

SUMMARY
The neural substrates and pathophysiological mechanisms underlying the onset of cognitive and motor deficits in autism spectrum disorders (ASDs) remain unclear. Mutations in ASD-associated SHANK3 in mice (Shank3B−/−) result in the accelerated maturation of corticostriatal circuits during the second and third postnatal weeks. Here, we show that during this period, there is extensive remodeling of the striatal synaptic proteome and a developmental switch in glutamatergic synaptic plasticity induced by cortical hyperactivity in striatal spiny projection neurons (SPNs). Behavioral abnormalities in Shank3B−/− mice emerge during this stage and are ameliorated by normalizing excitatory synapse connectivity in medial striatal regions by the downregulation of PKA activity. These results suggest that the abnormal postnatal development of striatal circuits is implicated in the onset of behavioral deficits in Shank3B−/− mice and that modulation of postsynaptic PKA activity can be used to regulate corticostriatal drive in developing SPNs of mouse models of ASDs and other neurodevelopmental disorders.
In Brief
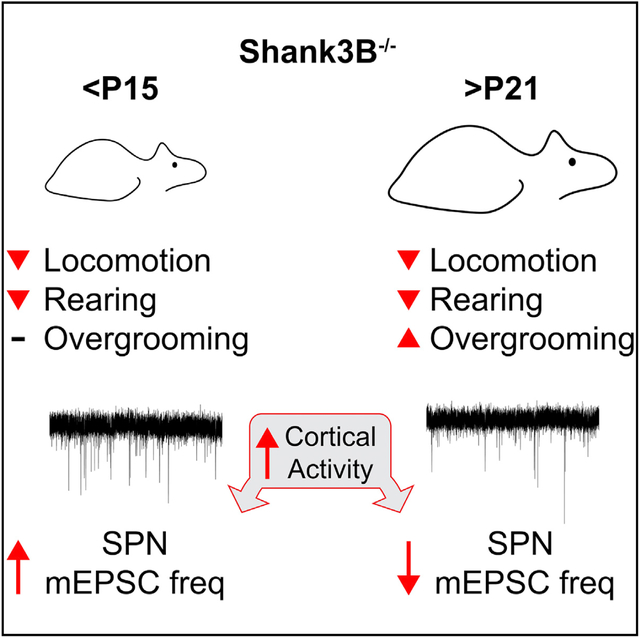
Peixoto et al. show that the onset of behavioral deficits in Shank3B−/− mice occurs during early postnatal development and that these can be ameliorated by reducing the glutamatergic synaptic drive in medial regions of the striatum by the downregulation of PKA activity.
Graphical Abstract
INTRODUCTION
Autism spectrum disorders (ASDs) are characterized by several cognitive and behavioral deficits that typically emerge during the second year of life (Lord et al., 2000; Watts, 2008). This distinctive developmental trajectory and selective degradation of particular motor and cognitive abilities suggest that ASDs arise from impaired developmental processes affecting the maturation of specific neural circuits. Recent evidence points to corticostriatal dysfunction as a common pathophysiology in ASDs (Fuccillo, 2016). Imaging studies in multiple autistic cohorts revealed hypertrophy and abnormal functional connectivity of striatal and frontal cortical regions (Langen et al., 2007, 2009, 2014; Turner et al., 2006; Wolff et al., 2013), and genomic and transcriptomic analyses have identified corticostriatal and striatal spiny projection neurons (SPNs) as neuronal populations with high expression of ASD risk genes (Chang et al., 2015; Willsey et al., 2013). In addition, autistic individuals often present classical neurological signs of basal ganglia (BG) dysfunction such as cognitive inflexibility, dystonia, bradykinesia, and stereotypies (Fuccillo, 2016; Shepherd, 2013).
Autism has a strong genetic basis and is associated with multiple de novo gene mutations and copy number variants (CNVs) (Geschwind, 2011). Several identified risk genes encode proteins involved in neuronal development and synaptogenesis (Parikshak et al., 2013; Pinto et al., 2014), consistent with the early age of onset of ASDs (Watts, 2008). Accordingly, many mouse models harboring ASD-associated mutations exhibit deficits in synaptic and neural circuit function and behavioral phenotypes reminiscent of core symptoms of autism (Dhamne et al., 2017; Jiang and Ehlers, 2013; Kabitzke et al., 2018; Kazdoba et al., 2016; Mei et al., 2016; Peça et al., 2011). However, longitudinal characterization of the neurobehavioral deficits in many of these animal models is still lacking (Del Pino et al., 2018), which, given the extensive molecular and circuit-level compensations occurring during brain development, precludes a conclusive estimate of the etiological significance of the different phenotypes observed at later developmental stages (Tien and Kerschensteiner, 2018; Turrigiano and Nelson, 2004).
Haploinsufficiency of SHANK3 is strongly associated with Phelan-McDermid syndrome (PMS) and idiopathic autism (De Rubeis et al., 2018; Monteiro and Feng, 2017). SHANK3 belongs to the SHANK family of postsynaptic scaffolding proteins that integrate an extensive protein complex at glutamatergic synapses, where they interact with multiple synaptic channels and receptors (Jiang and Ehlers, 2013). The deletion of exons 13–16 of Shank3 in mice (Shank3B−/−) leads to several behavioral deficits, including hypolocomotion, abnormal social interactions, and repetitive and anxiety-like behaviors (Dhamne et al., 2017; Guo et al., 2019; Jiang and Ehlers, 2013; Kabitzke et al., 2018; Mei et al., 2016; Peça et al., 2011; Wang et al., 2016). Adult Shank3B−/− mice have depressed corticostriatal transmission and altered patterns of cortical activity, potentially due to deficits in parvalbumin (PV) interneuron function (Dhamne et al., 2017; Gogolla et al., 2014; Guo et al., 2019; Peça et al., 2011). However, in contrast to adult stages, corticostriatal connectivity of Shank3B−/− mice during early postnatal periods is increased compared to wild type (WT), suggesting that the loss of Shank3 induces a multiphasic developmental perturbation of striatal circuit connectivity and function (Peixoto et al., 2016). Whether postnatal perturbations of corticostriatal circuitry are concurrent with and causal of the onset of behavioral abnormalities in Shank3B−/− mice remains unclear.
Here, we present a longitudinal behavioral characterization of Shank3B−/− mice, showing that many of their behavioral deficits emerge during the second and third postnatal weeks. Proteomic analyses of striatal synaptic fractions show extensive synaptic remodeling during this period, with pronounced expression changes of several ASD risk factors. The normalization of excessive glutamatergic connectivity in neurons of the medial striatum of Shank3B−/− mice by downregulating protein kinase A (PKA) activity during postnatal development reduces the severity of their phenotypes. These results suggest that disrupted postnatal development of striatal circuits is implicated in the onset of behavioral deficits in Shank3B−/− mice and highlight the need to characterize the developmental trajectory of behavioral abnormalities in animal models of autism and other neurodevelopmental disorders to identify the primary patterns of circuit dysfunctionassociated with the emergence of maladaptive behaviors.
