

Abstract
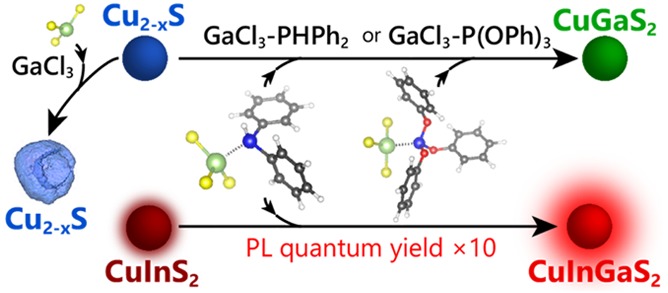
Nanoscale cation exchange (CE) has resulted in colloidal nanomaterials that are unattainable by direct synthesis methods. Aliovalent CE is complex and synthetically challenging because the exchange of an unequal number of host and guest cations is required to maintain charge balance. An approach to control aliovalent CE reactions is the use of a single reactant to both supply the guest cation and extract the host cation. Here, we study the application of GaCl3–L complexes [L = trioctylphosphine (TOP), triphenylphosphite (TPP), diphenylphosphine (DPP)] as reactants in the exchange of Cu+ for Ga3+ in Cu2–xS nanocrystals. We find that noncomplexed GaCl3 etches the nanocrystals by S2– extraction, whereas GaCl3–TOP is unreactive. Successful exchange of Cu+ for Ga3+ is only possible when GaCl3 is complexed with either TPP or DPP. This is attributed to the pivotal role of the Cu2–xS–GaCl3–L activated complex that forms at the surface of the nanocrystal at the onset of the CE reaction, which must be such that simultaneous Ga3+ insertion and Cu+ extraction can occur. This requisite is only met if GaCl3 is bound to a phosphine ligand, with a moderate bond strength, to allow facile dissociation of the complex at the nanocrystal surface. The general validity of this mechanism is demonstrated by using GaCl3–DPP to convert CuInS2 into (Cu,Ga,In)S2 nanocrystals, which increases the photoluminescence quantum yield 10-fold, while blue-shifting the photoluminescence into the NIR biological window. This highlights the general applicability of the mechanistic insights provided by our work.
Keywords: semiconductor nanocrystals, cation exchange, copper sulfide, copper gallium sulfide, copper indium sulfide, copper indium gallium sulfide
Compound copper chalcogenide nanocrystals (NCs) offer a promising alternative to the widely studied CdX and PbX NCs (X = S, Se, Te), as they consist of less toxic elements and display exciting optoelectronic properties, such as photoluminescence (PL) tunability in the visible to the near-infrared (NIR) spectral window and localized surface plasmon resonances (LSPR) in the NIR.1−4 Another advantage of these nanomaterials is the wide range of compositions that can be obtained, ranging from binary to multinary compounds with up to five different elements.1−3 This makes them a versatile class of materials, offering ample opportunities for property engineering.1−3,5 However, some morphologies and compositions are not easily accessible by direct approaches due to synthetic limitations, such as the lack of suitable precursors for multinary compounds.1 Nanoscale cation exchange (CE) has emerged as a promising postsynthetic tool to circumvent these limitations, providing pathways to NCs and hetero-NCs with compositions, morphologies, crystal structures, and heteroarchitectures that would otherwise remain unavailable.5−8 The extent of the CE (ranging from doping, e.g., Ag:CdSe NCs,9 Zn:CsPbBr3 NCs,10 Mn:ZnTe magic-size clusters,11 to full replacement)5−8 and the elemental distribution profile of the product NCs (i.e., homogeneous or gradient alloys, e.g., (Cd,Zn)Se NCs,12 or heterostructured NCs, e.g., ZnSe/CdSe,12 PbSe/CdSe13 core/shell quantum dots) depends on a number of physicochemical properties, such as the miscibility of the parent and product phases and the diffusion rates of the incoming and outgoing cations within the NCs and can be tailored by adjusting reaction variables, such as reaction temperature and concentration of guest cations in solution.5−13
In isovalent CE, such as Zn2+ for Cd2+ exchange in ZnSe NCs12 and Pb2+ for Cd2+ in PbX NCs,13−15 a direct place exchange reaction occurs at the surface of the template NCs, through which the outgoing host cation and the incoming guest cation swap places (i.e., the guest cation is incorporated in the NC, while the host cation forms a complex with the ligand molecule that was carrying the guest cation).12 This is in contrast with aliovalent CE, such as Cu+ for In3+ in Cu2–xS NCs16 or Cd2+ for Ag+ in CdSe,5,9,17 which typically occurs through two separate chemical pathways.5−8,16 This makes balancing the in- and outgoing diffusion rates challenging. An approach to better control aliovalent CE reactions is the use of a single precursor, which both provides the guest cation and extracts the host cation. In the case of the Cu+ for In3+ exchange, a stoichiometric InCl3–trioctylphosphine complex has been shown to be a suitable precursor, even allowing for topotactic sequential CE (viz., Cd2+ for Cu+, followed by partial Cu+ for In3+) in large and complex anisotropic NCs such as CdSe/CdS dot-in-rod nanorods, with preservation of the size, shape, and heterostructure of the template NCs in the product CuInSe2/CuInS2 dot-in-rod nanorods.18 The strict control over the CE rates provided by this approach has been recently exploited to fabricate air-stable CuInSe2 NC transistors by postdeposition sequential CE.19 Here, we extend the single precursor approach to the exchange of Cu+ for Ga3+ in Cu2–xS and CuInS2 NCs by using stoichiometric GaCl3–phosphine complexes to simultaneously deliver Ga3+ and extract Cu+. Our choice is motivated by the fact that CuGaS2 (CGS) and Cu(In,Ga)S2 (CIGS) exhibit interesting optoelectronic properties, which make them promising materials for applications in photocatalysis,20,21 light-emitting devices,22,23 and photovoltaics.24−27 Moreover, studies of Cu+ for Ga3+ and In3+ for Ga3+ cation exchange are scarce.28−30 Although Ga3+ and In3+ are both trivalent cations of group IIIA metals, they have significantly different ionic radii (rGa3+ = 47 pm, rIn3+ = 62 pm)31 and Lewis acidity (ηGa3+ = 17 eV, ηIn3+ = 13 eV)32 and are thus expected to behave differently in cation exchange reactions. Our study thus offers an ideal testbed to investigate the relationship between the chemistry of the metal chloride–phosphine complexes and the outcome of cation exchange reactions in Cu chalcogenide NCs.
We investigated different GaCl3–L Lewis acid–base adduct complexes [L = trioctylphosphine (TOP), triphenylphosphite (TPP), and diphenylphosphine DPP)] as reagents for the exchange of Cu+ for Ga3+ and found that the choice of ligand has a profound impact on the reaction outcome. Noncomplexed GaCl3 is a strong Lewis acid that yields hollow NCs through etching by sulfide extraction, whereas the complex GaCl3–trioctylphosphine leads to neither etching nor cation exchange. In contrast, both GaCl3–triphenylphosphite and GaCl3–diphenylphosphine successfully converted Cu2–xS NCs in CuGaS2 NCs through Cu+ for Ga3+ exchange. These different reactivities are attributed to the stability and geometry of the activated Cu2–xS–GaCl3–L complex that must be formed at the surface of the NCs prior to the cation exchange. Density functional theory (DFT) calculations show that, in these complexes, the Ga–P bond strength decreases with increasing electron-withdrawing nature of the side groups on the phosphine ligands (i.e., the Ga–P bond strength ε follows the trend εGaCl3–TOP > εGaCl3–DPP > εGaCl3–TPP). Complexing GaCl3 with DPP or TPP lowers the activation energy barrier for the CE reaction, while kinetically favoring the Cu+ for Ga3+ place exchange over the sulfide extraction. These insights were used to successfully convert CuInS2 NCs into Cu-poor (Cu,In,Ga)S2 NCs through Cu+ for Ga3+ exchange, yielding NCs with vastly improved photoluminescence quantum yields with respect to the parent NCs. The findings presented in this work are thus generally applicable, resulting in nanomaterials with combinations of size, shape, composition, and crystal structure that are not directly accessible by conventional synthesis routes.
Results and Discussion
Stoichiometric InCl3–TOP and GaCl3–TOP as Reactants for Cation Exchange in Cu2–xS NCs
Throughout this work, low-chalcocite 12 nm Cu2–xS (x < 0.04) bifrustum NCs are used as parent NCs to investigate the Cu+ for Ga3+ cation exchange reaction. For comparison, these NCs (Figure 1a,d) are converted into wurtzite CuInS2 (CIS) bifrustum NCs (Figure 1b,e) by partial, self-limited Cu+ for In3+ CE at 100 °C, using a stoichiometric InCl3–trioctylphosphine (InCl3–TOP) Lewis acid–base adduct as precursor, as previously reported by our group.18 The transformation from low chalcocite to wurtzite is evidenced by the electron diffraction (ED) patterns (Figure 1a,b,d,e). The powder ED (PED) pattern obtained after azimuthal integration of the 2D ED pattern of the product NCs (Figure 1e) corresponds well to the reference pattern of wurtzite CIS. The shape and size of the NCs are preserved after the reaction (Supporting Information, Figure S1), confirming the topotactic nature of the Cu+ for In3+ CE reaction. Energy-dispersive X-ray spectroscopy (EDS) measurements further confirm the conversion of Cu2–xS NCs into ternary CIS NCs (Cu/In = 1.00:0.79; Supporting Information, Figure S2). Compared to the Cu2–xS parent NCs, the product CIS NCs no longer exhibit a LSPR absorption band in the NIR (Supporting Information, Figure S3a,b). The absorption spectrum of the CIS NCs agrees well with spectra reported in the literature.33 In contrast, the use of the stoichiometric GaCl3–TOP Lewis acid–base adduct as a precursor does not lead to any significant change in the composition (Cu/Ga = 1.00:0.02, Supporting Information, Figure S4), crystal structure, size, and shape of the NCs (Figure 1c,f). The absorption spectrum of NCs obtained after reaction with GaCl3–TOP exhibits little to no LSPR band (Supporting Information, Figure S3c), which indicates the absence of excess free holes in these NCs, due to (limited) reduction at the NC surface.
Figure 1.

TEM images of (a) parent Cu2–xS bifrustum NCs, (b) product Cu1.03In0.81S2 (CIS) bifrustum NCs, after reaction with InCl3–TOP at 100 °C, and (c) Cu1.99Ga0.04S nanocrystals, after reaction with GaCl3–TOP at 100 °C. Top-right insets display the corresponding ED pattern. Bottom panels show azimuthally integrated ED (PED) patterns of (d) parent low-chalcocite Cu2–xS nanocrystals, (e) product wurtzite CIS nanocrystals, and (f) product low-chalcocite Cu1.99Ga0.04S nanocrystals. Reference bars in (d,f) are calculated patterns based on the low-chalcocite Cu2–xS crystal structure described in ref (34). Reference bars in (e) were obtained from JCPDS PDF-card 01-077-9459 for wurtzite CIS.
