

Abstract
Vascular endothelial growth factor receptor 2 (VEGFR2) is highly expressed in tumor‐associated endothelial cells, where it modulates tumor‐promoting angiogenesis, and it is also found on the surface of tumor cells. Currently, there are no Ab therapeutics targeting VEGFR2 approved for the treatment of prostate cancer or leukemia. Therefore, development of novel efficacious anti‐VEGFR2 Abs will benefit cancer patients. We used the Institute of Cellular and Organismic Biology human Ab library and affinity maturation to develop a fully human Ab, anti‐VEGFR2‐AF, which shows excellent VEGFR2 binding activity. Anti‐VEGFR2‐AF bound Ig‐like domain 3 of VEGFR2 extracellular region to disrupt the interaction between VEGF‐A and VEGFR2, neutralizing downstream signaling of the receptor. Moreover, anti‐VEGFR2‐AF inhibited capillary structure formation and exerted Ab‐dependent cell‐mediated cytotoxicity and complement‐dependent cytotoxicity in vitro. We found that VEGFR2 is expressed in PC‐3 human prostate cancer cell line and associated with malignancy and metastasis of human prostate cancer. In a PC‐3 xenograft mouse model, treatment with anti‐VEGFR2‐AF repressed tumor growth and angiogenesis as effectively and safely as US FDA‐approved anti‐VEGFR2 therapeutic, ramucirumab. We also report for the first time that addition of anti‐VEGFR2 Ab can enhance the efficacy of docetaxel in the treatment of a prostate cancer mouse model. In HL‐60 human leukemia‐xenografted mice, anti‐VEGFR2‐AF showed better efficacy than ramucirumab with prolonged survival and reduced metastasis of leukemia cells to ovaries and lymph nodes. Our findings suggest that anti‐VEGFR2‐AF has strong potential as a cancer therapy that could directly target VEGFR2‐expressing tumor cells in addition to its anti‐angiogenic action.
Keywords: angiogenesis, human antibody, phage display, targeted cancer therapy, VEGFR2
Affinity‐maturated anti‐vascular endothelial growth factor receptor 2 (VEGFR2) therapeutic Ab has strong clinical potential for cancer treatment due to its simultaneous inhibition of tumor angiogenesis in vascular endothelial cells and tumorigenesis in VEGFR2‐expressing tumor cells.

Abbreviations
- ADCC
antibody‐dependent cellular cytotoxicity
- Anti‐VEGFR2‐AF
affinity‐matured antivascular endothelial growth factor receptor 2 human antibody
- CDC
complement‐dependent cytotoxicity
- CDR
complementary‐determining region
- Fab
antigen‐binding fragment
- FAK
focal adhesion kinase
- Fc
fragment crystallizable region
- GEO
Gene Expression Omnibus
- hAb
human antibody
- ICOB
Institute of Cellular and Organismic Biology
- NHIgG
normal human IgG
- NSG
NOD/SCID gamma
- PBST
PBS containing 0.1% Tween 20
- PlGF
placenta growth factor
- scFv
single chain fragment variable
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- VH
variable heavy chain
- VL
variable light chain
1. INTRODUCTION
Angiogenesis is a tightly regulated multistage process of new blood vessel growth from preexisting vasculature and is widely recognized as one of the most important features of cancer progression.1, 2, 3 Several VEGF family members, including VEGF‐A, VEGF‐B, VEGF‐C, VEGF‐D, and PlGF, are known to participate in the regulation of angiogenesis. Vascular endothelial growth factors act by binding with high affinity to receptor tyrosine kinases, VEGFR1‐R3; among these VEGF binding events, VEGF‐A binding to VEGFR2 comprises the main activating signal for angiogenesis.4, 5 Importantly, VEGF‐A signaling through VEGFR2 is also the key driver for the neovascular growth known to support solid tumor progression.6 Angiogenesis rarely occurs in healthy adult tissues. As such, VEGFR2 is expressed infrequently and at low levels in normal endothelial cells compared to tumor‐associated endothelial cells.4, 7 Indeed, VEGFR2 expression is 3‐ to 5‐fold higher in tumor vessels than in normal vessels,8, 9 and immunohistochemistry of biopsies from cancer patients confirmed that VEGFR2 expression is significantly elevated in tumor vessels compared with the vascular endothelium of normal tissues adjacent to the tumor region.10 Notably, expression of VEGFR2 is also greater in the vessels of high‐metastatic tumors than in vessels of low‐metastatic tumors.11
Expression of VEGFR2 was originally thought to be restricted to the vessels of tumor tissues, however, increasing evidence suggests that it is also present in the cancer cells of lung, colorectal, and ovarian tumors.12, 13, 14, 15 The Pathology Atlas database indicates that VEGFR2 protein can be detected by immunohistochemical staining with verified Abs in cancer cells of 10%‐40% of surgical tumor sections, including urothelial, prostate, head and neck, cervical, and skin cancer.16 Interestingly, circulating tumor cells in the blood of breast cancer and small‐cell lung cancer patients were also found to express VEGFR2, and such expression is associated with tumor metastasis and poor prognosis.17, 18 Therefore, blocking VEGFR2‐mediated signaling in both tumor endothelial and malignant cells is considered to be a promising strategy for new cancer treatments.7
Since 2004, several drugs targeting VEGF signaling have been successfully applied in the treatment of various malignant diseases.19 Bevacizumab is a humanized mAb against VEGF and the first US FDA‐approved antiangiogenic drug for the treatment of metastatic colon, renal, ovarian, and nonsmall‐cell lung cancer.20, 21 Several pantyrosine kinase inhibitors that act primarily by suppressing VEGFR2 phosphorylation, such as sorafenib, sunitinib, and pazopanib, were approved by the US FDA for cancer treatment.22, 23, 24 Furthermore, mAbs that directly target VEGFR2 and specifically inhibit its signaling have been evaluated.25 To date, there is only one anti‐VEGFR2 Ab that has been approved by the US FDA. This human IgG1 mAb, ramucirumab, was originally discovered from a human Fab phage library by ImClone Systems Incorporated.26, 27 After the initial identification, the Ab affinity was matured (Kd, 50 pM) so that it strongly binds the extracellular domain of VEGFR2 to block VEGF‐A‐induced signaling.28 The efficacy of ramucirumab was established in clinical trials, and it was approved for the treatment of patients with advanced or metastatic gastric cancer, gastroesophageal junction adenocarcinoma,29 and metastatic nonsmall‐cell lung cancer in 2014, with approval for metastatic colorectal cancer coming in 2015.23, 30 Ramucirumab also succeeded in phase III trials as a monotherapy second‐line treatment for hepatocellular carcinoma31 and most recently was approved by the US FDA as a single agent for hepatocellular carcinoma on May 10, 2019. There are currently over 60 clinical trials investigating ramucirumab as a cancer monotherapy, or in combination with other Abs or small molecules (https://clinicaltrials.gov). In 2018, the worldwide revenue from ramucirumab reached US$800 million, and its market is gradually expanding with the approval of more indications.32 Together, the results from clinical studies show that VEGFR2 is a viable therapeutic target, and fully human Abs against VEGFR2 are safe and efficacious for cancer therapy. Targeting VEGFR2 shows promise as an emerging therapy for cancer and could act either by blocking tumor angiogenesis or directly on cancer cells. Hence, development of a novel anti‐VEGFR2 hAb with enhanced therapeutic efficacy is likely to benefit cancer patients.
Phage display is an efficient tool that allows rapid selection of ligands against various molecular targets.33 Using this method, we have previously identified several tumor‐targeting and tumor vasculature‐targeting peptides34, 35, 36, 37, 38; we have also identified hAbs that bind c‐Met, a receptor tyrosine kinase.39 Here, we identified and engineered a novel anti‐VEGFR2 fully human Ab by phage display techniques. This Ab could be useful as a cancer therapy that blocks the VEGF‐A/VEGFR2 interaction and antagonizes VEGFR2 signaling. Furthermore, we have completed proof‐of‐concept studies in endothelial and VEGFR2‐expressing malignant cancer cell models that suggest excellent therapeutic potential for our novel anti‐VEGFR2 hAb.
2. MATERIALS AND METHODS
2.1. Isolation of VEGFR2‐binding phages from a phage‐displayed scFv library
The ICOB hAb library is a phage‐displayed human naïve scFv library with 6 × 1010 complexity, which was previously established in our laboratory at ICOB, Academia Sinica (Taipei, Taiwan). This ICOB hAb library was used to screen scFv binding to VEGFR2 extracellular domain following affinity‐selection procedure. Briefly, nonspecific binding was subtracted from the library with protein G Dynabeads (Invitrogen), and the phages were subsequently incubated with VEGFR2‐Fc recombinant protein (R&D Systems) immobilized Dynabeads. After washing with PBST, phages bound to VEGFR2‐Fc were recovered by infection of Escherichia coli TG1 cells. After determination of phage titer, subsequent rounds of biopanning were carried out.
2.2. Competitive VEGF binding assay
Various concentrations of anti‐VEGFR2 scFvs were mixed with 3 nM human VEGF‐A (Peprotech), and the mixture was added to 96‐well plates coated with 1 μg/mL VEGFR2‐Fc and preblocked with 1% BSA. After incubation for 1 hour at room temperature and washing with PBST, the bound VEGF molecules were detected using anti‐VEGF mAb (GeneTex) and HRP‐labeled goat anti‐mouse IgG. Detection was accomplished with a mixture of o‐phenylenediamine dihydrochloride and H2O2, and the reaction was terminated with 3 N HCl. The absorbance was determined using a microplate reader at 490 nm.
