterns of pathogenic bacterial communities, we performed the variation partitioning analysis (VPA) (Borcard et al. 1992). VPA decomposed the total variation of community dynamics into four components, the fraction explained purely by environmental variables (Env.), the fraction explained purely by spatial variables (Spa.), the fraction shared by two types of variables (Env. & Spa.), and the unexplained variation (the residual). The total explained variation was calculated by the combination of forward selected environmental and spatial variables, and the fractions purely explained by each type was calculated with partial redundancy analysis (pRDA). In the analysis, Monte Carlo permutation test was conducted to produce the significance value. VPA was conducted using the R ‘varpart’ function (R core Team 2015).
Results
An environmental gradient along North Canal River
Based on our previous research, a significant environmental gradient was identified along the NCR, especially the nitrogen gradient (Yang et al. 2018, 2019a). The pollution level was highest in the upstream (Section I), moderate in the middle stream (Section II), and lowest in the downstream (Section III). The concentration of nutrients (e.g., TN, TP, and NH4-N) decreased from Section I towards Section III. For example, the concentration of TN was significantly higher in Section I (average: 80.642 mg/L) than those in Section II (average: 29.389 mg/L) and Section III (average: 3.621 mg/L). Detailed information of environmental variables was displayed with a heatmap (Supplementary Fig. S1).
Basic composition of the potential pathogen communities
According to our previous research of the whole bacterioplankton of North Canal River, a total of 69 functional groups were identified after consulting FAPROTAX annotations (Yang et al. 2019a). In order to assess the risks of potential pathogen to both human beings and aquatic wildlife (e.g., aquatic animals and plants), we focused on groups that were relevant to pathogens or parasites in this study. In total, nine (potential) pathogenic groups were identified, including intracellular parasites (IntCelP), animal parasites or symbionts (AniP), human pathogens (all, HPall), human pathogens (pneumonia, HPpneu), human pathogens (nosocomial, HPnos), human pathogens (gastroenteritis, HPgast), plant pathogens (PlaP), invertebrate parasites (InvertP), and fish parasites (FishP). In total, 407 records were obtained, and most of these records were assigned as IntCelP (159), AniP (123), and HPall (98). As one single OTU could be assigned to more than one functional group using FAPROTAX, we manually matched the functional OTU table with original OTU table and assigned those records into 200 OTUs.
Here, we separated those 200 OTUs from the original OTU table for downstream analyses. Comparing to the original dataset of whole bacterioplankton, the selected OTUs harbored 23.28% sequences (original data: 1 349 123 sequences, 4 567 OTUs; Yang et al. 2019a). Based on our previous results, the DNA extracts were of high quality and sequencing depth was sufficient to capture most biodiversity (Yang et al. 2019a). The high coverage of the 200 OTUs was mainly contributed by dominant OTUs in the original communities, where such dominant OTUs, including OTU1, OTU2, and OTU5, were potential pathogenic bacteria. Furthermore, seven OTUs occurred at all sampling sites, forming the core microbiome of potential pathogenic community. The seven OTUs were OTU1 (Gammaproteobacteria/Acinetobacter), OTU2 (Betaproteobacteria/Polynucleobacter), OTU5 (Gammaproteobacteria/Acinetobacter), OTU6185 (Gammaproteobacteria/Acinetobacter), OTU11 (Betaproteobacteria/GKS98 freshwater group), OTU20 (Gammaproteobacteria/Acinetobacter), and OTU6817 (Gammaproteobacteria/Acinetobacter). These seven OTUs contributed to 93.68% abundance of the potential pathogen communities, but they showed discrepant variation in abundance along the river (Fig. 2). For example, the relative abundance of OTU1 increased significantly from Section I to Section III (p < 0.05), while the others fluctuated in three sections.
Fig. 2.

The relative abundance of top seven OTUs in the communities of potential pathogenic bacteria. Sections I–III corresponded to the up-, middle, and downstream of NCR
By summarizing the taxonomic information of OTUs at the genus level, we found that the potential pathogen communities were composed of 48 genera. The dominant genera were Acinetobacter, Polynucleobacter, and GKS98 freshwater group. When linking the genus-level OTU composition with functional groups, we found that HPpneu, HPnos, HPall, and AniP were largely contributed by Acinetobacter, Polynucleobacter, and GKS98 freshwater group (Fig. 3). IntCelP, PlaP, InvertP, and HPgast were largely contributed by Legionella, Pseudomonas, Rickettsiella, and Arcobacter, respectively (Fig. 3).