Reaction of Noncomplexed GaCl3 with Cu2–xS NCs
Clearly, the Cu+ for Ga3+ CE does not significantly occur when GaCl3–TOP is used as the reactant under the same conditions that successfully lead to Cu+ for In3+ CE using InCl3–TOP. Therefore, other Ga precursors were examined, starting with the simplest possible case: GaCl3 dissolved in neat toluene. The reaction with GaCl3 at the same temperature used with GaCl3–TOP (100 °C) was found to severely deteriorate the NCs (Supporting Information, Figure S5). To minimize the degradation, the temporal evolution of this reaction was studied at a lower temperature (30 °C) (Figure 2b–e). The TEM images of the product NCs show that, despite the lower reaction temperature, a low-contrast spot appears in the NCs already after 15 min of reaction (Figure 2b, red arrows) and increases in size over time (Figure 2c–e). The PED pattern shows that the crystal structure of the NCs remains low-chalcocite Cu2–xS, even after 120 min of reaction (Figure 2f), despite the dramatic morphological changes (Figure 2e). After 120 min of reaction, these NCs no longer exhibit an LSPR absorption band (Supporting Information, Figure S3f), presumably due to their poorly defined shape.
Figure 2.

TEM images of (a) parent Cu2–xS NCs and (b–e) product NCs obtained after reacting the NCs shown in (a) with GaCl3 at 30 °C for (b) 15 min, (c) 30 min, (d) 60 min, and (e) 120 min. Red arrows indicate regions of low contrast within the nanocrystals (cartoons in the lower-left corner give an impression of the observed contrast in the NCs). Top-right inset in (e) depicts an electron diffraction pattern of the corresponding sample. Panel (f) is the azimuthally integrated ED pattern depicted in (e). Reference bars in (f) are calculated patterns based on the low-chalcocite Cu2–xS crystal structure described in ref (34). The blue curve in (f) corresponds to the PED pattern of the parent Cu2–xS nanocrystals. Dotted lines serve as guides for the eye.
To determine the nature of the low contrast region and its position within the particles, high-angle annular dark-field scanning transmission microscopy (HAADF-STEM) tomography measurements were performed. Visualizations of the 3D reconstructed volumes of NCs after different reaction times are shown in Figure 3. Animated versions of the tomograms are also provided in the web-enhanced movies 1, 2, and 3. The tomography measurements show that, after 15 min of reaction with GaCl3, a small cavity is present at the surface of some Cu2–xS NCs (Figure 3a) and increases in size as the reaction progresses (Figure 3b,c). Acquired orthoslices, which were used to reconstruct the 3D morphologies, are displayed in Figure 3d–f. These measurements indicate that the brighter regions observed in TEM images coincide with cavities at the surface of the NCs. The cavities appear to form at the surface of the NCs at early reaction stages and subsequently grow inward over time. A small portion of the NCs (indicated by red arrows in Figure 3) appears to be hollow, without an obvious opening at the surface.
Figure 3.

3D characterization of tomographic visualization of the Cu2–xS bifrustum NCs after reaction with GaCl3 at 30 °C for (a) 15 min, (b) 30 min, and (c) 60 min. After 15 min, only a small dimple is present at the surface of the NCs. After longer reaction times, these cavities are found to be larger. Red arrows indicate hollow NCs without an apparent opening at the surface. (d–f) Orthoslices used to reconstruct the 3D morphologies presented in (a–c). Animated (rotating) versions of the models depicted in panels (a–c) are provided.
These observations bear similarities with previous reports on hollow nanoparticles, whose formation has been ascribed to the nanoscale Kirkendall effect,35,36 which involves an imbalance between outward and inward diffusion fluxes during the chemical conversion of a NC into a different material through reaction with a reactant present in solution. The surface of the parent NC is first converted into a shell of the product material by reaction with a species present in solution, after which further conversion can only take place through solid-state diffusion of the reactant species through the NC.35 In case the outward diffusion rate is faster than the inward diffusion rate, there is a net flow of material going out of the NC, leaving vacancies behind.35 Condensation of these vacancies gives rise to voids that eventually collect in a central void in the NCs.35 The nanoscale Kirkendall process has also been recently proposed as an explanation for the conversion of Cu2–xS nanoplatelets into CuInS2 nanorings through partial Cu+ for In3+ CE.37 Our observations, however, are not consistent with the nanoscale Kirkendall effect, as the cavities form at the surface of the NCs and grow inward, in striking contrast with the outward growing inner voids that characterize the Kirkendall effect.
Surface erosion of the NCs and preservation of a low-chalcocite Cu2–xS shell after the reaction with GaCl3 indicate that a reaction other than cation exchange is taking place. In the successful Cu+ for In3+ cation exchange reaction using the InCl3–TOP Lewis acid–base adduct complex, TOP (a soft Lewis base) acts as Cu+ extractor,5,18 likely forming CuCl–TOP Lewis acid–base adduct complexes. In the present reaction using noncomplexed GaCl3, there are no species present to directly extract Cu+. However, GaCl3 is a strong Lewis acid38,39 and therefore can extract S2– from the NC as S2– is a Lewis base. This extraction destabilizes the crystal lattice, as the S2– anions form the framework of Cu2–xS NCs, resulting in etching of the NCs and formation of cavities at the surface. The collapse of the NC framework will release Cu+ ions, which are likely stabilized in solution by binding to the chloride ions released from GaCl3 after its reaction with the NC sulfides.
Although the NCs react with a strong Lewis acid, they do not dissolve completely, even after 300 min (Supporting Information, Figure S6). Interestingly, in micrographs obtained using HAADF-STEM, most particles exhibit a uniform contrast after reacting for 300 min (Supporting Information, Figure S6e). This uniform contrast indicates that the previously formed cavities are filled with new material. EDS elemental maps show that this material consists mostly of gallium. These observations suggest that Ga-containing monomers, formed upon extraction of S2– by GaCl3, precipitate inside the NC cavities after longer reaction times. This refilling of the cavities likely blocks further reactions on the NC surface, preventing further dissolution.
Tailoring the Ga Precursor Chemistry To Boost the Cu+ for Ga3+ Cation Exchange
We have discussed above two extreme cases of reactivity imbalance: whereas GaCl3–TOP is too inert, hardly leading to any change in the parent Cu2–xS NCs, noncomplexed GaCl3 is too reactive toward the sulfide anions, thereby etching the parent NCs without any significant cation exchange. In the following, we will tailor the chemistry of the Ga precursor complex to allow successful Cu+ for Ga3+ cation exchange.
The observation that the reactivity of the GaCl3–TOP Lewis acid–base adduct complex as a cation exchange precursor is much lower than that of the InCl3–TOP complex is surprising, considering that the metal–phosphorus bond in GaCl3–TOP should be weaker than that in InCl3–TOP as Ga3+ is a harder Lewis acid than In3+ (η = 17 and 13 eV, respectively),32 whereas TOP is a soft Lewis base (η ≈ 6 eV).5,40 This implies that other variables are more relevant in determining the Cu+ for Ga3+ cation exchange rates and prompted us to explore the influence of the nature of the phosphine ligand on the cation exchange reaction by using triphenylphosphite and diphenylphosphine as ligands for GaCl3 (Figure 4b,c). The coordination behavior of a phosphine ligand with respect to a metal center is determined by its steric and electronic properties.41,42 The most frequently used parameter to quantify the steric properties of phosphines is the Tolman cone angle, defined as the apex angle of a cylindrical cone with origin 2.28 Å from the center of the phosphorus atom, whose sides just touch the Van der Waals surfaces of the outermost atoms of the organic substituents.41 The three phosphine ligands used in the present work have significantly different electronic properties and total molecular volume, but their Tolman cone angles are essentially the same: 128° for both DPP and TPP and 132° for TOP.41 The value for TOP is taken to be the same as that reported by Tolman for other tertiary alkylphosphines, such as tri-n-butylphosphine, although a recent work has estimated it to be 128°.40 Steric effects are thus not expected to play a significant role in the cation exchange reactions investigated in our work. The electronic properties of phosphines are determined by their σ-donor ability and π-acceptor strength as they bind to metals by a combination of σ-donation from the lone pair of the P atom and π-back bonding from the d orbitals of the metal into their empty P–R σ* molecular orbital (R = organic substituent).41,42 More electronegative (i.e., electron-withdrawing) substituents on the phosphorus atom decrease the basicity and σ-donating ability of the phosphine, while increasing its π-acceptor strength.40−43 This effect is due to the decrease of the electronic density on the phosphorus atom and increase of the s-character of the lone electron pair.41,42 An increase in electronegativity of the substituents on the phosphorus atom should lead to weaker bonding to the metal, as should be the case for GaCl3–TPP and GaCl3–DPP compared to GaCl3–TOP.
Figure 4.
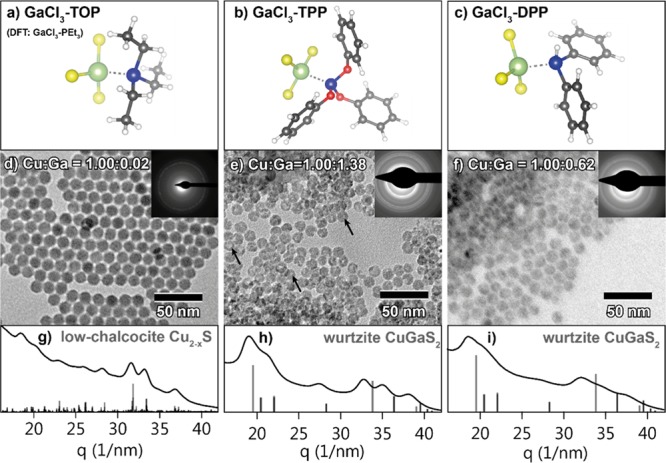
(Top row) Ground-state geometries of three GaCl3–phosphine complexes obtained from DFT calculations (see below): (a) GaCl3 complexed with triethylphosphine, used as model for GaCl3 complexed with trioctylphosphine (GaCl3–TOP), (b) GaCl3 complexed with triphenylphosphite (GaCl3–TPP), and (c) GaCl3 complexed with diphenylphosphine (GaCl3–DPP). (Middle row) TEM images of product NCs obtained after reaction of Cu2–xS NCs with (d) GaCl3–TOP at 100 °C, (e) GaCl3–TPP at 100 °C, and (f) GaCl3–DPP at 50 °C. Black arrows indicate intraparticle contrast in the NCs reacted with GaCl3–TPP. Top-right insets show the corresponding ED patterns. Bottom row (g–i) shows the corresponding azimuthally integrated ED patterns. Reference bars in (h,i) are calculated patterns based on the wurtzite CuGaS2 crystals structure,44 and reference bars in (g) are based on the low-chalcocite Cu2–xS crystal structure.34
Indeed, the use of both GaCl3–TPP and GaCl3–DPP as reactants results in successful CE reactions even after reaction times much shorter than those used for GaCl3–TOP (1 h instead of 12 h, at the same temperature), as clearly demonstrated by EDS analysis of the product NCs obtained after reaction with GaCl3–TPP and GaCl3–DPP at 100 °C, which reveals Cu/Ga ratios of 1.00:1.38 and 1.00:1.48, respectively (Supporting Information, Figures S7 and S8). The NCs that reacted with GaCl3–TPP are partially deformed and exhibit intraparticle contrast in TEM images (Figure 4e, indicated by black arrows), similar to the NCs that reacted with GaCl3 (see Figures 2 and 3). The NCs that reacted with GaCl3–DPP form agglomerates (Supporting Information, Figure S9), which is likely due to displacement of the original organic capping ligands (dodecanethiol) with DPP during the CE reaction. DPP is a relatively small molecule and thus poorly suitable as a steric stabilizer. In comparison, the NCs cluster less after reaction with GaCl3–TPP as triphenylphosphite is bulky enough to provide steric stabilization. To increase the colloidal stability of the NCs reacted with GaCl3–DPP, oleylamine (OLAM) was added to the reaction mixture and a lower reaction temperature (50 °C) was employed, which indeed inhibited NC agglomeration (Figure 4f). However, under these milder reaction conditions, the Cu+ for Ga3+ exchange proceeds to a lesser extent, as shown by EDS analysis (Cu/Ga ratio of 1.00:0.62, Supporting Information, Figure S10). The PED pattern (Figure 4e,h) of the NCs reacted with GaCl3–TPP corresponds to the wurtzite crystal structure, confirming that Cu+ for Ga3+ CE has indeed taken place. Interestingly, the peaks in the ED pattern are shifted to lower q values compared to those of the reference wurtzite CGS, indicating that the product CGS NCs have a larger lattice constant than the reference. This observation will be discussed in more detail below. The PED pattern (Figure 4f,i) of the NCs reacted with GaCl3–DPP exhibits broad, poorly defined peaks, indicating that these particles have partially lost their crystallinity during the CE procedure, which precludes a definitive assignment of the crystal structure. The NCs reacted with GaCl3–TPP and GaCl3–DPP are both smaller than the parent Cu2–xS NCs (diameters = 10.8 ± 1.0, 11.1 ± 1.1, and 12.3 ± 1.0 nm, respectively, Supporting Information, Figure S1). The NCs reacted with GaCl3–TPP no longer exhibit an LSPR absorption band, whereas those reacted with GaCl3–DPP exhibit an LSPR absorption band with a reduced intensity (Supporting Information, Figure S3d,e). In the latter NCs, Cu+ was exchanged for Ga3+ to a lesser extent; the presence of an LSPR absorption band thus indicates that these NCs remain cation-deficient, requiring excess holes for charge neutrality.