2.3. Human tumor vasculature staining with anti‐VEGFR2 scFvs
Human lung cancer surgical specimens were obtained from the Department of Pathology, National Taiwan University Hospital (Taipei, Taiwan). Slides with frozen sections were washed with PBS and then fixed with paraformaldehyde. After another wash with PBS, slides were blocked with normal horse serum (Vector), and incubated with the scFv. After washing with PBST, a mixture of rabbit anti‐E tag Ab (Bethyl Laboratories) and mouse anti‐human CD31 mAb (BD Biosciences) was added. The slides were then incubated for 1 hour with FITC‐labeled anti‐mouse IgG, rhodamine‐labeled goat anti‐rabbit IgG, and DAPI. Samples were then imaged on an inverted fluorescence microscope (Axiovert 200M; Zeiss).
2.4. Tube formation assay
Matrigel (BD Biosciences) was thawed at 4°C overnight, after which 10 aL Matrigel was added to each well of a prechilled μ‐Slide Angiogenesis (Ibidi); the slide was then incubated at 37°C for 15 minutes. Starved HUVECs (4 × 104 cells) were added to EBM‐2 containing 0.2% serum with or without 40 ng/mL VEGF‐A and anti‐VEGFR2 Abs. After 24 hours of incubation, endothelial cell tube formation was assessed with an inverted microscope and digital camera (DP‐12; Olympus). Tubular lengths and branching points were quantitatively evaluated with ImageJ software. Inhibition percentage by Abs was expressed as a percentage of that in VEGF‐A‐treated wells without competitor.
2.5. Clinical dataset analyses
Raw microarray data were downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/). Raw data were normalized. The GEO profile GDS2545/ 1954_at/ KDR was used for metastatic prostate cancer analysis. Expression of VEGFR2 mRNA was analyzed for patients with low, medium, and high risk of prostate cancer using the SurvExpress Webserver (http://bioinformatica.mty.itesm.mx/SurvExpress).
2.6. Construction and expression of anti‐VEGFR2 hAb
The VH regions of R2S12, R2S12‐AF, and ramucirumab (clone name, IMC‐1121B)28 were each cloned separately in‐frame into a modified expression vector with a signal peptide and human IgG1 constant region. The VL regions of R2S12, R2S12‐AF, and ramucirumab were also separately cloned into modified expression vectors. Heavy and light chain gene‐containing plasmids were then combined into a biscistronic vector to generate a single vector system. The plasmids were transfected into FlpIn‐CHO cells (Invitrogen), which then were selected for hygromycin B resistance; the resistant clones were cultured in SFM4CHO media (Thermo Fisher Scientific) to produce hAbs. The culture supernatant from the stable clones was filtered through a 0.45‐μm membrane and then subjected to protein G column chromatography (GE Healthcare) for purification of human IgG. After dialysis of eluents with PBS, the concentration of Ab was assessed using Bradford reagent (Thermo Fisher Scientific) and spectrophotometry.
2.7. Affinity maturation of anti‐VEGFR2 human IgG
Affinity maturation was carried out as previously described.40 Briefly, we constructed a synthetic phage‐displayed scFv library comprised of the VH and VL gene repertoire of R2S12, with random mutations introduced at 7 amino acid residues of VL‐CDR3. This synthetic library was used to undertake biopanning for VEGFR2‐Fc‐immobilized Dynabeads. After 4‐5 rounds of stringent in vitro biopanning, positive clones were screened and identified by ELISA. Superior VEGFR2‐binding clones were identified by comparing affinity to that of the respective parental clone.
2.8. Measurement of binding kinetics
The binding affinity and kinetics of anti‐VEGFR2 Abs were measured by surface plasmon resonance in a BIAcore T100 (GE Healthcare). The VEGFR2‐Fc protein was coupled to an EDC‐ and NHS‐activated CM5 sensor chip in a BIAcore flow cell and then blocked with ethanolamine according to the manufacturer’s directions. Associated and dissociated phases were monitored for 5 minutes under continuous flow of 30 μL/min, using Ab concentrations ranging from 0.1 to 100 nM. Regeneration was carried out by injection of regenerate buffer (0.2 M NaCl, 10 mM glycine, pH 2.7). To determine binding constants, the sensorgrams were fitted globally to a 1:1 interaction model using BIAevaluation software (GE Healthcare).
2.9. Animal models
Animal care and procedures involving animals were carried out according to the guidelines of the Academia Sinica Institutional Animal Care and Utilization Committee. The NOD/SCID mice were purchased from the National Laboratory Animal Center (Taiwan). A human prostate cancer xenograft tumor model was created by s.c. injecting 2 × 106 PC‐3 cells into the dorsal flank of a 6‐week‐old male mouse. Animals were monitored daily for general health, and body weights were measured twice each week. Tumor size was measured with slide calipers and calculated as length × width2 × 0.52. Mice with size‐matched tumors (50 mm3) were randomly assigned to different treatment groups (n = 9 per group) and i.v. injected with NHIgG (Jackson ImmunoResearch), ramucirumab, anti‐VEGFR2‐AF, or an equivalent volume of PBS in the tail vein. An Ab dose of 20 mg/kg was injected twice every week for 4 weeks. For combination therapy, docetaxel (ScinoPharm, Taiwan) was also i.v. injected at a dose of 5 mg/kg once every week for 3 weeks. At the end of the experiment, tumor tissue and visceral organs were removed and fixed for histological analysis.
For systemic leukemia engraftment studies, 6‐week‐old NSG female mice were i.v. injected with 5 × 106 HL‐60 cells in the tail vein. Three days after tumor inoculation, mice were randomly selected (n = 9 per group) for i.v. injection with 20 mg/kg NHIgG, ramucirumab, anti‐VEGFR2‐AF, or an equivalent volume of PBS, twice every week. Mice were observed daily for signs of toxicity, and times of survival were recorded. At the end of treatment, the visceral organs of each mouse were removed and fixed for histological examination. Please see supplemental materials and methods for details (Appendix S1).
3. RESULTS
3.1. Identification of phage‐displayed scFvs that bind to VEGFR2
To identify VEGFR2‐binding scFvs, we used a phage‐displayed human naïve scFv library to isolate phages that bind to recombinant VEGFR2 extracellular domain protein. After 4 rounds of affinity selection (biopanning), the titer of bound phage increased by as much as 3455‐fold (Figure 1A). Through ELISA screening and DNA sequencing, we identified 5 distinct phage clones (Table S1) that bind tightly to VEGFR2‐Fc, but not to Fc control protein (Figure 1B). We then used flow cytometry with HUVECs to confirm that all 5 clones have the ability to bind to VEGFR2 on the cell surface. Among the 5 clones, R2PC12 showed the greatest reactivity (Figure 1C). We next generated soluble scFv proteins from the 5 VEGFR2‐binding phage clones, which were designated as R2S8, R2S12, R2S28, R2S29, and R2S45 (Figure 1D). The binding of these anti‐VEGFR2 scFvs to tumor vascular endothelium in human lung cancer surgical specimens was investigated by immunofluorescence. We observed colocalization of fluorescent signals from anti‐VEGFR2 scFvs with endothelial cell marker CD31 (Figure 1E), suggesting that these scFvs are able to specifically recognize tumor vasculature.
Figure 1.
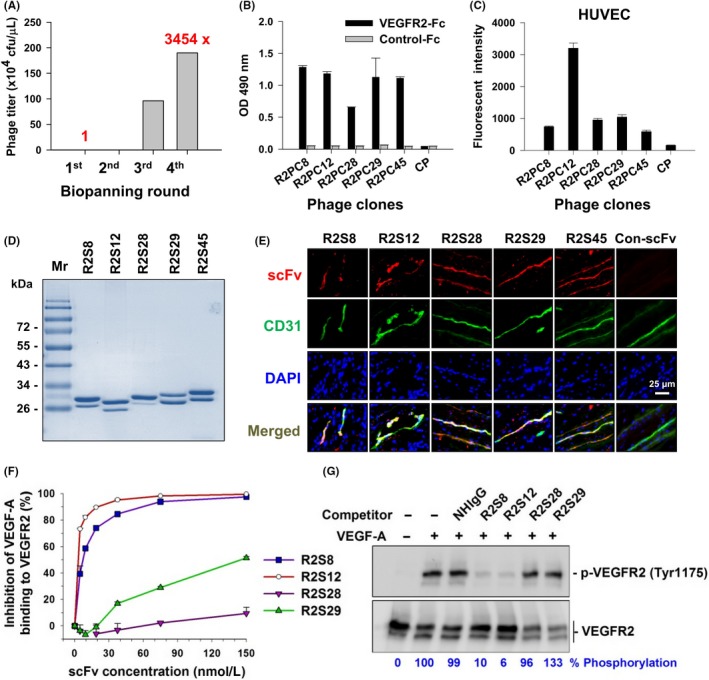
Selection and identification of vascular endothelial growth factor receptor‐2 (VEGFR2)‐binding single chain fragment variables (scFvs). A, After 4 rounds of biopanning for VEGFR2‐Fc recombinant protein, the phage recovery rate was increased by 3454‐fold over that of the first round. cfu, colony‐forming units. B, Comparison of the binding of selected phage clones to VEGFR2‐Fc protein by ELISA with a 1 × 109 cfu phage titer. CP, control phage used as a negative control; OD, optical density. C, Binding affinity of phage clones to cellular VEGFR2 was evaluated with flow cytometry of HUVECs and 1 × 1010 cfu phage titer. D, Soluble anti‐VEGFR2 scFvs were purified and analyzed by SDS‐PAGE with Coomassie blue staining. Mr, molecular weight. E, Immunofluorescence for human tumor vasculature. Frozen sections of surgical specimens of lung cancer patients were probed with anti‐VEGFR2 scFvs, followed by anti‐E tag Ab and rhodamine‐conjugated secondary Ab. Vascular endothelium was stained with anti‐human CD31 Ab and FITC‐conjugated secondary Ab. Nuclei were stained with DAPI. Con‐scFv, control scFv. F, Competitive binding of anti‐VEGFR2 scFv and vascular endothelial growth factor‐A (VEGF‐A) was analyzed by ELISA. VEGF‐A binding to immobilized VEGFR2 in the absence of competitors was considered to be 100%. G, Phosphorylated VEGFR2 (p‐VEGFR2) expression in HUVECs treated with VEGF‐A and scFv competitors was detected by western blot. Quantification of p‐VEGFR2 was based on luminescence intensity and normalized to total VEGFR2. NHIgG, normal human IgG. Error bars, SE
3.2. Anti‐VEGFR2 scFvs attenuate VEGF‐A/VEGFR2 interaction and VEGF‐A‐induced VEGFR2 phosphorylation
To determine whether the anti‐VEGFR2 scFvs can block VEGF‐A binding to VEGFR2, we carried out a plate‐based competitive binding assay, in which increasing concentrations of scFvs competed with VEGF‐A for binding to immobilized VEGFR2. The interaction of VEGF‐A with VEGFR2 was strongly suppressed by R2S8 and R2S12, with IC50 values of 7.03 and 3.26 nM, respectively; in contrast, R2S28 and R2S29 showed comparatively weak competitive binding (Figure 1F). We next investigated whether the scFvs could antagonize VEGF‐A‐mediated activation of VEGFR2 in HUVECs. We found that addition of R2S8 and R2S12 inhibited tyrosine phosphorylation of VEGFR2 by VEGF‐A, with R2S12 showing the strongest inhibitory activity (Figure 1G).
