

Abstract
A substrate-directed enantioselective anti-carboboration reaction of alkenes has been developed, wherein a carbon-based nucleophile and a boron moiety are installed across the C=C bond through a 5-membered palladacycle intermediate. A preliminary result also shows it is possible to extend this reaction to alkenes that are more distal from the directing group and react via a 6-membered palladacycle. The reaction is promoted by a palladium(II) catalyst and a monodentate oxazoline ligand. A range of enantioenriched secondary alkylboronate products were obtained with moderate to high enantioselectivity that could be further upgraded by recrystallization. This work represents an efficient method to synthesize versatile and valuable alkylboronate building blocks. Building on an earlier mechanistic proposal by Peng, He, and Chen, a revised model is proposed to account for the stereoconvergent nature of this transformation.
Keywords: palladium, directing group, carboboration, enantioselective catalysis, MOX ligand
Graphical Abstract
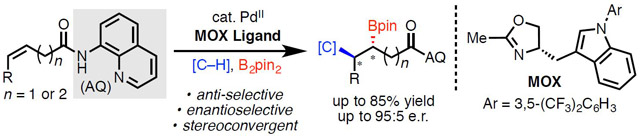
Substrate-directed nucleopalladation offers a powerful platform for generating multiple consecutive stereocenters (Scheme 1A). The key feature of these transformations is the formation of a stabilized 5- or 6-membered metallacycle intermediate, which enables trapping with an additional reaction partner, such as a proton, an electrophile, or a nucleophile, under various redox manifolds. Recently, our group1 and others2 have developed numerous alkene hydrofunctionalization and 1,2-difunctionalization reactions by exploiting this approach. A frontier in this area of research is controlling the absolute stereochemistry of the products by developing enantioselective variants of these transformations. In 2016, we reported a non-stereoselective γ-selective hydrocarbofunctionalization,1b which was subsequently rendered enantioselective in 2018 by He, Peng, and Chen through the elegant design and development of a monodentate oxazoline (MOX) ligand.2c In a complementary line of inquiry using prochiral carbon nucleophiles, our laboratory1f and the group of Zhang and Gong2e have achieved enantioselective transformations with a chiral phosphoric acid (CPA) ligand and a chiral amine catalyst respectively. Although these strategies have been successful in enantioselective hydrocarbofunctionalization, enantioselective alkene 1,2-difunctionalization within this family of reactions has not been reported to date. Herein, we describe the first asymmetric alkene anti-carboboration reaction via directed nucleopalladation.3 As in our previously reported racemic version,4 the reaction proceeds smoothly via either 5- or 6-membered palladacycle intermediates. The resulting enantioenriched organoboron compounds are highly valuable in organic synthesis5,6 and naturally map onto various bioactive compounds, such as β-homotryptophan. 7
Scheme 1.

Background and Project Synopsis
To begin our study, we selected N-methylindole (2a) as the nucleophile, B2pin2 (3a) as boron coupling partner, and 8-aminoquinoline (AQ)-masked8 (Z)-3-hexenoic acid (1d) as our pilot alkene substrate. In order to render this reaction enantioselective, we carried out extensive screening of base, solvent, and reaction temperature, while at the same time examining a variety of chiral ligands (Table 1). During optimization of the solvent, we found that HFIP provided high yield but low ee, while THF delivered moderate ee but lower yield. Interestingly, using a mixture of HFIP and THF (1:1) as solvent improved both the yield and ee (see the Supporting Information (SI) for optimization details). Adding DMF as a third solvent component further improved the ee. Commonly used bi- and tridentate oxazoline-based ligands as well as Yu’s APAO9 and MPAAM10 ligands (L2–L10) only induced low to moderate enantioselectivity with attenuated reactivity. Initial screening of monodentate ligands such as CPA (L1) and electron-deficient olefins (L11–L13) also failed to exert any chiral induction. To our delight, screening of monodentate imidazoline (MIM)(L15–L17) and MOX ligands (L18–L39) gave us reasonable levels of enantioselectivity, showing that these ligand scaffolds are privileged in this reaction. We decided to choose the MOX scaffold for further optimization based on synthetic accessibility considerations. The effect of different MOX ligands on the alkene carboboration reaction is similar to the trends in He, Peng, and Chen’s report.2c Tryptophan-derived ligands showed higher levels of chiral induction than other amino acid derivatives in terms of chiral induction. Systematically varying the N-aryl group revealed that the steric and electronic nature of the substituents impacts both yield and er (albeit to a minor extent), with electron-withdrawing groups proved to be advantageous. Ultimately, our optimization efforts converged with earlier findings from the aforementioned study2c in identifying L34, which bears a 3,5-bis-CF3-phenyl substituent on the indole nitrogen, as the optimal ligand, providing up to 94:6 er and 71% yield. Interestingly, 3,5-bis-nitro-phenyl substituted ligand L36 also offered the same enantioselectivity, albeit with slightly lower yield. Additionally, several oxazolines bearing a phenyl or methyl group at C-5 (L37–L39) were prepared based on the idea that increased steric hindrance could further improve the enantioselectivity. Unfortunately, none of these ligands performed better than L34.