The intraparticle contrast observed in the NCs after reaction with GaCl3–TPP was studied with HAADF-STEM and EDS elemental mapping. In micrographs obtained using HAADF-STEM, the intraparticle contrast appears similar to that observed in TEM images (compare Figure 5a to Figure 4e). In the elemental maps, the regions of low contrast coincide with regions of low signal for both cations (Figure 5, indicated by orange arrows). As the Cu and Ga distributions correlate well (Figure 5b,c), the intraparticle contrast is clearly not due to an inhomogeneous composition. Instead, it appears that the low contrast regions in HAADF-STEM and TEM images simply contain less material. Likely, surface erosion by GaCl3 is not completely suppressed when GaCl3–TPP is used as reactant, resulting in the formation of small cavities at the NC surface.
Figure 5.

(a) HAADF-STEM images and EDS elemental maps of (b, blue) Cu and (c, red) Ga of product NCs obtained by reacting Cu2–xS NCs with GaCl3–TPP at 100 °C. Orange arrows indicate regions of low contrast (HAADF-STEM) coinciding with regions of low signal (EDS maps).
Similar to the previously reported conversion of low-chalcocite Cu2–xS into wurtzite CIS NCs by Cu+ for In3+ CE,16,18,33 the exchange of Cu+ for Ga3+ in Cu2–xS NCs using GaCl3–DPP or GaCl3–TPP as precursors is also partial and self-limited as CGS, rather than Ga2S3, NCs are produced. In the case of the Cu+ for In3+ CE in Cu2–xS NCs, the topotactic, partial, and self-limited character of the exchange was attributed to the high energy barrier associated with the conversion of the hexagonal close-packed anionic sublattice of the parent low-chalcocite Cu2–xS NCs into the face-centered cubic sublattice of the spinel structure of In2S3, which requires a substantial reorganization of the sulfide anions (by 58% of a S–S interatomic distance).16 In contrast, the hexagonal wurtzite structure of CuInS2 requires only minor rearrangements of the sulfide sublattice with respect to that of the template NCs (1.4 and 6.4% contraction within and between the hexagonal S2– layers, respectively).16 The topotactic nature of the cation exchange in Cu2–xS NCs has been recently exploited to achieve structure-selective conversion of Cu2–xS NC templates into ZnS, CdS, and CuInS2, with hexagonal roxbyite Cu1.78S and cubic digenite Cu1.8S yielding product NCs with the wurtzite and the zinc blende structures, respectively.45
In the present case, however, the structural differences between the possible products and the template NCs are less pronounced because low-chalcocite Cu2–xS, wurtzite CuGaS2, and the stable form of bulk Ga2S3, monoclinic α′-Ga2S3, all have a hexagonal close-packed sulfide sublattice,34,44,46 with hexagonally structured layers stacked in an ...ABAB... fashion (Figure 6a,b). However, we observe that, in all three dimensions, the wurtzite CuGaS2 structure exhibits a significant lattice contraction compared to that of low-chalcocite Cu2–xS (Figure 6c,d). These contractions are 5.6, 5.7, and 9.5% in x, y, and z, respectively. On the other hand, the anion positions of wurtzite CuGaS2 and α′-Ga2S3 are very similar: transforming the former into the latter, the lattice has to contract by only 0.6, 0.5, and 2.5% in x, y, and z, respectively (the definition of x, y, and z directions in terms of crystallographic directions are provided in the caption of Figure 6). It is thus clear that, based on the bulk crystal structure parameters, the sulfide sublattice of the template low-chalcocite Cu2–xS would have to undergo similar degrees of reconstruction, regardless of whether the cation exchange would be partial, yielding wurtzite CuGaS2, or complete, yielding α′-Ga2S3. However, we note that the product wurtzite CGS NCs (with Cu/Ga ratios ranging from 1:1.48 to 1:0.62) obtained in our work exhibit a larger lattice constant than the wurtzite CuGaS2 reference (Figure 4h,i), indicating that the degree of contraction was smaller than expected based on the bulk lattice parameters, and therefore, that the product CGS NCs are under tensile strain. This suggests that the mild reaction conditions used in our work do not allow the anion sublattice to undergo full reconstruction in order to relax the strain induced by the CE.
Figure 6.

(a,b) Hexagonal close-packed structure viewed from two different orientations: (a) characteristic arrangement into hexagonally constructed layers, defined here as the x,y-plane, and (b) characteristic ...ABAB... stacking of these hexagonal layers, defined here as the x,z-plane. (c,d) Anion positions of (black) low-chalcocite Cu2–xS, (cyan) wurtzite CuGaS2, and (purple) α′-Ga2S3 overlaid onto each other. (c) Hexagonal ordering in the x,y-plane and (d) ...ABAB... stacking in the x,z-plane. Δx, Δy, and Δz, denoted in (a,b), correspond to the lattice distances used to determine the lattice contractions mentioned in the main text. The x,y,z-directions correspond to the following crystallographic directions: (low-chalcocite Cu2–xS) [201],[010],[001]; (wurtzite CuGaS2) [11̅0],[110],[001]; (monoclinic α′-Ga2S3) [010],[100],[102]. Anion coordinates were obtained from the crystal structures of low-chalcocite Cu2–xS,34 wurtzite CuGaS2,44 and monoclinic α′-Ga2S3.46
Recently, lattice strain was shown to play an important role in the conversion of djurleite Cu2–xS nanorods (NRs) into Cu2–xS/CGS hetero-NRs by partial Cu+ for Ga3+ CE.30 Shim and co-workers showed that lattice strain prevented the homogeneous conversion of Cu2–xS NRs (∼170 nm long, 20 nm diameter) into CGS NRs, resulting instead in hetero-NRs containing one or multiple wurtzite CGS domains.30 The authors argued that the strain induced by the lattice mismatch between the parent djurleite Cu2–xS phase and the product wurtzite CGS phase would make Ga3+ diffusion into Cu2–xS unfavorable, thereby limiting the extent of the CE reaction and leading to wurtzite–djurleite superlattice formation as a way to minimize the resulting strain. However, we note that in the present work Cu2–xS NCs were successfully converted into homogeneous CGS NCs, despite the tensile strain resulting from insufficient contraction to the lattice parameters of fully relaxed bulk CuGaS2. This difference can be attributed to the fact that the NCs used here are much smaller than those used by Shim and co-workers (viz. 12 nm diameter vs ∼170 nm long, 20 nm diameter rods). The smaller volume of the template NCs used in our work favors conversion to homogeneous CGS NCs because smaller NCs can more easily accommodate lattice strain. This also offers a possible explanation for the self-limiting character of the Cu+ for Ga3+ exchange in Cu2–xS NC templates: the α′-Ga2S3 structure is probably unable to accommodate as much tensile strain as wurtzite CGS, preventing full CE.
Density Functional Theory Calculations
To corroborate our assumption of weaker Ga–P bond strengths in GaCl3–DPP and GaCl3–TPP with respect to GaCl3–TOP, we conducted DFT calculations at the B3LYP/6-311+G(d,p);Ga:SDD level to estimate both the ground-state geometry of the complexes and the Ga–P bond enthalpy. In the DFT calculations, we modeled TOP as triethylphosphine to avoid an extensive conformational search due to the many degrees of freedom of the long alkyl chains. We expect that only the first few CH2 groups of TOP significantly influence the Ga–P bond, making PEt3 an acceptable surrogate for TOP. To verify whether the outcome of these calculations is physically reasonable, we compare calculated geometries to experimental structures obtained from X-ray crystallography. We include an additional complex (GaCl3–PPh3), as, to the best of our knowledge, the structures of GaCl3–DPP and GaCl3–TPP have not been resolved experimentally. The results of the DFT calculations (Ga–P bond lengths and dissociation enthalpies) and the experimentally determined Ga–P bond lengths are shown in Table 1. The Ga–P bond lengths determined by DFT deviate from the experimental ones by only 4–5%. Moreover, the DFT results reproduce the experimental trend in Ga–P bond length from GaCl3–PEt3 to GaCl3–PPh3. Thus, although the absolute values obtained from the DFT calculations are 4–5% longer than the experimental ones, the trends in the DFT results are reliable. We observe that, with increasing electron-withdrawing nature of the side groups on the phosphine ligand, the Ga–P bond length L increases (viz., LGaCl3–TOP < LGaCl3–DPP < LGaCl3–TPP). The dissociation enthalpy follows the reverse trend. These results show that, indeed, the Ga–P bonds in GaCl3–DPP and GaCl3–TPP are weaker than those in GaCl3–TOP.
Table 1. DFT-Calculated (B3LYP/6-311+G(d,p);Ga:SDD) and Experimental Ga–P Bond Lengths and DFT-Calculated Dissociation Enthalpies of GaCl3–PEt3, GaCl3–PPh3, GaCl3–DPP, and GaCl3–TPP.