3.3. Identification of B‐cell epitopes of anti‐VEGFR2 Ab
To map the binding domain recognized by anti‐VEGFR2 scFv, we generated a series of VEGFR2 deletion mutants, consisting of the signal peptide and transmembrane domain (Figure 2A). The protein mutants were ectopically expressed in 293T cells, and the cells were probed with R2S8, R2S12, and R2S28 for immunofluorescence (Table S2). We found that R2S8 and R2S12 bound to 293T cells expressing VEGFR2(1‐7) and VEGFR2(2‐7), but not to the cells that expressed constructs lacking domain 3, for example, VEGFR2(4‐7), VEGFR2(△2‐3), or VEGFR2(△3), indicating that the binding epitopes for these two Abs are located within domain 3.
Figure 2.
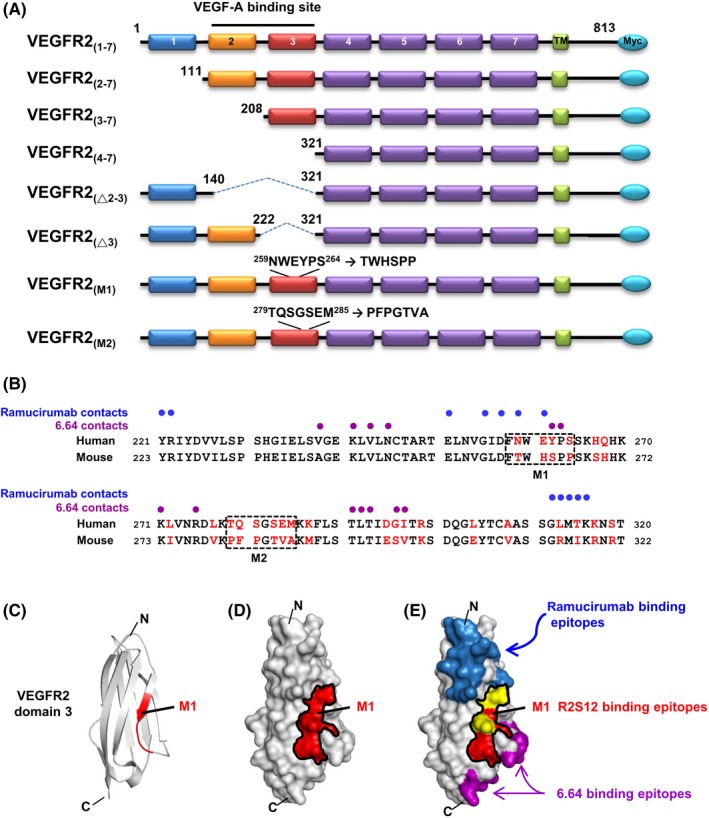
B‐cell epitope mapping of antivascular endothelial growth factor receptor‐2 (VEGFR2) single chain fragment variable R2S12. A, Schematic presentation of the human VEGFR2 domains and its deletion or substitution mutants used in this study is shown. There are 7 Ig‐like domains in the extracellular region of VEGFR2, labeled 1‐7. TM, transmembrane domain. B, Sequence alignment of human and mouse VEGFR2 domain 3. Residues that differ between the 2 species are highlighted in red. M1 and M2 clusters are marked with dashed‐line boxes. Blue and purple dots indicate human VEGFR2 domain 3 residues in contact with ramucirumab and 6.64 Abs, respectively. C, Ribbon diagram for the human VEGFR2 domain 3 structure; NWEYPS residues (M1) responsible for R2S12 binding are highlighted in red. D, Model of the surface of VEGFR2 domain 3. NWEYPS residues are indicated in red, and the M1 area is delineated by a black line. E, Residues that make contact with ramucirumab and 6.64 on the surface of VEGFR2 domain 3 are shown in blue and purple, respectively. The contacting residues for ramucirumab that are localized in the M1 area are shown in yellow. C, C terminus; N, N terminus
Furthermore, we found that neutralizing neither scFv (R2S8 and R2S12) showed detectable cross‐reactivity with murine VEGFR2 protein (data not shown). Alignment of the human and murine amino acid sequences of VEGFR2 revealed that domain 3 is only 67% identical and the most diverse of the 7 domains in the extracellular region of VEGFR2. Thus, we speculated that the distinct epitopes recognized by R2S8 and R2S12 are not present in murine VEGFR2. Thirty residues in domain 3 of human and mouse VEGFR2 are different, and these residues are grouped into clusters.41 To identify the amino acid residues in domain 3 that are critical for R2S8 and R2S12 binding, we selected 2 major clusters (M1 and M2) for mutagenesis. The human NWEYPS and TQSGSEM residues were substituted with mouse TWHSPP and PFPGTVA residues, respectively (Figure 2B). Immunostaining of 293T cells expressing VEGFR2‐M1 and VEGFR2‐M2 was carried out, and neither R2S8 nor R2S12 recognized cells expressing VEGFR2‐M1 mutant protein. In contrast, the M2 mutant of VEGFR2 showed no diminishment of R2S8 and R2S12 binding (Table S2). We also found that M1 and M2 mutants did not affect phosphorylation of VEGFR2 after VEGF‐A treatment, suggesting that neither cluster is involved with VEGF‐A binding (Figure S1).
We built a molecular model of VEGFR2 domain 3 from a previously reported crystal structure41 and our mutagenesis data. The ribbon and surface models show that the NWEYPS residues (M1 region) are contained within a β‐strand on the middle of the VEGFR2 domain 3 surface (Figure 2C,D). Moreover, the contact residues and binding surfaces of other neutralizing anti‐VEGFR2 Abs, ramucirumab and 6.64, are also known to be located on the VEGFR2 domain 3 (Figure 2B,E).41 We observed that the M1 region is near the binding epitopes for the ramucirumab and 6.64 Abs. Two ramucirumab‐recognized residues, Asn259 and Glu261, were found to be contained within the M1 region. Therefore, our results suggest that the binding epitopes of R2S8 and R2S12 are most likely different from those recognized by ramucirumab and 6.64 Abs.
3.4. Affinity maturation of R2S12 to generate an anti‐VEGFR2‐AF hAb with higher binding activity
Evidence from clinical trials in cancer patients has revealed that high affinity of neutralizing Abs is important for therapeutic efficacy.42 As R2S12 showed the highest binding and antagonistic activity among the identified scFvs, we chose to further improve its binding through phage display‐based affinity maturation. After 4 rounds of stringent in vitro selection, a clone with superior binding activity (R2S12‐AF) was identified (Figure S2). Four residues within VL‐CDR3 of R2S12‐AF are different from the parental R2S12 clone (Figure 3A). Because the scFv format has limited clinical utility due to short serum half‐life (approximately 3.5 hours) and an inability to trigger human effector functions,43 we inserted the VH and VL coding sequences of R2S12 and R2S12‐AF scFv into a human IgG1 backbone to create 2 fully human Abs, anti‐VEGFR2 and anti‐VEGFR2‐AF.
Figure 3.