Table 1.
Screening of Chiral Ligandsa
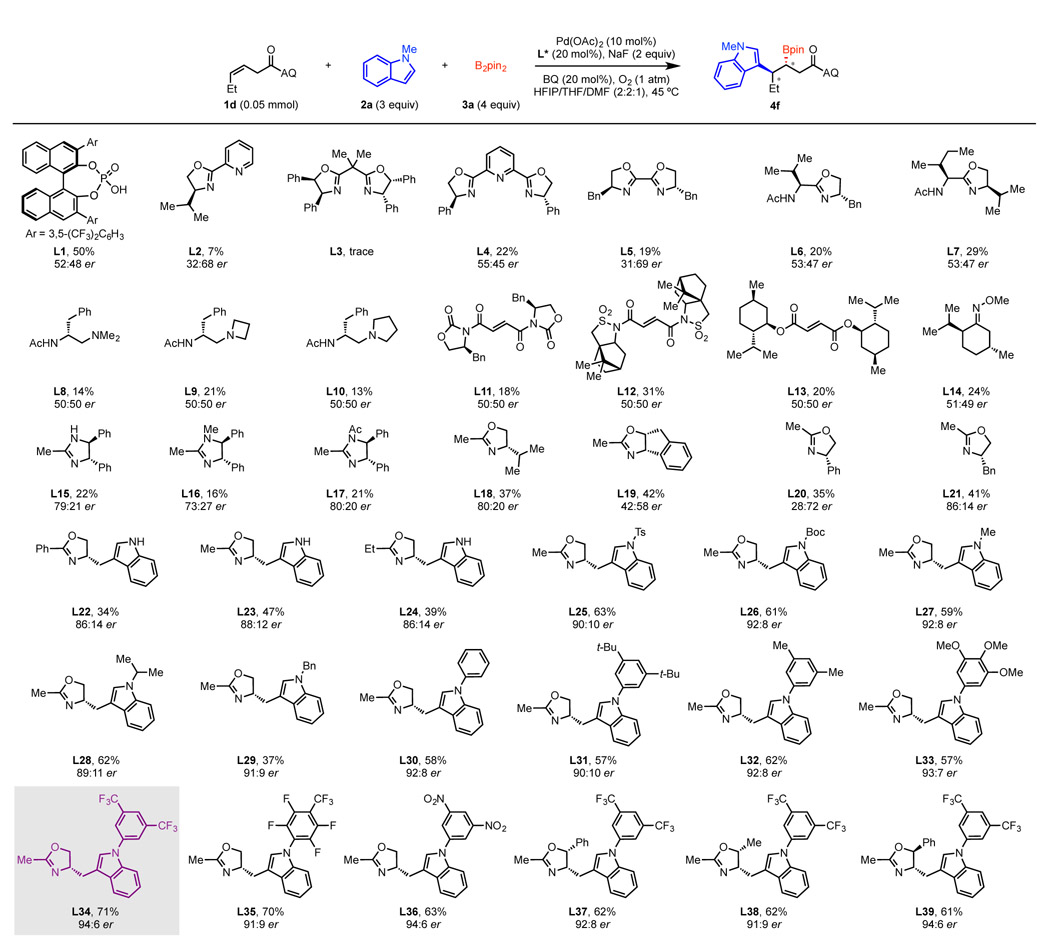 |
Reaction conditions: 1d (0.05 mmol), 2a (3 equiv), 3a (4 equiv), Pd(OAc)2 (10 mol%), chiral ligand (20 mol%), BQ (20 mol%), NaF (2 equiv), HFIP/THF/DMF (2:2:1, 0.1 mL), 45 °C, O2 (1 atm), 5 d. Percentages refer to 1H NMR yields with CH2Br2 as internal standard. The enantioselectivity was determined by SFC analysis.