RESULTS
Early Onset of Behavioral Deficits in Shank3B−/− Mice
To determine the developmental stage at which Shank3B−/− mice begin manifesting behavioral deficits, we tested their performance in multiple behavioral assays across development (postnatal days [P] P15–P90). In the open field, Shank3B−/− mice moved less compared to control WT littermates at all of the developmental ages tested, traversing less distance during the 30-min recording session (Figures 1A and 1B; mean ± SEM; P15–P90 WT: 20.1 ± 2, 26.4 ± 2, 29.6 ± 2.5, 36.9 ± 3.1, 32.1 ± 2.8 m; Shank3B−/−: 9.0 ± 1.3, 14.6 ± 1.8, 17.5 ± 1.4, 25.3 ± 3, 22.2 ± 1.9 m, n = 10,13; repeated measures ANOVA [RMANOVA] with Bonferroni multiple comparison test *p < 0.05, **p < 0.005, ***p < 0.001). Moreover, Shank3B−/− animals explored the center of the arena less compared to WT after P21 and spent less time in the center of arena at P15 (Figures 1C and 1D; mean ± SEM; total distance in center of arena P15–P90 WT: 4.4 ± 0.7, 6.3 ± 0.9, 6.1 ± 0.9, 12.7 ± 1.7, 11.3 ± 1.8 m; Shank3B−/−: 1.3 ± 0.2, 2.4 ± 0.5, 2.3 ± 0.3, 6.8 ± 1.0, 5.7 ± 0.7 m, n = 10,13; RMANOVA with Bonferroni multiple comparison test *p < 0.05, ***p < 0.001; mean ± SEM center/arena time P15–P90 WT: 0.07 ± 0.01, 0.14 ± 0.03, 0.14 ± 0.02, 0.47 ± 0.12, 0.45 ± 0.12 m; Shank3B−/−: 0.02 ± 0.01, 0.07 ± 0.02, 0.06 ± 0.01, 0.23 ± 0.05, 0.22 ± 0.05, n = 10,13, RMANOVA with Bonferroni multiple comparison test *p < 0.05). Reduced rearing of Shank3B−/− mice in relation to WT was observed at P15–P60 and became similar between genotypes at P90 (Figure 1E; mean ± SEM; wall rearing bouts per session P15–P90 WT: 43.6 ± 5.8, 61.2 ± 6.4, 56.6 ± 4.9, 69.3 ± 11.9, 45.8 ± 8.33 bouts; Shank3B−/−: 18 ± 3.5, 37 ± 7.2, 31.4 ± 3.2, 42.17 ± 8.9, 32.2 ± 4.5 bouts, n = 10,13; RMANOVA with Bonferroni multiple comparison test *p < 0.05). Consistent with previous reports, Shank3B−/− mice groomed for longer periods, although increased grooming only emerged during the P15–P21 stage, when the maximal phenotypic difference across genotypes was reached (Figure 1G; mean ± SEM; total grooming time P15–P90 WT: 418.6 ± 34.5, 363.4 ± 45.3, 319.5 ± 63.8, 238.2 ± 50.6, 190.6 ± 45.2 s; Shank3B−/−: 497.7 ± 72.9, 730.2 ± 59.8, 567.8 ± 47, 433.1 ± 63.7, 324.3 ± 56.6 s, n = 10,13; RMANOVA with Bonferroni multiple comparison test *p < 0.05, ***p < 0.001). The increase in total grooming time occurred predominantly due to longer duration of individual grooming bouts, which remained similar in number across groups (Figures 1G and 1H; mean ± SEM; average groom bout duration P15–P90 WT: 63.6 ± 8.3, 64.3 ± 7.2, 43.5 ± 6.5, 24.1 ± 3.3, 30 ± 3.7 s; Shank3B−/−: 69.6 ± 9.2, 96.4 ± 9.1, 62.4 ± 6.4, 52.6 ± 12.3, 64.6 ± 6.9 s, n = 10,13; grooming bouts per 30-min session P15–P90 WT: 7.05 ± 0.57, 6.05 ± 0.74, 8.7 ± 2.09, 9.25 ± 1.53, 6.0 ± 1.33 bouts; Shank3B−/−: 7.19 ± 1.2, 8.39 ± 0.56, 10.65 ± 1.39, 10.33 ± 1.13, 5.46 ± 0.67 bouts, n = 10,13; RMANOVA with Bonferroni multiple comparison test *p < 0.05). No sex dimorphism was observed in the open field tests as both male and female Shank3B−/− mice exhibited similar behavioral trends compared to WT (Figure S1), which is consistent with previous findings (Kabitzke et al., 2018). In addition, the performance of Shank3B−/− mice in the accelerating rotarod was worse compared to WT at P17–P20 (Figure 1I; mean ± SEM; time to fall in seconds P17 WT: 60.5 ± 8.6, 72.4 ± 9.1, 80.5 ± 13.4; Shank3B−/−: 37.1 ± 9, 54.8 ± 9.6, 49.9 ± 7.8; P18 WT: 76.6 ± 11, 99.6 ± 12.9, 114.6 ± 10.8; Shank3B−/−: 53.7 ± 7.7, 63.8 ± 12.1, 66.3 ± 11.7; P19 WT: 97.5 ± 11.1, 141 ± 18.3, 122.6 ± 12.4; Shank3B−/−: 71.8 ± 9, 84.2 ± 10.5, 88.9 ± 10.2; P20 WT: 113.2 ± 22.4, 151.9 ± 17.2, 172.6 ± 23.2; Shank3B−/−: 79.8 ± 9.2, 100.8 ± 16.1, 107.3 ± 14.6, n = 10,15; RMANOVA Fisher’s least significant difference [LSD] test *p < 0.05, **p < 0.005, ***p < 0.0005). Thus, behavioral abnormalities in Shank3B−/− begin during early post-natal development, a period during which extensive activity-dependent striatal circuit remodeling occurs.
Figure 1. Early Onset of Behavioral Deficits in Shank3B−/− Mice.

(A) Total distance traveled for 30 min in the open field test by P21 and P60 WT and Shank3B−/− mice. Heatmap represents distance values normalized to P60 WT.
(B and C) Mean ± SEM of distance traveled by P15–P90 WT and Shank3B−/− mice during 30 min (B) in the open field arena or (C) in the center region of the arena.
(D) Mean ± SEM of fraction of distance traveled in the center versus total arena.
(E) Mean ± SEM number of wall rearing bouts per session.
(F) Mean ± SEM total time spent grooming.
(G) Mean ± SEM duration of individual grooming bouts.
(H) Mean ± SEM number of grooming bouts per session.
(I) Mean ± SEM latency to fall in the accelerating rotarod test.
Dynamic Regulation of the Striatal Synaptic Proteome during Postnatal Development
To gain insight into the molecular mechanisms and signaling pathways regulating the postnatal maturation of striatal circuits, we analyzed synaptosomal fractions of the dorsal striatum of P8–P18 mice by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 2). The biochemical isolation of synaptosomes enriched synaptic proteins (Figure S2) and permitted the identification of 7,405 protein species that exhibited different expression patterns across development (Figures 2A–2C; Data S1). The relative abundance of several markers of synaptic and neuronal maturation such as doublecortin (DCX), synaptic vesicle glycoprotein 2 (SV2A), postsynaptic density protein 95 (PSD95), Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα), excitatory amino acid transporter 2 (EAAT2), and the myelin-associated enzyme 2′,3′-cyclic nucleotide 3′ phosphodiesterase (Cnp) changed markedly during this developmental period (Figures 2B and 2C), consistent with previous reports (Gonzalez-Lozano et al., 2016). An intra-sample comparison across development identified a cluster of 142 proteins with significant changes in relative abundance during P8–P18 (Figures 2D and 2E; Table S1; p < 0.001, Friedman test with Benjamini-Hochberg multiple test correction with p < 0.001), 92 of which formed a highly interconnected network defined by protein-protein interactions (Figure 2D). Notably, 35 of these proteins are encoded by ASD risk genes and 28 others are homologs of other identified risk alleles (https://gene.sfari.org), pointing to a high degree of functional convergence and interaction of many ASD risk factors during this stage of striatal development. Furthermore, regulatory and catalytic subunits of cyclic AMP (cAMP)-dependent PKA and modulators of PKA activity are dynamically regulated during this period (Figures 2F–2I), with a progressive reduction in the synaptic levels of PKA subunits and of several A-kinase anchoring proteins (AKAPs), similar to what is observed in developing cortical circuits (Gonzalez-Lozano et al., 2016) and consistent with the altered regulation of PKA signaling during this period of striatal maturation (Kozorovitskiy et al., 2015; Yang et al., 2009; Yasuda et al., 2003). These results point to an extensive remodeling of the striatal synaptic proteome during this developmental period, which is consistent with ongoing neuronal maturation and synaptogenesis.
Figure 2. Dynamic Regulation of PKA and ASD Risk Factors during Postnatal Striatal Development.

(A) Electron microscopy images of synaptosomal fractions isolated from mouse dorsal striatum at P8–P18.
(B) Developmental trajectory of the relative abundance of several proteins associated with synaptic and neural circuit maturation identified by LC-MS/MS.
(C) Immunoblot analysis of proteins in whole extracts or isolated synaptosome fractions of dorsal striatum from P8–P18 mice.
(D) Protein-protein interaction network of proteins of striatal synaptosomes with significant abundance change between P8 and P18. The color of the node center represents changes in protein abundance during this period with proteins (red, increased expression; blue, lower expression; log2 normalized to P8 levels). Green and orange circles around the nodes label ASD risk factors identified in SFARI gene list or closely related homologs, respectively. Nodes with numbers and letters represent the SFARI gene scoring criteria (1–5, confidence categories; S, syndromic). Edges represent protein-protein interactions pooled from Genemania, STRING, and BioGRID online databases.
(E) Proteins of striatal synaptosomes with significant change in abundance between P8 and P18 with no known protein-protein interactions. Labeling of nodes similar to that represented in (D).
(F–I) Normalized relative change in abundance from P8 to P18 of (F) catalytic and regulatory PKA subunits, (G) adenylyl-cyclases, (H) AKAPs, and (I) negative regulators of PKA signaling protein-phosphatase-1 and phosphodiesterases 1B and 10A.