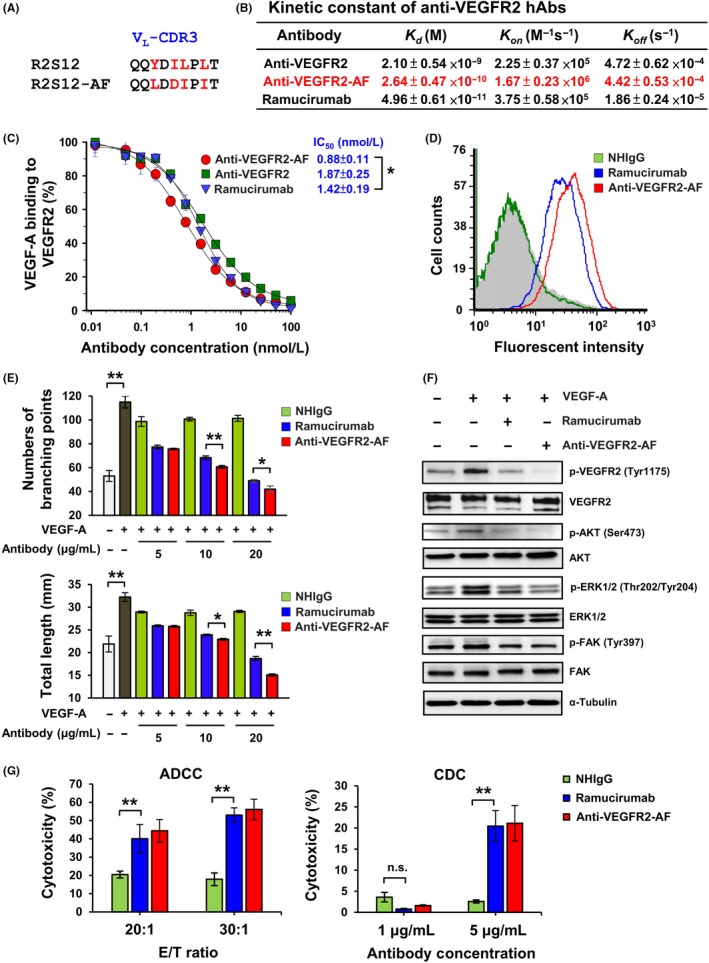
Affinity maturation of antivascular endothelial growth factor receptor‐2 (VEGFR2) human Ab (hAb), and analysis of anti‐VEGFR2‐AF activity. A, Amino acids of the light chain variable domain of CDR3 (VL‐CDR3). Residues that differ between R2S12 and R2S12AF are shown in red. B, Kinetic constants were determined using BIAcore T100 and calculated by BIAcore T100 evaluation software. C, Competitive ELISA was carried out to examine dose‐dependent inhibition of VEGF‐A binding to VEGFR2 by hAb. A value of 100% was attributed to the binding of 4 nM VEGF‐A to immobilized VEGFR2 in the absence of competitors. n = 4. D, Determination of the binding activity of anti‐VEGFR2 Ab to HUVECs by flow cytometry analysis. Ab concentration, 0.1 μg/mL. E, Capillary structure formation assays were carried out using Matrigel‐coated μ‐Slides. HUVECs treated with 40 ng/mL VEGF‐A alone or together with normal human IgG (NHIgG), ramucirumab, or anti‐VEGFR2‐AF for 5 h at 37°C. Relative sprout length and branching points were quantitatively measured. F, HUVECs were treated with 4 nM vascular endothelial growth factor‐A (VEGF‐A) or 100 nM anti‐VEGFR2‐AF or ramucirumab for 10 min at 37°C. Total protein was prepared from treated HUVECs and examined by western blot analysis. α‐Tubulin was used as a loading control. G, For the Ab‐dependent cellular cytotoxicity assay, fresh PBMCs were used as effector cells (E) and HUVECs were as target cells (T). The ratio of effector vs target cells (E:T) is 20:1 and 30:1 with treatment of 10 μg/mL ramucirumab, anti‐VEGFR2‐AF, and NHIgG. For the complement‐dependent cytotoxicity assay, HUVECs were treated with 1 or 5 μg/mL NHIgG, ramucirumab, and anti‐VEGFR2‐AF Ab with 10% rabbit complement at 37°C overnight. Specific cell lysis was measured by lactate dehydrogenase release. One percent of Triton X‐100 treatment was defined as 100% of cell lysis. Error bars, SD. *P < .05; **P < .01. n.s., no significant
Measurement of the Ab‐antigen binding kinetics showed that anti‐VEGFR2‐AF possesses a subnanomolar affinity constant (Kd = 0.264 nM), which represents an 8‐fold increase in binding affinity to VEGFR2 over its parental clone, anti‐VEGFR2 hAb (Kd = 2.1 nM; Figure 3B. We then undertook solid‐phase competitive binding assays to quantitatively evaluate the disruption of VEGF‐A/VEGFR2 binding by individual Abs. As shown in Figure 3C, the IC50 for inhibition of VEGF‐A binding to VEGFR2 was 0.88 nM for anti‐VEGFR2‐AF and 1.42 nM for ramucirumab. These data indicate that anti‐VEGFR2‐AF is superior to ramucirumab at blocking the VEGF‐A/VEGFR2 interaction. Flow cytometry with HUVECs was then carried out to confirm that anti‐VEGFR2‐AF shows stronger binding than ramucirumab to VEGFR2 on the cell surface (Figures 3D, S3A,B).
3.5. Anti‐VEGFR2‐AF inhibits activation of VEGFR2‐mediated signaling and disrupts formation of capillary structures by HUVECs
To explore the antiangiogenic potential of anti‐VEGFR2‐AF, we analyzed the effects of anti‐VEGFR2‐AF on HUVEC growth, migration, and tube formation. We found that anti‐VEGFR2‐AF suppressed VEGF‐A‐induced HUVEC proliferation and migration using MTT and wound‐healing assays, respectively (Figure S3C,D). To investigate the effect of anti‐VEGFR2‐AF on endothelial cell tube formation (a critical step in angiogenesis), HUVECs on Matrigel layers were incubated in the absence or presence of VEGF‐A and anti‐VEGFR2‐AF (Figure 3E). Anti‐VEGFR2‐AF was effective at preventing the VEGF‐A‐triggered formation of capillary‐like structures, based on measurements of tubule length and branch point number (Figure 3E).
To investigate the molecular mechanism underlying the antiangiogenic properties of anti‐VEGFR2‐AF, we examined the activation of signaling molecules and pathways by western blotting (Figure 3F) We found that anti‐VEGFR2‐AF efficiently diminished VEGF‐A‐induced phosphorylation of VEGFR2 and its downstream signaling molecules, AKT, ERK1/2, and FAK. Moreover, the level of phosphorylated VEGFR2 was lower in the anti‐VEGFR2‐AF‐treated cells compared to ramucirumab‐treated cells, whereas FAK phosphorylation was only marginally reduced by treatment with either Ab (Figure 3F). To determine the effector cell function of anti‐VEGFR2‐AF, we undertook ADCC and CDC analyses (Figure 3G). Anti‐VEGFR2‐AF and ramucirumab revealed similar HUVEC killing activity. Collectively, these results indicate that anti‐VEGFR2‐AF significantly inhibits several essential steps of vascular endothelial cell angiogenesis, suggesting that this Ab could have antiangiogenic potential in vivo.
3.6. Anti‐VEGFR2‐AF inhibits VEGF‐A‐induced cellular function in VEGFR2‐expressing human prostate cancer cells
In addition to the angiogenic actions of VEGFR2 protein in endothelial cells, the receptor is also known to be expressed in various cancer cells, where it is associated with tumor malignancy.12, 14, 44 A previous study reported that the antitumor activity of a VEGFR2 kinase inhibitor is correlated with the expression level of VEGFR2 in human prostate tumor xenografts.45 However, the role of VEGFR2 in prostate tumorigenesis is not well understood. PC‐3 is a well‐characterized human prostate cancer cell line and has been reported to express VEGFR2 protein.46 Therefore, we chose PC‐3 as a model to evaluate the therapeutic efficacy of anti‐VEGFR2‐AF at inhibiting cellular activities associated with tumor growth. We first carried out quantitative RT‐PCR to investigate the endogenous expression of VEGFR2 in PC‐3 cells (Figure 4A). We found that VEGFR2 mRNA was readily detected in PC‐3 cells but was barely detectable in negative control 293T cells. We also undertook western blotting to confirm that PC‐3 cells express endogenous VEGFR2 protein (Figure S4A). Treatment of PC‐3 cells with VEGF‐A caused phosphorylation of VEGFR2 and its downstream effectors, AKT and ERK1/2; however, these phosphorylation events were inhibited by ramucirumab and anti‐VEGFR2‐AF treatment (Figure S4B).
Figure 4.