After identifying the optimal ligand and reaction conditions, we next investigated the scope of this palladium(II)-catalyzed asymmetric alkene anti-1,2-carboboration reaction (Table 2). A variety of AQ-masked alkenyl carbonyl compounds (1a–1h) were competent substrates in this transformation, providing the desired products in moderate to good yields with satisfactory er. The absolute configurations of products 4f and 4g were assigned as (R, R) by X-ray crystallography analysis, and other products were assigned by analogy.11 Increasing the size of R group on the alkene substrate improved the er while slightly lowering the yield. For example, terminal alkene 1a (R = H) showed only a moderate er of 86:14, while switching the R group to methyl (4d and 4e) and ethyl (4f), led to higher enantiomeric ratios of 91:9 and 94:6, respectively. More sterically bulky groups on the alkene, however, shut down the reaction. For instance, in the case of 4q, only trace amount of product was formed under the standard reaction conditions. Generally, Z-alkene substrates are more reactive than E-alkenes. Under the standard conditions, E-alkene 1b was carboborylated in less than 50% conversion. Nevertheless, with more forcing conditions, 4d could be generated from 1b in high yield and good enantioselectivity. In all the cases where internal alkene substrates were used, >20:1 dr was observed,12 demonstrating the excellent stereocontrol of this carboboration reaction. To our surprise, both E- and Z-alkene substrates (1b and 1c) resulted in the same major diastereomer after carboboration (4d and 4e), which is inconsistent with the stereoinduction and -convergence model proposed by He, Peng, and Chen.2c We instead believe the E-alkene is first isomerized to Z-configuration upon Pd(II) coordination, followed by anti-nucleopalladation to give a common alkylpalladium intermediate in both cases (vide infra). Gratifyingly, AQ-masked 4-pentenoic acid substrate 1i underwent this transformation smoothly through a putative 6-membered palladacycle delivering 4p in high yield and moderate er. However, internal alkene 1k was incompatible with this difunctionalization reaction.
Table 2.
Enantioselective Alkene Carboboration Scopea
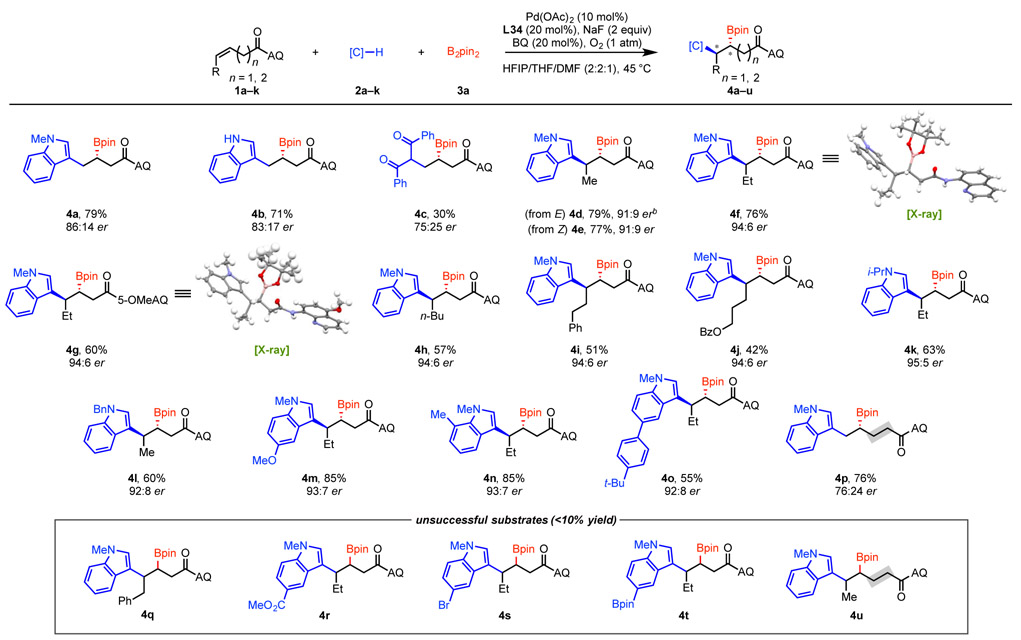 |
Reaction conditions: 1a–k (0.1 mmol), 2a–k (3 equiv), 3a (4 equiv), Pd(OAc)2 (10 mol%), L34 (20 mol%), BQ (20 mol%), NaF (2 equiv), HFIP/THF/DMF (2:2:1, 0.2 mL), 45 °C, O2, 5 d. Percentages refer to isolated yields. Diastereomeric ratios (dr) are >20:1 in all cases. The enantioselectivity was determined by SFC analysis. bKF (2 equiv), HFIP/THF (1:1, 0.2 mL), 60 °C, 2 d.