Developmental Switch in SPN Adaptation to Cortical Hyperactivity
Compared to WT controls, Shank3B−/− SPNs have an elevated frequency of miniature excitatory postsynaptic currents (mEPSCs) at P14, suggesting an increase in the number, or accelerated maturation, of excitatory synapses (Peixoto et al., 2016). However, in adult stages, this phenotype is reversed, and corticostriatal transmission and connectivity are reduced (Peça et al., 2011; Wang et al., 2017). The extensive postnatal changes in striatal synaptic composition suggested that these two phenotypes could be due to distinct plasticity mechanisms induced by elevated cortical activity in developing and mature SPNs. To test this hypothesis, we recorded mEPSCs in SPNs after different periods of cortical hyperactivity induced across development (Figure 3). To increase cortical activity, we blocked synaptic GABA release from cortical interneurons by conditional deletion of the vesicular GABA transporter (VGAT) in Slc32a1f/f transgenic mice (Peixoto et al., 2016). Unilateral injection of adeno-associated viruses (AAVs) expressing Cre recombinase under control of the neuron-specific synapsin promoter (AAV-Syn-Cre-EGFP) allowed restricted Cre recombination in neurons of the anterior cingulate cortex (ACC), a major source of corticostriatal projections to dorsomedial striatum (DMS) (Figure 3A). Mice were injected at P8 to improve stereotaxic precision and avoid disrupting GABA-mediated developmental processes during the first postnatal week. AAV-Syn-EGFP injections were used as controls. Whole-cell recordings of DMS SPNs at P14 (7 days post-injection) revealed that cortical hyperactivity during P8–P14 significantly increased SPN mEPSC frequency (Figure 3B; mean ± SEM; SPN mEPSC frequency P14, GFP 0.71 ± 0.07 Hz; Cre 1.39 ± 0.16 Hz, n = 27,26 neurons from 4 mouse pairs, unpaired Mann-Whitney test p = 0.0002) and mEPSC amplitude (Figure 3C; mean ± SEM; SPN mEPSC amplitude P14, GFP 18.41 ± 0.59 pA; Cre 20.93 ± 0.82 Hz, unpaired Mann-Whitney test p = 0.039). However, VGAT deletion from P8 to P60 resulted in a reversed phenotype such that the SPN mEPSC frequency of AAV-Cre-EGFP mice was reduced compared to controls (Figure 3D; mean ± SEM; SPN mEPSC frequency P60, GFP 2.37 ± 0.25 Hz; Cre 1.89 ± 0.20 Hz, n = 52, 54 neurons from 5 mouse pairs, Mann-Whitney test p = 0.033). mEPSC amplitude remained elevated compared to SPNs in AAV-GFP-injected control mice (Figure 3E; mean ± SEM; SPN mEPSC amplitude P60, GFP 14.66 ± 0.33 pA; Cre 15.97 ± 0.43 Hz, unpaired Mann-Whitney test p = 0.036).
Figure 3. Developmental Switch in SPN Response to Cortical Hyperactivity.

(A) Coronal brain section of Slc32a1f/f mouse injected with AAV-Cre-EGFP in ACC. Scale bar, 1 mm. CTX, cortex; STR, striatum.
(B) Whole-cell voltage-clamp recordings in SPNs of DMS in acute brain slices of P14 or P60 Slc32a1f/f mice injected at P8 with AAV-EGFP (Ctrl) or AAV-Cre-EGFP (Cre) in the ACC.
(C and D) Mean ± SEM of AMPAR mEPSC frequency (left) and amplitude (right) of (C) P14 or (D) P60 SPNs of Slc32a1f/f mice injected at P8.
(E) Experimental diagram depicting whole-cell voltage-clamp recordings in SPNs of DMS in acute brain slices of P21, P28, or P60 Slc32a1f/f mice injected with AAV-EGFP (Ctrl) or AAV-Cre-EGFP (Cre) 7 days before recordings. Bottom traces are representative mEPSC recordings performed at P21 of mice injected at P15.
(F–H) Mean ± SEM of AMPAR mEPSC frequency (left) and amplitude (right) of (F) P21, (G) P28, and (H) P60 SPNs of Slc32a1f/f mice injected 7 days before recordings.
(I and J) Normalized values of SPN AMPAR (I) mEPSC frequency and (J) mEPSC amplitude of AAV-Cre-injected animals compared to AAV-EGFP controls across development. Note the developmental shift in the change of mEPSC frequency after AAV-Cre-EGFP injections at ~P15.
(K) Example traces of eEPSCs in SPNs of P60 Slc32a1f/f mice injected with AAV-EGFP (Ctrl) or AAV-Cre-EGFP (Cre) in response to paired electrical pulses with 50 ms inter-stimulus interval (ISI).
(L) Mean ± SEM ratio of eEPSC amplitude in response to the two electrical stimuli in (K).
To identify at which developmental stage cortical hyperactivity starts inducing a reduction in SPN mEPSC frequency, we performed similar experiments manipulating cortical activity at different developmental time points (Figure 3E). In contrast to injections at P8, injections of AAV-Cre at P15, P21, or P53 resulted in a lower mEPSC frequency 7 days post-injection (Figures 3F–3H, left plot; mean ± SEM; SPN mEPSC frequency P15–P21 GFP 3.64 ± 0.46 Hz; Cre 2.29 ± 0.26 Hz, n = 18,19 neurons from 3 mouse pairs; P21–P28 GFP 4.46 ± 0.74 Hz; Cre 2.5 ± 0.35 Hz, n = 18,18 neurons from 3 mouse pairs; P53–P60 GFP 2.75 ± 0.25 Hz; Cre 1.92 ± 0.20 Hz, n = 30,30 neurons from 4 mouse pairs; Mann-Whitney test *p < 0.05, **p < 0.01) with an increase in mEPSC amplitude observed only in the P53–P60 group (Figures 3F–3H, right plot; mean ± SEM; SPN mEPSC amplitude: P15–P21 GFP 14.79 ± 0.33 pA; Cre 14.75 ± 0.38 Hz, n = 18,17 neurons from 3 mouse pairs; P21–P28 GFP 15.29 ± 0.59 pA; Cre 14.83 ± 0.36 Hz, n = 18,18 neurons from 3 mouse pairs; P53–P60 GFP 13.28 ± 0.21 pA; Cre 14.27 ± 0.33 Hz, n = 30,30 neurons from 4 mouse pairs, unpaired Mann-Whitney test *p < 0.05). There was no significant difference in the pair-pulse ratio (PPR) of electrically evoked EPSCs (eEPSC) in SPNs of AAV-Cre-EGFP-injected animals at P53 compared to AAV-GFP-injected controls (Figures 3K and 3L; mean ± SEM; eEPSC PPR, AAV-GFP 1.09 ± 0.04; AAV-Cre-EGFP 1.01 ± 0.07, n = 14,12 neurons from 3 mouse pairs, unpaired t test p = 0.37), suggesting the decrease in mEPSC frequency is likely not caused by lower vesicular release probability. These results indicate that cortical hyperactivity induces distinct forms of synaptic plasticity in developing and mature SPNs, consistent with previous ex vivo studies in acute slices (Partridge et al., 2000).
PKA Inhibition in SPNs during Postnatal Development Reduces Corticostriatal Connectivity
Postnatal maturation of glutamatergic synapses in SPNs is regulated by PKA activity (Kozorovitskiy et al., 2015), but whether postsynaptic PKA signaling is required for this process remains unclear. To address this, we infected the striata of P8 mice with AAVs expressing protein kinase inhibitor peptide (PKIα) under the neuron-specific synapsin promoter (AAV8-hSyn-PKIα-IRES-mRuby2 [internal ribosome entry site, IRES]) and performed whole-cell recording in PKIα+ and neighboring uninfected SPNs 7 days post-injection (Figures 4A and 4B). PKIα expression decreased the SPN mEPSC frequency (Figures 4C and 4D; mean ± SEM; control SPN 1.28 ± 0.17 Hz, PKIα SPN 0.64 ± 0.07 Hz, n = 19,23 neurons from 5 mice, unpaired Mann-Whitney test p = 0.001), with no significant change in mEPSC amplitude (Figures 3C and 3E; mean ± SEM; control SPN 14.5 ± 0.5 pA, PKIα SPN 13.9 ± 0.4 pA, n = 19,23 neurons from 5 mice, unpaired Mann-Whitney test p = 0.21). PKIα expression increased SPN input resistance (Ri) and induced a small reduction in membrane capacitance (Figure S3; mean ± SEM; Ri: control SPN 173.2 ± 15.52 MΩ; PKIα SPN 315.7 ± 27.49 MΩ; unpaired Mann-Whitney test p < 0.0001; membrane capacitance: control SPN 51.3 ± 2.3 pF; PKIα SPN 47.5 ± 2.2 pF, n = 19,23 neurons from 5 mice). To determine whether PKIα expression specifically reduces corticostriatal connectivity, we compared optogenetically evoked postsynaptic currents (oEPSC) in PKIα+ and control SPNs of Rbp4-Cre;Ai32 (Rbp4-Cre;ChR2f/f) transgenic mice that express Cre recombinase under the retinal binding protein 4 (Rbp4) promoter, restricting channelrhodopsin-2 (ChR2) expression predominantly to layer 5 cortical neurons (Figure 4E). The optical stimulation of ChR2+ fibers in DMS was induced by 1-ms pulses of 473 nm light delivered through the objective while performing voltage-clamp whole-cell recordings in SPNs. α-Amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)-type and N-methyl-D-aspartate (NMDA)-type glutamate receptor-mediated currents were isolated by inhibiting GABAA receptors and recording at holding potentials (Vh) of −70 and +40 mV, respectively. The expression of PKIα from P8 to P14 strongly reduced Rbp4+ AMPA receptor (AMPAR) oEPSC amplitude (Figures 4F and 4G; mean ± SEM; control −956.1 ± 187.9; PKIα −416 ± 84.7 pA; n = 13,16 neurons from 3 mice, unpaired Mann-Whitney test p = 0.013) without a significant change in the NMDAR/AMPAR current amplitude ratio (Figure 4H; mean ± SEM; control 1.42 ± 0.16; PKIα 1.55 ± 0.15; n = 9,10 neurons from 3 mice, unpaired Mann-Whitney test p = 0.28).