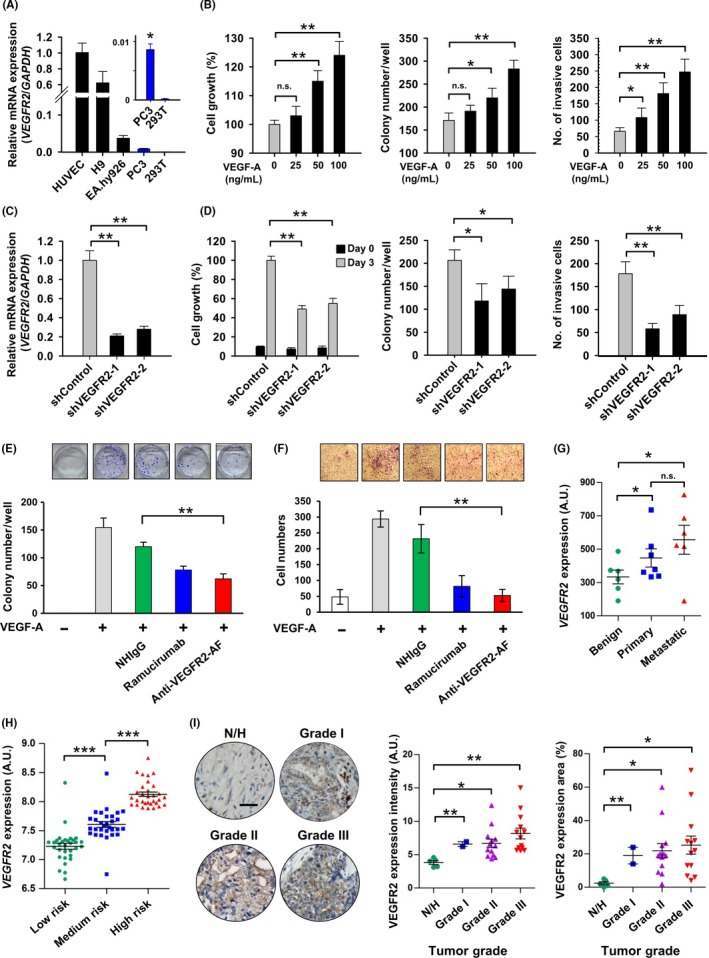
Characterization of vascular endothelial growth factor receptor‐2 (VEGFR2) activity in human prostate cancer cells. A, Analysis of VEGFR2 expression in the indicated cell lines was assessed by quantitative RT‐PCR. HUVECs, EA.hy926, and hESC‐H9 cells are known to show high expression of VEGFR2 mRNA and were thus used as positive controls. 293T cells were used as a negative control. Expression of VEGFR2 was normalized to that of GAPDH. B, PC‐3 cells treated with 25, 50, or 100 ng/mL vascular endothelial growth factor‐A (VEGF‐A) were subjected to colony formation, MTT, and invasion assays. n = 6, each group. C, PC‐3 cells were treated with 2 VEGFR2‐targeted shRNAs (shVEGFR2‐1 and shVEGFR2‐2), and VEGFR2 expression was analyzed by quantitative RT‐PCR. The shRNA targeting luciferase gene (shControl) was used as a negative control. D, MTT, colony formation, and Transwell invasion assays were carried out on VEGFR2‐knockdown PC‐3 cells while treated with 100 ng/mL VEGF‐A. E, F, Colony formation (E) and Transwell assays (F) of PC‐3 cells were carried out with treatment of 100 ng/mL VEGF‐A and 10 μg/mL NHIgG, ramucirumab, or anti‐VEGFR2‐AF. n = 3, each group. G, Scatter plots show relative VEGFR2 mRNA expression in metastatic prostate tumors as compared with benign and primary tumors. Raw normalized microarray data were obtained from the NCBI Gene Expression Omnibus database (benign, n = 6; primary, n = 7; metastatic, n = 6). H, VEGFR2 mRNA expression was analyzed in samples from patients with low to high risk of prostate cancer using the SurvExpress database (low risk, n = 47; medium risk, n = 46; high risk, n = 47). I, Immunohistochemical staining with an anti‐VEGFR2 Ab was undertaken on a human prostate cancer tissue array. The intensity and area of VEGFR2 distribution in the stained tissue sections was quantified using ImageJ. Normal prostate (N)/hyperplasia (H), n = 5; grade I, n = 2; grade II, n = 13; grade III, n = 13. Error bars, SE. *P < .05; **P < .01. n.s., not significant
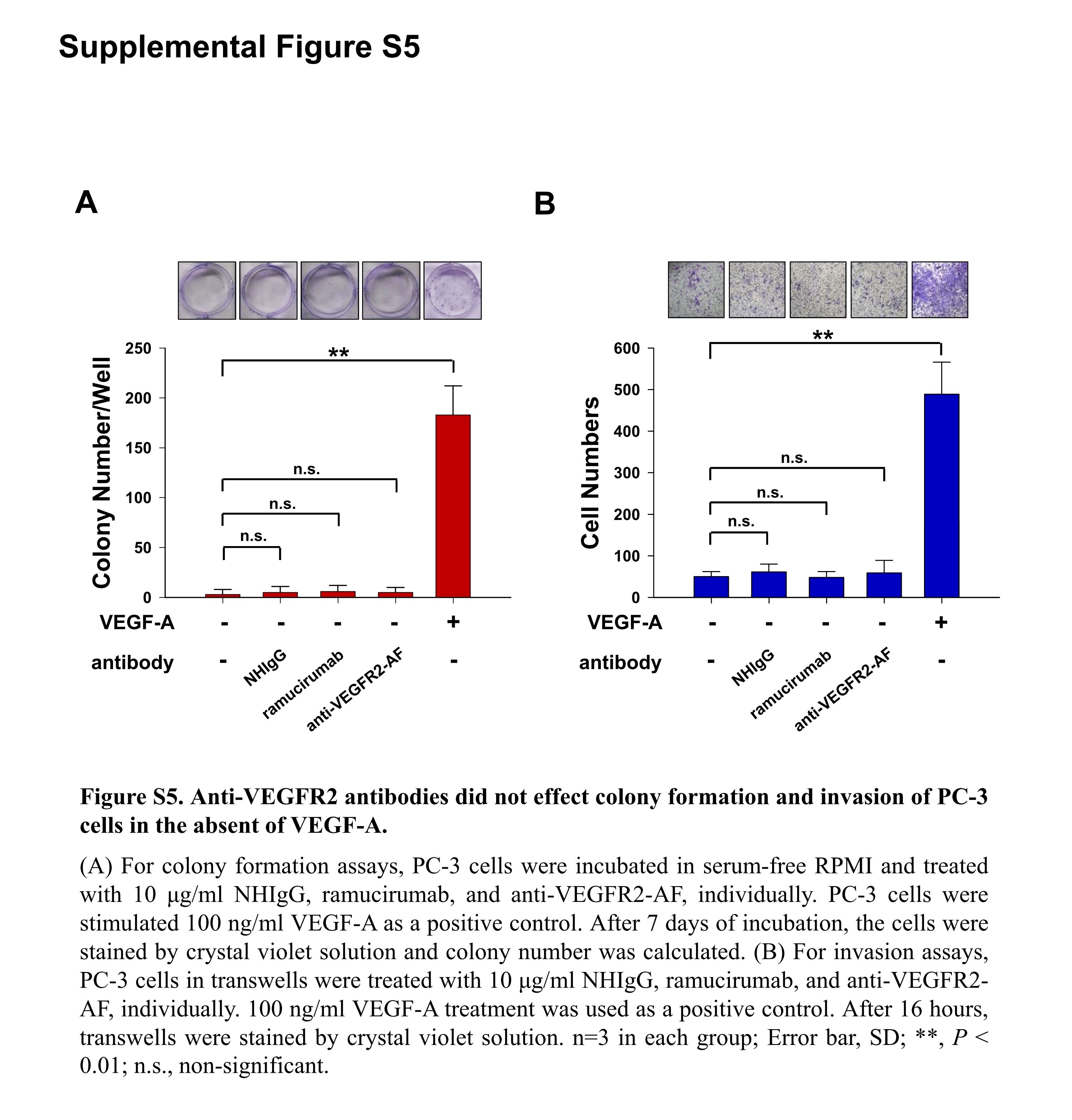
To characterize the functional significance of VEGFR2 expression in PC‐3 cells, we analyzed several activities of PC‐3 cells following VEGF‐A treatment. Treatment with VEGF‐A enhanced various PC‐3 cellular activities, including proliferation, colony formation, and invasiveness (Figure 4B). We proceeded to establish VEGFR2‐knockdown PC‐3 cells with lentivirus‐mediated shRNAs (shControl, Figure 4C). Knockdown of VEGFR2 reduced proliferation, colony formation, and invasiveness compared to control shRNA (Figure 4D). We then treated PC‐3 cells with anti‐VEGFR2‐AF, confirming that VEGF‐A‐induced cellular activities could be suppressed by anti‐VEGFR2‐AF (Figures 4E,F, S4C). However, anti‐VEGFR2 Ab did not affect colony formation or invasion of PC‐3 cells without VEGF‐A treatment (Figure S5). Hence, the VEGF‐A/VEGFR2 axis appears to be crucial for clonogenic and tumorigenic activities in PC‐3 cells.
To explore whether our findings are clinically relevant to human prostate cancer, we investigated VEGFR2 mRNA expression patterns in publicly available databases. We found that the level of VEGFR2 transcripts in metastatic prostate tumors was higher than that in primary tumors from the GEO database by NCBI (Figure 4G). By querying another clinical cancer outcome database, SurvExpress,47 we discovered that the amount of VEGFR2 positively correlates with the risk of human prostate cancer (Figure 4H). Therefore, we used a commercially available well‐defined anti‐VEGFR2 Ab (55B11) to stain a human prostate cancer tissue array (Figure S6). The level of VEGFR2 was elevated in 3 grades of prostate adenocarcinoma, compared to normal/hyperplasia prostate tissues (Figure 4I).
3.7. Therapeutic efficacy of anti‐VEGFR2‐AF in human prostate cancer xenografts
We next used the PC‐3 xenograft prostate tumor model to evaluate the in vivo antitumor activity of anti‐VEGFR2‐AF in comparison to ramucirumab. Docetaxel is a first‐line chemotherapeutic agent for patients with metastatic castration‐resistant prostate cancer,48 and thus we also investigated the therapeutic effects of combined docetaxel and anti‐VEGFR2‐AF or ramucirumab. NOD/SCID mice bearing PC‐3 xenografts were treated with ramucirumab, anti‐VEGFR2‐AF, docetaxel, anti‐VEGFR2‐AF plus docetaxel, or ramucirumab plus docetaxel (Figure 5A). By day 38, the reduction in tumor growth reached 90% for mice treated with a combination of anti‐VEGFR2‐AF plus docetaxel, 82% for mice treated with a combination of ramucirumab plus docetaxel, 70% for mice treated with docetaxel, 52% for mice treated with anti‐VEGFR2‐AF, and 45% for mice treated with ramucirumab (Figure 5B). Body weight was used as a surrogate indicator of health status (Figure 5C). The anti‐VEGFR2‐AF and ramucirumab groups showed no significant changes in body weight compared to the NHIgG group throughout the treatment period. Treatment with docetaxel alone caused a marked loss of body weight (~20%). Mice treated with docetaxel in combination with Abs showed similar body weight loss compared to mice treated with docetaxel alone, which suggests that anti‐VEGFR2‐AF and ramucirumab do not enhance docetaxel‐induced toxicity.
Figure 5.