We have also studied the scope of indole nucleophiles under the standard reaction conditions. Various indole derivatives bearing substituents at different positions on the indole ring were well tolerated in this transformation (4m–4o). Indoles with electron-withdrawing substituents were incompatible coupling partners (2i–2k), potentially due to diminished nucleophilicity (4r–4t). Different groups on indole nitrogen also have a significant effect on reaction outcomes. With larger substituents (4k and 4l), the enantioselectivity of the reaction was improved, albeit with diminished yields. We have also attempted nucleophiles that were demonstrated to be compatible in our previously published hydrocarbofunctionalization method,1b such as 1,3-cyclopentadione and 3-(dimethylamino)phenol. However, these nucleophiles were ineffective in 1,2-carboboration, even under non-stereoselective conditions (See SI). To our delight, 1,3-dicarbonyl compound 2c was found to be a suitable nucleophile and provided the corresponding product (4c) in low yield and moderate er.
Subsequently, this Pd(II)-catalyzed asymmetric alkene carboboration method was performed on gram-scale to demonstrate its operational simplicity and practicality (Scheme 2). Compound 4f was prepared from Z-alkene 1d, N-methylindole (2a), and B2pin2 (3a) on gram scale, with the yield and er consistent with the smaller scale experiment. Furthermore, recrystallization of the final product from Et2O or EtOH could provide nearly enantiopure secondary boronate 4f in satisfying overall yield.
Scheme 2.

Gram-Scale Synthesis and Recrystallization
A series of derivatizations were next conducted to further illustrate the synthetic utility of the carboborylated products (Scheme 3). Treatment of compound 4f with aqueous KHF2 generated trifluoroborate salt 5 in high conversion. Initial attempts of boronate oxidation with strong oxidants, such as H2O2 solution, proved to be unsuccessful, presumably because of undesired oxidation of the electron-rich indole ring. Finally, using a mild oxidant NaBO3•4H2O,13 boronate 4f was efficiently converted into chiral alcohol 6 with almost complete stereoretention. Ni(tmhd)2-mediated methanolysis14,15 and a two-step transamidation deprotection16 of the AQ protecting group were also performed to deliver the corresponding methyl ester 7 and amide 8 with retention of the C–B bond, which could potentially be carried forward into further transformations.
Scheme 3.