Figure 4. Postnatal Inhibition of PKA Reduces Glutamatergic Synapse Maturation in DMS SPNs.

(A) Coronal brain slice of P15 mouse infected with AAV8-Syn-PKIα-mRuby2 in DMS at P8. The inset shows a magnified view of the striatum with PKIα+ and neighboring uninfected (control) SPNs. Scale bar, 1 mm. CTX, cortex; STR, striatum.
(B) Representative mESPC recordings from DMS control or PKIα+ SPNs at P15.
(C and D) Mean ± SEM (C) mEPSC frequency and (D) mEPSC amplitude in control or PKIα+ DMS SPNs.
(E) Coronal brain slice of P15 Rbp4-Cre;ChR2-EYFPf/f mouse infected with AAV8-Syn-PKIα-mRuby2 in DMS at P8. Scale bar, 1 mm. CTX, cortex; STR, striatum.
(F) Example AMPAR and NMDAR oEPSCs recorded in control and PKIα+ DMS SPNs in response to 1-ms 473 nm light pulses.
(G) Mean ± SEM. AMPAR oEPSC amplitude in control and PKIα+ DMS SPNs.
(H) Mean ± SEM ratio of NMDAR/AMPAR oEPSC amplitude in control and PKIα+ DMS SPNs.
Postnatal Expression of PKIα in DMS Neurons Ameliorates Behavioral Abnormalities in Shank3B−/− Mice
Repeated corticostriatal stimulation in ventromedial striatum generates persistent overgrooming in mice (Ahmari et al., 2013), whereas reduction of corticostriatal connectivity in individuals with obsessive-compulsive disorder (OCD) significantly improves compulsive behaviors (Dunlop et al., 2016). Moreover, focal disinhibition of striatal circuits in mice causes dystonia and motor stereotypies (Bronfeld et al., 2013; Israelashvili and Bar-Gad, 2015), suggesting that hyperactivity of striatal circuits may underlie core behavioral deficits in ASDs (Fuccillo, 2016). Given the critical role of PKA in regulating SPN glutamatergic synapse maturation (Kozorovitskiy et al., 2015), we tested whether reducing the excessive postnatal striatal connectivity found in Shank3B−/− mice by postsynaptic inhibition of PKA would decrease the severity of their behavioral deficits. We expressed PKIα in medial striatal neurons from P8 to P21 by injecting AAV8-hSyn-PKIα-IRES-mRuby2 (Chen et al., 2017; Lee et al., 2019) and compared behavioral performance with control AAV-GFP-injected littermates at P21. As observed in WT SPNs, expression of PKIα in Shank3B−/− SPNs during postnatal development reduces both mEPSC frequency (Figures 5A and 5B; mean ± SEM; control 1.54 ± 0.14 Hz; PKIα 0.51 ± 0.09 Hz; n = 17,16, unpaired Mann-Whitney test p < 0.0001) and amplitude (Figure 5C; mean ± SEM; control 13.9 ± 0.3 pA; PKIα 12.7 ± 0.4 pA; n = 17,16 neurons from 3 mouse pairs, unpaired Mann-Whitney test p = −0.023). Expression of PKIα in medial striatum had a profound impact on behavior, resulting in overall decreased grooming time and grooming bout duration (Figures 5D–5G; mean ± SEM; total grooming time: control 481.8 ± 62, PKIα 320.4 ± 34 s; groom bout duration: control 108.4 ± 14.5, PKIα 76.2 ± 5.8 s; groom bout number per session: control 4.9 ± 0.5, PKIα 4.2 ± 0.4; n = 16,16 mice, unpaired t test *p < 0.05). Moreover, striatal PKIα expression increased locomotor activity, increasing the distance traversed in total and in the arena center, as well as the number of wall rearings (Figures 5H–5L; mean ± SEM; total distance in arena: control 14.9 ± 1.7, PKIα 25 ± 2.5 m; center:arena distance ratio: control 0.19 ± 0.02, PKIα 0.24 ± 0.01; total time spent in center of arena: control 188.4 ± 39.5, PKIα 0.24 ± 0.01; number of wall rearings per session: control 37.9 ± 4.2, PKIα 64.8 ± 7.8; n = 16,16 mice, unpaired t test *p < 0.05, **p < 0.005), both metrics being associated with increased exploratory activity and reduced anxiety in mice. By contrast, PKIα expression in the medial striatum of WT mice had no influence on animal behavior in the open field (Figure S4).
Figure 5. Postnatal Expression of PKIα in DMS Rescues Behavioral Abnormalities in Shank3B−/− Mice.

(A) Representative mESPC recordings from control or PKIα+ DMS SPNs of Shank3B−/− mice at P13.
(B and C) Mean ± SEM (B) mEPSC frequency and (C) mEPSC amplitude in control or PKIα+ DMS SPNs.
(D) Coronal section of P21 Shank3B−/− mouse infected with AAV8-Syn-PKIα-mRuby2 in DMS and ventromedial striatum (VMS) at P8. Scale bar, 1 mm.
(E) Mean ± SEM total grooming time of P21 Shank3B−/− mice expressing GFP or PKIα in the open field.
(F and G) Mean ± SEM of (F) duration and (G) number of grooming bouts per session.
(H) The total distance traveled for 20 min in the open field test by P21 Shank3B−/− mice injected with AAV-EGFP or AAV-PKIα. Heatmap represents distance normalized to AAV-PKIα.
(I) Mean ± SEM of total distance traveled in arena.
(J) Mean ± SEM of the ratio of the distance traveled in the center of the arena over the total distance traveled.
(K) Mean ± SEM of time spent in the center of the arena.
(L) Mean ± SEM number of wall rearing bouts per session.
DISCUSSION
Despite increasing knowledge of the genetic underpinnings of autism risk, the specific neural substrates and pathophysiological mechanisms underlying the emergence of motor and cognitive deficits in ASDs remain unclear. Studies in transgenic mouse models have begun to address how different genetic mutations associated with ASDs disrupt neuron physiology, circuit organization, and animal behavior (Fuccillo, 2016; Kazdoba et al., 2016). However, many of these studies often focused on the characterization of adult animals, which, given the extensive molecular and circuit-level adaptations occurring during brain development, limits our understanding of the etiological significance of the phenotypes observed during these adult stages (Tien and Kerschensteiner, 2018; Turrigiano and Nelson, 2004). Here, by longitudinally tracking mouse behavior, we found that several of the behavioral deficits previously reported in adult Shank3B−/− mice are already observed at P15–P21. This early onset indicates that the relevant pathogenic processes implicated in the emergence of maladaptive behaviors in these mice occur either during or before this developmental stage. Notably, the ~P12–P14 period of brain maturation in the mouse is homologous to the ~1–2 years of human development (Semple et al., 2013), which is the typical age of symptom onset in ASDs. The emergence of behavioral deficits during this particular period strengthens the face validity of Shank3B−/− mice as a model system of ASD pathogenesis and highlights the need to characterize early postnatal circuit maturation in mouse models of autism and other neurodevelopmental disorders to distinguish primary pathophysiological mechanisms from secondary developmental adaptations.
Whereas deficits in locomotor and exploratory behavior in Shank3B−/− mice were observed as early as P15, overgrooming only emerged during P15–P21. This slightly delayed onset is consistent with previous reports showing unaltered grooming patterns of Shank3B−/− mice at P15 (Kabitzke et al., 2018) and indicates that this maladaptive behavioral pattern is established during the third postnatal week, a period of extensive functional maturation of striatal circuits (Peixoto et al., 2016; Tepper et al., 1998). Moreover, this developmental trajectory of overgrooming resembles the gradual emergence of repetitive and ritualistic patterns of behavior in autistic children during the first years of life (Goldman and Greene, 2013; Wolff et al., 2014) and may account for the conflicting reports of the extent of overgrooming in Shank3B−/− mice across different studies (Dhamne et al., 2017; Kabitzke et al., 2018; Peça et al., 2011). By contrast, the early onset of hypolocomotion in Shank3B−/− mice is consistent with the observed hypotonia in infants diagnosed with PMS (De Rubeis et al., 2018). However, no early deficits in geotaxis were found in Shank3B−/− pups during the first 2 postnatal weeks, suggesting that this early phenotype is likely due to central and not peripheral motor deficits (Kabitzke et al., 2018). In addition, we observed no sexual dimorphism in the behavior of <P21 Shank3B−/− mice, which is consistent with previous reports (Kabitzke et al., 2018) and potentially due to sexual immaturity.