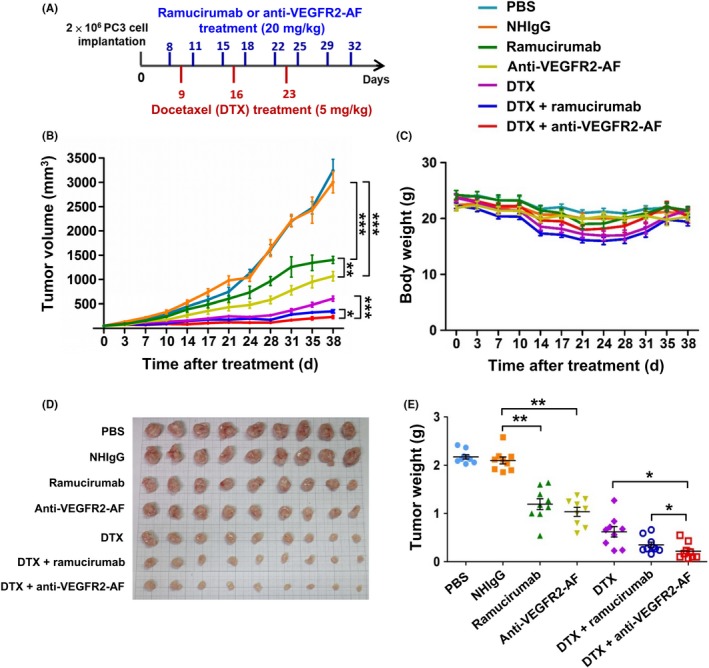
Analysis of the therapeutic efficacy of antivascular endothelial growth factor receptor‐2 (VEGFR2)‐AF in a PC‐3 xenograft mouse model. A, Treatment schedule shows mice bearing PC‐3‐derived xenografts were treated with 20 mg/kg Abs and 5 mg/kg docetaxel (DTX). B, Mice with an average tumor size of 50 mm3 were treated with Abs, DTX, or antibodies plus DTX. Tumor growth was measured in each group. C, Body weight of mice in each group was monitored during treatment. D, At the end of the treatment period, tumors were dissected from each mouse and tumor mass was determined. E, Tumor weight was measured at the end of the treatment period. All data are shown as the mean of 9 mice per group. Error bars, SE. *P < .05
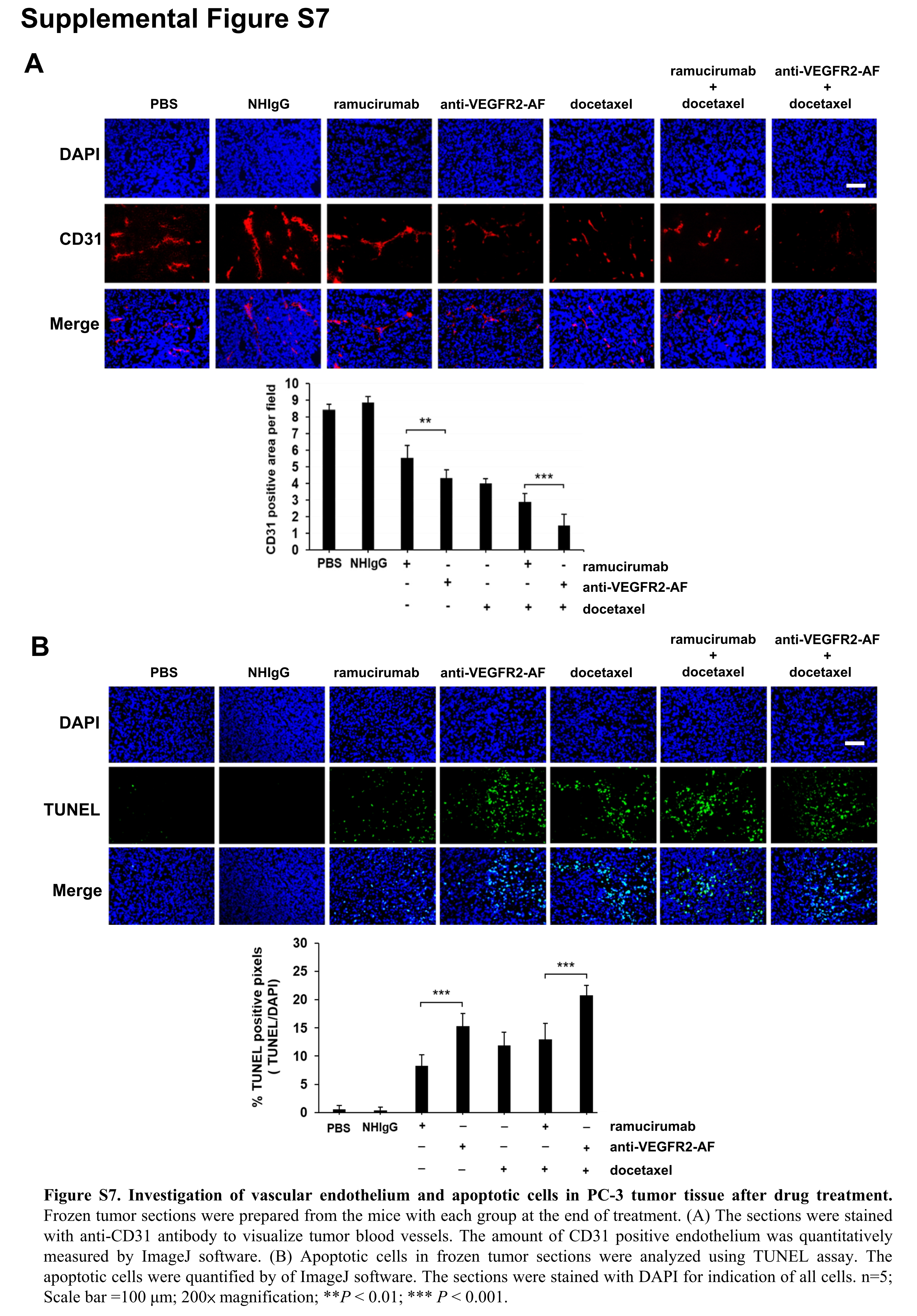
At the end of the treatment period, tumor weights were measured and found to consistently reflect tumor volume (Figure 5D,5). We further examined tumor tissues, using anti‐CD31 Ab to detect tumor blood vessels and TUNEL assay to identify apoptotic cells. We found that anti‐VEGFR2‐AF and ramucirumab prominently reduced tumor vascular density and enhanced cancer cell apoptosis (Figure S7). From these results, we conclude that anti‐VEGFR2‐AF is as effective and safe as ramucirumab at attenuating tumor growth, and that anti‐VEGFR2‐AF significantly enhances the effectiveness of docetaxel in the treatment of human prostate tumors in mice.
3.8. Anti‐VEGFR2‐AF prolongs survival of mice bearing HL‐60 leukemia xenografts
The VEGF‐A/VEGFR2 pathway has important functions not only in solid tumors but also in liquid tumors, such as leukemia or lymphoma.49, 50 We also found that the HL‐60 human leukemia cell line mildly expressed VEGFR. Moreover, VEGF‐A treatment led to the phosphorylation of VEGFR2 and its downstream signaling effectors, such as ERK1/2 and AKT (Figure S8A). Treatment of HL‐60 cells with VEGF‐A also enhanced cell proliferation (Figure S8B). Therefore, we utilized an HL‐60 leukemia xenograft model in NSG mice to evaluate the antileukemia effect of anti‐VEGFR2‐AF. Mice received i.v. injections of 5 × 106 HL‐60 cells, and were treated 3 days later with ramucirumab, anti‐VEGFR2‐AF, NHIgG, or PBS. As shown in Figure 6A, all PBS‐ or NHIgG‐treated mice died within 36 days; however, leukemia‐bearing mice treated with Abs against VEGFR2 showed a marked extension in survival time. Mice treated with anti‐VEGFR2‐AF survived longer (70 days; median, 53 days) than those treated with ramucirumab (56 days; median, 43 days). No significant changes in body weight were observed between groups (Figure 6B).
Figure 6.