Fig. 3.

Association of bacterial genera with potential pathogenic groups predicted by FAPROTAX. Blue indicates higher abundance, while black indicates lower abundance
Beta diversity and correlation with explanatory variables
The plot of NMDS showed that intra-section variation among samples was smaller than inter-section variation (Fig. 4). Further statistical tests indicated that the community composition of Section I was significantly different from that of Section II (p = 0.001) and Section III (p < 0.01). No significance was detected between Sections II and III (p > 0.05). Through diverse statistical analyses, four environmental variables (TN, Cond, pH, and TP) and three spatial variables (V2, V5, and V9) were selected as the crucial variables to affect diversification of community composition (Fig. 5; Supplementary Table S1). All the four environmental variables explained 48.5% variation of community composition, with TN alone contributing to 34.7%. All the three spatial variables explained 25.1% variation of community composition, with V2 alone contributing to 16.8%.
Fig. 4.
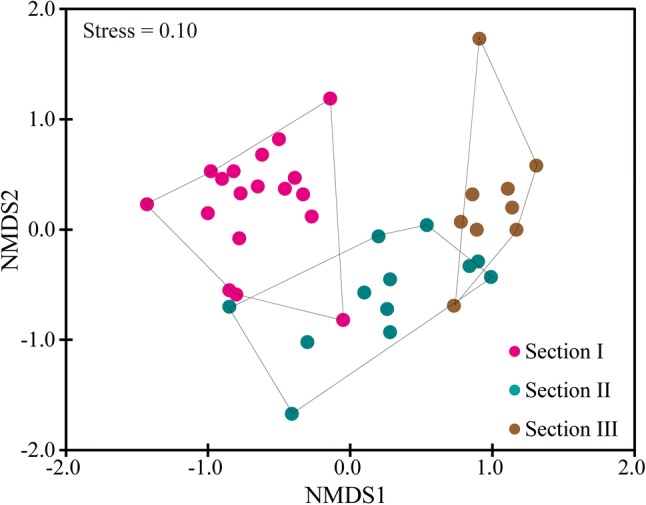
Non-metric multidimensional scaling (NMDS) of OTU-level community composition based on Bray-Curtis distance
Fig. 5.
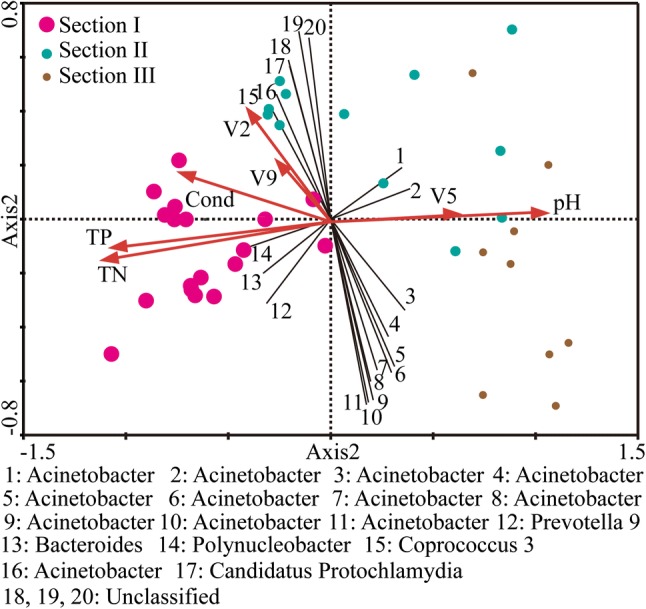
The ordination plot of redundancy analysis (RDA) of community composition at all sites. OTUs that were weakly associated with the first two axes (with fitness < 50%) were omitted for clarity. The larger and smaller sites corresponded to higher and lower concentration of total nitrogen, respectively. Red arrows represent environmental and spatial variables, while black lines represent OTUs
Dissecting the influences of environmental filtering and dispersal limitation
As both environmental filtering and dispersal limitation were crucial, we employed variation partitioning analysis (VPA) to dissect the relative importance of each process. Our results revealed that a large fraction (62%) of total variation was explained by these two processes. The fraction explained purely by environmental variables (36.9%; p < 0.001) was larger than that by spatial variables (13.5%; p < 0.01).
Discussion
Combing the technique of high-throughput sequencing and the framework of metacommunity, we analyzed the composition and structure of potential pathogenic bacterial communities in a typical wastewater-receiving river—the North Canal River (NCR) in China. Our study identified a broader spectrum of pathogen bacteria than previous studies based on traditional methods, especially the prevalent distribution of Acinetobacter. Similar to other aquatic organisms, pathogenic bacteria communities presented apparent spatial distribution patterns (Fig. 4). More importantly, environmental filtering was identified to be the dominant process to drive community variation, although dispersal also affected the community structure to some extent. Overall, the results obtained in this study highlight the necessity and strength to comprehensively investigate potential pathogenic bacteria from the metacommunity perspective, and to further determine the environmental drivers for their geographical distributions.
There used to be a long-term debate over whether microbes were distributed randomly or not (as reviewed by Martiny et al. 2006). Recent comprehensive investigations have reached an agreement that microbes, similar to plants and animals, exhibit specific temporal–spatial distribution patters (Martiny et al. 2006; Zeglin 2015). For instance, our previous study revealed significant dissimilarity of bacterioplankton communities in different sections of NCR, corresponding to the significant environmental gradient (Yang et al. 2019a). Similarly, we observed significant geographical variation of potential pathogenic bacterial communities along NCR (Fig. 4). Such significant variation in community was largely attributed to the occurrence frequency of dominant OTUs, such as OTU1 (Gammaproteobacteria/Acinetobacter), OTU2 (Betaproteobacteria/Polynucleobacter), and OTU5 (Gammaproteobacteria/Acinetobacter), rather than the presence–absence of species (Fig. 2).
Using high-throughput sequencing, we provided broad context of potential pathogen risk in NCR, and a total of 48 genera were successfully recovered from the communities. Indeed, this spectrum of pathogen bacteria was broader than most of previous studies based on traditional methods (Pianetti et al. 1998; Wilkes et al. 2013) or Sanger sequencing technique (Jiang and Chu 2004; Ibekwe et al. 2013). Therefore, risks of pathogenic bacteria in river ecosystems can be largely underestimated, even in this study where we used FAPROTAX which relies only on records of cultivable species (Louca et al. 2016). Regarding the community composition of NCR, the most dominant genus was Acinetobacter, belonging to Gammaproteobacteria/Moraxellaceae. Being typical opportunistic pathogens, Acinetobacter spp. are normally harmless to healthy individuals, but could be implicated in nosocomial infections in deliberated individuals and patients with impaired immune system (Rathinavelu et al. 2003). Consequently, pathogenic Acinetobacter has been identified as a cause of diverse diseases such as chronic gastritis, septicemia, pneumonia, meningitis, urinary tract infections, and skin and wound infections (Regalado et al. 2009). With the explosion of various epidemics and their resistance to multi-antibiotics, Acinetobacter has become a widespread concern in a variety of hospitals worldwide (Almasaudi 2018). Outside of hospitals, recent studies have identified environmental reservoirs of Acinetobacter, such as surface waters, polluted waters, and wastewater treatment plants (Al Atrouni et al. 2016; Yang et al. 2017; Jin et al. 2018), suggesting the increasing risk to public health. Besides, the multi-antibiotic resistance of Acinetobacter could be triggered and enhanced by the large abundance of antibiotics found in polluted rivers, especially in China, the biggest producer and consumer of antibiotics (Zhu et al. 2013). Despite this significance, previous studies based on traditional methods paid little attention to Acinetobacter (Yang et al. 2012; Chen et al. 2017; Wang et al. 2017). Our research suggests that the community level survey based on high-throughput sequencing could provide more comprehensive information on risk assessment of potential pathogens.