Proteomic analysis of striatal synaptosome fractions revealed extensive remodeling of synaptic protein composition from P8 to P18 (Figure 2). The expression of DCX, a microtubule-associated protein involved in dendritic growth that is downregulated as synaptic connectivity is stabilized (Donato et al., 2017), decreased markedly during this period, consistent with the extensive activity-dependent maturation of striatal synapses (Peixoto et al., 2016). Of the 140 proteins with the most pronounced developmental expression changes, 92 form a cluster of identified protein-protein interactions. Notably, 35 of the 92 are products of identified ASD risk genes and 28 others of closely related homologs. These results point to convergent and dynamic regulation of several ASD risk factors during this particular period of striatal development, which may have important implications for understanding how genetic variants of limited penetrance interact and contribute to ASD pathogenesis (Geschwind, 2011; Klei et al., 2012). Furthermore, we detected a developmental reduction in both the catalytic and regulatory subunits of PKA and an increase in the negative regulators of PKA activity from P8 to P18, suggesting a change in PKA signaling regulation during this period. Induction of long-term potentiation (LTP) in hippocampal CA3-CA1 synapses is PKA dependent before but not after P14 (Yasuda et al., 2003), whereas mice lacking the PKA regulatory subunit Prkar2b show impaired hippocampal LTP at P10–P14 but not P21–P28 (Yang et al., 2009). Our data show a pronounced reduction in Prkar2b expression in striatal synapses from P8 to P18, and in AKAPs that bind and recruit Prkar2b such as AKAP12 (Havekes et al., 2012) and the ASD-associated neurobeachin (Wang et al., 2000). Moreover, we have previously shown that in vivo systemic administration of PKA inhibitor H89 abrogates the activity-dependent potentiation of SPN corticostriatal synapses at P8–P9 (Kozorovitskiy et al., 2015), suggesting a critical role for PKA signaling in early SPN synaptic plasticity. In addition, caged-glutamate-induced spinogenesis in both striatal SPN and cortical pyramidal neurons is also blocked by H89 and shows a progressive developmental decrease in induction efficiency up to ~P18, after which point it is no longer inducible (Kozorovitskiy et al., 2015; Kwon and Sabatini, 2011). Here, we extend these results and show that the postsynaptic inhibition of PKA in SPNs during P8–P15 greatly reduces corticostriatal transmission. PKIα expressing SPNs also exhibited higher membrane resistance, suggesting that PKA signaling may also regulate the maturation of SPN-intrinsic properties, which is consistent with the dopamine-dependent maturation of SPN excitability described recently (Lieberman et al., 2018).
In Shank3B−/− mice corticostriatal connectivity is increased during postnatal development and decreased during adult stages (Peça et al., 2011; Peixoto et al., 2016; Wang et al., 2017). The mechanisms underlying this biphasic developmental trajectory remain unclear, but reducing cortical activity during early postnatal periods in Shank3B−/− mice normalizes the number of excitatory synapses onto SPNs. The loss of Shank3 also increases cortical (Dhamne et al., 2017) and striatal (Wang et al., 2016) activity during adult stages and upregulates the expression and post-translational processing of activity-dependent proteins such as P-CREB (Amal et al., 2018). Based on this evidence, we hypothesized that the different SPN mESPC phenotypes observed in young and adult Shank3B−/− mice may reflect distinct adaptations of these neurons to cortical hyperactivity during different developmental periods. Consistent with this idea, both chronic and transient elevations of cortical activity during adulthood decrease SPN mEPSC frequency (Figures 3D and 3H). This reduction in glutamatergic transmission in response to elevated cortical activity is in line with the induction of long-term depression (LTD) of striatal synapses by high-frequency stimulation (HFS) in ex vivo slice preparations (Kreitzer and Malenka, 2008). This potential mechanism may also explain why the overall decrease in mEPSC frequency observed in adult Shank3B−/− SPNs is predominantly due to the loss of inputs in striatopallidal SPNs (Wang et al., 2017), as some studies reported more robust induction of HFS-LTD in this subclass of SPNs compared to striatonigral SPNs (Kreitzer and Malenka, 2008). Nevertheless, we found that increased cortical activity before ~P15 increases SPN mEPSC frequency instead (Figure 3I). This developmental switch is consistent with the transition from HFS-induced LTP to LTD in rat corticostriatal synapses ex vivo at ~P15–P16 (Partridge et al., 2000) and may underlie the distinct SPN mEPSC phenotypes observed in developing versus adult Shank3B−/− mice. The mechanisms underlying these different developmental responses remain unclear, but developmental changes in synaptic composition, intrinsic excitability, and neuromodulatory signals (Kozorovitskiy et al., 2015; Lieberman et al., 2018; Peixoto et al., 2016; Tepper et al., 1998; Dehorter et al., 2011; Krajeski et al., 2019) may alter spike-timing-dependent plasticity rules in corticostriatal synapses, similar to what is observed during the development of cortical circuits (Kimura and Itami, 2019).
Postsynaptic expression of PKIα in striatal neurons during postnatal development ameliorated the severity of multiple behavioral deficits in Shank3B−/− mice. It is important to note that adult restoration of Shank3 expression or chemogenetic increase of striatopallidal SPN excitability in Shank3B−/− mice can also normalize some abnormal behaviors such as over-grooming (Mei et al., 2016; Wang et al., 2017). However, such manipulations had no effect on hypolocomotion and rearing, two of the most robust and replicable phenotypes in Shank3 knockout (KO) animals (Dhamne et al., 2017; Kabitzke et al., 2018; Mei et al., 2016; Peça et al., 2011; Wang et al., 2016). The strategy reported here is, to our knowledge, the first manipulation that effectively improved these behavioral parameters in Shank3B−/− mice. It will be important for future studies to identify the specific patterns of striatal circuit malformation induced by early cortical hyperactivity and to address whether early insults during this postnatal period result in irreversible circuit dysfunction. The increased activity of medial striatal circuits is associated with overgrooming in mice (Ahmari et al., 2013; Burguière et al., 2013), and structural and functional deficits between frontal cortical regions and DMS have been found in multiple autistic individuals (Langen et al., 2007, 2009, 2014; Turner et al., 2006; Wolff et al., 2013). The behavioral improvements observed in response to PKIα expression in medial striatum may thus be due to a reduction in frontal cortical-DMS corticostriatal activity or connectivity. However, it is important to note that medial striatal regions also receive extensive inputs from the thalamus, the basolateral amygdala (BLA), and the bed nucleus of the stria terminalis (BNST) (Lago et al., 2017; Smith et al., 2016; Wall et al., 2013). Although it remains unclear how the development of these subcortical projection systems is regulated by early neural activity, the positive effect of PKIα expression may be due to the downregulation of these inputs. Alternatively, a net reduction in the excitatory drive into medial SPN populations may normalize the activity converging onto BG output nuclei and allow for proper integration of sensorimotor information processed by lateral striatal circuits (Hintiryan et al., 2016; Shepherd, 2013). Indeed, excessive BG activity is associated with motor deficits in numerous movement disorders (Wichmann and Dostrovsky, 2011), and surgical ablation of BG output nuclei often results in marked behavioral improvements in some of these conditions (Lozano et al., 2018). By contrast, no available treatments exist for ASDs and all of the clinical trials performed to date have had minimal success (Jeste and Geschwind, 2016). PKA activity is modulated by a myriad of upstream signaling pathways that are differentially expressed and regulated in different neuronal populations across development (Greengard, 2001). The modulation of PKA signaling thus offers the potential for cell-type-specific tuning of synaptogenesis, providing a potential avenue for correcting the abnormal developmental trajectories of striatal circuits in neurodevelopmental disorders with early imbalances in neural activity.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rui Peixoto (rup14@pitt.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experimental manipulations on mice were performed in accordance with protocols approved by the Harvard Standing Committee on Animal Care and guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were housed on a 12/12hr light/dark cycle with chow and water provided ad libitum. Mice were weaned at P21–23 and separated by sex in cages of 2–4 animals of mixed genotypes. For electrophysiology studies, Rbp4-Cre transgenic mice (GENSAT #RP24–285K21) were bred to conditional channelrhodopsin-2 (ChR2) expressing mice expressing ChR2(H134R)-EYFP under control of an upstream loxP-flanked STOP cassette (Ai32; referred to as ChR2f/f; The Jackson Laboratory #012569). For conditional deletion of the vesicular GABA transporter in cortical cells we used Slc32a1f/f mice (The Jackson Laboratory #012897). Shank3B−/− knock-out mutant mice were described previously (Peça et al., 2011) and obtained from The Jackson Laboratory (#017688). Longitudinal characterization of Shank3B−/− mouse behavior was performed in age-matched littermates from breeding pairs of Shank3B+/− heterozygous animals. For PKIα rescue experiments described in Figure 5 Shank3B−/− mice were bred in a homozygous scheme to increase cohort size. Mixed cohorts of male and female mice were used in all experiments as no sexual dimorphism was ever detected in the mouse lines used and behavioral assays tested (Figure S1) as reported in previous studies (Kabitzke et al., 2018).
METHOD DETAILS
Behavioral assays
For all behavioral assays, mice were habituated to the test room for at least one hour prior to experiments. For tests in pre-weaned mice (P15-P21) the cage with the dam and the litter was taken to the testing room where pups were taken out of the cage and placed in a new cage with water and food available until all pups were tested. Dams remained in the home cage for the duration of the experiment. Open field test was performed in a 22×22cm plexiglass chamber with white opaque walls (center of arena - 13×13cm inner region). Uniform illumination was kept consistent across experiments with 1500lux 5500k diffused light (Genaray Octalux OLED-55). Behavior was video recorded with a CCD camera (Thorlabs DCU22M) at 22fps using Thorcam software.