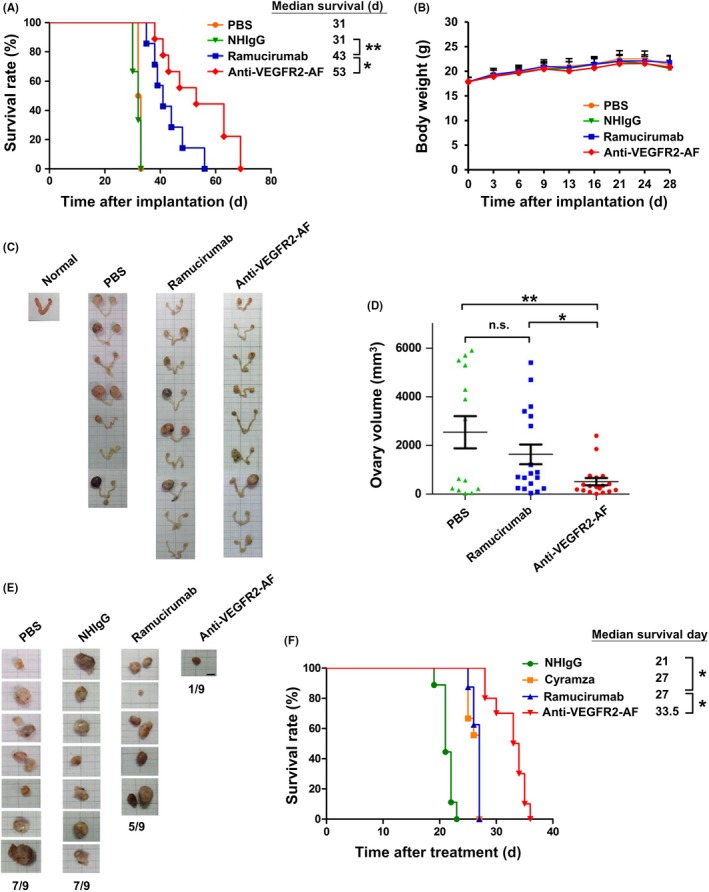
Antivascular endothelial growth factor receptor‐2 (VEGFR2)‐AF shows greater antitumor activity than ramucirumab in the HL60‐derived xenograft mouse models. A, Kaplan‐Meier survival analysis of each group. Survival was significantly prolonged in the anti‐VEGFR2‐AF group compared with the ramucirumab group (n = 9 per group). B, Body weight of mice in each group. C, Ovaries were dissected from PBS, ramucirumab, or anti‐VEGFR2‐AF groups after mouse death. Ovaries from an NSG mouse without leukemia served as a normal control. D, Ovary volume in mice treated with PBS, ramucirumab, or anti‐VEGFR2‐AF was measured. E, Morphometric analysis of lymph node changes in leukemia‐tumor bearing mice. Leukemia cells that had metastasized to lymph nodes were identified from mice of the indicated groups (n = 9, each group). F, Kaplan‐Meier survival analysis of mice of each group (n = 9). Anti‐VEGFR2‐AF treatment significantly extended survival compared to commercially available Cyramza and ramucirumab. Error bars, SE. *P < .05; **P < .01. NHIgG, normal human IgG; n.s., not significant
We carried out post‐mortem histopathological examinations of all mice. No obvious pathological changes were observed in the liver, spleen, heart, or kidney of mice in any treatment group (data not shown). Interestingly, we observed that the ovaries of leukemia‐bearing mice were swollen compared to control mice (Figure 6C), and H&E staining revealed that the ovaries had been infiltrated by metastatic leukemia cells (Figure S9). The average ovarian volume in the anti‐VEGFR2‐AF‐treated group was significantly smaller than that in the ramucirumab‐treated group (Figure 6D). Furthermore, lymph nodes with leukemia cell infiltration were identified in 1 of 9 mice treated with anti‐VEGFR2‐AF, whereas lymph nodes with leukemia cell infiltration were present in 5 of 9 mice treated with ramucirumab (Figure 6E). To confirm the anticancer activity of anti‐VEGFR2‐AF, a third party repeated the HL‐60 leukemia xenograft experiment, this time undertaken a head‐to‐head comparison of efficacy between anti‐VEGFR2‐AF, ramucirumab, and commercially available Cyramza (brand drug of ramucirumab, Eli Lilly and Company) (Figure 6F). Anti‐VEGFR2‐AF treatment significantly extended the median survival of mice (33.5 days) compared to ramucirumab (27 days), Cyramza (27 days), and NHIgG (21 days).
4. DISCUSSION
Angiogenesis is a pivotal process in tumor growth, dissemination, invasion, and metastasis; hence, destroying or suppressing the formation of tumor vasculature can cause disease regression and prevent metastasis.7, 51, 52 Antiangiogenic therapy using targeting agents, such as small molecules (sunitinib and sorafenib) or mAbs (bevacizumab and ramucirumab), can block the VEGF/VEGFR2 signaling to provide effective cancer treatment.22, 23, 29, 53
The tumor vessel was traditionally thought to be an especially attractive target tissue because it is formed from nonmalignant endothelial cells with relatively stable genetic backgrounds compared to tumor cells.54 Therefore, tumor vessels should be less likely to acquire therapy‐induced mutations than cancer cells.55 There is a growing body of studies elucidating the mechanisms underlying acquired resistance to antiangiogenic therapy.56 In animal models, long‐term blockage of VEGF‐mediated signaling induced a compensatory response through upregulation of other angiogenic factors, such as hepatocyte growth factor,57 fibroblast growth factor‐2,58 and PlGF.59 Additionally, an in vitro study reported that tumor‐associated endothelial cells could acquire resistance to paclitaxel by potentiating multidrug resistance 1 expression through the VEGF‐A signaling pathway.60 Interestingly, cytogenetic abnormalities (such as aneuploidy or abnormal centrosomes) have been found in tumor‐associated endothelial cells from human renal carcinoma.61 Tumor‐associated endothelial cells in high‐metastatic tumors show higher VEGFR2 expression levels and resistance to paclitaxel than those in low‐metastatic tumors.11 These observations might at least partially explain the modest clinical benefits of anti‐VEGF therapies, that is, progression‐free survival and overall survival generally do not extend beyond a few months, and tumor relapse often results in metastasis.56, 62 In this study, anti‐VEGFR2‐AF‐mediated targeting of VEGFR2 on tumor endothelium not only disrupts VEGF‐A‐induced signaling, but also triggers ADCC or CDC to directly kill the tumor‐associated endothelial cells and targeted cancer cells. Thus, anti‐VEGFR2‐AF therapy might be a strategy to overcome the anti‐VEGF therapy‐induced resistance and could improve the efficacy of current antiangiogenic therapy.
Anti‐VEGFR2‐AF might be able to exert dual targeting and inhibition effects on both tumor vascular and malignant cells as tumor cells also express VEGFR2. This dual targeting ability has been discussed in previous reports, as it could produce synergistic effects in cancer therapy. For example, the use of iRGD peptide and anti‐Met Ab in preclinical studies presented compelling evidence that dual targeting can strongly inhibit tumor growth and eliminate angiogenesis.63, 64 Placenta growth factor is a pleiotropic cytokine that stimulates angiogenesis and tumor cell growth directly by binding to VEGFR1. Fischer and colleagues showed that PlGF‐neutralizing Ab treatment inhibited angiogenesis, lymphangiogenesis and tumor cell motility, in addition to its enhancement of anti‐VEGFR2 Ab therapy.59
In the present study, we developed a fully human Ab, anti‐VEGFR2‐AF, which shows superior binding to VEGFR2, antagonizing the activity of the receptor. Similar to ramucirumab, anti‐VEGFR2‐AF specifically bound to human VEGFR2, but not to murine VEGFR2. Therapeutic efficacy of anti‐VEGFR2‐AF was elucidated in solid and liquid xenograft mouse models. Anti‐VEGFR2‐AF is currently under study in preclinical trials. In conclusion, our findings suggest that anti‐VEGFR2‐AF could be useful as a therapeutic Ab for cancer treatment that simultaneously inhibits angiogenesis by vascular endothelial cells and tumorigenesis by VEGFR2‐expressing tumor cells.
DISCLOSURE
Yaw‐Jen Liu is vice president of the R&D Center at United Biopharma, Hsinshu, Taiwan. Ruei‐Min Lu, Chiung‐Yi Chiu, I‐Ju Liu, Yu‐Ling Chang, and Han‐Chung Wu are employees of Academia Sinica. Academia Sinica has licensed out this patent to United Biopharma, Taiwan and is currently conducting preclinical studies on the anti‐VEGFR2 Ab described herein.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENT
We thank the Core Facility of the Institute of Cellular and Organismic Biology in Academia Sinica (Taipei, Taiwan) for technical support, Dr Chang‐Yi Wang (United Biopharma Inc, Taiwan), and Dr Wen‐Jiun Peng (UBI Pharma Inc, Taiwan) for kindly providing valuable suggestions. This research was supported by Academia Sinica and the Ministry of Science and Technology (106‐0210‐01‐15‐02 and 107‐0210‐01‐19‐01), and the Program for Translational Innovation of Biopharmaceutical Development – Technology Supporting Platform Axis (106‐0210‐01‐10‐01 and 107‐0210‐01‐19‐04).
Lu R‐M, Chiu C‐Y, Liu I‐J, Chang Y‐L, Liu Y‐J, Wu H‐C. Novel human Ab against vascular endothelial growth factor receptor 2 shows therapeutic potential for leukemia and prostate cancer. Cancer Sci. 2019;110:3773–3787. 10.1111/cas.14208
Liu and Chang contributed equally to this work.
Funding information
This research was supported by Academia Sinica and Ministry of Science and Technology (106‐0210‐01‐15‐02 and 107‐0210‐01‐19‐01), and the Program for Translational Innovation of Biopharmaceutical Development – Technology Supporting Platform Axis (106‐0210‐01‐10‐01 and 107‐0210‐01‐19‐04).