Mechanism for the Cu+ for Ga3+ Exchange in Cu2–xS NCs
Nanoscale CE reactions consist of a number of linked elementary kinetic steps:5,6,8,12 (i) extraction of the host cations from the NC surface, (ii) incorporation of guest cations at the NC surface, (iii) solid-state diffusion of the guest cations into the NC, and (iv) solid-state diffusion of the native cations to the NC surface. The thermodynamic driving force for the reaction is determined by the energy balance of the overall reaction.5,6,8 However, the energy input or output involved in each step is important as all steps are kinetically linked and must proceed in a concerted manner. Therefore, the outcome of nanoscale CE reactions (i.e., extent of the exchange, homogeneous or heterostructured nanocrystals, etc.) is dictated by the kinetic balance between a number of competing processes taking place in solution, at the NC surface and within the NC.
Aliovalent CE reactions are inherently more complex than isovalent ones as preservation of charge balance is in this case kinetically more challenging because a different number of host and guest cations must be exchanged to keep charge neutrality (e.g., three Cu+ cations must be extracted to allow the incorporation of one In3+ or Ga3+ cation). Aliovalent CE involving Cu+ has been extensively investigated because the small charge and high solid-state diffusion rates of Cu+ ions make both their incorporation into and extraction from NCs facile and fast. This makes copper chalcogenide NCs attractive not only as templates for other binary, multinary, or hetero-NCs that cannot be easily obtained by direct routes (e.g., CuInS2,16,28,30,33,37,45 CuInSe2,8,49,50 Cu4SnS4,19,51 Cu2–xSe/ZnSe,52 ZnS/Cu1.8S,28,53etc.) but also as intermediates in sequential CE reactions, making it possible to synthesize NCs and hetero-NCs that would otherwise remain inaccessible (e.g., PbS NRs from CdS NRs by Cd2+ for Cu+ followed by Cu+ for Pb2+ CE,5 wurtzite ZnSe/ZnS and CuInSe2/CuInS2 core/shell NRs from CdSe/CdS core/shell NRs18,54). As a result of this extensive research, the thermodynamics and kinetics of aliovalent CE reactions involving Cu+ are reasonably well-understood and known to depend on many variables: presence of Cu-extracting ligands in solution,5,6,8 leaving ability of the ligands bound to the incoming guest Mn+ cation,37 concentration of Cu+ vacancies,52,55 valence and diffusion rate of the incoming guest Mn+ cation,8,28,30 crystal structure of the template Cu2–xS NCs,8,45 and miscibility of the parent and product phases.8 As we will discuss below, the results presented above unravel one more crucial parameter governing the kinetics of CE reactions: the activated complex that must form at the surface of the template NC when a direct place exchange reaction is used (i.e., when the Cu-extracting ligand is bound to the guest heterocation).
As previously demonstrated by Buhro and co-workers,37 the aliovalent Cu+ for In3+ exchange in Cu2–xS NCs is in the reaction-limited regime. The incorporation of In3+ is preceded by the adsorption of the InX3 precursor on the NC surface. The displacement of the X– ligand is proposed to occur by nucleophilic attack of lattice S2– ions on the adsorbed InX3 precursor.37 The height of the energy barrier for this reaction is given by the leaving-group ability of the X– ligand.37 We add that when a direct place exchange reaction is promoted by using a TOP–InCl3 complex, the energy barrier is lowered by the formation of an incipient bond between the P donor group and a Cu+ ion in the vicinity of the S2– ion responsible for the nucleophilic attack on the In–Cl bond. This makes the reaction rates faster and allows for a direct place exchange between the incoming In3+ guest cation and one outgoing Cu+ cation, while facilitating extraction of two additional Cu+ ions as neutral CuCl units. However, the formation of such an activated complex imposes constraints on the orientation of the binding TOP–InCl3 with respect to the surface Cu+ and S2–. Nevertheless, in the case of Cu+ for In3+ CE, these constraints are not very strict because the activated complex can follow only two possible reaction pathways: CE or desorption without reaction, in which case it may bind again in the proper orientation later.
In the case of Cu+ for Ga3+ CE, the constraints imposed on the activated complex are more severe because GaCl3 is a much stronger Lewis acid than InCl3, therefore being capable of extracting lattice S2– from the template Cu2–xS NCs (Ga–S = 393 kJ/mol, In–S = 313–316 kJ/mol),56 leading to etching rather than CE (Figure 7a), as demonstrated above (Figures 2 and 3). Complexation of GaCl3 with TOP suppresses the NC etching but does not lead to CE. This demonstrates that the activated complex formed upon adsorption of GaCl3–TOP on the NC surface effectively weakens the interaction between Ga and the lattice S2– and also prevents interaction between the P donor head and the Cu+ ions (Figure 7b). In contrast, both GaCl3–TPP and GaCl3–DPP result in successful Cu+ for Ga3+ CE. All three ligands (TOP, TPP, DPP) are bound to GaCl3 through the lone pair on the phosphorus atom and an empty sp3 orbital of gallium.48,57,58 To extract Cu+, the phosphine acts as Lewis base and binds to Cu+ through the same lone pair. In order for the phosphine to bind to Cu+, while still preventing etching by attack of the Ga3+ to lattice S2– ions, the activated complex at the NC surface (Figure 7c) must be such that the Cu–P bond formation and the Ga–P bond breaking occur simultaneously (Figure 7c, steps 1–3) and are followed by the immediate place exchange between Cu+ and Ga3+. We attribute the higher CE reactivities of the GaCl3–TPP and GaCl3–DPP to a more favorable geometry of the activated complex formed upon their adsorption on the surface of the template Cu2–xS NCs, as well as a weaker Ga–P bond compared to GaCl3–TOP. In GaCl3–TPP and GaCl3–DPP, the Ga–P bond is weaker than that in GaCl3–TOP (see Table 1), fulfilling the conditions for a fast place exchange between Ga3+ and Cu+, while still making the competing reaction between GaCl3 and the lattice S2– ions less likely (Figure 7c, steps 1–3). We note that the Cu+ for Ga3+ place exchange is thermodynamically favored as it results in the formation of two new bonds that are stronger than the initial ones (viz., the Cu–P bond in the Cu–phosphine Lewis acid–base adduct, which is favored because Cu+ is a much softer Lewis acid than Ga3+, η = 6.28 and 17 eV, respectively,32 and the Ga–S bond, which has a bond strength of 393 kJ/mol while the enthalpy of formation of Cu2S is only −79.5 kJ/mol59). Nevertheless, some degree of etching still occurs as the NCs reacted with GaCl3–TPP and GaCl3–DPP are smaller than the template NCs (viz., 10.8 ± 1.0, 11.1 ± 1.1, and 12.3 ± 1.0 nm, respectively, Supporting Information, Figure S1). The observed shrinkage (12.2 and 9.76%, respectively) is too large to be attributed to a decrease in unit cell volume alone (see discussion on crystal lattice contraction above). The difference in reactivity between InCl3–TOP and GaCl3–TOP as CE reactants may be due to the fact that In3+ exhibits three-, four-, five-, and six-coordination, whereas Ga3+ is limited to three- and four-coordination.57 InCl3–TOP has thus more degrees of freedom to rearrange itself at the nanocrystal surface to form the Cu2–xS–InCl3–TOP activated complex, whereas GaCl3–TOP does not, making the formation of the Cu2–xS–GaCl3–TOP activated complex subjected to more stringent orientational constraints and therefore less likely.
Figure 7.

Schematic depiction of the Cu+ for Ga3+ cation exchange reaction in Cu2–xS nanocrystals. (a) Free GaCl3 reacts as a Lewis acid, extracting S2–, leading to etching of the nanocrystals. (b) Extraction of S2– is inhibited when GaCl3 is complexed with a phosphine ligand. (c) Successful cation exchange involves four steps: (i) cleavage of the Ga–P bond at the Cu2–xS nanocrystal surface, at which point Ga–S and P–Cu bonds are made simultaneously; (ii) extraction of Cu+ by the phosphine; (iii) subsequent incorporation of Ga3+ into the nanocrystal; and (iv) reaction of remaining Cl– with Cu+, extracting two additional Cu+ ions, completing the exchange.
As mentioned above, the steric demands of all three phosphines used in the present work are essentially the same (viz., 128° for DPP and TPP, 132° for TOP),41 and therefore, steric effects are not expected to play a significant role in the outcome of the cation exchange reactions. However, Tolman cone angles are just a first approximation as they do not take into account the effect of different M–P bond lengths, different metal centers, and the overall interaction with other ligands coordinated to the same metal center.43,60,61 Therefore, actual cone angles obtained from X-ray structural data (the so-called solid cone angles) typically deviate from the Tolman cone angles, varying over a wide range depending on the nature of the metal center and bond length (shorter bonds result in larger cone angles).60 For example, the solid cone angles determined for triphenylphosphine complexes range from 129 to 168°,61 whereas the Tolman cone angle of triphenylphosphine is 145°.41,43 In the case of Ni complexes, a slightly larger solid cone angle has been reported for TPP with respect to DPP complexes (135 and 124°, respectively),43 consistent with the bulkier nature of the former (containing three phenyl groups) with respect to the latter (which contains only two phenyl groups). If the impact of steric effects on the outcome of the cation exchange reactions were significant, one would expect a difference between the reactivity of the complexes using TPP and DPP as ligands, in contrast with our experimental data, which show that GaCl3–TPP and GaCl3–DPP have similar reactivities. This implies that the kinetics of the cation exchange reactions studied in our work are primarily dictated by the electronic properties of the phosphine ligands used, which determine the Ga–P bond strength of the resulting Lewis acid–base adduct complexes with GaCl3 and the geometry of the activated Cu2–xS–GaCl3–L complex that forms at the surface of the Cu2–xS NC at the onset of the cation exchange reaction. After the place exchange between the outgoing Cu+ and the incoming Ga3+ cations has taken place, the two chlorides still bound to the incorporated Ga3+ cation will react with two neighboring Cu+ cations, extracting them from the NC surface as neutral CuCl units, which are highly soluble in the apolar reaction medium used in our experiments (Figure 7c, step 4). This likely increases the thermodynamic driving force of the overall CE reaction both entropically and enthalpically as two more chemical species are released in solution and solvated.5,6,8 The two Cu+ vacancies left will further promote the CE reaction by allowing more Cu+ cations to diffuse to the surface, while allowing the incorporated Ga3+ guest cation to diffuse inward. As previously shown for CE reactions in copper chalcogenide NCs, the CE rates increase with increasing concentration of Cu+ vacancies.52,62
Cu+ for Ga3+ Exchange in Luminescent CuInS2 NCs
To illustrate the general applicability of the findings presented above, we used GaCl3–DPP to convert luminescent CuInS2 (CIS) NCs into (Cu,In,Ga)S2 (CIGS) NCs with much brighter photoluminescence (see Methods for details). The parent CIS NCs have an indium-rich composition with a Cu/In ratio of 0.68 ± 0.008:1. EDS analysis of the product NCs gives a Cu/Ga/In ratio of 0.20 ± 0.001:0.16 ± 0.001:1, indicating that Cu+ ions are exchanged for Ga3+. These cation ratios imply that the charge balance is preserved through the CE reaction, as the relative concentration of Ga ions multiplied by their charge of +3 exactly corresponds to the decrease in Cu concentration (with charge +1; 0.16 × 3 = 0.48; 0.68–0.20 = 0.48). Interestingly, the size (∼5.5 nm) and pyramidal shape of the CIS NCs are preserved in the product CIGS NCs (see Figure 8a,d), despite the removal of a significant amount of cations from the NC. Fast Fourier transform (FFT) analysis of the high-resolution TEM images allows determination of the lattice spacings, being 0.18, 0.21, and 0.34 nm before and after the reaction (i.e., they are not significantly changed by the CE reaction). However, analysis of the ED patterns shows a shift to higher q values (Figure 8e), indicating a small contraction of the unit cell. This contraction is consistent with the exchange of the relatively large Cu+ for the relatively small Ga3+ (rCu+ = 60 pm, rGa3+ = 47 pm).31
Figure 8.
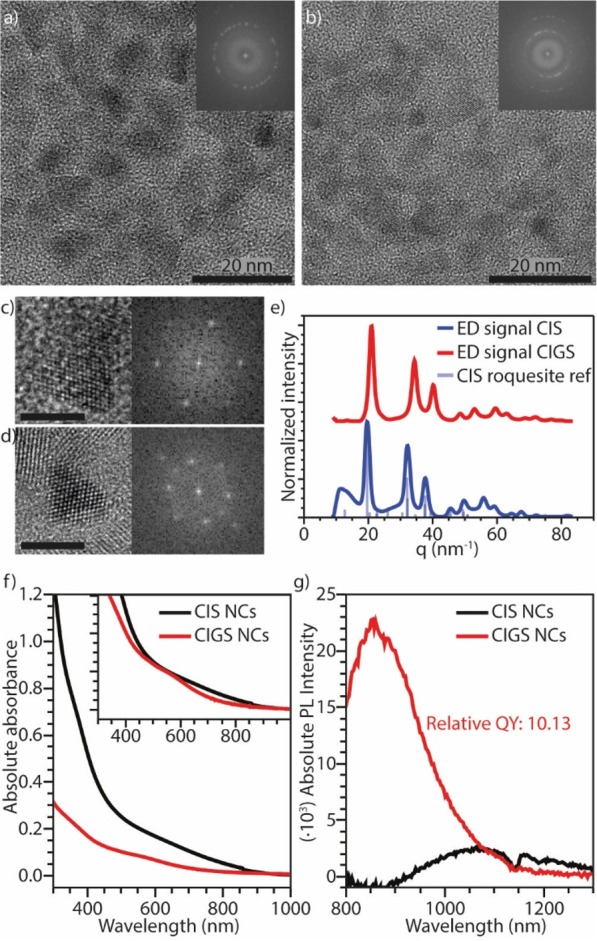
Cu+ for Ga3+ cation exchange in luminescent CIS NCs. (a) High-resolution TEM image of the parent CIS NCs. The inset shows the FFT of the image. (b) High-resolution TEM image of the product CIGS NCs after reaction; the inset shows the FFT of the image. (c,d) Enlarged TEM image of single NCs before (c) and after (d) the Ga3+ for Cu+ CE reaction, with the corresponding FFTs. Both scale bars correspond to 5 nm. (e) Azimuthally integrated ED patterns of the parent CIS NCs and product CIGS NCs, and bulk roquesite CIS X-ray diffraction reference pattern (JPCDS PDF-card 00-047-1372). (f) Absorption and (g) PL spectra of the parent CIS NCs and product CIGS NCs. The inset in (f) shows the normalized absorption spectra. The PL quantum yield of the product CIGS NCs is at least 10 times higher than that of the parent CIS NCs. The dip in the PL spectrum around 1150 nm is caused by absorption of emitted light by toluene, which was used as solvent.
The optical properties of the template CIS NCs and the product Ga-containing (CIGS) NCs are shown in Figure 8f,g. Both the absorption (Figure 8f) and photoluminescence (PL, Figure 8g) spectra show a blue shift after the CE reaction, indicating a widening of the band gap. This can be attributed to either an increase in quantum confinement due to a smaller effective CIS core size,2,33 in case a CIS/CIGS core/shell NC is formed, or by the intrinsic increase in band gap,63,64 in case a homogeneous CIGS alloy is formed, as the band gap of bulk CuGaS2 is larger than that of CuInS2 (2.50 and 1.55 eV, respectively).1−3 Our results, however, do not allow a distinction between the two cases to be made. The PL quantum yield (QY) of the product CIGS NCs increases by 1 order of magnitude in comparison to that of the parent CIS NCs (Figure 8g). Such an increase in PLQY could originate from a strong enhancement of the radiative recombination rates, from elimination of nonradiative recombination pathways, or from a combination of both. Considering that similar PLQY enhancements have been previously observed upon shelling of CIS NCs with both ZnS and CdS, and attributed to passivation of nonradiative recombination centers at the surface,1,65−67 it is likely that the higher PLQY of the CIGS NCs obtained by cation exchange is also due to improved surface passivation. It is interesting to note that PL of CIGS NCs is generally not reported,26,68 suggesting that the exciton recombination in these materials tends to be dominated by nonradiative decay pathways, in striking contrast to the (Cu-poor) CIGS NCs prepared in our work by postsynthetic Cu+ for Ga3+ cation exchange in CIS NCs. The scarce literature on the PL of CIGS NCs makes it difficult to unambiguously identify the radiative recombination mechanism in these materials. However, their large global Stokes shift and broad PL peak suggest that, similar to CIS and other I–III–VI2 NCs, the PL in these nanomaterials most likely originates from the radiative recombination of a delocalized conduction band electron with a hole localized at a Cu(I) ion.69 Time-resolved spectroscopic measurements could provide additional insights into the origin of the radiative recombination in these nanomaterials but are beyond the scope of the present work. It should be noted that the PL of these Pb- and Cd-free NCs is centered in the NIR biological window.65 This, in combination with their high PLQY, makes them promising candidates for bioimaging applications. Most importantly, the results discussed above show that the CE mechanism proposed in the present work is widely applicable.
Conclusions
Our work shows the importance of reactant chemistry in aliovalent cation exchange reactions in Cu2–xS nanocrystals. Noncomplexed GaCl3 reacts as Lewis acid, extracting S2– from the nanocrystals, thereby etching them. Etching is inhibited when GaCl3 is bound to a phosphine ligand in a GaCl3–L complex (L = trioctylphosphine, triphenylphosphite, and diphenylphosphine). GaCl3–TOP was found to be unreactive, whereas GaCl3–TPP and GaCl3–DPP lead to successful exchange of Cu+ for Ga3+, converting the parent Cu2–xS nanocrystals into CuGaS2 nanocrystals. This is rationalized by considering that the activated Cu2–xS–GaCl3–L complex that forms at the surface of the nanocrystal upon adsorption of the GaCl3–L complex must allow for simultaneous Ga3+ insertion and Cu+ extraction, while preventing S2– extraction. This requirement is only fulfilled by activated complexes in which the Ga–P bond is sufficiently weak. DFT calculations show that the Ga–P bond strength decreases with increasing electron-withdrawing nature of the side groups of the phosphine ligand. Our findings highlight several factors (viz., Lewis acidity, influence of ligand electronic properties on reactivity) which need be taken into account when designing cation exchange protocols. Finally, we showed that the mechanism proposed here is generally applicable by using GaCl3–DPP to convert CuInS2 nanocrystals into Cu-poor (Cu,Ga,In)S2 nanocrystals with an increased photoluminescence quantum yield in the NIR spectral window.
Methods
Materials
Copper(II) sulfate pentahydrate (CuSO4·5H2O, 99.995%), oleic acid (OA, tech., 90%), 1-dodecanethiol (DDT, ≥98%), oleylamine (tech., 70%), gallium(III) chloride (GaCl3, anhydrous, beads), trioctylphosphine (90%), diphenylphosphine (98%), triphenylphosphite (97%), indium(III) acetate (In(Ac)3, 99.99%), copper(I) iodide (CuI, 98%), anhydrous methanol, anhydrous butanol, and anhydrous toluene were all purchased from Sigma-Aldrich and used as supplied, except for OA and OLAM, which were degassed prior to use.
Synthesis of Cu2–xS Bifrustum Nanocrystals
Cu2–xS bifrustum nanocrystals were prepared according to the method described in the literature.16 Typically, 0.203 g of CuSO4·5H2O, 6 mL of OA, and 7.5 mL of DDT were brought into a three-neck round-bottom flask, connected to a Schlenk line. Under constant stirring and nitrogen atmosphere, the mixture was heated to 200 °C at a rate of about 30 °C/min. After reaction for 2 h at 200 °C, the mixture was allowed to cool to approximately 110 °C, when it was quenched by injection of 5 mL of toluene. The product was precipitated by addition of a mixture of methanol and butanol as nonsolvent and isolated by centrifugation at 2500 rpm for 10 min. The supernatant was removed, and the residue was redispersed in toluene. The mass of product was estimated by weighing the empty sample vials prior to sample retrieval and after removal of the supernatant. The amount of toluene used to redisperse the particles was chosen such that a concentration of approximately 20 g/L was obtained.
Preparation of Cation Exchange Precursors
Ga– and In–phosphine precursors were prepared in a way similar to our previously reported method.18
InCl3–TOP was prepared in a N2-filled glovebox, by addition of 1.250 mL (2.8 mmol) of TOP to an equimolar amount (0.620 g) of InCl3, followed by heating to 50 °C under constant stirring. The resulting turbid, white, viscous liquid was diluted by addition of 1.250 mL of toluene.
GaCl3–TOP was prepared in a N2-filled glovebox, by addition of 0.500 mL (1.12 mmol) TOP to an equimolar amount (0.197 g) of GaCl3, followed by heating to 50 °C under constant stirring, yielding a transparent, orange, viscous liquid.
GaCl3–DPP was prepared in a N2-filled glovebox, by addition of 0.815 mL (4.683 mmol) DPP to an equimolar amount (0.8246 g) of GaCl3, followed by heating to 50 °C under constant stirring. After a few minutes, an opaque, white, viscous liquid formed. Over the course of several hours, the liquid changed into an opaque, white, crystalline solid, which was dissolved in 10 mL of toluene at 100 °C.
GaCl3–TPP was prepared in a N2-filled glovebox, by addition of 1.125 mL (4.281 mmol) TPP to an equimolar amount (0.7538 g) of GaCl3, followed by heating to 50 °C under constant stirring. After a few minutes, an opaque, white, crystalline solid formed, which was dissolved in 10 mL of toluene at 100 °C.
GaCl3 cation exchange precursor was prepared in a N2-filled glovebox, by making a 1 mol/L (176.08 g/L) solution of GaCl3 in toluene, resulting in a transparent, bright green liquid. It was found that the solution degraded over time, even when stored under inert atmosphere. Degradation was indicated by a change of color from bright green to dark red. In all reactions with GaCl3 as precursor, a fresh batch of precursor was used.
Cation Exchange in Cu2–xS Bifrustum Nanocrystals
The cation exchange reactions were performed following the method reported in ref (18) inside a N2-filled glovebox. The equivalent of 0.2 mmol cation exchange precursor (0.200 mL of InCl3–TOP, 0.100 mL of GaCl3–TOP, 0.425 mL of GaCl3–DPP, 0.465 mL of GaCl3–TPP, 0.200 mL of GaCl3 solution) was placed in a vial. Toluene was added to make the total reaction volume after addition of the NC dispersion 2.700 mL (added volume: with InCl3–TOP, 2.000 mL; with GaCl3–TOP, 2.100 mL; with GaCl3–DPP, 1.775 mL; with GaCl3–TPP, 1.735 mL; with GaCl3, 2.000 mL). The mixture of toluene and cation exchange precursor was heated to the reaction temperature (100 °C with InCl3–TOP, GaCl3–TOP, GaCl3–DPP, GaCl3–TPP and 30 °C with GaCl3) under constant stirring at 400 rpm. Once the desired temperature was reached, 0.500 mL of parent NC dispersion (equivalent to approximately 10 mg of parent particles) was swiftly injected. The reaction was stopped (reaction time: with InCl3–TOP, overnight; with GaCl3–TOP, overnight; with GaCl3–TPP, 60 min; with GaCl3–DPP, 60 min; with GaCl3, 15, 30, 60, 120, and 300 min) by removing the vial from the heater and quenched by the addition of methanol and butanol as nonsolvents. All samples were washed at least twice prior to analysis, by addition of methanol and butanol as nonsolvent, centrifugation and removal of the supernatant. Finally, the particles were redispersed in toluene.
Cation Exchange under Milder Reaction Conditions with GaCl3–DPP
To prevent particle agglomeration, the reaction of Cu2–xS nanocrystals with GaCl3–DPP was also performed under milder reaction conditions. This reaction was performed at half the scale of the reactions discussed above, inside a N2-filled glovebox. To this end, 0.1 mmol (0.210 mL) GaCl3–DPP, 0.860 mL of toluene, and 0.025 mL of OLAM were placed in a vial. The mixture was heated to 50 °C, under constant stirring at 400 rpm. Once the desired temperature was reached, 0.250 mL of parent NC dispersion (equivalent to approximately 5 mg of parent particles) was swiftly injected. The reaction was stopped by removing the vial from the heater and quenched by the addition of methanol and butanol as nonsolvents. The product was washed twice prior to analysis, by addition of methanol and butanol as nonsolvent, centrifugation, and removal of the supernatant. Finally, the particles were redispersed in toluene.
Synthesis of CuInS2 NCs
For the synthesis of luminescent CuInS2 (CIS) NCs a protocol reported earlier by Li et al.(66) was adapted. Briefly, 297 mg of In(Ac)3, 191 mg of CuI, and 5 mL of DDT were mixed and degassed under vacuum for 1 h at 80 °C. The reaction mixture was then heated under N2 flow to 230 °C and allowed to react for 100 min. The reaction was stopped by removing the heating mantle. The NCs were precipitated with a methanol/butanol 1/1 mixture and, after centrifugation and decanting the supernatant, dispersed in 10 mL of toluene.
Cation Exchange in CuInS2 NCs
CIS NCs (0.250 mL) in toluene were diluted in a vial with 1 mL of toluene and mixed with 0.1 mL of DDT (extra ligand) and 0.1 mL of GaCl3–DPP (prepared as described above, however, not dissolved in toluene), which was melted at a heating plate with a plate temperature of 100 °C. The vial containing this mixture was placed on the heating plate with Tplate = 100 °C for 30 min, after which the vial was removed and the NCs were precipitated with a methanol/butanol 1/1 mixture and, after centrifugation and decanting the supernatant, dispersed in 0.2 mL of toluene.
Optical Spectroscopy
Samples were prepared by placing a diluted dispersion of nanocrystals in toluene in a quartz cuvette with 10 mm path length. Absorption spectra were measured using a PerkinElmer Lambda 16 UV–vis–NIR spectrometer. Photoluminescence spectra were measured on an Edinburgh Instruments FLS920 spectrofluorimeter equipped with a Hamamatsu R5509-72 PMT with a monochromator grated at 1200 nm. A 900 W Xe lamp was used as the excitation source.
Transmission Electron Microscopy
TEM images were recorded on Thermo Fisher Scientific (formerly Philips/FEI) Tecnai 10, Tecnai 12, and Tecnai 20 electron microscopes. TEM samples were prepared by drop-casting a dispersion of particles in toluene onto a copper TEM grid, with a carbon-coated polymer support film.
Energy-Dispersive X-ray Spectroscopy
EDS measurements were performed on Thermo Fisher Scientific Tecnai 20F and Talos F200X electron microscopes using Tecnai Imaging and Analysis software. Sample preparation was similar to the preparation of TEM samples. In all cases, aluminum TEM grids were used. A low-background sample holder was used. Measurements were performed on areas with >500 nanoparticles. Discussed elemental compositions are normalized to the stoichiometric sulfur contents.
Elemental Mapping
Elemental mapping was performed on a Talos F200X electron microscope equipped with a 200 kV XFEG. X-ray detection was done using four symmetrically placed SuperX-EDX detectors. Imaging was performed in high-angle annular dark-field scanning transmission electron microscopy mode, with a current of 700 pA using a Fischione Instruments HAADF detector. Obtained data were analyzed using Bruker Esprit software.
HAADF-STEM Tomography
Tilt series for HAADF-STEM tomography were acquired with a FEI Tecnai Osiris operated at 200 kV in combination with an advanced tomography holder from Fischione Instruments and the FEI XPlore3D acquisition software. Tilt series consisting of 31 HAADF-STEM images were acquired with tilt increments of 5° over a range of ±75°. Alignment of the data was carried out using the FEI Inspect3D software package. The reconstruction was performed using the simultaneous iterative reconstruction technique, with 25 iterations, implemented in Inspect3D. Amira (Visage Imaging GmbH) was used for the visualization of the reconstructed volume.
Electron Diffraction
Due to the large amount of sample required for X-ray diffraction (XRD) measurements and low signal-to-noise ratio in XRD measurements for the samples presented in this work, ED was used instead to determine the crystal structure and crystallinity of the samples. ED patterns were measured on a Tecnai 12 electron microscope with an acceleration voltage of 120 kV at a camera length of 1 m.
Data processing of ED patterns measured on Cu2–xS NCs and the particles obtained after reaction of these parent NCs with cation exchange precursors was performed as follows: measured two-dimensional patterns were reduced to one dimension by azimuthal integration of the patterns around the center, which was manually defined for every pattern. The measured ED patterns were calibrated to a gold reference sample (Agar S106). The calibration procedure is described in detail in the Supporting Information.
Data processing of ED patterns measured on CIS NCs and the particles obtained after reaction of these parent NCs with cation exchange precursors, was performed using the Crystallographic Tool Box (CrysTBox) software,70 which automatically performed the required steps of center localization, azimuthal integration, and calibration to a gold reference sample.
Calculated Diffraction Patterns
Diffraction patterns for the low-chalcocite Cu2–xS and wurtzite CuGaS2 crystal structures were calculated from the relevant crystal structures (for low-chalcocite Cu2–xS, the crystal structure reported in ref (34); for wurtzite CGS, the crystal structure reported in ref (44)) using the Visualization for Electronic and Structural Analysis (VESTA) software program (version 3.3.9),71 using the default settings. The calculated lattice distances in real space, d, were transformed into distances in reciprocal space, q, using the equation q = 2π/d.
Comparison of the Anion Sublattices
Using the VESTA software program,71 all cations were removed from the
crystal structures of low-chalcocite Cu2–xS,34 wurtzite CGS,44 and monoclinic
α′-Ga2S3,46 and a supercell
(consisting of multiple unit cells) was constructed. Data corresponding to a single
hexagonal layer and a single ...ABAB... layer was exported for each crystal structure. The
anion coordinates were translated and rotated to orient the three data sets in the same
manner. In this common orientation, the x, y, and
z directions correspond to the following crystallographic directions:
low-chalcocite Cu2–xS: [201],[010],[001]; wurtzite
CuGaS2: [11̅0],[110],[001]; monoclinic
α′-Ga2S3: [010],[100],[102]. Lattice distances
Δx, Δy, and Δz were
measured over 5, 8, and 5 anion–anion distances in x,
y, and z, respectively. Relative lattice contraction
values, corresponding to a transformation of lattice A into
B, are defined as 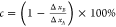 (and similarly for the y
and z directions).
(and similarly for the y
and z directions).
DFT Calculations
All DFT calculations were performed using the Gaussian 09, revision D.01, software package.72 Geometry optimizations were performed using the B3LYP (Becke, three-parameter, Lee–Yang–Parr) functional, the relativistic Stuttgart–Dresden (SDD) pseudopotential and basis set on Ga and In and the 6-311+G(d,p) basis set on all other atoms. The structures were optimized without any symmetry restraints. Frequency analyses at the same level were performed on all calculations to verify that the obtained stationary points are in fact energy minima. Thermochemical analysis was performed using a temperature of 298.15 K. Depicted visualizations of the geometry-optimized structures were prepared using the VESTA software program.71
Acknowledgments
S.O.M.H., W.v.d.S., A.C.B., and C.d.M.D. acknowledge financial support from the division of Chemical Sciences (CW) of The Netherlands Organization for Scientific Research (NWO) under Grant Nos. ECHO.712.012.0001 and ECHO.712.014.001. S.B. acknowledges financial support from the European Research Council (ERC Consolidator Grant No. 815128-REALNANO). S.O.M.H. is supported by The Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation Programme funded by the Ministry of Education, Culture and Science of the government of The Netherlands. DFT calculations were carried out on the Dutch national e-infrastructure with the support of SURF Cooperative. This work was sponsored by NWO Physical Sciences for the use of supercomputer facilities. The authors thank Jessi van der Hoeven for EDS and TEM measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b05337.
NC size histograms, EDS of NCs before and after CE reaction, absorption spectra of NCs before and after CE, TEM images of NCs after reaction with GaCl3 and GaCl3–DPP at 100 °C, HAADF-STEM images and EDS elemental maps of Cu2–xS NCs after reaction with GaCl3, detailed description of PED calibration procedure (PDF)
File showing the results of the DFT calculations (ground-state geometries and bond enthalpies for the phosphine complexes used in our work (XYZ)
Animated versions of the 3D tomographic reconstructions shown in Figure 3.
Author Present Address
⊥ S.O.M.H.: Soft Condensed Matter/Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, P.O. Box 80000, 3508 TA Utrecht, The Netherlands.
Author Present Address
¶ A.C.B.: Seaborough Research B.V., Science Park 106, 1098XG Amsterdam, The Netherlands.
Author Present Address
# M.K.: Foseco Nederland B.V., Pantheon 30, 7521PR Enschede, The Netherlands.
Author Present Address
▲ W.v.d.S.: Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, P.O. Box 80000, Universiteitsweg 99, 3584CG Utrecht, The Netherlands.
The authors declare no competing financial interest.
Supplementary Material
References
- Van der Stam W.; Berends A. C.; de Mello Donegá C. Prospects of Colloidal Copper Chalcogenide Nanocrystals. ChemPhysChem 2016, 17, 559–581. 10.1002/cphc.201500976. [DOI] [PubMed] [Google Scholar]
- Kolny-Olesiak J.; Weller H. Synthesis and Application of Colloidal CuInS2 Semiconductor Nanocrystals. ACS Appl. Mater. Interfaces 2013, 5, 12221–12237. 10.1021/am404084d. [DOI] [PubMed] [Google Scholar]
- Coughlan C.; Ibáñez M.; Dobrozhan O.; Singh A.; Cabot A.; Ryan K. M. Compound Copper Chalcogenide Nanocrystals. Chem. Rev. 2017, 117, 5865–6109. 10.1021/acs.chemrev.6b00376. [DOI] [PubMed] [Google Scholar]
- Van der Stam W.; Gudjonsdottir S.; Evers W. H.; Houtepen A. J. Switching between Plasmonic and Fluorescent Copper Sulfide Nanocrystals. J. Am. Chem. Soc. 2017, 139, 13208–13217. 10.1021/jacs.7b07788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beberwyck B. J.; Surendranath Y.; Alivisatos A. P. Cation Exchange: A Versatile Tool for Nanomaterials Synthesis. J. Phys. Chem. C 2013, 117, 19759–19770. 10.1021/jp405989z. [DOI] [Google Scholar]
- Rivest J. B.; Jain P. K. Cation Exchange on the Nanoscale: An Emerging Technique for New Material Synthesis, Device Fabrication, and Chemical Sensing. Chem. Soc. Rev. 2013, 42, 89–96. 10.1039/C2CS35241A. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Kershaw S. V.; Rogach A. L. 25th Anniversary Article: Ion Exchange in Colloidal Nanocrystals. Adv. Mater. 2013, 25, 6923–6944. 10.1002/adma.201302400. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; Manna L. Forging Colloidal Nanostructures via Cation Exchange Reactions. Chem. Rev. 2016, 116, 10852–10887. 10.1021/acs.chemrev.5b00739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A.; Kang M. S.; Kompch A.; Notthoff C.; Wills A. W.; Deng D.; Winterer M.; Frisbie C. D.; Norris D. J. Electronic Impurity Doping in CdSe Nanocrystals. Nano Lett. 2012, 12, 2587–2594. 10.1021/nl300880g. [DOI] [PubMed] [Google Scholar]
- Van der Stam W.; Geuchies J. J.; Altantzis T.; Van den Bos K. H. W.; Meeldijk J. D.; Van Aert S.; Bals S.; Vanmaekelbergh D.; de Mello Donegá C. Highly Emissive Divalent Ion Doped Colloidal CsPb1–xMxBr3 Perovskite Nanocrystals through Cation Exchange. J. Am. Chem. Soc. 2017, 139, 4087–4097. 10.1021/jacs.6b13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers J.; Groeneveld E.; de Mello Donegá C.; Meijerink A. Optical Properties of Mn-Doped ZnTe Magic Size Nanocrystals. J. Phys. Chem. Lett. 2012, 3, 1663–1667. 10.1021/jz300300g. [DOI] [PubMed] [Google Scholar]
- Groeneveld E.; Witteman L.; Lefferts M.; Ke X.; Bals S.; Van Tendeloo G.; de Mello Donegá C. Tailoring ZnSe-CdSe Colloidal Quantum Dots via Cation Exchange: From Core/Shell to Alloy Nanocrystals. ACS Nano 2013, 7, 7913–7930. 10.1021/nn402931y. [DOI] [PubMed] [Google Scholar]
- Grodzińska D.; Pietra F.; Van Huis M. A.; Vanmaekelbergh D.; de Mello Donegá C. Thermally Induced Atomic Reconstruction of PbSe/CdSe Core/Shell Quantum Dots into PbSe/CdSe Bi-Hemisphere Hetero-Nanocrystals. J. Mater. Chem. 2011, 21, 11556–11565. 10.1039/c0jm04458j. [DOI] [Google Scholar]
- Fan Z.; Lin L.-C.; Buijs W.; Vlugt T. J. H.; Van Huis M. A. Atomistic Understanding of Cation Exchange in PbS Nanocrystals Using Simulations with Pseudoligands. Nat. Commun. 2016, 7, 11503. 10.1038/ncomms11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewavitharana I. K.; Brock S. L. When Ligand Exchange Leads to Ion Exchange: Nanocrystal Facets Dictate the Outcome. ACS Nano 2017, 11, 11217–11224. 10.1021/acsnano.7b05534. [DOI] [PubMed] [Google Scholar]
- Van der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; de Mello Donegá C. Luminescent CuInS2 Quantum Dots by Partial Cation Exchange in Cu2-xS Nanocrystals. Chem. Mater. 2015, 27, 621–628. 10.1021/cm504340h. [DOI] [Google Scholar]
- Jharimune S.; Sathe A. A.; Rioux R. M. Thermochemical Measurements of Cation Exchange in CdSe Nanocrystals Using Isothermal Titration Calorimetry. Nano Lett. 2018, 18, 6795–6803. 10.1021/acs.nanolett.8b02661. [DOI] [PubMed] [Google Scholar]
- Van der Stam W.; Bladt E.; Rabouw F. T.; Bals S.; de Mello Donegá C. Near-Infrared Emitting CuInSe2/CuInS2 Dot Core/Rod Shell Heteronanorods by Sequential Cation Exchange. ACS Nano 2015, 9, 11430–11438. 10.1021/acsnano.5b05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Butler D. J.; Straus D. B.; Oh N.; Wu F.; Guo J.; Xue K.; Lee J. D.; Murray C. B.; Kagan C. R. Air-Stable CuInSe2 Nanocrystal Transistors and Circuits via Post-Deposition Cation Exchange. ACS Nano 2019, 13, 2324–2333. 10.1021/acsnano.8b09055. [DOI] [PubMed] [Google Scholar]
- Regulacio M. D.; Ye C.; Lim S. H.; Zheng Y.; Xu Q.-H.; Han M.-Y. Facile Noninjection Synthesis and Photocatalytic Properties of Wurtzite-Phase CuGaS2 Nanocrystals with Elongated Morphologies. CrystEngComm 2013, 15, 5214–5217. 10.1039/c3ce40352a. [DOI] [Google Scholar]
- Guijarro N.; Prévot M. S.; Yu X.; Jeanbourquin X. A.; Bornoz P.; Bourée W.; Johnson M.; Le Formal F.; Sivula K. A Bottom-Up Approach toward All-Solution-Processed High-Efficiency Cu(In,Ga)S2 Photocathodes for Solar Water Splitting. Adv. Energy Mater. 2016, 6, 1501949. 10.1002/aenm.201501949. [DOI] [Google Scholar]
- Lee K.-H.; Kim J.-H.; Jang H. S.; Do Y. R.; Yang H. Quantum-Dot-Based White Lighting Planar Source through Downconversion by Blue Electroluminescence. Opt. Lett. 2014, 39, 1208–1211. 10.1364/OL.39.001208. [DOI] [PubMed] [Google Scholar]
- Kim J.-H.; Lee K.-H.; Jo D.-Y.; Lee Y.; Hwang J. Y.; Yang H. Cu-In-Ga-S Quantum Dot Composition-Dependent Device Performance of Electrically Driven Light-Emitting Diodes. Appl. Phys. Lett. 2014, 105, 133104. 10.1063/1.4896911. [DOI] [Google Scholar]
- Moon S. H.; Park S. J.; Kim S. H.; Lee M. W.; Han J.; Kim J. Y.; Kim H.; Hwang Y. J.; Lee D.-K.; Min B. K. Monolithic DSSC/CIGS Tandem Solar Cell Fabricated by a Solution Process. Sci. Rep. 2015, 5, 8970. 10.1038/srep08970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Chen Q.; Hsieh Y.-T.; Song T.-B.; De Marco N.; Zhou H.; Yang Y. Multilayer Transparent Top Electrode for Solution Processed Perovskite/Cu(In,Ga)(Se,S)2 Four Terminal Tandem Solar Cells. ACS Nano 2015, 9, 7714–7721. 10.1021/acsnano.5b03189. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Zhang J.; Wang W.; Wang P.; Li F.; Ren D.; Si H.; Sun X.; Ji F.; Hao Y. Facile Synthesis of CuInGaS2 Quantum Dot Nanoparticles for Bilayer-Sensitized Solar Cells. Dalton Trans 2014, 43, 16588–16592. 10.1039/C4DT02150A. [DOI] [PubMed] [Google Scholar]
- Dilena E.; Xie Y.; Brescia R.; Prato M.; Maserati L.; Krahne R.; Paolella A.; Bertoni G.; Povia M.; Moreels I.; Manna L. CuInxGa1-xS2 Nanocrystals with Tunable Composition and Band Gap Synthesized via a Phosphine-Free and Scalable Procedure. Chem. Mater. 2013, 25, 3180–3187. 10.1021/cm401563u. [DOI] [Google Scholar]
- Liu Y.; Yin D.; Swihart M. T. Valence Selectivity of Cation Incorporation into Covellite CuS Nanoplatelets. Chem. Mater. 2018, 30, 1399–1407. 10.1021/acs.chemmater.7b05198. [DOI] [Google Scholar]
- Song J.; Zhang Y.; Dai Y.; Hu J.; Zhu L.; Xu X.; Yu Y.; Li H.; Yao B.; Zhou H. Polyelectrolyte-Mediated Nontoxic AgGaxIn1-xS2 QDs/Low-Density Lipoprotein Nanoprobe for Selective 3D Fluorescence Imaging of Cancer Stem Cells. ACS Appl. Mater. Interfaces 2019, 11, 9884–9892. 10.1021/acsami.9b00121. [DOI] [PubMed] [Google Scholar]
- Zhai Y.; Flanagan J. C.; Shim M. Lattice Strain and Ligand Effects on the Formation of Cu2–xS/I-III-VI2 Nanorod Heterostructures through Partial Cation Exchange. Chem. Mater. 2017, 29, 6161–6167. 10.1021/acs.chemmater.7b02392. [DOI] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Pearson R. G. Absolute Electronegativity and Hardness: Application to Inorganic Chemistry. Inorg. Chem. 1988, 27, 734–740. 10.1021/ic00277a030. [DOI] [Google Scholar]
- Xia C.; Wu W.; Yu T.; Xie X.; Van Oversteeg C.; Gerritsen H. C.; de Mello Donegá C. Size-Dependent Band-Gap and Molar Absorption Coefficients of Colloidal CuInS2 Quantum Dots. ACS Nano 2018, 12, 8350–8361. 10.1021/acsnano.8b03641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans H. T. Crystal Structure of Low Chalcocite. Nature, Phys. Sci. 1971, 232, 69–70. 10.1038/physci232069a0. [DOI] [Google Scholar]
- Wang W.; Dahl M.; Yin Y. Hollow Nanocrystals through the Nanoscale Kirkendall Effect. Chem. Mater. 2013, 25, 1179–1189. 10.1021/cm3030928. [DOI] [Google Scholar]
- Yin Y.; Rioux R. M.; Erdonmez C. K.; Hughes S.; Somorjai G. A.; Alivisatos A. P. Formation of Hollow Nanocrystals through the Nanoscale Kirkendall Effect. Science 2004, 304, 711–714. 10.1126/science.1096566. [DOI] [PubMed] [Google Scholar]
- Mu L.; Wang F.; Sadtler B.; Loomis R. A.; Buhro W. E. Influence of the Nanoscale Kirkendall Effect on the Morphology of Copper Indium Disulfide Nanoplatelets Synthesized by Ion Exchange. ACS Nano 2015, 9, 7419–7428. 10.1021/acsnano.5b02427. [DOI] [PubMed] [Google Scholar]
- Ogawa A.; Fujimoto H. Lewis Acidity of Gallium Halides. Inorg. Chem. 2002, 41, 4888–4894. 10.1021/ic020268m. [DOI] [PubMed] [Google Scholar]
- Hogg J. M.; Coleman F.; Ferrer-Ugalde A.; Atkins M. P.; Swadźba-Kwaśny M. Liquid Coordination Complexes: A New Class of Lewis Acids as Safer Alternatives to BF3 in Synthesis of Polyalphaolefins. Green Chem. 2015, 17, 1831–1841. 10.1039/C4GC02080D. [DOI] [Google Scholar]
- Ruberu T. P. A.; Albright H. R.; Callis B.; Ward B.; Cisneros J.; Fan H.-J.; Vela J. Molecular Control of the Nanoscale: Effect of Phosphine–Chalcogenide Reactivity on CdS–CdSe Nanocrystal Composition and Morphology. ACS Nano 2012, 6, 5348–5359. 10.1021/nn301182h. [DOI] [PubMed] [Google Scholar]
- Tolman C. A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- Kühl O. Predicting the Net Donating Ability of Phosphines - Do We Need Sophisticated Theoretical Methods?. Coord. Chem. Rev. 2005, 249, 693–704. 10.1016/j.ccr.2004.08.021. [DOI] [Google Scholar]
- Brown T. L.; Lee K. J. Ligand Steric Properties. Coord. Chem. Rev. 1993, 128, 89–116. 10.1016/0010-8545(93)80025-Z. [DOI] [Google Scholar]
- Xiao N.; Zhu L.; Wang K.; Dai Q.; Wang Y.; Li S.; Sui Y.; Ma Y.; Liu J.; Liu B.; Zou G.; Zou B. Synthesis and High-Pressure Transformation of Metastable Wurtzite-Structured CuGaS2 Nanocrystals. Nanoscale 2012, 4, 7443–7447. 10.1039/c2nr31629c. [DOI] [PubMed] [Google Scholar]
- Fenton J. L.; Steimle B. C.; Schaak R. E. Structure-Selective Synthesis of Wurtzite and Zincblende ZnS, CdS, and CuInS2 Using Nanoparticle Cation Exchange Reactions. Inorg. Chem. 2019, 58, 672–678. 10.1021/acs.inorgchem.8b02880. [DOI] [PubMed] [Google Scholar]
- Pardo M. P.; Guittard M.; Chilouet A.; Tomas A. Diagramme de Phases Gallium-Soufre et Études Structurales Des Phases Solides. J. Solid State Chem. 1993, 102, 423–433. 10.1006/jssc.1993.1054. [DOI] [Google Scholar]
- Nogai S.; Schmidbaur H. Dichlorogallane (HGaCl2)2: Its Molecular Structure and Synthetic Potential. Inorg. Chem. 2002, 41, 4770–4774. 10.1021/ic0203015. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Codgbrook H. L.; Hector A. L.; Levason W.; Reid G.; Webster M.; Zhang W. Gallium(III) Halide Complexes with Phosphines, Arsines and Phosphine Oxides – a Comparative Study. Polyhedron 2007, 26, 4147–4155. 10.1016/j.poly.2007.05.008. [DOI] [Google Scholar]
- Yarema O.; Yarema M.; Wood V. Tuning the Composition of Multicomponent Semiconductor Nanocrystals: The Case of I-III-VI Materials. Chem. Mater. 2018, 30, 1446–1461. 10.1021/acs.chemmater.7b04710. [DOI] [Google Scholar]
- Berends A. C.; Van der Stam W.; Akkerman Q. A.; Meeldijk J. D.; Van der Lit J.; de Mello Donegá C. Anisotropic 2D Cu2-xSe Nanocrystals from Dodecaneselenol and Their Conversion to CdSe and CuInSe2 Nanoparticles. Chem. Mater. 2018, 30, 3836–3846. 10.1021/acs.chemmater.8b01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J. M. R.; Scott M. C.; Hao W.; Baikie T.; Nelson C. T.; Pedireddy S.; Tao R.; Ling X.; Magdassi S.; White T.; Li S.; Minor A. M.; Zheng H.; Wong L. H. Revealing Cation-Exchange-Induced Phase Transformations in Multielemental Chalcogenide Nanoparticles. Chem. Mater. 2017, 29, 9192–9199. 10.1021/acs.chemmater.7b03029. [DOI] [Google Scholar]
- Lesnyak V.; Brescia R.; Messina G. C.; Manna L. Cu Vacancies Boost Cation Exchange Reactions in Copper Selenide Nanocrystals. J. Am. Chem. Soc. 2015, 137, 9315–9323. 10.1021/jacs.5b03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton J. L.; Steimle B. C.; Schaak R. E. Tunable Intraparticle Frameworks for Creating Complex Heterostructured Nanoparticle Libraries. Science 2018, 360, 513–517. 10.1126/science.aar5597. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Krahne R.; Bertoni G.; Alcocer M. J. P.; D’Andrea C.; Scotognella F.; Tassone F.; Zanella M.; De Giorgi M.; Manna L. Blue-UV-Emitting ZnSe(Dot)/ZnS(Rod) Core/Shell Nanocrystals Prepared from CdSe/CdS Nanocrystals by Sequential Cation Exchange. ACS Nano 2012, 6, 1637–1647. 10.1021/nn204601n. [DOI] [PubMed] [Google Scholar]
- Chakraborty P.; Jin Y.; Barrows C. J.; Dunham S. T.; Gamelin D. R. Kinetics of Isovalent (Cd2+) and Aliovalent (In3+) Cation Exchange in Cd1-xMnxSe Nanocrystals. J. Am. Chem. Soc. 2016, 138, 12885–12893. 10.1021/jacs.6b05949. [DOI] [PubMed] [Google Scholar]
- Zuckerman J. J.; Hagen A. P.. Inorganic Reactions and Methods: Formation of Bonds to O, S, Se, Te, Po (Part 1), 1st ed.; VCH Publishers, Inc.: New York, 1992; Vol. 5, p 217. [Google Scholar]
- Burt J.; Levason W.; Reid G. Coordination Chemistry of the Main Group Elements with Phosphine, Arsine and Stibine Ligands. Coord. Chem. Rev. 2014, 260, 65–115. 10.1016/j.ccr.2013.09.020. [DOI] [Google Scholar]
- Chen F.; Ma G.; Bernard G. M.; Wasylishen R. E.; Cavell R. G.; McDonald R.; Ferguson M. J. An Investigation of 1:1 Adducts of Gallium Trihalides with Triarylphosphines by Solid-State 69/71Ga and 31P NMR Spectroscopy. Chem. - Eur. J. 2013, 19, 2826–2838. 10.1002/chem.201202954. [DOI] [PubMed] [Google Scholar]
- Haynes W. M.CRC Handbook of Chemistry and Physics, 95th ed.; CRC Press: Boca Raton, FL, 2014; section 5–11. [Google Scholar]
- Clavier H.; Nolan S. P. Percent Buried Volume for Phosphine and N-Heterocyclic Carbene Ligands: Steric Properties in Organometallic Chemistry. Chem. Commun. (Cambridge, U. K.) 2010, 46, 841–861. 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- Snelders D. J. M.; Van Koten G.; Klein Gebbink R. J. M. Steric, Electronic, and Secondary Effects on the Coordination Chemistry of Ionic Phosphine Ligands and the Catalytic Behavior of Their Metal Complexes. Chem. - Eur. J. 2011, 17, 42–57. 10.1002/chem.201002508. [DOI] [PubMed] [Google Scholar]
- Chakraborty P.; Jin Y.; Barrows C. J.; Dunham S. T.; Gamelin D. R. Kinetics of Isovalent (Cd2+) and Aliovalent (In3+) Cation Exchange in Cd1-xMnxSe Nanocrystals. J. Am. Chem. Soc. 2016, 138, 12885–12893. 10.1021/jacs.6b05949. [DOI] [PubMed] [Google Scholar]
- Choi Y.-M.; Lee Y.-I.; Kim S.; Choa Y.-H. Metallic Alloy Nanoparticle-Based Fabrication and Optical Properties of a Cu(In1-xGax)S2 Absorber Layer for Solar Cells. J. Alloys Compd. 2014, 615, 496–500. 10.1016/j.jallcom.2014.06.174. [DOI] [Google Scholar]
- Chang S.-H.; Chiang M.-Y.; Chiang C.-C.; Yuan F.-W.; Chen C.-Y.; Chiu B.-C.; Kao T.-L.; Lai C.-H.; Tuan H.-Y. Facile Colloidal Synthesis of Quinary CuIn1-xGax(SySe1-y) (CIGSSe) Nanocrystal Inks with Tunable Band Gaps for Use in Low-Cost Photovoltaics. Energy Environ. Sci. 2011, 4, 4929–4932. 10.1039/c1ee02341a. [DOI] [Google Scholar]
- Xia C.; Meeldijk J. D.; Gerritsen H. C.; de Mello Donegá C. Highly Luminescent Water-Dispersible NIR-Emitting Wurtzite CuInS2/ZnS Core/Shell Colloidal Quantum Dots. Chem. Mater. 2017, 29, 4940–4951. 10.1021/acs.chemmater.7b01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Pandey A.; Werder D. J.; Khanal B. P.; Pietryga J. M.; Klimov V. I. Efficient Synthesis of Highly Luminescent Copper Indium Sulfide-Based Core/Shell Nanocrystals with Surprisingly Long-Lived Emission. J. Am. Chem. Soc. 2011, 133, 1176–1179. 10.1021/ja108261h. [DOI] [PubMed] [Google Scholar]
- Berends A. C.; Rabouw F. T.; Spoor F. C. M.; Bladt E.; Grozema F. C.; Houtepen A. J.; Siebbeles L. D. A.; De Mello Donegá C. Radiative and Nonradiative Recombination in CuInS2 Nanocrystals and CuInS2-Based Core/Shell Nanocrystals. J. Phys. Chem. Lett. 2016, 7, 3503–3509. 10.1021/acs.jpclett.6b01668. [DOI] [PubMed] [Google Scholar]
- Bai X.; Purcell-Milton F.; Gun’ko Y. Optical Properties, Synthesis, and Potential Applications of Cu-Based Ternary or Quaternary Anisotropic Quantum Dots, Polytypic Nanocrystals, and Core/Shell Heterostructures. Nanomaterials 2019, 9, 85. 10.3390/nano9010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berends A. C.; Mangnus M. J. J.; Xia C.; Rabouw F. T.; De Mello Donegá C. Optoelectronic Properties of Ternary I-III-VI2 Semiconductor Nanocrystals: Bright Prospects with Elusive Origins. J. Phys. Chem. Lett. 2019, 10, 1600–1616. 10.1021/acs.jpclett.8b03653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M.; Jäger A. Crystallographic Tool Box (CrysTBox): Automated Tools for Transmission Electron Microscopists and Crystallographers. J. Appl. Crystallogr. 2015, 48, 2012–2018. 10.1107/S1600576715017252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momma K.; Izumi F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. 10.1107/S0021889811038970. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.