Stereotaxic intracranial injections
Intracranial injections were performed in P8, P15, P21 or P53 mice anesthetized with isoflurane placed into a stereotaxic apparatus (Kopf Instruments model 1900). Viruses were delivered by injecting 50–200nl at a maximum rate of 100 nl/min using a Harvard Apparatus PHD ULTRA CP injector. For deletion of VGAT in cortical neurons, virus expressing Cre-EGFP under the neuron specific Synapsin promoter (AAV9-hSyn-Cre-EGFP, Penn vector core AV-9-PV1848) were injected in Slc32a1f/f mice using coordinates: AP +1.5 mm; ML +0.6 mm; Depth −1 mm from dura. For PKIα infection of striatal neurons, WT and Shank3B−/− P8 mice were injected bilaterally with AAV8-hSyn-PKI-ires-mRuby or AAV9-hSyn-EGFP (Penn vector core AV-9-PV1696) using the following coordinates: AP +0.5 mm; ML +1.25 mm; Depth −2.5 mm from dura. Following injections and wound closure, mice received ketoprofen for analgesia and were returned to home cages for at least 7 days. Injection sites and viral expression were confirmed post hoc by whole brain sectioning and imaging and mice with erroneous injections were discarded from analysis.
Acute slice preparation and electrophysiology
Acute brain slices and whole-cell recordings from SPNs were performed using standard methods as described previously (Kozorovitskiy et al., 2012). Briefly, mice (6–60 days old) were anesthetized by isofluorane inhalation and perfused transcardially with ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4 and 25 glucose (310 mOsm per kg). Cerebral hemispheres were removed and sliced in cold ACSF (275um coronal slices in Leica VT1200S vibratome). Coronal slices containing striatum were recovered for 15–20 minutes at 34°C in choline-based recovery solution (in mM): 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 25 glucose, 11.6 ascorbic acid, and 3.1 pyruvic acid), and then transferred to a holding chamber with 34°C ACSF that progressively cooled down to room temperature (20–22°C). All recordings were obtained within 1–6 h after slicing and solutions were constantly bubbled with 95% O2/5% CO2. Individual slices were transferred to a recording chamber mounted on an upright microscope (Olympus BX51WI) and continuously perfused (1–2 mL per minute) with ACSF at room temperature. Cells were visualized using a 40 × water-immersion objective with infrared DIC optics. Whole-cell voltage-clamp recordings were made from SPNs in dorsomedial regions of striatum. Patch pipettes (2–4 MU) pulled from borosilicate glass (BF150–86-7.5, Sutter Instruments) were filled with a Cs+-based internal solution containing (in mM): 130 CsMeSO4, 10 HEPES, 1.8 MgCl2, 4 Na2ATP, 0.3 NaGTP, and 8 Na2-phosphocreatine, 10 CsCl2, 3.3 QX-314 (Cl− salt), (pH 7.3 adjusted with CsOH; 295 mOsm per kg). For all voltage-clamp experiments, errors due to voltage drop across the series resistance (< 20 MΩ) were left uncompensated. In all experiments, GABAR currents were blocked with 20 μM SR95531 hydrobromide to block inhibition from local interneuron or SPNs collaterals. For optogenetic light-evoked AMPAR oEPSC recordings ACSF contained 10 μM (R)-CPP to block NMDA currents and recordings were performed at −70 mV holding potential. For mEPSC recordings, ACSF also included 1 μM TTX. Voltage-clamp recordings were performed at room temperature (20–22°C). To activate ChR2-expressing neuronal fibers, light from a 473 nm laser (Opto engine LLC) was focused on the back aperture of the microscope objective to produce wide-field illumination of the recorded cell. TTL triggered pulses of light (1 ms duration; 6 mW/mm2 under the objective) were delivered at the recording site at 30 s inter-stimulus intervals. To calculate paired-pulse ratio (PPR), recordings were performed in current-clamp mode using a K+ based low chloride internal solution composed of (in mM) 130 KMeSO3, 3 KCl, 10 HEPES, 1 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 8Na2-phosphocreatine (pH 7.3 adjusted with KOH; 295 mOsm). Current-clamp recordings were perfomed at 32°C and electrical stimulation was performed with bipolar tungsten electrodes inserted in the callosal tract dorsal to DMS. Membrane currents and potentials were amplified and low-pass filtered at 3 kHz using Multiclamp 700B amplifier (Molecular Devices), digitized at 10 kHz and acquired using National Instruments acquisition boards and a custom version of ScanImage written in MATLAB (Mathworks).
Brain tissue processing and imaging
Mice were deeply anesthetized with isofluorane and perfused transcardially with 4% paraformaldehyde in 0.1 M sodium phosphate buffer. Brains were fixed for 24 hours at 4°C, washed in phosphate buffer saline (PBS) and sectioned (50 μm) coronally using a vibratome (Leica VT1000s). Brain sections were mounted on glass slides, dried and mounted with ProLong antifade reagent containing DAPI (Molecular Probes). Whole brain sections were imaged with an Olympus VS110 slide-scanning microscope.
Biochemical isolation of synaptosomes
Mice of different ages were anesthetized with isoflurane and perfused with ice cold Hank’s Balanced Salt solution (HBSS). Brains were immediately extracted and dorsal striatal regions were dissected in ice cold HEPES buffered sucrose buffer (0.32 M sucrose, 4 mM HEPES pH 7.4) containing protease inhibitor cocktail (Roche) and 2mM EDTA. Striatal tissue from 6 mice (3M/3F) was pooled in 10 volumes of HEPES-buffered sucrose and homogenized in a motor driven glass-teflon homogenizer at ~900 rpm (12 strokes). Homogenate was centrifuged at 1000 g at 4°C for 10 min to remove the pelleted nuclear fraction and supernatant was centrifuged at 15,000 g for 15 min to yield the crude synaptosomal pellet. This pellet was resuspended in 1 mL of HEPES-buffered sucrose buffer, layered on top of a discontinuous gradient containing 5mM HEPES and 0.8 to 1.0 to 1.2 M sucrose (top to bottom; equals 27%/34%/41%) in a ultracentrifuge tube, and centrifuged at 150,000 g for 2 hr in a swinging bucket rotor. Synaptosomes were recovered at the interface of 1.0 and 1.2 M sucrose layers, diluted to 0.32 M sucrose by adding 2.5 volumes of 5 mM HEPES pH 7.5 and pelleted by centrifugation at 15,000 g for 20 min.
Immunoblotting and Electron Microscopy
For immunoblotting, whole brain extracts and synaptosomes were normalized for total protein content using the BCA Assay (Pierce). Following normalization, samples were denatured by the addition of 5 × Laemmli sample buffer (237 mM Tris-HCl, pH 6.8, 10% (wt/vol) SDS, 25% (vol/vol) 2-mercaptoethanol, 25% (vol/vol) glycerol, and 0.056% (wt/vol) bromophenol blue). For AKAP12 blots, aliquots of denatured synaptosomes were boiled for 5 minutes. All samples were resolved by 8%–16% SDS-PAGE, and analyzed by immunoblotting. Antibodies used in this study include anti-DCX (Santa Cruz sc-271390), anti-SV2A (SySy 119002), anti-AKAP12 (Abcam ab49849), anti-Prkacb (Sigma-Aldrich sab2104516), anti GAPDH (CST 2118S), anti-Synaptotagmin (SySy 105011), anti-GluN1 (SySy 114011), anti-VDAC (CST 4661S), anti-Calreticulin (CST 12238S). For electron microscopy, synaptosomes were fixed overnight in 1.25% paraformaldehyde, 2.5% glutaraldehyde, 0.03% picric acid, 0.1M Cacodylate buffer, pH 7.4. Fixed synaptosomes were pelleted and washed in 0.1M cacodylate buffer and postfixed with 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 hour. Samples were then washed twice in water, once in Maleate buffer (MB) and incubated in 1% uranyl acetate in MB for 1 hr. This was followed by two washes in water and subsequent dehydration for 10 minutes each in the following grades of alcohol: 50%, 70%, 90%, 2×10min 100%. The samples were then placed in propyleneoxide for 1 hr and infiltrated overnight in a 1:1 mixture of propyleneoxide and TAAB Epon (Marivac Canada Inc. St. Laurent, Canada). The following day the samples were embedded in TAAB Epon and polymerized at 60C for 48 hr. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, transferred to copper grids stained with lead citrate, and visualized in a JEOL 1200EX Transmission electron microscope or a TecnaiG2 Spirit BioTWIN. Images were recorded with an AMT 2k CCD camera.