Product Diversification
Both in He, Peng, and Chen’s asymmetric hydrocarbonation reaction2c and this work, stereoconvergence was observed, with Z- and E-alkene substrates giving the same absolute and relative stereochemistry (Scheme 4A). In the previously proposed stereoinduction model (Scheme 4B),2c with an E-alkene, Int-E-I-down was computed to be more stable than Int-E-II-up, and with a Z-alkene, Int-Z-II-up was computed to be more stable than Int-Z-I-down. These trends also hold in the corresponding transition state energies, where nucleopalladation from Int-E-I-down (to give Int-E-III) and from Int-Z-II-up (to give Int-Z-III) is favored for E and Z alkenes, respectively. This model predicts that the C(sp3)–Pd stereocenter at C2 would have opposite configuration with E and Z alkenes, which is not in agreement with our results.17 This discrepancy prompted us to consider an alternative explanation (Scheme 4C). Specifically, we envisioned a different scenario in which alkene isomerization to interconvert Int-E-I-down and Int-Z-II-up takes place under the reaction conditions, followed by nucleopalladation, which occurs preferentially through the lower-energy pathway involving Int-Z-II-up. Although the precise mechanism of alkene isomerization remains unclear at this stage,18 evidence suggests that it occurs in the present system only after alkene complexation with palladium(II). To test the viability of this alternative mechanism, several mechanistic experiments were performed (Scheme 4D). At room temperature, treatment of Z-alkene 1d with stoichiometric Pd(OAc)2 both with and without L34 led to formation of the corresponding palladium(II) complex Pd-I with the retention of alkene geometry. In contrast, E-alkene 1l underwent E/Z isomerization in the presence and absence of MOX ligand. Interestingly, L34 seems to promote E-to-Z isomerization, possibly by creating a more sterically hindered environment around the palladium(II) center (See SI for additional mechanistic experiments).19 These results are consistent with the model depicted in Scheme 4C, where isomerization of Int-E-I-down to Int-Z-II-up takes place before the nucleopalladation step for the E-alkene substrates. which accounts for the observed stereoconvergence in the described 1,2-difunctionalization.
Scheme 4.

Investigation of Stereoconvergent Nucleopalladation
In conclusion, we have developed an enantioselective carboboration reaction of unactivated alkenyl carbonyl compounds using a chiral monodentate oxazoline (MOX) ligand. This reaction proceeded smoothly through 5- and 6-membered palladacycle intermediates to install a secondary boron group to the β or γ positions relative to the carbonyl group. A variety of carbon nucleophiles and alkene substrates were demonstrated to be compatible, with moderate to good yields and enantioselectivity. Recrystallization of the final product was shown to greatly improve the product er. The reaction was scalable, and cleavage of the 8-aminoquinoline (AQ) directing group was demonstrated. To explain the stereoconvergence of Z- and E-alkenes, a revised mechanism based on Chen’s original stereoinduction model was proposed. Future work will focus on the development of new chiral ligands and expanding the scope of substrated-directed palladium(II)-catalyzed 1,2-difunctionalization to other types of reactions and substrates. These results will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by TSRI, Pfizer, Inc., Bristol-Myers Squibb (Unrestricted Grant), and the National Institutes of Health (5R35GM125052-02). We gratefully acknowledge the Nankai University College of Chemistry for an International Research Scholarship (X. L.). We thank Dr. De-Wei Gao, Van T. Tran, Mingyu Liu, and Wei Hao (TSRI) for donation of chiral ligands. Dr. Jason Chen and Brittany Sanchez (TSRI) are acknowledged for SFC and HRMS analysis. John A. Gurak, Jr. (TSRI) is also acknowledged for growing a single crystal of Pd-I. We thank Andrew Romine (TSRI) for his assistance of proofreading the manuscript. We further thank Prof. Arnold L. Rheingold and Dr. Milan Gembicky (UCSD) for X-ray crystallographic analysis.