We also detected common dominant genera with many other studies (Maugeri et al. 2004; Vital et al. 2010; Ibekwe et al. 2013; Pandey et al. 2014), including Polynucleobacter, Pseudomonas, Arcobacter, Legionella, and Rickettsiella. These genera could infect not only human beings, but also plants, fishes, invertebrates, and other animals (Fig. 3). Therefore, integrating these taxa into monitoring and warning programs is necessary to improve our understanding of pathogen risks to human beings and water ecosystems, as well as associated fauna and flora. In our previous analysis of the whole microbial community, we also tracked fecal indicator bacteria such as E. coli with low abundance (detailed information could be found in Yang et al. 2019a). This finding was different from results of quantitative microbial risk assessment of NCR (Chen et al. 2017; Wang et al. 2017). Interestingly, the abundance of E. coli decreased from upstream towards downstream, contrasting to the increase of Acinetobacter. These results were consistent with previous studies and supported the decoupling of indicator bacteria and pathogenic bacteria (Field and Samadpour 2007; Santo Domingo et al. 2007). Considering the economic and ecological significance of NCR, we suggest to improve traditional monitoring strategies to enhance more comprehensive monitoring and warning programs.
In metacommunity ecology, it remains a great challenge to disentangle the dominant ecological processes and corresponding dominant variables within each process, which drives the specific spatial–temporal patterns of communities (Downes 2010; Göthe et al. 2013). For pathogen bacterial communities in this study, we found environmental filtering overwhelmed dispersal limitation to be the dominant ecological process, with total nitrogen (TN) as the most important environmental factor (Fig. 5). These findings reflected the underlying interactions between pathogen bacteria and environmental conditions, especially local environmental factors which shaped specific microenvironment. For example, poor sanitation, insufficient treatment of wastewater, and catastrophic floods could introduce pathogenic bacteria into rivers, especially in highly populated megacities (Abraham 2010). Furthermore, riverine pathogen bacteria were facing the pressure of environmental selection. For instance, our study identified the significant influence of TN on pathogen bacterial communities, which was similar to previous analyses of whole bacterioplankton communities in NCR (Yang et al. 2019a). This was mainly due to the 100-fold + variation of TN along NCR. Combined with other factors, the sufficiently long gradient of TN impacted the survival, reproduction, and distribution of pathogen bacteria. As reported, the relatively high concentration of TN might function as an efficient antiseptic reagent, killing those pathogens (Yang et al. 2019a). On the other hand, the distribution variation of different pathogens was also related to their inherent tolerance and adaptive capability (Li et al. 2017; Yang et al. 2019a). For instance, the increasing abundance of OTU1 (Gammaproteobacteria/Acinetobacter) from the highly polluted upstream towards the lightly polluted downstream might result from its adaptive potentials. According to previous studies, Acinetobacter could endure, even favor less-nutrient habitats, with the ability to survive desiccation for a long period and a wide range of temperature (Rathinavelu et al. 2003).
Finally, the results obtained here have significant implications for early warning and risk assessment of pathogen bacteria, as well as restoration practices in river ecosystems. On one hand, the identification of influential factors is crucial for pollution control and management. For example, the enrichment of nutrient (e.g., N and P) has been widely identified in polluted rivers (Ibekwe et al. 2013; Xiong et al. 2017; Yang et al. 2019a). The enrichment was mainly derived from wastewater discharge or surface flow which could introduce exogenous pathogen into rivers (Abraham 2010; Chen et al. 2017). Therefore, cutting down the exogenous inputs represents a direct and efficient approach to reduce the risks of pathogen. On the other hand, the significant correlation between environmental factors and pathogen bacteria indicates that it is possible to predict the potential spread routes and explosion ranges of pathogens by monitoring environmental variation (Martiny et al. 2006; Li et al. 2017). Accordingly, efficient warning signatures could be obtained to guide timely disturbance, preventing potential impacts of pathogen explosion. As it is difficult to accurately classify short reads to the species level, our research could possibly lead to somewhat overestimation of the spectrum of potential pathogens. Future studies are needed to incorporate high-resolution primers, especially for pathogenic bacteria. Considering the worldwide increase of water pollution and microbe pollution (Dudgeon et al. 2006; Abraham 2010), especially the severe environmental situation of China, we suggest that more attention should be paid to the potential health risks from severely polluted rivers.
Conclusions
By incorporating the metacommunity concept, this study provided a relatively full profile of potential pathogen bacteria in North Canal River (NCR), a highly disturbed and polluted rive running through two megacities (Beijing and Tianjin). The total number of 48 potential pathogen genera in nine categories was larger than that found by most of previous studies, suggesting the severe health risks of NCR to human beings, as well as aquatic flora and fauna. Reducing direct exposure to polluted water was crucial to self-protection, especially for persons with impaired immune systems. The identification of significant correlation between environmental variables and potential pathogen communities was crucial to future warning, monitoring, and management of highly polluted rivers. For example, the identification of specific environmental factors provided useful information to cut off exogenous inputs of potential pathogen. By monitoring the variation of those factors, it is possible to predict potential spread routes and explosion ranges of pathogens to prevent health risk. Altogether, the integration of high-throughput sequencing technique and metacommunity concept could enhance understanding of dynamics and risks of potential pathogen bacteria. This approach could be expanded to other polluted water systems to improve future monitoring and management strategies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Great thanks to Yangchun Gao and Xuena Huang for help during the field sampling, and Ping Ni for assistance in the measurement of water parameters. This work was supported by the National Nature Science Foundation of China [Grant Nos.: 31800419; 31572228], the Water Pollution Control and Treatment Special Project [Grant No.: 2018ZX07105-001], National Key R&D Program of China [Grant No.: 2016YFC0500406], the Innovation in Cross-functional Team Program of the Chinese Academy of Sciences [Grant No.: No.: 2015], Chinese Academy of Science [Grant No.: ZDRW-ZS-2016-5], and the State Key Joint Laboratory of Environment Simulation and Pollution Control [RCEES, Chinese Academy of Sciences; Grant No.: 15K01ESPCR].
Biographies
Yuzhan Yang
is a postdoctoral researcher at the Aquatic Molecular Ecology and Evolution Laboratory led by Prof. Aibin Zhan in the Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences. Her research interests include microbial ecology, pollution ecology, and community ecology.
Yang Hou
works at the Beijing Dongcheng District Food and Drug Safety Monitoring Center. His research interests center on safety monitoring of food, drug, and associated products.
Min Ma
works at the Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences. Her research interests include Chinese medicinal herbs and health assessment.
Aibin Zhan
is a professor at the Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences. He has broad research interests in molecular and evolutionary ecology in aquatic organisms (both invasive and non-invasive species) and functioning of human activities-influenced aquatic ecosystems.
Data accessibility
The high-throughput sequencing data have been deposited into NCBI Sequence Read Archive (SRA) (accession no.: PRJNA504486).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yuzhan Yang, Email: yyzhan@mail.ustc.edu.cn.
Yang Hou, Phone: +86-10-67670820, Email: dc_syakzx@163.com.
Min Ma, Email: mamin@rcees.ac.cn.
Aibin Zhan, Phone: +86-10-62849882, Email: azhan@rcees.ac.cn, Email: zhanaibin@hotmail.com.
References
- Abraham WR. Megacities as sources for pathogenic bacteria in rivers and their fate downstream. International Journal of Microbiology. 2010;2011:798292. doi: 10.1155/2011/798292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Atrouni A, Joly-Guillou ML, Hamze M, Kempf M. Reservoirs of non-baumannii Acinetobacter species. Frontiers in Microbiology. 2016;7:49. doi: 10.3389/fmicb.2016.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasaudi SB. Acinetobacter spp. as nosocomial pathogens: Epidemiology and resistance features. Saudi Journal of Biological Sciences. 2018;25:586–596. doi: 10.1016/j.sjbs.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet FG, Legendre P, Borcard D. Forward selection of explanatory variables. Ecology. 2008;89:2623–2632. doi: 10.1890/07-0986.1. [DOI] [PubMed] [Google Scholar]
- Borcard D, Legendre P. All-scale spatial analysis of ecological data by means of principal coordinates of neighbor matrices. Ecological Modelling. 2002;153:51–68. doi: 10.1016/S0304-3800(01)00501-4. [DOI] [Google Scholar]
- Borcard D, Legendre P, Drapeau P. Partialling out the spatial component of ecological variation. Ecology. 1992;73:1045–1055. doi: 10.2307/1940179. [DOI] [Google Scholar]
- Cabral JP. Water microbiology. Bacterial pathogens and water. International Journal of Environmental Research and Public Health. 2010;7:3657–3703. doi: 10.3390/ijerph7103657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Wang M, Wang J, Tan S, Cheng R, Zheng X, Wei Y, Wang Y. Health risk assessment based on indicator microorganisms in Wenyu River. Acta Scientiae Circumstantiae. 2017;37:3177–3184. [Google Scholar]
- Chen C, Li L, Zhi X, Zhang P, Dai Y, Xiao Y, Shen Z. Pollution characteristics and health risk assessment of microorganism pollutions in the Beiyun River. Environmental Science. 2019;40:633–639. doi: 10.13227/j.hjkx.201806197. [DOI] [PubMed] [Google Scholar]
- Downes BJ. Back to the future: Little-used tools and principles of scientific inference can help disentangle effects of multiple stressors on freshwater ecosystems. Freshwater Biology. 2010;55:60–79. doi: 10.1111/j.1365-2427.2009.02377.x. [DOI] [Google Scholar]
- Dudgeon D, Arthington AH, Gessner MO, Kawabata ZI, Knowler DJ, Lévêque C, Naiman RJ, Prieur-Richard AH, et al. Freshwater biodiversity: Importance, threats, status and conservation challenges. Biological Reviews. 2006;81:163–182. doi: 10.1017/S1464793105006950. [DOI] [PubMed] [Google Scholar]
- Edberg SCL, Rice EW, Karlin RJ, Allen MJ. Escherichia coli: The best biological drinking water indicator for public health protection. Journal of Applied Microbiology. 2000;88:106S–116S. doi: 10.1111/j.1365-2672.2000.tb05338.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 2013;10:996. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Field KG, Samadpour M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Research. 2007;41:3517–3538. doi: 10.1016/j.watres.2007.06.056. [DOI] [PubMed] [Google Scholar]
- Göthe E, Angeler DG, Sandin L. Metacommunity structure in a small boreal stream network. Journal of Animal Ecology. 2013;82:449–458. doi: 10.1111/1365-2656.12004. [DOI] [PubMed] [Google Scholar]
- Heeb F, Singer H, Pernet-Coudrier B, Qi W, Liu H, Longrée P, Müller B, Berg M. Organic micropollutants in rivers downstream of the megacity Beijing: Sources and mass fluxes in a large-scale wastewater irrigation system. Environmental Science and Technology. 2012;46:8680–8688. doi: 10.1021/es301912q. [DOI] [PubMed] [Google Scholar]
- Heino J, Melo AS, Bini LM. Reconceptualising the beta diversity-environmental heterogeneity relationship in running water systems. Freshwater Biology. 2015;60:223–235. doi: 10.1111/fwb.12502. [DOI] [Google Scholar]
- Ibekwe AM, Leddy M, Murinda SE. Potential human pathogenic bacteria in a mixed urban watershed as revealed by pyrosequencing. PLoS ONE. 2013;8:e79490. doi: 10.1371/journal.pone.0079490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James E, Joyce M. Assessment and management of watershed microbial contaminants. Critical Reviews in Environmental Science and Technology. 2004;34:109–139. doi: 10.1080/10643380490430663. [DOI] [Google Scholar]
- Jiang SC, Chu W. PCR detection of pathogenic viruses in southern California urban rivers. Journal of Applied Microbiology. 2004;97:17–28. doi: 10.1111/j.1365-2672.2004.02269.x. [DOI] [PubMed] [Google Scholar]
- Jin D, Kong X, Cui B, Jin S, Xie Y, Wang X, Deng Y. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Scientific Reports. 2018;8:13368. doi: 10.1038/s41598-018-31706-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P. Studying beta diversity: Ecological variation partitioning by multiple regression and canonical analysis. Journal of Plant Ecology. 2008;1:3–8. doi: 10.1093/jpe/rtm001. [DOI] [Google Scholar]
- Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, Holt RD, Shurin JB, et al. The metacommunity concept: A framework for multi-scale community ecology. Ecology Letters. 2004;7:601–613. doi: 10.1111/j.1461-0248.2004.00608.x. [DOI] [Google Scholar]
- Li X, Meng D, Li J, Yin H, Liu H, Liu X, Cheng C, Xiao Y, et al. Response of soil microbial communities and microbial interactions to long-term heavy metal contamination. Environmental Pollution. 2017;231:908–917. doi: 10.1016/j.envpol.2017.08.057. [DOI] [PubMed] [Google Scholar]
- Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–1277. doi: 10.1126/science.aaf4507. [DOI] [PubMed] [Google Scholar]
- Martiny JBH, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, et al. Microbial biogeography: Putting microorganisms on the map. Nature Reviews Microbiology. 2006;4:102. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- Maugeri TL, Carbone M, Fera MT, Irrera GP, Gugliandolo C. Distribution of potentially pathogenic bacteria as free living and plankton associated in a marine coastal zone. Journal of Applied Microbiology. 2004;97:354–361. doi: 10.1111/j.1365-2672.2004.02303.x. [DOI] [PubMed] [Google Scholar]
- Oksanen, J., F.G. Blanchet, R. Kindt, P. Legendre, P.R. Minchin, R.B. O’hara, G.L. Simpson, P. Solymos, et al. 2013. Package ‘vegan’. Community ecology package, version, 2.9.
- Pandey PK, Kass PH, Soupir ML, Biswas S, Singh VP. Contamination of water resources by pathogenic bacteria. AMB Express. 2014;4:51. doi: 10.1186/s13568-014-0051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Xiong W, Zhan A. Fine-scale environmental gradients formed by local pollutants largely impact zooplankton communities in running water ecosystems. Aquatic Biology. 2018;27:43–53. doi: 10.3354/ab00695. [DOI] [Google Scholar]
- Pernet-Coudrier B, Qi W, Liu H, Müller B, Berg M. Sources and pathways of nutrients in the semi-arid region of Beijing-Tianjin, China. Environmental Science and Technology. 2012;46:5294–5301. doi: 10.1021/es3004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianetti A, Baffone W, Bruscolini F, Barbieri E, Biffi MR, Salvaggio L, Albano A. Presence of several pathogenic bacteria in the Metauro and Foglia rivers (Pesaro-Urbino, Italy) Water Research. 1998;32:1515–1521. doi: 10.1016/S0043-1354(97)00340-0. [DOI] [Google Scholar]
- R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- Rathinavelu S, Zavros Y, Merchant JL. Acinetobacter lwoffii infection and gastritis. Microbes and Infection. 2003;5:651–657. doi: 10.1016/S1286-4579(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Regalado NG, Martin G, Antony SJ. Acinetobacter lwoffii: Bacteremia associated with acute gastroenteritis. Travel Medicine and Infectious Disease. 2009;7:316–317. doi: 10.1016/j.tmaid.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Santo Domingo JW, Bambic DG, Edge TA, Wuertz S. Quo vadis source tracking? Towards a strategic framework for environmental monitoring of fecal pollution. Water Research. 2007;41:3539–3552. doi: 10.1016/j.watres.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Soininen J, Heino J, Wang J. A meta-analysis of nestedness and turnover components of beta diversity across organisms and ecosystems. Global Ecology and Biogeography. 2018;27:96–109. doi: 10.1111/geb.12660. [DOI] [Google Scholar]
- Tolonen KT, Cai Y, Vilmi A, Karjalainen SM, Sutela T, Heino J. Environmental filtering and spatial effects on metacommunity organization differ among littoral macroinvertebrate groups deconstructed by biological traits. Aquatic Ecology. 2018;52:119–131. doi: 10.1007/s10452-018-9649-4. [DOI] [Google Scholar]
- Vannote RL, Minshall GW, Cummins KW, Sedell JR, Cushing CE. The river continuum concept. Canadian Journal of Fisheries and Aquatic Sciences. 1980;37:130–137. doi: 10.1139/f80-017. [DOI] [Google Scholar]
- Vital M, Stucki D, Egli T, Hammes F. Evaluating the growth potential of pathogenic bacteria in water. Applied and Environmental Microbiology. 2010;76:6477–6484. doi: 10.1128/AEM.00794-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kang M, Zheng X, Chen C, Wang M, Xiao B, Zhou Z, Wei Y, et al. Occurrence and temporal-spatial distribution of fecal indicator microorganisms in three rivers of the Haihe River Basin. Acta Scientiae Circumstantiae. 2017;37:138–145. [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes G, Brassard J, Edge TA, Gannon V, Jokinen CC, Jones TH, Neumann N, Pintar KDM, et al. Bacteria, viruses, and parasites in an intermittent stream protected from and exposed to pasturing cattle: Prevalence, densities, and quantitative microbial risk assessment. Water Research. 2013;47:6244–6257. doi: 10.1016/j.watres.2013.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes G, Edge TA, Gannon VPJ, Jokinen C, Lyautey E, Neumann NF, Ruecker N, Scott A, et al. Associations among pathogenic bacteria, parasites, and environmental and land use factors in multiple mixed-use watersheds. Water Research. 2011;45:5807–5825. doi: 10.1016/j.watres.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Xiong W, Li J, Chen Y, Shan B, Wang W, Zhan A. Determinants of community structure of zooplankton in heavily polluted river ecosystems. Scientific Reports. 2016;6:22043. doi: 10.1038/srep22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Ni P, Chen Y, Gao Y, Shan B, Zhan A. Zooplankton community structure along a pollution gradient at fine geographical scales in river ecosystems: The importance of species sorting over dispersal. Molecular Ecology. 2017;26:4351–4360. doi: 10.1111/mec.14199. [DOI] [PubMed] [Google Scholar]
- Yang F, Huang L, Li L, Yang Y, Mao D, Luo Y. Discharge of KPC-2 genes from the WWTPs contributed to their enriched abundance in the receiving river. Science of the Total Environment. 2017;581:136–143. doi: 10.1016/j.scitotenv.2016.12.063. [DOI] [PubMed] [Google Scholar]
- Yang Y, Deng Y, Cao L. Characterising the interspecific variations and convergence of gut microbiota in Anseriformes herbivores at wintering areas. Scientific Reports. 2016;6:32655. doi: 10.1038/srep32655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gao Y, Huang X, Ni P, Wu Y, Deng Y, Zhan A. Adaptive shifts of bacterioplankton communities in response to nitrogen enrichment in a highly polluted river. Environmental Pollution. 2019;245:290–299. doi: 10.1016/j.envpol.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li S, Gao Y, Chen Y, Zhan A. Environment-driven geographical distribution of bacterial communities and identification of indicator taxa in Songhua River. Ecological Indicators. 2019;101:62–70. doi: 10.1016/j.ecolind.2018.12.047. [DOI] [Google Scholar]
- Yang Y, Ni P, Gao Y, Xiong W, Zhao Y, Zhan A. Geographical distribution of zooplankton biodiversity in highly polluted running water ecosystems: Validation of fine-scale species sorting hypothesis. Ecology and Evolution. 2018;8:4830–4840. doi: 10.1002/ece3.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wei Y, Zheng X, Wang Y, Yu M, Xiao Q, Yu D, et al. Investigation of microbial contamination in Wenyu River of Beijing. Acta Scientiae Circumstantiae. 2012;32:9–18. [Google Scholar]
- Zeglin LH. Stream microbial diversity in response to environmental changes: Review and synthesis of existing research. Frontiers in Microbiology. 2015;6:454. doi: 10.3389/fmicb.2015.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan A, Bailey SA, Heath DD, MacIsaac HJ. Performance comparison of genetic markers for high-throughput sequencing-based biodiversity assessment in complex communities. Molecular Ecology Resources. 2014;14:1049–1059. doi: 10.1111/1755-0998.12254. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Johnson TA, Su J, Qiao M, Guo G, Stedtfeld RD, Hashsham SA, Tiedje JM. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proceedings of the National Academy of Sciences. 2013;110:3435–3440. doi: 10.1073/pnas.1222743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The high-throughput sequencing data have been deposited into NCBI Sequence Read Archive (SRA) (accession no.: PRJNA504486).