ERLIC separation
Proteomic analysis of striatal synaptosomes was performed by TMTplex experiment using samples of different ages (P8, P12, P14, P18). Each sample contained a pool of dorsal striatal tissue collected from 6 mice (3M/3F) at the different ages. Each sample was submitted for single LC-MS/MS experiment that was performed on a LTQ Orbitrap Elite (Thermo Fischer) equipped with Waters (Milford, MA) NanoAcquity HPLC pump. Peptides were separated onto a 100 μm inner diameter microcapillary trapping column packed first with approximately 5 cm of C18 Reprosil resin (5 μm, 100Å, Dr. Maisch GmbH, Germany) followed by analytical column ~20 cm of Reprosil resin (1.8 μm, 200Å, Dr. Maisch GmbH, Germany). Separation was achieved through applying a gradient from 5%–27% ACN in 0.1% formic acid over 90 min at 200 nL min−1. Electrospray ionization was enabled through applying a voltage of 1.8 kV using a home-made electrode junction at the end of the microcapillary column and sprayed from fused silica pico tips (New Objective, MA). The LTQ Orbitrap Elite was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Orbitrap in the range of 395 –1,800 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions (TOP20) for CID-MS2 fragmentation in the Ion trap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 200 ms. Singly charged ion species were not subjected to CID fragmentation. Normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 60 s. The same TOP20 ions were subjected to HCD MS2 event in Orbitrap part of the instrument. The fragment ion isolation width was set to 0.7 m/z, AGC was set to 50,000, the maximum ion time was 200 ms, normalized collision energy was set to 27V and an activation time of 1 ms for each HCD MS2 scan.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of mouse behavior
Automated analysis of locomotor behavior was performed in Ethovision XT 11.5 software (Noldus). Wall rearing and grooming behavior was scored manually analyzing the videos offline with observers blinded to genotype. Grooming includes all sequences of face wiping, scratching/rubbing of head and ears, and full-body grooming. Grooming segments less than 3 s apart were considered part of the same grooming bout. For longitudinal characterization of behavior the same animals were analyzed in the open field at different developmental stages. For each developmental time point two 30-minute open field sessions were recorded in consecutive days and results averaged per mouse. All cages and chambers were thoroughly cleaned with Quatricide (Pharmacal Research Labs) and water in between all subjects. Accelerating rotarod test was performed in a different cohort of animals and tested in four consecutive days (P17-P20) with three daily sessions per animal 20–30 minutes apart (4–40rpm in Med Associates ENV-577). Each run was terminated when the animal fell or was unable to maintain locomotion on top of the rotarod (full spin). All experiments described in Figure 1 were analyzed using repeated-measures ANOVA across groups with Benferroni multiple comparison test. Behavioral experiments in Figures 5 and S4 were analyzed using unpaired t test. Data for all behavioral experiments are represented per age or experimental group as Mean ± standard error of the mean (SEM). Sample sizes (number of mice per group) and statistical significance for each comparison are indicated in the results section of each individual panel. All statistical tests were performed in GraphPad PRIZM 8 software (GraphPad).
Analysis of electrophysiology data
Detection and analysis of mEPSCs was performed using custom routines in Igor Pro (Wavemetrics) using the following criteria: Minimum amplitude 7pA, Minimum refractory period 2ms, maximum rise/decay separation 4ms, maximum decay tau 20ms, minimum decay tau 0.7 ms, minimum decay time 2ms, minimum rise time 1ms, minimum rise rate 3pA/ms. Frequency and amplitude of mEPSCs were averaged per cell, and further compared using non-parametric Mann Whitney test due to non-Gaussian distribution of values. Plots represented in Figures 3I and 3J represent mEPSC frequency and amplitude values of AAV-GFP and AAV-Cre groups normalized to the average of the AAV-GFP group for each developmental time point. Analysis of eEPSC amplitude peak for calculation of PPR in Figures 3K and 3L were based on detection of minimum value after 20ms of stimulation subtracted to average baseline 10ms before stimulation. Calculation of input resistance and membrane capacitance in Figure S3 were performed by fitting evoked currents in response to −5mV voltage steps in the first seconds after break-in. AMPAR oEPSC amplitude peak in Figure 4 was determined by calculating the minimum within 20ms after stimulation subtracted to average 50ms baseline prior to optical pulse. NMDAR oEPSC amplitude was determined as value 50ms following optical stimulation. Sample sizes for all electrophysiology experiments (representing number of neurons recorded per group) and statistical significance for each comparison are indicated in the results section of each individual panel. All statistical analyses were done in GraphPad PRIZM 8 software (GraphPad).
Mass spectrometry analysis
Raw data were submitted for analysis in Proteome Discoverer 2.1.0.81 (Thermo Scientific) software. Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database including all entries from the Human Uniprot database (SwissProt 16,768 and TrEMBL 62,460 total of 79,228 protein forms, 2015) and other known contaminants such as human keratins and common lab contaminants. Sequest HT searches were performed using a 20 ppm precursor ion tolerance and requiring each peptides N-/C termini to adhere with Trypsin protease specificity, while allowing up to two missed cleavages. 6-plex TMT tags on peptide N termini and lysine residues (+229.162932 Da) was set as static modifications while methionine oxidation (+15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of 1% on protein level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator (64bit version) (Käll et al., 2008). For quantification, a 0.02 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were exported in result file of Proteome Discoverer 2.1 search engine as an excel tables. The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially uneven TMT labeling and/or sample handling variance for each labeled channel. Statistical significance of protein enrichment changes across development (P8–P18) were determined by analyzing variance of individual peptides across samples (different age) using Friedman test with Benjamini-Hochberg multiple test correction with p < 0.00134 cutoff in Scaffold 4.5 (Proteome Software). Shank3 showed increased enrichment during P8–18 with p = 0.011 but was included for analysis due to previous validation of increased postnatal expression (Wang et al., 2014). Protein expression changes in Figures 2D and 2E are represented as log2 of the max or min abundance values normalized to P8 for proteins with increased or decreased expression across development, respectively. Putative protein-protein interactions of Shank3 and proteins with significant differential enrichment at P8–P18 were analyzed in STRING, GeneMANIA and BioGRID and curated in Cytoscape 3.6. Only data from protein-protein interaction studies were considered as positive interactions. SFARI gene database (https://gene.sfari.org) was used to identify ASD risk genes in the identified protein cluster.
DATA AND CODE AVAILABILITY
This study did not generate new code. MATLAB and IgorPro procedures used for analysis of electrophysiology data are available upon request (rup14@pitt.edu). The accession number for the mass spectrometry data reported in this paper is MassIVE: MSV000084435.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Doublecortin | Santa Cruz | Cat# sc-271390, RRID:AB_10610966 |
| anti-SV2A | Sysy | Cat# 119 002, RRID:AB_887802 |
| Anti-AKAP12 | Abcam | Cat# ab49849, RRID:AB_2225608 |
| anti-Prkacb | Sigma-Aldrich | Cat# SAB2104516, RRID:AB_10669496 |
| Anti-GAPDH | CST | Cat# 2118, RRID:AB_561053 |
| anti-Synaptotagmin | Sysy | Cat# 105 011, RRID:AB_887832 |
| anti-GluN1 | Sysy | Cat# 114 011, RRID:AB_887750 |
| anti-VDAC | CST | Cat# 4661, RRID:AB_10557420 |
| anti-Calreticulin | CST | Cat# 12238, RRID:AB_2688013 |
| Bacterial and Virus Strains | ||
| AAV8-hSyn-PKI-ires-mRuby | Yao Chen | yaochen@wustl.edu |
| AAV9-hSyn-Cre-EGFP | Penn vector core | AV-9-PV1848 |
| AAV9-hSyn-EGFP | Penn vector core | AV-9-PV1696 |
| Deposited Data | ||
| Mass spectrometry data | MassIVE: MSV000084435 | |
| Experimental Models: Organisms/Strains | ||
| Ai32(RCL-ChR2(H134R)/EYFP) | JAX | 012569 |
| Slc32a1f/f | JAX | 012897 |
| Rbp4-Cre | GENSAT | #RP24–285K21 |
| pAAV-hSyn-PKI-ires-mRuby | Yao Chen | yaochen@wustl.edu |
Highlights.
Early onset of behavioral deficits in Shank3B−/− mice during postnatal development
Developmental switch in SPN synaptic adaptation to cortical hyperactivity during P15–P21
Dynamic regulation of multiple ASD risk factors during striatal synapse maturation
PKA inhibition in medial striatal regions ameliorates Shank3B−/− behavioral deficits
ACKNOWLEDGMENTS
We thank Susana da Silva and the Sabatini lab for helpful comments on the manuscript, and Scott Soderling and Tyler Bradshaw for help with Cytoscape programming and protein database curation. The PKIα-IRES-mRuby2 construct was a kind gift from Yao Chen. We thank Maria Ericsson and the Harvard Medical School electron microscopy (EM) facility for help in preparing and imaging the synaptosomes. This work was supported by the Nancy Lurie Marks Family Foundation (B.L.S. and R.T.P.), the William Randolph Hearst Foundation Fund (R.T.P.), the HHMI Hanna H. Gray Fellowship (L.C.), and NINDS R37NS046579 (B.L.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.021.
DECLARATION OF INTERESTS
B.B. is a scientific advisor to Merck & Co. All of the other authors declare no competing interests.
REFERENCES
- Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, Gordon JA, and Hen R (2013). Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science 340, 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amal H, Barak B, Bhat V, Gong G, Joughin BA, Wang X, Wishnok JS, Feng G, and Tannenbaum SR (2018). Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry. 10.1038/s41380-018-0113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronfeld M, Yael D, Belelovsky K, and Bar-Gad I (2013). Motor tics evoked by striatal disinhibition in the rat. Front. Syst. Neurosci 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguière E, Monteiro P, Feng G, and Graybiel AM (2013). Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science 340, 1243–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Gilman SR, Chiang AH, Sanders SJ, and Vitkup D (2015). Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci 18, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Granger AJ, Tran T, Saulnier JL, Kirkwood A, and Sabatini BL (2017). Endogenous Gαq-Coupled Neuromodulator Receptors Activate Protein Kinase A. Neuron 96, 1070–1083.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Siper PM, Durkin A, Weissman J, Muratet F, Halpern D, Trelles MDP, Frank Y, Lozano R, Wang AT, et al. (2018). Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Michel FJ, Marissal T, Rotrou Y, Matrot B, Lopez C, Humphries MD, and Hammond C (2011). Onset of Pup Locomotion Coincides with Loss of NR2C/D-Mediated Cortico-Striatal EPSCs and Dampening of Striatal Network Immature Activity. Front. Cell. Neurosci 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pino I, Rico B, and Marín O (2018). Neural circuit dysfunction in mouse models of neurodevelopmental disorders. Curr. Opin. Neurobiol 48, 174–182. [DOI] [PubMed] [Google Scholar]
- Dhamne SC, Silverman JL, Super CE, Lammers SHT, Hameed MQ, Modi ME, Copping NA, Pride MC, Smith DG, Rotenberg A, et al. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Jacobsen RI, Moser MB, and Moser EI (2017). Stellate cells drive maturation of the entorhinal-hippocampal circuit. Science 355, eaai8178. [DOI] [PubMed] [Google Scholar]
- Dunlop K, Woodside B, Olmsted M, Colton P, Giacobbe P, and Downar J (2016). Reductions in Cortico-Striatal Hyperconnectivity Accompany Successful Treatment of Obsessive-Compulsive Disorder with Dorsomedial Prefrontal rTMS. Neuropsychopharmacology 41, 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo MV (2016). Striatal circuits as a common node for autism pathophysiology. Front. Neurosci 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci 15, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, Takesian AE, Feng G, Fagiolini M, and Hensch TK (2014). Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron 83, 894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S, and Greene PE (2013). Stereotypies in autism: a video demonstration of their clinical variability. Front. Integr. Nuerosci 6, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lozano MA, Klemmer P, Gebuis T, Hassan C, van Nierop P, van Kesteren RE, Smit AB, and Li KW (2016). Dynamics of the mouse brain cortical synaptic proteome during postnatal brain development. Sci. Rep 6, 35456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P (2001). The neurobiology of slow synaptic transmission. Science 294, 1024–1030. [DOI] [PubMed] [Google Scholar]
- Guo B, Chen J, Chen Q, Ren K, Feng D, Mao H, Yao H, Yang J, Liu H, Liu Y, et al. (2019). Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat. Neurosci 22, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Havekes R, Canton DA, Park AJ, Huang T, Nie T, Day JP, Guercio LA, Grimes Q, Luczak V, Gelman IH, et al. (2012). Gravin orchestrates protein kinase A and β2-adrenergic receptor signaling critical for synaptic plasticity and memory. J. Neurosci 32, 18137–18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, et al. (2016). The mouse cortico-striatal projectome. Nat. Neurosci 19, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelashvili M, and Bar-Gad I (2015). Corticostriatal divergent function in determining the temporal and spatial properties of motor tics. J. Neurosci 35, 16340–16351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste SS, and Geschwind DH (2016). Clinical trials for neurodevelopmental disorders: At a therapeutic frontier. Sci. Transl. Med 8, 321fs1. [DOI] [PubMed] [Google Scholar]
- Jiang YH, and Ehlers MD (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabitzke P, Brunner D, He D, Fazio PA, Cox K, Sutphen J, Thiede L, Sabath E, Hanania T, Alexandrov V, et al. (2018). Comprehensive analysis of two Shank3 and the Cacna1c mouse models of autism spectrum disorder. Genes Brain Behav. 17, 4–22. [DOI] [PubMed] [Google Scholar]
- Käll L, Storey JD, and Noble WS (2008). Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 24, i42–i48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Yang M, Silverman JL, Solomon M, and Crawley JN (2016). Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr. Top. Behav. Neurosci 28, 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura F, and Itami C (2019). A Hypothetical Model Concerning How Spike-Timing-Dependent Plasticity Contributes to Neural Circuit Formation and Initiation of the Critical Period in Barrel Cortex. J. Neurosci 39, 3784–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, Geschwind D, et al. (2012). Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, and Sabatini BL (2012). Recurrent network activity drives striatal synaptogenesis. Nature 485, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Peixoto R, Wang W, Saunders A, and Sabatini BL (2015). Neuromodulation of excitatory synaptogenesis in striatal development. eLife 4, e10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajeski RN, Macey-Dare A, van Heusden F, Ebrahimjee F, and Ellender TJ (2019). Dynamic postnatal development of the cellular and circuit properties of striatal D1 and D2 spiny projection neurons. J. Physiol 597, 5265–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, and Sabatini BL (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago T, Davis A, Grillon C, and Ernst M (2017). Striatum on the anxiety map: small detours into adolescence. Brain Res. 1654 (Pt B), 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen M, Durston S, Staal WG, Palmen SJMC, and van Engeland H (2007). Caudate nucleus is enlarged in high-functioning medication-naive subjects with autism. Biol. Psychiatry 62, 262–266. [DOI] [PubMed] [Google Scholar]
- Langen M, Schnack HG, Nederveen H, Bos D, Lahuis BE, de Jonge MV, van Engeland H, and Durston S (2009). Changes in the developmental trajectories of striatum in autism. Biol. Psychiatry 66, 327–333. [DOI] [PubMed] [Google Scholar]
- Langen M, Bos D, Noordermeer SDS, Nederveen H, van Engeland H, and Durston S (2014). Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76, 405–411. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Chen Y, Lodder B, and Sabatini BL (2019). Monitoring Behaviorally Induced Biochemical Changes Using Fluorescence Lifetime Photometry. Front. Neurosci 13, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman OJ, McGuirt AF, Mosharov EV, Pigulevskiy I, Hobson BD, Choi S, Frier MD, Santini E, Borgkvist A, and Sulzer D (2018). Dopamine Triggers the Maturation of Striatal Spiny Projection Neuron Excitability during a Critical Period. Neuron 99, 540–554.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Cook EH, Leventhal BL, and Amaral DG (2000). Autism spectrum disorders. Neuron 28, 355–363. [DOI] [PubMed] [Google Scholar]
- Lozano CS, Tam J, and Lozano AM (2018). The changing landscape of surgery for Parkinson’s disease. Mov. Disord 33, 36–47. [DOI] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim J-A, Gao X, Fu Z, and Feng G (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P, and Feng G (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci 18, 147–157. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, and Lovinger DM (2000). Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J. Neurophysiol 84, 1422–1429. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, and Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat. Neurosci 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am.J. Hum. Genet 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, and Noble-Haeusslein LJ (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol 106–107, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci 14, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JB, Klug JR, Ross DL, Howard CD, Hollon NG, Ko VI, Hoffman H, Callaway EM, Gerfen CR, and Jin X (2016). Genetic-Based Dissection Unveils the Inputs and Outputs of Striatal Patch and Matrix Compartments. Neuron 91, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koós TZ, and Trent F (1998). Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev. Neurosci 20, 125–145. [DOI] [PubMed] [Google Scholar]
- Tien N-W, and Kerschensteiner D (2018). Homeostatic plasticity in neural development. Neural Dev. 13, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KC, Frost L, Linsenbardt D, McIlroy JR, and Müller R-A (2006). Atypically diffuse functional connectivity between caudate nuclei and cerebral cortex in autism. Behav. Brain Funct. 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, and Nelson SB (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci 5, 97–107. [DOI] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, and Kreitzer AC (2013). Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron 79, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herberg FW, Laue MM, Wullner C, Hu B, Petrasch-Parwez E, and Kilimann MW (2000). Neurobeachin: a protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J. Neurosci 20, 8551–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu Q, Bey AL, Lee Y, and Jiang YH (2014). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li C, Chen Q, Van Der Goes MS, Hawrot J, Yao AY, Gao X, Lu C, Zang Y, Zhang Q, et al. (2017). Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest 127, 1978–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts TJ (2008). The pathogenesis of autism. Clin. Med. Pathol 1, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, and Dostrovsky JO (2011). Pathological basal ganglia activity in movement disorders. Neuroscience 198, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Hazlett HC, Lightbody AA, Reiss AL, and Piven J (2013). Repetitive and self-injurious behaviors: associations with caudate volume in autism and fragile X syndrome. J. Neurodev. Disord 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Botteron KN, Dager SR, Elison JT, Estes AM, Gu H, Hazlett HC, Pandey J, Paterson SJ, Schultz RT, et al. ; IBIS Network (2014). Longitudinal patterns of repetitive behavior in toddlers with autism. J. Child Psychol. Psychiatry 55, 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Bennett MVL, and Zukin RS (2009). Developmental switch in requirement for PKA RIIbeta in NMDA-receptor-dependent synaptic plasticity at Schaffer collateral to CA1 pyramidal cell synapses. Neuropharmacology 56, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, and Malenka RC (2003). A developmental switch in the signaling cascades for LTP induction. Nat. Neurosci 6, 15–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new code. MATLAB and IgorPro procedures used for analysis of electrophysiology data are available upon request (rup14@pitt.edu). The accession number for the mass spectrometry data reported in this paper is MassIVE: MSV000084435.