Footnotes
Supporting Information
Experiment details, spectra data, copies of 1H and 13C NMR spectra, and X-ray crystallographic data. These materials are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Gurak JA Jr.; Yang KS; Liu Z; Engle KM Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc 2016, 138, 5805–5808. [DOI] [PubMed] [Google Scholar]; (b) Yang KS ; Gurak JA Jr.; Liu Z; Engle KM Catalytic, Regioselective HydrocarbofUnctionalization of Unactivated Alkenes with Diverse C–H Nucleophiles. J. Am. Chem. Soc 2016, 138, 14705–14712. [DOI] [PubMed] [Google Scholar]; (c) Liu Z; Zeng T ; Yang KS; Engle KM β,γ-Vicinal Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Directed Nucleopalladation. J. Am. Chem. Soc 2016, 138, 15122–15125. [DOI] [PubMed] [Google Scholar]; (d) Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc 2017. 139, 11261–11270. [DOI] [PubMed] [Google Scholar]; (e) Zeng T; Liu Z; Schmidt MA; Eastgate MD; Engle KM Directed, Palladium(II)-Catalyzed Intermolecular Aminohydroxylation of Alkenes Using a Mild Oxidation System. Org. Lett 2018. 20, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nimmagadda SK; Liu M; Karunananda MK; Gao D-W; Apolinar O; Chen JS; Liu P; Engle KM Catalytic, Enantioselective α-Alkylation of Azlactones with Non-Conjugated Alkenes via Directed Nucleopalladation. Angew. Chem. Int. Ed 2019, DOI: 10.1002/anie.201814272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).Neufeldt SR; Sanford MS Asymmetric Chiral Ligand-Directed Alkene Dioxygenation. Org. Lett 2013, 15, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Talbot EPA; Fernandes T. d. A.; McKenna JM; Toste FD Asymmetric Palladium-Catalyzed Directed Intermolecular Fluoroarylation of Styrenes. J. Am. Chem. Soc 2014, 136, 4101–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang H; Bai Z; Jiao T; Deng Z; Tong H; He G; Peng Q; Chen G Palladium-Catalyzed Amide-Directed Enantioselective Hydrocarbofunctionalization of Unactivated Alkenes Using a Chiral Monodentate Oxazoline Ligand. J. Am. Chem. Soc 2018, 140, 3542–3546. [DOI] [PubMed] [Google Scholar]; (d) Wang C; Xiao G; Guo T; Ding Y; Wu X; Loh T-P Palladium-Catalyzed Regiocontrollable Reductive Heck Reaction of Unactivated Aliphatic Alkenes. J. Am. Chem. Soc 2018, 140, 9332–9336. [DOI] [PubMed] [Google Scholar]; (e) Shen H-C; Zhang L; Chen S-S; Feng J; Zhang B-W; Zhang Y; Zhang X; Wu Y-D; Gong L-Z Enantioselective Addition of Cyclic Ketones to Unactivated Alkenes Enabled by Amine/Pd(II) Cooperative Catalysis. ACS Catal. 2019, 9, 791–797. For an example of 1,2-diarylation of a chelating vinyl ether, see: [Google Scholar]; (f) Trejos A; Fardost A; Yahiaoui S; Larhed M Palladium(II)-catalyzed coupling reactions with a chelating vinyl ether and arylboronic acids: a new Heck/Suzuki domino diarylation reaction. Chem. Commun 2009, 7587–7589. [DOI] [PubMed] [Google Scholar]
- (3).(a) For selected examples of other approaches to catalytic 1,2-carboboration of alkenes, see: Semba K; Nakao Y Arylboration of Alkenes by Cooperative Palladium/Copper Catalysis. J. Am. Chem. Soc 136, 7567–7570. [DOI] [PubMed] [Google Scholar]; (b) Meng F; Haeffner F; Hoveyda AH Diastereo- and Enantioselective Reactions of Bis(pinacolato)diboron, 1,3-Enynes, and Aldehydes Catalyzed by an Easily Accessible Bisphosphine–Cu Complex. J. Am. Chem. Soc 2014, 136, 11304–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Logan KM; Smith KB; Brown MK Copper/Palladium Synergistic Catalysis for the syn- and anti-Selective Carboboration of Alkenes. Angew. Chem. Int. Ed 2015, 54, 5228–5231. [DOI] [PubMed] [Google Scholar]; (d) Su W; Gong T-J, Lu X; Xu M-Y; Yu C-G; Xu Z-Y; Yu H-Z; Xiao B; Fu Y Ligand-Controlled Regiodivergent Copper-Catalyzed Alkylboration of Alkenes. Angew. Chem. Int. Ed 2015, 54, 12957–12961. [DOI] [PubMed] [Google Scholar]; (e) Semba K; Ohtagaki Y; Nakao Y Arylboration of 1-Arylalkenes by Cooperative Nickel/Copper Catalysis. Org. Lett 2016, 18, 3956–3959. [DOI] [PubMed] [Google Scholar]; (f) Yang K; Song Q Pd-Catalyzed Regioselective Arylboration of Vinylarenes. Org. Lett 2016, 18, 5460–5463. [DOI] [PubMed] [Google Scholar]; (g) Logan KM; Sardini SR; White SD; Brown MK Nickel-Catalyzed Stereoselective Arylboration of Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 159–162. For an example of transition metal-free 1,2-carboboration of alkenes, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Cheng Y; Mück-Lichtenfeld C; Studer A Transition Metal-Free 1,2-Carboboration of Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 6221–6225. For an example of 1,1-arylborylation of alkenes, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Nelson HM; Williams BD; Miró J; Toste FD Enantioselective 1,1-Arylborylation of Alkenes: Merging Chiral Anion Phase Transfer with Pd Catalysis. J. Am. Chem. Soc 137, 3213–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu Z; Ni H-Q; Zeng T; Engle KM Catalytic Carbo- and Aminoboration of Alkenyl Carbonyl Compounds via Five- and Six-Membered Palladacycles. J. Am. Chem. Soc 2018, 140, 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) For a representative review on asymmetric conjugate addition of boron nucleophiles, see: Hartmann E; Vyas DJ; Oestreich M Enantioselective formal hydration of α, β--unsaturated acceptors: asymmetric conjugate addition of silicon and boron nucleophiles. Chem. Commun 2011, 47, 7917–7932. For examples of asymmetric hydroboration to access chiral β-boronates, see: [DOI] [PubMed] [Google Scholar]; (b) Smith SM; Thacker NC; Takacs JM Efficient Amide-Directed Catalytic Asymmetric Hydroboration. J. Am. Chem. Soc 2008, 130, 3734–3735. [DOI] [PubMed] [Google Scholar]; (c) Gao T-T; Zhang W-W; Sun X; Lu H-X; Li B-J Stereodivergent Synthesis through Catalytic Asymmetric Reversed Hydroboration. J. Am. Chem. Soc 2019, DOI: 10.1021/jacs.8b13520. For an example of asymmetric β-C–H borylation, see: [DOI] [PubMed] [Google Scholar]; (d) He J; Shao Q; Wu Q; Yu J-Q Pd(II)-Catalyzed Enantioselective C(sp3)–H Borylation. J. Am. Chem. Soc 2017, 139, 3344–3347. [DOI] [PubMed] [Google Scholar]
- (6).For a review describing methods to transform enantioenriched organoboron compounds, see: Sandford C; Aggarwal VK Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem. Commun 2017, 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- (7).Pahlke DM; Diederichsen U Synthesis and characterization of β-peptide helices as transmembrane domains in lipid model membranes. J. Pept. Sci 2016, 22, 636–641. [DOI] [PubMed] [Google Scholar]
- (8).For a representative review, see: Daugulis O; Do H-Q; Shabashov D Palladium- and Copper-Catalyzed Arylation of Carbon–Hydrogen Bonds. Acc. Chem. Res 2009, 42, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wu Q-F; Shen P-X; He J; Wang X-B; Zhang F; Shao Q; Zhu R-Y; Mapelli C; Qiao JX; Poss MA; Yu J-Q Formation of α-chiral centers by asymmetric β-C(sp3)–H arylation, alkenylation, and alkynylation. Science 2017, 355, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q Pd(II)-Catalyzed Enantioselective C(sp3)–H Arylation of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).The absolute stereochemistry of compound 4a was determined by 1H NMR analysis of its Mosher ester (see SI).
- (12).The dr values in these cases are notably higher than those observed under non-stereoselective conditions (Ref. 4), which merits brief comment. Based on the experimental data in Scheme 4, we surmise that E-alkenes will isomerize to the corresponding Z-alkenes under the reaction conditions, both with and without the MOX ligand, and furthermore that Z-alkenes react much faster in carbopalladation than E-alkenes. Under the enantioselective conditions, the MOX ligand appears to facilitate rapid E-to-Z isomerization, which results in good diastereocontrol (see examples 4d and 4e). However, under the non-stereoselective conditions (Ref. 4), where the reaction temperature is higher and there is no ancillary ligand to promote alkene isomerization, E-alkenes can react via carboboration at a rate that is competitive with that of the Z-isomers (and that is similar to the rate of E-to-Z isomerization), which results in low levels of diastereocontrol.
- (13).Bonet A; Gulyás H; Fernández E Metal-Free Catalytic Boration at the β-Position of α,β-Unsaturated Compounds: A Challenging Asymmetric Induction. Angew. Chem. Int. Ed 2010, 49, 5130–5134. [DOI] [PubMed] [Google Scholar]
- (14).Deguchi T; Xin H-L; Morimoto H; Ohshima T Direct Catalytic Alcoholysis of Unactivated 8-Aminoquinoline Amides. ACS Catal. 2017, 7, 3157–3161. [Google Scholar]
- (15).Use of a well-sealed reaction vessel and extended reaction time (6 days) are required to achieve high conversion in this reaction (see SI for experimental details).
- (16).Verho O; Lati MP; Oschmann MJ A Two-Step Procedure for the Overall Transamidation of 8-Aminoquinoline Amides Proceeding via the Intermediate N-Acyl-Boc-Carbamates. J. Org. Chem 2018, 83, 4464–4476. [DOI] [PubMed] [Google Scholar]
- (17).In hydrocarbofunctionalization (Ref. 2c) this C(sp3)–Pd stereocenter is ablated in the next elementary step (protodepalladation), but in the present transformation with B2Pin2, the C(sp3)–Pd is transferred to the C(sp3)–B stereocenter in the product.
- (18).(a) For related references, see: Henry PM Palladium(II)-Catalyzed Exchange and Isomerization Reactions. VII. Isomerization and Exchange of Enol Propionates in Acetic Acid Catalyzed by Palladium(II) Chloride. J. Am. Chem. Soc 1972, 94, 7316–7322. [Google Scholar]; (b) Sen A; Lai T-W Catalytic isomerization of alkenes by palladium (II) compounds. An alternative mechanistic view. Inorg. Chem 1981, 20, 4036–4038. [Google Scholar]; (c) Solin N; Szabó KJ Mechanism of the η3-η1-η3 Isomerization in Allylpalladium Complexes: Solvent Coordination, Ligand, and Substituent Effects. Organometallics 2001, 20, 5464–5471. [Google Scholar]; (d) Yu J-Q; Gaunt MJ; Spencer JB Convenient Preparation of trans-Arylalkenes via Palladium(II)-Catalyzed Isomerization of cis-Arylalkenes. J. Org. Chem 2002, 67, 4627–4629. [DOI] [PubMed] [Google Scholar]; (e) Zawisza AM; Bouquillon S; Muzat J Palladium(II)-Catalyzed Isomerization of (Z)-1,4-Diacetoxy-2-Butene: Solvent Effects. Eur. J. Org. Chem 2007, 3901–3904. [Google Scholar]; (f) Tan EHP; Lloyd-Jones GC; Harvey JN; Lennox AJJ; Mills BM [(RCN)2PdCl2]-Catalyzed E/Z Isomerization of Alkenes: A Non-Hydride Binuclear Addition–Elimination Pathway. Angew. Chem. Int. Ed 2011, 50, 9602–9606. [DOI] [PubMed] [Google Scholar]
- (19).In Ref. 2c the authors note that isomerized alkene was not observed in solution during the course of the reaction, but the authors did not examine the E/Z-configuration of the corresponding complex. Stoichiometric experiments in MeOH (the solvent used in Ref. 2c) suggest that E-to-Z isomerization of the complex is rapid at room temperature (see SI).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.