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7:1016‐1036. [PMC free article] [PubMed] [Google Scholar]
- 3. Folkman J. Tumor angiogenesis: therapeutic implications. New Engl J Med. 1971;285:1182‐1186. [DOI] [PubMed] [Google Scholar]
- 4. Simons M, Gordon E, Claesson‐Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611‐625. [DOI] [PubMed] [Google Scholar]
- 5. Peach CJ, Mignone VW, Arruda MA, et al. Molecular pharmacology of VEGF‐A isoforms: binding and signalling at VEGFR2. Int J Mol Sci. 2018;19:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17:457. [DOI] [PubMed] [Google Scholar]
- 7. Nasir A. Angiogenic signaling pathways and anti‐angiogenic therapies in human cancer In: Badve S, Kumar GL, eds. Predictive Biomarkers in Oncology: Applications in Precision Medicine. Cham: Springer International Publishing; 2019:243‐262. [Google Scholar]
- 8. Ferrara N, Gerber HP. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 2001;106:148‐156. [DOI] [PubMed] [Google Scholar]
- 9. Oh P, Testa JE, Borgstrom P, Witkiewicz H, Li Y, Schnitzer JE. In vivo proteomic imaging analysis of caveolae reveals pumping system to penetrate solid tumors. Nat Med. 2014;20:1062‐1068. [DOI] [PubMed] [Google Scholar]
- 10. Smith NR, Baker D, James NH, et al. Vascular endothelial growth factor receptors VEGFR‐2 and VEGFR‐3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res. 2010;16:3548‐3561. [DOI] [PubMed] [Google Scholar]
- 11. Ohga N, Ishikawa S, Maishi N, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high‐ and low‐metastatic tumors. Am J Pathol. 2012;180:1294‐1307. [DOI] [PubMed] [Google Scholar]
- 12. Spannuth WA, Nick AM, Jennings NB, et al. Functional significance of VEGFR‐2 on ovarian cancer cells. Int J Cancer. 2009;124:1045‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donnem T, Al‐Saad S, Al‐Shibli K, et al. Inverse prognostic impact of angiogenic marker expression in tumor cells versus stromal cells in non small cell lung cancer. Clin Cancer Res. 2007;13:6649‐6657. [DOI] [PubMed] [Google Scholar]
- 14. Chatterjee S, Heukamp LC, Siobal M, et al. Tumor VEGF:VEGFR2 autocrine feed‐forward loop triggers angiogenesis in lung cancer. J Clin Invest. 2013;123:1732‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giatromanolaki A, Koukourakis MI, Sivridis E, et al. Activated VEGFR2/KDR pathway in tumour cells and tumour associated vessels of colorectal cancer. Eur J Clin Invest. 2007;37:878‐886. [DOI] [PubMed] [Google Scholar]
- 16. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 17. Pizon M, Zimon DS, Pachmann U, Pachmann K. Insulin‐like growth factor receptor I (IGF‐IR) and vascular endothelial growth factor receptor 2 (VEGFR‐2) are expressed on the circulating epithelial tumor cells of breast cancer patients. PLoS ONE. 2013;8:e56836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Messaritakis I, Politaki E, Plataki M, et al. Heterogeneity of circulating tumor cells (CTCs) in patients with recurrent small cell lung cancer (SCLC) treated with pazopanib. Lung Cancer. 2017;104:16‐23. [DOI] [PubMed] [Google Scholar]
- 19. Baka S, Clamp AR, Jayson GC. A review of the latest clinical compounds to inhibit VEGF in pathological angiogenesis. Expert Opin Ther Targets. 2006;10:867‐876. [DOI] [PubMed] [Google Scholar]
- 20. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti‐VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391‐400. [DOI] [PubMed] [Google Scholar]
- 21. Gerriets V, Kasi A. Bevacizumab. StatPearls. Treasure Island, FL: StatPearls Publishing StatPearls Publishing LLC., 2018. [Google Scholar]
- 22. Stitzlein L, Rao P, Dudley R. Emerging oral VEGF inhibitors for the treatment of renal cell carcinoma. Expert Opin Investig Drugs. 2018;1–10. [DOI] [PubMed] [Google Scholar]
- 23. Lopez A, Harada K, Vasilakopoulou M, Shanbhag N, Ajani JA. Targeting angiogenesis in colorectal carcinoma. Drugs. 2019;79:63‐74. [DOI] [PubMed] [Google Scholar]
- 24. Wu P, Nielsen TE, Clausen MH. FDA‐approved small‐molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422‐439. [DOI] [PubMed] [Google Scholar]
- 25. Falcon BL, Chintharlapalli S, Uhlik MT, Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR‐2) as anti‐angiogenic agents. Pharmacol Ther. 2016;164:204‐225. [DOI] [PubMed] [Google Scholar]
- 26. Spratlin J. Ramucirumab (IMC‐1121B): monoclonal antibody inhibition of vascular endothelial growth factor receptor‐2. Curr Oncol Rep. 2011;13:97‐102. [DOI] [PubMed] [Google Scholar]
- 27. Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int J Cancer. 2002;97:393‐399. [DOI] [PubMed] [Google Scholar]
- 28. Lu D, Shen J, Vil MD, et al. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J Biol Chem. 2003;278:43496‐43507. [DOI] [PubMed] [Google Scholar]
- 29. Javle M, Smyth EC, Chau I. Ramucirumab: successfully targeting angiogenesis in gastric cancer. Clin Cancer Res. 2014;20:5875‐5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arrieta O, Zatarain‐Barron ZL, Cardona AF, Carmona A, Lopez‐Mejia M. Ramucirumab in the treatment of non‐small cell lung cancer. Expert Opin Drug Saf. 2017;16:637‐644. [DOI] [PubMed] [Google Scholar]
- 31. Chau I, Peck‐Radosavljevic M, Borg C, et al. Ramucirumab as second‐line treatment in patients with advanced hepatocellular carcinoma following first‐line therapy with sorafenib: patient‐focused outcome results from the randomised phase. III REACH study. Eur J Cancer. 1990;2017(81):17‐25. [DOI] [PubMed] [Google Scholar]
- 32. Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018;36:1136‐1145. [DOI] [PubMed] [Google Scholar]
- 33. Wu CH, Liu IJ, Lu RM, Wu HC. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016;23:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee TY, Lin CT, Kuo SY, Chang DK, Wu HC. Peptide‐mediated targeting to tumor blood vessels of lung cancer for drug delivery. Cancer Res. 2007;67:10958‐10965. [DOI] [PubMed] [Google Scholar]
- 35. Chi YH, Hsiao JK, Lin MH, Chang C, Lan CH, Wu HC. Lung cancer‐targeting peptides with multi‐subtype indication for combinational drug delivery and molecular imaging. Theranostics. 2017;7:1612‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yeh CY, Hsiao JK, Wang YP, Lan CH, Wu HC. Peptide‐conjugated nanoparticles for targeted imaging and therapy of prostate cancer. Biomaterials. 2016;99:1‐15. [DOI] [PubMed] [Google Scholar]
- 37. Wu CH, Kuo YH, Hong RL, Wu HC. alpha‐Enolase‐binding peptide enhances drug delivery efficiency and therapeutic efficacy against colorectal cancer. Sci Transl Med. 2015;7:290ra91. [DOI] [PubMed] [Google Scholar]
- 38. Lu R‐M, Chen M‐S, Chang D‐K, et al. Targeted drug delivery systems mediated by a novel peptide in breast cancer therapy and imaging. PLoS ONE. 2013;8:e66128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu R‐M, Chang Y‐L, Chen M‐S, Wu H‐C. Single chain anti‐c‐Met antibody conjugated nanoparticles for in vivo tumor‐targeted imaging and drug delivery. Biomaterials. 2011;32:3265‐3274. [DOI] [PubMed] [Google Scholar]
- 40. Nielsen UB, Marks JD. Affinity maturation of phage antibodies In: Clackson T, Lowman HB, eds. Phage Display: A Practical Approach. New York, NY: Oxford University Press; 2004;289–314. [Google Scholar]
- 41. Franklin Matthew C, Navarro Elizabeth C, Wang Y, et al. The structural basis for the function of two anti‐VEGF receptor 2 antibodies. Structure. 2011;19:1097‐1107. [DOI] [PubMed] [Google Scholar]
- 42. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278‐287. [DOI] [PubMed] [Google Scholar]
- 43. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126‐1136. [DOI] [PubMed] [Google Scholar]
- 44. Silva SR, Bowen KA, Rychahou PG, et al. VEGFR‐2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int J Cancer. 2011;128:1045‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pratheeshkumar P, Budhraja A, Son YO, et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR‐2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS ONE. 2012;7:e47516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dev I, Dornsife R, Hopper T, et al. Antitumour efficacy of VEGFR2 tyrosine kinase inhibitor correlates with expression of VEGF and its receptor VEGFR2 in tumour models. Br J Cancer. 2004;91:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aguirre‐Gamboa R, Gomez‐Rueda H, Martínez‐Ledesma E, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE. 2013;8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maluf FC, Smaletz O, Herchenhorn D. Castration‐resistant prostate cancer: systemic therapy in 2012. Clinics (Sao Paulo). 2012;67:389‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation of VEGFR‐2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Piechnik A, Dmoszynska A, Omiotek M, et al. The VEGF receptor, neuropilin‐1, represents a promising novel target for chronic lymphocytic leukemia patients. Int J Cancer. 2013;133:1489‐1496. [DOI] [PubMed] [Google Scholar]
- 51. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967‐974. [DOI] [PubMed] [Google Scholar]
- 52. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alshangiti A, Chandhoke G, Ellis PM. Antiangiogenic therapies in non‐small‐cell lung cancer. Curr Oncol. 2018;25:S45‐s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med. 2003;3:643‐651. [DOI] [PubMed] [Google Scholar]
- 55. Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404. [DOI] [PubMed] [Google Scholar]
- 56. Dahan N, Magidey K, Shaked Y. Resistance to inhibitors of angiogenesis In: Yarden Y, Elkabets M, eds. Resistance to Anti‐Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Cham: Springer International Publishing; 2018:211‐236. [Google Scholar]
- 57. Shojaei F, Lee JH, Simmons BH, et al. HGF/c‐Met acts as an alternative angiogenic pathway in sunitinib‐resistant tumors. Cancer Res. 2010;70:10090‐10100. [DOI] [PubMed] [Google Scholar]
- 58. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late‐stage pancreatic islet tumors. Cancer Cell. 2005;8:299‐309. [DOI] [PubMed] [Google Scholar]
- 59. Fischer C, Jonckx B, Mazzone M, et al. Anti‐PlGF inhibits growth of VEGF(R)‐inhibitor‐resistant tumors without affecting healthy vessels. Cell. 2007;131:463‐475. [DOI] [PubMed] [Google Scholar]
- 60. Akiyama K, Ohga N, Hida Y, et al. Tumor endothelial cells acquire drug resistance by MDR1 up‐regulation via VEGF signaling in tumor microenvironment. Am J Pathol. 2012;180:1283‐1293. [DOI] [PubMed] [Google Scholar]
- 61. Akino T, Hida K, Hida Y, et al. Cytogenetic abnormalities of tumor‐associated endothelial cells in human malignant tumors. Am J Pathol. 2009;175:2657‐2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hida K, Akiyama K, Ohga N, Maishi N, Hida Y. Tumour endothelial cells acquire drug resistance in a tumour microenvironment. J Biochem. 2013;153:243‐249. [DOI] [PubMed] [Google Scholar]
- 63. Vigna E, Comoglio PM. Targeting the oncogenic Met receptor by antibodies and gene therapy. Oncogene. 2014;34:1883. [DOI] [PubMed] [Google Scholar]
- 64. Sugahara KN, Teesalu T, Karmali PP, et al. Coadministration of a tumor‐penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials