

TNFα is critical for epigenetic DC maturation, DC phenotype stabilization, and driving protective TH1 immunity to fungal infection.
Abstract
Epigenetic modifications play critical roles in inducing long-lasting immunological memory in innate immune cells, termed trained immunity. Whether similar epigenetic mechanisms regulate dendtritic cell (DC) function to orchestrate development of adaptive immunity remains unknown. We report that DCs matured with IFNγ and TNFα or matured in the lungs during invasive fungal infection with endogenous TNFα acquired a stable TNFα-dependent DC1 program, rendering them resistant to both antigen- and cytokine-induced alternative activation. TNFα-programmed DC1 had increased association of H3K4me3 with DC1 gene promoter regions. Furthermore, MLL1 inhibition blocked TNFα-mediated DC1 phenotype stabilization. During IFI, TNFα-programmed DC1s were required for the development of sustained TH1/TH17 protective immunity, and bone marrow pre-DCs exhibited TNFα-dependent preprogramming, supporting continuous generation of programmed DC1 throughout the infection. TNFα signaling, associated with epigenetic activation of DC1 genes particularly via H3K4me3, critically contributes to generation and sustenance of type 1/17 adaptive immunity and the immune protection against persistent infection.
INTRODUCTION
Epigenetic modifications, apart from their well-known role in cell differentiation and cancer pathogenesis, are increasingly reported to be crucial in regulation of the immune responses. The involvement of epigenetic modifications during primary immune responses is emerging, with much attention focused on the T and B lymphocyte differentiation [reviewed in (1)]. In contrast, relatively few studies focus on the role of epigenetic modifications to innate immune cells during primary infection. Innate immune cell epigenetic modifications may be important axes in controlling initial immune polarization in vitro (2) and effector immune responses of myeloid cells in vivo (3). In parallel, the mainstream understanding of immunological memory has expanded by recent demonstration of innate immune cells exhibiting immunological memory-type responses, which enhance protection against reinfection (4–7). This phenomenon of innate immunological memory (trained immunity) was shown to be mediated by specific changes to epigenetic modifications and thereby the metabolic and transcriptional programs of innate effector cells such as macrophages, monocytes, and natural killer cells (8–10). However, it remains unknown whether epigenetic modifications to innate cells, particularly dendritic cells (DCs), could be important in not only responding to but also shaping the immune responses to primary infections in vivo.
Myeloid cells demonstrate diverse functional capabilities and, apart from acting as antimicrobial effector cells, are the major subset responsible for antigen processing and generation of antigen-specific effector T cells (11, 12). Their functions are modulated by the local inflammatory environment, which dictates myeloid cell classical (DC1/M1) or alternative (DC2/M2) polarization profiles. Classically activated (DC1/M1-like) myeloid cells are induced via STAT1 (signal transducer and activator of transcription 1), IRF1 (interferon regulatory factor 1), and/or IRF5 pathways, triggered by stimulation of cells with lipopolysaccharide (LPS), interferon-γ (IFNγ), and/or granulocyte-macrophage colony-stimulating factor (GM-CSF). These cells induce inducible nitric oxide synthase (iNOS) and a variety of important proinflammatory factors. Conversely, alternatively activated (DC2/M2-like) cells develop in the environment of interleukin-4 (IL-4), IL-10, or M-CSF and are known to activate Stat3/6, Klf2/4, and IRF3/4 pathways. A DC2 phenotype is characterized by arginase1-dependent metabolism of arginine and is associated with secretion of a spectrum of anti-inflammatory factors. The DC1 phenotype is essential for priming T helper 1 (TH1) responses, required for killing of intracellular bacteria, such as Mycobacterium tuberculosis, and other persistent intracellular parasites, including fungi such as Cryptococcus neoformans (C. neo) (13). DCs also actively contribute to T cell restimulation in the infection sites and serve as effector cells destroying microorganisms (14–16), and thus, DC polarization may affect antimicrobial defenses at multiple levels.
Myeloid cell polarization is thought to be highly plastic in response to changes in stimulation by different group of mediators (17). On the other hand, M2-polarized macrophages were shown to display unique histone modification signature, suggesting that some level of transcriptional stability is present in these cells (18). If DC could acquire this DC1/DC2 program stabilization during infections, one would expect this to affect TH1/TH2 priming and/or possibly influence the long-term maintenance of the polarized TH phenotype. However, DC1/DC2 polarization plasticity versus stability has not been investigated, and thus, it remains unknown how transcriptional stability of DC polarization program could affect DC functions during immune responses.
Cryptococcosis is a relevant example of highly fatal and persistent infection, which requires stable TH1/TH17 immune response for clearance, in contrast with TH2 responses that support the development of chronic infection and latency (19–21). Studies by our group demonstrated that manipulation of one of the central inflammatory mediator, tumor necrosis factor α (TNFα), deranges protective TH1 responses to C. neo (7). Consistent with our models, epidemiology studies show that cryptococcosis is increasingly seen in patients undergoing anti-TNFα antibody therapies (22–24).
Here, we show that TNFα induces a state of DC1 phenotypic memory or programming, which is required for TH1/TH17 immune protection during cryptococcosis. Mechanistically, our study overwhelmingly suggests that TNFα signaling facilitates methylation of histone 3 lysine 4 (H3K4) at crucial DC1 gene promoter regions and affects expression and function of mixed-lineage leukemia protein-1 (MLL1) methyltransferase activity. Our study demonstrates that this programming of DC can translate into the fate of adaptive immunity and that TNFα is the central mediator required to trigger these effects.
RESULTS
TNFα results in prolonged stable DC1 gene expression and DC2 gene suppression
Acute-phase production of TNFα is critical for control and clearance of the model invasive fungal infection (IFI) C. neo (7). CBA/J mice with intact TNFα signaling (control) clear C. neo within 28 days post-infection (dpi), but those that were TNFα depleted (anti-TNFα) with a single injection of TNFα-blocking antibody at the time of infection do not clear C. neo (fig. S1A). We show that control mice with intact TNFα signaling induce sustained classical activation of DCs (DC1) and strongly polarized TH1/TH17 protective immunity upon infection with C. neo (fig. S1, B and C) (7). The absence of TNFα results in a suppressed DC1 signature [iNOS, IL-12b, and major histocompatibility complex II (MHCII)]; up-regulation of DC2 markers (Fizz1, IL-13, and CD206) in pulmonary DC; and nonprotective, dysregulated immune responses with marked up-regulation of TH2 hallmarks (fig. S1, B and C) (7). While the recovery of serum TNFα levels in anti-TNFα–treated mice begins to occur around 14 dpi (fig. S1D), this does not result in a recovery of DC1 phenotype in the infected anti-TNFα mice or restoration of fungal clearance (fig. S1A) (25, 26).
Our in vivo data linked TNFα priming with the induction of long-lasting DC1 programming and preventing DC2 skewing during C. neo infection. To study this yet-unknown mechanism of acute priming with TNFα and its possible role in generating DC1 and DC1-type persistence throughout infection, we conducted a series of studies in vitro. First, we demonstrated plasticity of DC by challenging DC1 generated from bone marrow (BMDCs) treated with IFNγ with a DC2-driving cytokine, IL-4 (see fig. S2A for schematic). DCs treated with IFNγ had high expression of DC1 markers over baseline [all quantitative polymerase chain reactions (qPCRs) were performed relative to untreated DCs], which was suppressed by challenge with IL-4 at both the mRNA (Fig. 1, A to C) and protein (Fig. 1, D to F) levels. While TNFα on its own did not induce DC1 activation, DC1 pretreated with TNFα during their initial IFNγ-induced polarization resisted subsequent IL-4–mediated change to DC2, maintaining robust DC1 gene expression at both the mRNA (Fig. 1, A to C) and protein (Fig. 1, D to F) levels. Similarly, DC1s challenged with IL-4 up-regulated DC2 markers at both the mRNA (Fig. 1, G to I) and protein (Fig. 1, J to L) levels, showing strong DC1-DC2 plasticity; however, pretreatment with TNFα prevented DC2 magnitude up-regulation of these factors in an IL-4 environment. Fizz1 and CD206 mRNA increased slightly but significantly with IL-4 treatment; however, the levels induced are still at (Fizz1) or below (CD206) the level seen in nonstabilized DC1 with a P value of <0.001. No DCs from any culture conditions made an appreciable amount of TNFα when assessed at the mRNA level (fig. S2B). We performed these experiments using both 24- and 48-hour incubation periods (fig. S2, C and D) for programming and challenge, achieving similar results at 48 hours. Together, these data demonstrate that TNFα stimulation during the initial DC1 activation resulted in a stabilized DC1 phenotype (TNFα-programmed DC1) capable of sustaining a DC1 phenotype in a DC2-skewing environment.
Fig. 1. TNFα stabilizes DC1 programming in vitro to pro-DC2 cytokine challenge.
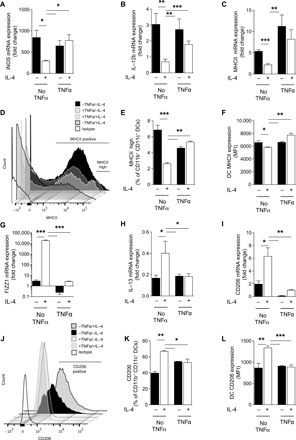
The effect of TNFα on plasticity versus stability of BMDC polarization in response to IFNγ (type 1) followed by IL-4 (type 2) polarizing conditions was tested in vitro. BMDCs were treated initially with IFNγ to become DC1 polarized in the absence (No TNFα) or presence of supplemental TNFα (TNFα) and subsequently challenged with IL-4. DC1 and DC2 marker mRNA expression for DC1 markers (iNOS, IL-12b, and MHCII) and DC2 markers (Fizz1, IL-13, and CD206) was quantified by quantitative reverse transcription (qRT)–PCR. MHCII and CD206 surface expression was further evaluated by flow cytometry. Note that BMDCs, which are DC1 prepolarized in the absence of TNFα, are highly plastic and rapidly acquire DC2 characteristics in response to IL-4, but those pretreated with TNFα are stabilized and sustain DC1 phenotype in the presence of IL-4. (A to C and G to I) n = 18 from three separate matched experiments; (D to F and J to L) n = 6 from two separate matched experiments. MFI, mean fluorescence intensity. *P < 0.05, **P < 0.01, and ***P < 0.001 between samples indicated. Statistical analysis was performed using two-way analysis of variance (ANOVA) with multiple comparisons test.
To determine the DC1 specificity of TNFα-mediated effect, we also assessed whether TNFα could stabilize DC2 polarization. DC2s were generated using IL-4 in the absence or presence of TNFα with a subsequent challenge with IFNγ. Both untreated and TNFα-treated DC2s challenged with IFNγ up-regulated DC1 markers (fig. S2D) and down-regulated DC2 markers (fig. S2E), indicating that TNFα did not promote DC2-type stabilization and TNFα-treated DC2 did not resist changes to DC1 phenotype. Thus, TNFα programming exclusively supports DC1 stabilization but does not alter plasticity of DC2 phenotype.
TNFα signals through two canonical receptors, TNFRI and TNFRII, both of which are expressed on DCs (27–29). We tested which receptor was required for the TNFα-associated stabilization of DC1 programming by inhibiting the receptors with blocking antibodies individually and in combination during the programming phase. When each receptor was blocked individually (fig. S3, A to C), DC1 and DC2 genes partially recovered plasticity. DC1 and DC2 gene plasticity completely recovered only when both TNFRI and TNFRII were simultaneously blocked (fig. S3D), suggesting that signaling through both receptors contributes to TNFα-mediated DC1 programming.
TNFα promotes DC1 stability and TH1 priming in immunomodulatory environments
We next sought to determine whether TNFα could promote DC1 stability in the presence of C. neo antigen, which is known to exhibit strong DC2 skewing capacity (30–32). Heat-killed C. neo (HKC) suppressed iNOS in all types of cytokine treatments relative to non-HKC–treated samples (Fig. 2, A to C). However, TNFα-programmed DCs maintained higher expression of iNOS (Fig. 2A) and IL-12b (Fig. 2B) and lower expression of IL-13 (Fig. 2C) relative to their HKC-treated DC2 counterparts. However, the cytokine patterns of DCs treated with IFNγ alone in the presence of HKC resembled HKC-treated DC2: They maintained low iNOS and IL-12b expression and high IL-13 expression. Thus, C. neo antigen–mediated DC2 skewing can compromise DC1 activation, but the presence of TNFα during activation can intercept this immunomodulation to stabilize the DC1 programming.
Fig. 2. TNFα programming maintains DC1 programming and TH1 priming in response to immune skewing by immunomodulatory antigen.
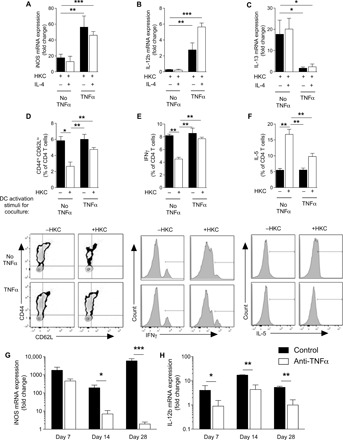
DC1-polarized BMDCs generated by IFNγ in the presence (TNFα) or absence (No TNFα) of supplemental TNFα were treated with heat-killed C. neo (HKC). (A and B) DC1 (iNOS and IL-12b) and (C) DC2 (IL-13) genes from BMDCs were quantified by qPCR. *P < 0.05, **P < 0.01, and ***P < 0.001 between indicated sample and “HKC-treated No TNFα” sample. Note that there were no significant differences between HKC-treated No TNFα samples regardless of IL-4 challenge. n = 18 from three separate matched experiments. (D to F) BMDCs generated from OT-II mice were stimulated to become DC1 in the presence (TNFα) or absence (No TNFα) of TNFα, and HKC were incubated with naïve splenic CD4 T cells and OVA peptide for 5 days. (D) CD4 T cell activation (CD44hiCD62Llo) and frequency of (E) IFNγ-producing and (F) IL-5–producing T cells were assessed by flow cytometry as seen in representative flow plots and cumulative data. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 between the indicated sample and HKC-treated No TNFα sample. (G and H) Magnetically sorted CD11c+ cells isolated from mouse lung at 7 and 14 dpi and qPCR performed for iNOS (G) and IL-12b (H). n = 8 per group from two separate matched experiments conducted months apart and from different cages. *P < 0.05, **P < 0.01, and ***P < 0.001 between the indicated samples.
To test whether TNFα-programmed DC1s were functional DC1s in immunomodulatory environments, we next asked whether TNFα-programmed DC1 could prime TH1 responses in the presence of C. neo antigen. DC1 cocultured with CD4 T cells over 5 days triggered increased numbers of activated CD44hiCD62Llo CD4 T cells, robust T cell IFNγ production, and low T cell IL-5 production (Fig. 2, D to F). The addition of HKC to DC1-CD4 T cell cocultures resulted in diminished abundance of CD44hiCD62Llo CD4 T cells, lower T cell IFNγ production, and higher T cell IL-5 production compared to HKC-free conditions. However, in the presence and absence of HKC, TNFα-programmed DC1s cocultured with CD4 T cells had uniformly increased CD44hiCD62Llo CD4 T cells, robust T cell IFNγ production, and low T cell IL-5 production (Fig. 2, D to F). Thus, TNFα stabilized DC1 programming and TH1 priming functionality, especially helping to overcome the immunomodulatory environment of cryptococcal antigen.
TNFα stabilizes DC1 programming in vivo and induces major changes in the epigenetic landscape of H3K4me3 histone modification
We next tested whether TNFα could induce sustained DC1 polarization in our C. neo mouse model of IFI. Throughout the course of infection, CD11c+ cells from the lungs of C. neo–infected control mice had robust induction of iNOS and IL-12b at the mRNA level (Fig. 2, G and H), indicative of DC1 programming, in contrast to CD11c+ cells from the lungs of infected anti-TNFα mice. To verify DC1 programming at the protein level, flow-gated CD11b+/CD11c+ DCs from infected control mice also had high MHCII and CD86 expression, while DCs from infected anti-TNFα mice had lower expression of both MHCII and CD86 (fig. S4A); furthermore, infected control mice had high levels of IL-12p70 in the serum, while very low amounts of IL-12p70 were found in the serum of infected anti-TNFα–treated mice throughout the infection (fig. S4B). Early TNFα is thus necessary for sustained DC1 gene induction in the presence of C. neo antigen at the mRNA and protein levels in vivo, and DCs from infected control mice, which clear C. neo infection, are phenotypically similar to the in vitro TNFα-programmed DC1.
Epigenetic modifications of chromatin play an important role in immunological memory-type responses of innate cells. We performed an RNA screening of a large panel of histone and DNA modification enzymes that could be associated with DC1 activation from our in vitro BMDC model and in vivo mouse model (fig. S5). While histone deacetylases (HDACs) were broadly up-regulated during in vivo infection in the TNFα-competent animals, TNFα-programmed DC1 both in vitro and in vivo featured up-regulation of MLL1, an enzyme responsible for adding methyl groups to H3K4. The trimethylation mark on H3K4 (H3K4me3) is associated with open chromatin and functionally corresponds with stable up-regulation of the gene expression downstream of H3K4me3 promoter regions. Increased H3K4 signature at a large group of specific gene promoter regions can be detected as a global increase in H3K4me3 signature in monocytes, which is a hallmark and proposed mechanism of trained innate immunity (9). To determine whether endogenous TNFα contributes to changes in H3K4me3 signature in DCs in vivo, we analyzed the level of H3K4me3 within DC by flow cytometry: DCs from infected control mice had the elevated H3K4me3 signature relative to DCs from infected anti-TNFα mice (fig. S6), providing a clue that the H3K4me3 signatures could be differentially affected by the infection process in the presence or absence of TNFα signaling.
To examine the impact of endogenous TNFα on epigenetic regulation of pulmonary DC during C. neo infection, specifically the H3K4me3 landscape, we performed chromatin immunoprecipitation sequencing (ChIP-seq) on the infected mice (day 14) with or without TNFα depletion. DC isolated from C. neo–infected, anti-TNF–treated mice showed less pronounced coverage in H3K4me3 than that from C. neo–infected control mice (Fig. 3A). A principal components analysis of lung DC H3K4me3 normalized read counts consistently showed infected control, and TNFα-depleted groups formed separate clusters along principal components 1 and 2 (Fig. 3B), providing evidence of their distinct H3K4me3 patterns. Subsequent analysis of H3K4me3 on or around known genes based on peak occupancy between each experimental group (detailed in Supplemental file 1 C.neo H3K4me3 Peaks.xlsx) revealed enrichment of both unique and shared H3K4me3 peaks in DC from the C. neo and C. neo anti-TNF samples. These genes with differential H3K4me3 enrichment peaks are listed in Supplemental file S2 (52D Infected Unique and Shared Peaks.xlsx). Gene ontology pathways with different H3K4me3 occupancy in C. neo and C. neo anti-TNF samples were primarily involved in basic cell survival and function, including cellular metabolism, response to stress, cell cycle, and transcription (Supplemental file S3 52D Infected Gene Ontology.xlsx). We found that C. neo samples had different peak occupancy in H3K4me3 at the promoter site or inside DC1 genes, including NOS2 and IL12B, when compared to C. neo anti-TNF samples (Fig. 3, C and D). To solidify and quantify these findings, we next performed ChIP-PCR and showed that DCs from infected control mice had significant enrichment of H3K4me3 at iNOS and IL-12b gene regions, while anti-TNFα mice showed only a background levels of H3K4me3 at those genes at 14 dpi (Fig. 3, E and F). In contrast, the β-actin gene showed similar levels of H3K4me3 in control and anti-TNFα mice (Fig. 3G). Collectively, our data suggest that TNFα is required for epigenetic stabilization of key DC1 genes in vivo during IFI.
Fig. 3. TNFα depletion at the time of C. neo infection altered lung DC H3K4me3 at crucial DC1 genes.
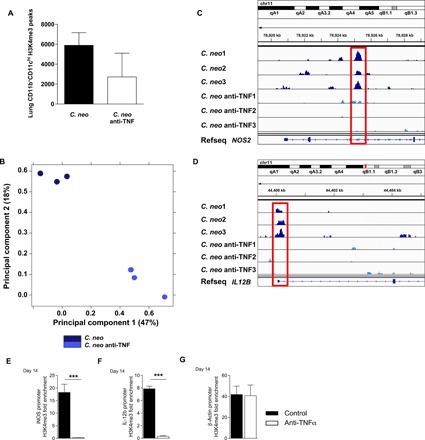
CBA/J mice were infected with C. neo intratracheally, and half were injected intraperitoneally with anti-TNFα antibodies on day 0 and were harvested at 14 dpi. (A) Total CD11b+CD11chi lung DC H3K4me3 peaks. Box represents mean; bar represents SEM. (B) Principal components analysis of total CD11b+CD11chi lung DC H3K4me3 normalized read counts. The principal components are based on H3K4me3 binding site location and affinity for those locations. C. neo–infected cells cluster together along principal components 1 and 2, and C. neo anti-TNF–treated cells cluster together along principal components 1 and 2 with significant separation from each other. (C) C. neo samples have an enriched H3K4me3 peak inside the DC1 gene NOS2 that is absent in C. neo anti-TNF samples. (D) C. neo samples have an H3K4me3 peak at the promoter site of the DC1 gene IL12B, which is absent in the C. neo anti-TNF samples. Input-subtracted H3K4me3 bigwig peak files were visualized using the Integrated Genomics Viewer (IGV). C. neo: C. neoformans infected; C. neo anti-TNF: C. neoformans infected, anti-TNF antibody treated; 1, 2, 3: replicates. (D to F) ChIP-qPCR showed DC1 gene promoter enrichment for H3K4me3. CD11c+ cells were harvested from the lungs of infected control and anti-TNFα mice at day 14, and ChIP was performed using H3K4me3 antibodies. qPCR was performed on recovered DNA for promoter regions of iNOS (D), IL-12b (E), and β-actin (F). n = 3 replicates per group pooled from three separate experiments of 20 million DCs. ***P < 0.001 between the indicated samples; no pairing was performed.
TNFα produces epigenetically stabilized DC1 associated with MLL1-mediated H3K4 methylation
We next assessed whether our in vitro model of TNFα-mediated DC1 programming, in addition to being phenotypically similar to DCs from TNFα-competent mice, also had a similar epigenetic makeup. Analysis of the broad H3K4me3 signature of DC1, DC2, and TNFα-programmed DC1 revealed that in the absence of TNFα, both DC1 and DC2 had lower global H3K4me3 signature, but TNFα-programmed DC1 had a higher H3K4me3 signature, even with secondary IL-4 challenge (Fig. 4, A to C). TNFα treatment alone did not increase H3K4me3 signature (not shown).
Fig. 4. TNFα-programmed DC1 phenotype and gene promoters associated with increased H3K4me3 and MLL1 are necessary for TNFα programming of DC1.
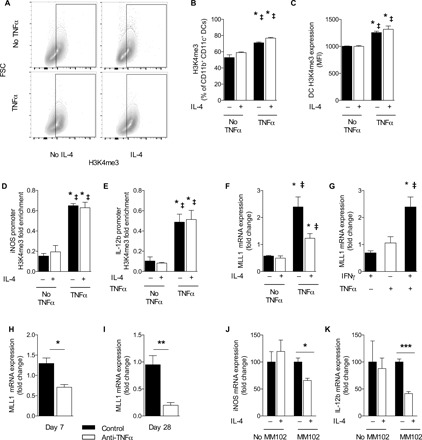
DC1-polarized BMDCs were treated initially with IFNγ in the presence (TNFα) or absence (No TNFα) of supplemental TNFα. (A to C) Global H3K4me3 signature was assessed by intranuclear flow cytometry between DC1, DC2, and TNFα-programmed DC1. (D and E) ChIP was performed with H3K4me3 antibodies on DC1, DC2, and TNFα-programmed DC1 with subsequent PCR using iNOS and IL-12b promoter region–specific primers. Data are presented as already normalized to β-actin. n = 3 replicates pooled from three separate experiments of 6 million DCs. For (B) to (E), *P < 0.05 between the indicated sample and DC1 and ‡P < 0.05 between the indicative sample and DC2. (F) DC1-polarized BMDCs were treated initially with IFNγ in the presence (TNFα) or absence (No TNFα) of supplemental TNFα and then challenged with IL-4, and qPCR for MLL1 was performed. *P < 0.05 relative to DC1 and ‡P < 0.05 relative to DC2. n = 18 from three separate matched experiments. (G) MLL1 was quantified by mRNA expression in DC1, in unpolarized BMDC treated only with TNFα, and in BMDC treated with TNFα and IFNγ combined. *P < 0.05 relative to DC1 and ‡P < 0.05 relative to unpolarized DC treated with TNFα alone. n = 18 from three separate matched experiments. (H and I) MLL1 mRNA was quantified from magnetically sorted CD11c+ pulmonary cells at early (H) and late (I) points of infection. *P < 0.05, **P < 0.01, and ***P < 0.001 between indicated samples; no pairing was performed. (J and K) TNFα-programmed DC1s were generated in the presence or absence of MLL1 inhibitor MM102 and then challenged with IL-4. *P < 0.05 and ***P < 0.001 between the indicated samples.
We next asked whether the high H3K4me3 signature showed preferential association of key DC1 gene promoters with the activating histone mark H3K4me3. We performed ChIP-qPCR to assess the association between H3K4me3 and the promoter regions of iNOS and IL-12b in DC1, DC2, and TNFα-programmed DC1. TNFα-programmed DC1 had increased association of both iNOS and IL-12b promoters with H3K4me3, while DC1 and DC2 did not (Fig. 4, D and E).
Given the increased H3K4me3 global signature and association with DC1 gene promoters in TNFα-programmed DC1, we next screened the expression of methyltransferases and demethylases acting on H3K4. MLL1, which adds activating methyl groups to H3K4, was uniquely up-regulated in TNFα-treated DC1s, unlike non-TNFα–treated DC1 or in DC2 (Fig. 4F). Moreover, TNFα alone or IFNγ alone was insufficient to induce MLL1 expression; only the combination of TNFα and IFNγ induced MLL1 expression over baseline (Fig. 4G). We also studied whether the presence or absence of TNFα affected the level of MLL1 expression in infected mice in vivo. Consistent with the in vitro data, DC from infected control mice maintained MLL1 expression, while anti-TNFα mice had suppressed MLL1 expression (Fig. 4, H and I). Last, to test whether MLL1-mediated H3K4 methylation (dependent on the enzymatic activity of MLL1) was required for TNFα-mediated DC stabilization, we inhibited MLL1 during the TNFα programming phase using the specific small-molecule inhibitor of MLL1, MM102 (33). TNFα-programmed DC1s had uniformly high iNOS and IL-12b expression, regardless of IL-4 challenge in the absence of MM102 (Fig. 4, J and K). However, in the presence of MM102, IL-4 challenge decreased iNOS and IL-12b, restoring DC plasticity to TNFα-programmed DC1s (Fig. 4, J and K). Thus, our data indicate that TNFα-treated DC1 stability depends on MLL1-mediated H3K4 methylation.
TNFα-programmed DC1s promote TH1 immunity in vivo
To determine whether sustained DC1 programming was linked to the maintenance of protective TH1/TH17 immune responses during C. neo infection, we compared the polarization of pulmonary CD4 T cells in control and αTNFα mice. Significantly more CD4 T cells from control mice made IFNγ and IL-17, while fewer made IL-5 at 14 dpi relative to CD4 T cells from αTNFα mice (Fig. 5, A to C). To mechanistically link the TH1/TH17 phenotype with stable TNFα-programmed DC1s in infected mice, we tested whether TNFα-programmed DC1s were sufficient to rescue TH1 polarization in TNFα-depleted mice relative to transfer of non-TNFα–programmed DC1s. As detailed in Fig. 5D, all mice were TNFα-depleted and infected on day 0 and then received intravenous transfer of either 1 million conventional DC1s (no TNFα) or TNFα-programmed DC1s at 1 and 8 dpi. Transfer of non-TNFα–programmed conventional DC1s was insufficient to induce TH1 polarization in C. neo–infected αTNFα mice, resulting in low numbers of IFNγ-producing CD4 T cells and higher numbers of IL-5–producing CD4 T cells compared to mice receiving transfer of TNFα-programmed DC1 (Fig. 5, D to F). Transfer of TNFα-programmed DC1s significantly increased IFNγ-producing CD4 T cells and decreased IL-5–producing CD4 T cells in αTNFα mice at 14 dpi (Fig. 5, E and F). Thus, TNFα-programmed DC1s, but not conventional DC1s, can enhance TH1 polarization in C. neo–infected αTNFα mice and overcome the effects of early TNFα depletion.
Fig. 5. Adoptive transfer of TNFα-programmed DC1 rescues TH1 polarization in C. neo–infected TNFα-depleted mice.
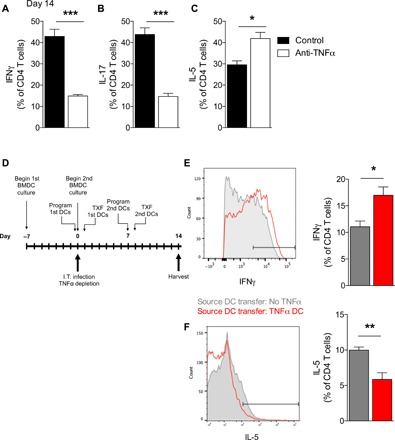
(A to C) Mice were infected as described in Fig. 3. Intracellular cytokine staining was performed at day 14 after infection for TH1 (IFNγ), TH17 (IL-17), and TH2 (IL-5) markers in CD4 T cells from infected control and anti-TNFα mice. (D) Schematic of adoptive transfer experiments. DC1-polarized BMDCs were generated in advance for transfer on days 1 and 8 after infection. Transferred DCs were either conventional DC1(No TNFα; gray) or TNFα-programmed DC1 (TNFα DC; red). (E and F) Intracellular cytokine staining for IFNγ (E) and IL-5 (F) from CD4 T cells isolated from the lungs of infected mice with differently polarized adoptively transferred DCs. n = 5. *P < 0.05 and **P < 0.01 between the indicated samples.
TNFα is required for DC1 preprogramming in the BM of C. neo–infected mice
We expected that many DCs would be replaced during the 4-week duration of cryptococcal clearance, yet they maintained the protective DC1 phenotype. Thus, we hypothesized that myeloid DC precursors in the BM of C. neo–infected mice may become preprogrammed by TNFα for future differentiation into stable DC1s. TNFα depletion did not appreciably change the frequencies of total BM cells, general myeloid precursor cells (MPCs) (CD3−/GR1−/CD11b−/B220−/Ter119−/SCA1−/Flt3+/CD115+), or specific pre-DCs (CD3−/GR1−/CD11b−/B220−/Ter119−/SCA1−/Flt3+/CD115+/c-kit−) in the presence or absence of infection (fig. S7). However, immunofluorescence microscopy of magnetically sorted pre-DCs revealed statistically significant increases in nuclear MLL1 and H3K4me3 intensity in pre-DCs of control mice relative to αTNFα mice (Fig. 6A). Intranuclear flow cytometry staining revealed a higher frequency of MPCs in infected control mice displaying elevated H3K4me3 signature relative to those MPCs from αTNFα mice (Fig. 6B). This indicated that the enhanced H3K4me3 signature found in pulmonary myeloid cells during C. neo infection was paralleled by similar signatures in DC precursors, supporting the idea that TNFα preprograms DC precursors during infection.
Fig. 6. TNFα is required for prepolarization of DC1 from BM throughout C. neo infection.
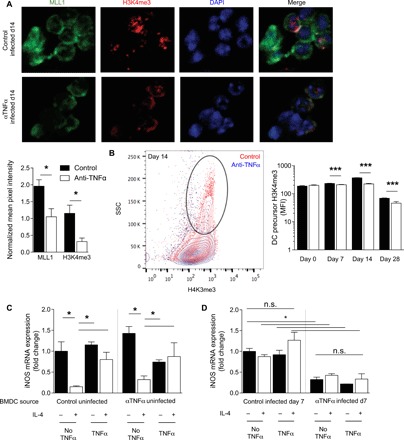
BM was harvested from control or αTNFα mice at the indicated times after infection. (A) Magnetically separated BM DC precursors were stained for MLL1 (green), H3K4me3 (red), and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Representative images were taken at 40×. Fluorescence intensity was measured by imageJ and normalized to DAPI between images. n = 6 independent wells from separate matched experiments; three random fields were analyzed from each independent well. (B) FACS (fluorescence-activated cell sorting)–sorted DC precursors were stained for intranuclear H3K4me3; representative histograms of 14 dpi with cumulative bar graph of MFI throughout infection are shown. n = 8 from separate matched experiments. ***P < 0.001. (C and D) BMDCs were matured for 7 days ex vivo with GM-CSF from BM of uninfected control and αTNFα mice (C) or 7 dpi control and αTNFα mice (D) and then treated as in Fig. 1. DC1 gene stability was assessed. Data are normalized to control DC1 values within each graph. n = 6 from separate matched experiments. *P < 0.05 by ANOVA within uninfected control and uninfected αTNFα and ‡P < 0.05 by ANOVA between infected control and αTNFα. n.s., not significant.
To determine whether MPCs from BM during infection were predisposed to DC1 or DC2 polarization before their arrival to the lung, we collected BM from four cohorts of mice: (i) uninfected control mice, (ii) uninfected αTNFα mice 7 days after TNFα depletion, (iii) isotype-treated mice at 7 dpi, and (iv) αTNFα mice at 7 dpi. BM obtained from each cohort was cultured for 7 days with GM-CSF, treated with IFNγ in the presence or absence of TNFα (programming phase), and then challenged with IL-4 (challenge phase). BMDCs from uninfected control and αTNFα mice were receptive to TNFα-mediated programming: Cells treated with IFNγ up-regulated DC1 marker iNOS, IFNγ-treated DCs challenged with IL-4 suppressed iNOS, and the addition of TNFα to the initial IFNγ treatment resulted in stable DC1 that resisted IL-4–mediated iNOS suppression (Fig. 6C). However, DCs from infected control or αTNFα mice did not respond to TNFα programming or IFNγ or IL-4 challenges regardless of cytokine stimulation. BMDCs matured from infected control mice maintained high expression of DC1 marker iNOS regardless of ex vivo programming or challenge after differentiation into DCs (Fig. 6D, left), indicating that DCs from the BM of infected mice with intact TNFα signaling were preprogrammed for DC1 polarization. In contrast, BMDCs matured from infected αTNFα mice displayed uniform down-regulation of iNOS regardless of ex vivo programming or challenge after differentiation into DCs (Fig. 6D, right), suggesting that the DCs from αTNFα mice were not prepolarized to DC1 and remained unresponsive to a subsequent DC1-polarizing stimulation.
DISCUSSION
Monoclonal antibody (MAB) therapies targeting central mediators of inflammatory responses, such as TNFα, have become a common immunotherapy approach in contemporary clinical practice. Unfortunately, TNFα MAB therapy comes at a cost of significantly increased risk for the development of devastating infections (22–24, 34). Following our previous work, which established the central role of TNFα for the development of protective TH1 immunity in the relevant model of IFI (7, 25, 26, 35), here, we defined a previously unknown mechanism by which TNFα exerts its profound and lasting effect on host defenses. We provide evidence that (i) TNFα contributes to the generation of a uniquely stable DC1 phenotype, which is resistant to DC2 repolarization; (ii) TNFα-stabilized DCs arising during the afferent phase of the immune response are required for generation and sustenance of protective TH1/TH17 immunity; (iii) TNFα-mediated DC1 stabilization is associated with the H3K4me3 histone modification at promoter regions of several thousand specific genes, including those responsible for type 1 DC polarization; and (iv) histone modification and the downstream changes in DC1 stability are linked to the histone methyltransferase MLL1 activity.
Mononuclear myeloid cells, at least in their naïve stage, are highly plastic, readily changing their phenotype following the environmental cues (36, 37). Our recent study demonstrated that TNFα depletion at the onset of the immune response resulted in permanent alteration of DC phenotype in the lymph nodes that lasts through extended infection with the persistent fungal pathogen C. neo (7). These findings suggested that TNFα-pulsed DC may also undergo a similar type of programming, which required the upstream process to properly direct the immune polarization. Our series of in vitro and in vivo studies now demonstrate directly that TNFα is required for generation of programmed DC1 and is crucial for initiating and sustaining TH1/TH17 responses and driving the clearance of IFI.
In vitro, a similar DC1 program could be sufficiently induced by joint DC stimulation with TNFα and IFNγ, but not by either of these cytokines alone, or with TNFα with the DC2-polarizing IL-4, which is consistent with the established role of TNFα-IFNγ synergism in generation of TH1 responses (38–40). However, the unique importance of TNFα in generation of stable DC1 phenotype is underscored by insufficiency of DC1 generated with IFNγ alone to improve TH1 polarization in the context of C. neo infection of TNFα-depleted mice. IFNγ alone rapidly, but only transiently, up-regulates DC1 gene expression, which differs from TNFα-stabilized DC1. Our data further show that this DC1 phenotypic stability helps to overcome the effects of both cytokines and C. neo antigen to suppress DC1 phenotype throughout the extended period needed to eliminate C. neo from the infected host. Last, both TNFRI and TNFRII appear to contribute to this TNFα-mediated DC1 programming (fig. S3); while TNFα is the major player, it is possible that other TNFR ligands (i.e., LTα) could potentially be involved.
The acquiescence of phenotypic stability by myeloid cells via epigenetic histone modification has been reported and is an area of intense investigation (2, 18, 41). The most recognized example is the phenomenon of “trained immunity,” when myeloid cells (monocytes and macrophages) acquire properties compatible with immunological memory. As a result, the trained myeloid cells show enhanced cytokine expression and enhanced microbicidal functions upon reexposure to pathogens. Trained immunity shows many parallels with TNFα-induced DC1 programming, including enhanced expression of proinflammatory cytokines and a broadly enhanced H3K4me3 signature on a large group of specific gene promoters and enhanced expression of these genes. Innate immune memory has been demonstrated to depend on similar H3K4me3 at proinflammatory gene promoter regions of monocytes during sepsis or upon challenge with LPS (18, 41–43), tuberculosis vaccine bacillus Calmette-Guerin (44–49), and fungal β-glucans (6, 9, 10). Although our work is the first, as far as we know, to identify that IFNγ and TNFα induce special DC stability, we hypothesize that this effect is very broad and similar response may be found in human primary monocytes and macrophages. It has been recognized for a long time that IFNγ and TNFα co-regulate immune responses in a synergistic manner (38–40). Furthermore, the TNFα and IFNγ/STAT1 signaling pathways are shown to play important roles in trained immunity in monocytes, macrophages, and peripheral blood mononuclear cell models (8, 9, 50, 51). Whether TNFα and IFNγ pathways are required and how they integrate into other essential programs, such as metabolic changes, are currently unclear and warrant future studies.
Unlike classically viewed trained immunity, TNFα-mediated DC1 programming occurs at the onset of the immune response and serves to direct the development of primary adaptive host responses. Our in vitro modeling shows that the early presence of TNFα during IFNγ-driven DC1 differentiation induces DC1-associated gene and protein expression profiles, which recapitulate the DC phenotype observed in vivo. The TNFα-stabilized DC1 phenotype can no longer be erased by pro-DC2 cytokine challenge or the immunomodulatory antigen. Considering these similarities and differences, it remains to be determined whether TNFα-induced DC1 program stabilization represents an initial step in the development of trained DCs or instead these two processes are independent from each other, despite sharing similar epigenetic mechanisms. Regardless, our report shows that epigenetic stabilization is involved (the MLL1 inhibitor studies; Fig. 4, J and K) in DC1 programming and is a necessary step for the DCs to promote the protective TH1/TH17 polarization required for clearance of C. neo and other important IFI and persistent bacterial infections, which can occur as a side effect of anti-TNFα immunotherapy (24, 52–54).
Our study provides evidence that the molecular basis for stabilizing DC1 programming during TNFα treatment involves specific changes in epigenetic enzyme machinery, in particular machinery involved in methylation of H3K4 (a gene expression activating histone modification) in a large group of DC genes, among which is the family of classical activation/DC1 polarization markers. This includes the IL-12B gene, coding for the p40 component of IL-12 and IL-23 cytokines, which promote TH1 and TH17 immune polarizations (55), and NOS2. The increased trimethylation of H3K4me3 residues at critical DC1 gene promoters was linked with more robust MLL1 expression in the presence of TNFα in vivo. In vitro, only the combined effect of TNFα and IFNγ enhanced MLL1 expression (Fig. 4G), unlike IFNγ alone, which was sufficient to induce transient DC1 phenotype but insufficient to induce stable DC1 or elevate the H3K4me3 signature in DC (Fig. 3). This, together with the effects of specific MLL1 inhibitor blocking the effect of TNFα on DC1 programming, strongly suggests that MLL1 mechanistically contributes to the development of stable DC1 by generation of H3K4me3 signature DC1 gene promoters. However, involvement of other epigenetic histone or DNA modifications cannot be ruled out.
Our study also opens new questions that still need to be addressed, including additional components of epigenetic machinery that target MLL1 to the specific promoter sites upon TNFα stimulation. Furthermore, analysis of other factors contributing to generation of the stable DC1 such as modifications of H3K27 residues and changes in DC metabolic status will be necessary, similar to those documented as a feature of trained immunity (10, 56). Likewise, broad investigation of histone acetylation patterns during infection in the presence of TNFα is necessary, as numerous HDACs were not induced in the αTNFα in vivo samples during infection. Further studies dissecting the epigenetic landscape of histone modifications, enzymes involved in these process, and metabolic phenotyping of TNFα-programmed DC1 are needed to address these points.
One of the commonalities between monocytes and DCs is their BM ontogeny, their relatively rapid turnover in vivo, and de novo differentiation from BM precursors. A similar epigenetic mechanism is used in the developing myeloid cells to stabilize the DC1 phenotype and drive protective TH1 responses, and the effect is seen at the level of the BM precursor DCs. Corresponding differences in levels of H3K4me3 signatures of the BM pre-DC population harvested from infected control hosts and anti-TNFα hosts translate into ex vivo differential maturation of BMDC to stable DC1 cells from the infected control BM donors versus DC2 cells from αTNFα-treated mice (Fig. 6D). We demonstrate that the DC1 and DC2 preprogramming can occur in the BM pre-DC population during cryptococcal infection depending on the presence or absence of endogenous TNFα. These results are consistent with the recent study showing that modulation of myeloid progenitors in the BM plays an important role conferring lasting memory-type effects on innate cells, although we found no increase in progenitor cell numbers (57). Together, our data support the idea that preprogramming of myeloid precursors at the BM is an important mechanism providing preprogramming capabilities to innate immune cells with high turnover rate, which, in turn, can support sustenance of protective immune response by sustained generation of DC1.
Clinically, and in direct relation to persistent infections, our work elucidates the cellular and molecular basis behind previously described long-term immune dysregulation resultant from anti-TNFα MAB therapies in humans (24, 53, 54, 58, 59) or in experimental models in mice (25, 26). Anti-TNFα MAB therapy, while beneficial for autoimmune disorders, results in a high susceptibility to fungal and mycobacterial infections. We propose that this could be partly due to the absence of proper DC programming when patients initially become infected with opportunistic fungi and bacteria. Last, TNFα-induced DC1 programming occurs in vitro both in the absence of any antigen and in the presence of highly immunomodulatory antigen, suggesting broad applicability of our findings to many types of pathogens for which TNFα is a necessary part of the immune response. Thus, we propose that DC (and potentially macrophage and monocyte) programming, such as those epigenetically induced by TNFα signaling, could be harnessed for immunotherapies. These therapies could be particularly important in situations when these myeloid cells need to tolerate immunomodulatory or suppressive environments (chronic infections or tumors) to trigger and sustain protective immunity. For example, immunostimulatory treatments like IFNγ are emerging as possible adjunctive treatments for immunoparalysis induced by sepsis (60, 61).
METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by M.A.O. (olszewm@umich.edu).
Experimental model and subject details
Mice
Six- to 8-week-old female CBA/J mice were obtained from The Jackson Laboratory and housed at the Veterinary Medicine Unit at the Ann Arbor Veterans Administration Hospital under specific pathogen–free conditions. Mice were 8 to 10 weeks old at the time of infection and housed with four mice per cage. At the time of data collection, mice were humanely euthanized by CO2 inhalation followed by severance of the portal vein. All experiments were approved by the University Committee on the Use and Care of Animals and the Veterans Administration Institutional Animal Care and Use Committee.
Cryptococcus neoformans
C. neo serotype D strain 52D (American Type Culture Collection 24067) was recovered from 10% glycerol frozen stocks stored at −80°C. Cultures were grown at 37°C in Sabouraud dextrose broth (1% neopeptone and 2% dextrose; Difco, Detroit, MI) on a shaker. When cultures reached mid–log phase growth (day 3), an aliquot of culture was washed in sterile nonpyrogenic saline (Travenol, Deerfield, IL), counted on a hemocytometer, and diluted to 3.3 × 105 yeast cells/ml in sterile nonpyrogenic saline.
Intratracheal inoculation of C. neo
Mice were anesthetized with intraperitoneal injection of ketamine/xylazine (100/6.8 mg/kg body weight) and secured onto a clean foam board. Hair was removed from over the trachea, and skin was sterilized with iodine and ethanol. A small incision was made over the trachea, and the underlying muscle and glands were separated to expose the trachea. A 30-gauge needle was inserted into the trachea, and 30 μl [104 colony-forming units (CFUs)] of the washed yeast (3.3 × 105 yeast cells/ml in sterile nonpyrogenic saline) was injected intratracheally from a 1-ml tuberculin syringe fitted to a stepper pipette. After inoculation, the incision was closed with cyanoacrylate adhesive, and mice were kept warm and monitored during recovery from anesthesia.
Adoptive transfer
At day −7, BMDCs were generated as described below and differentiated over 7 days, and on day 0, the loosely adherent fraction was plated as described above and stimulated for 24 hours with TNFα and IFNγ in combination (γα) to generate TNFα-programmed DC1s or with IFNγ alone (γ) to generate unprogrammed DC1s. On day 0, CBA/J mice were inoculated intratracheally as above with C. neo 52D and TNFα-depleted by intraperitoneal injection of TNFα-blocking antibody. On day 0, another set of BMDCs was generated and differentiated as described below for 7 days. On day 1, 1 million γα DCs or γ-only DCs in plain phosphate-buffered saline (PBS) were injected intravenously through the retro-orbital route into the C. neo–infected, TNFα-depleted mice. On day 7, the loosely adherent fraction of the second set of BMDCs was harvested, plated, and stimulated for 24 hours with γα or γ alone. On day 8, a second retro-orbital injection of 1 million γα or γ-alone DCs was transferred as described above. Mice were harvested at 14 dpi, and the CD4 T cells were assessed for TH polarization by intracellular flow cytometry.
In vitro experiments
All in vitro experiments were performed with primary BM-derived macrophages from femurs and tibias of female CBA/J mice, unless otherwise specified. Uninfected mice were humanely euthanized, and death was confirmed by exsanguination. Two femurs and two tibias were isolated from each mouse and washed in 70% ethanol, and then epiphyses were removed and marrow was flushed with D20 medium [Dulbecco’s modified Eagle’s medium + 20% fetal bovine serum, penicillin and streptomycin (1 U/ml; Invitrogen, Grand Island, NY), 1× sodium pyruvate, 1× GlutaMAX, 1× nonessential amino acids, 2-mercaptoethanol, 20 nM GM-CSF] using 26-gauge needles. Cell suspension was filtered through sterile 100-μm Nitex. Cells were pelleted, resuspended in D20, and grown in sterile, non–tissue culture–treated 150-mm petri dishes for 7 days. Medium was replenished with D20 on day 3. After 7 days, the loosely adherent fraction was harvested in PBS, pelleted, counted, and plated in non–tissue culture–treated six-well dishes at a concentration of 2 × 106 cells per well.
For DC–T cell coculture, BMDCs were generated from C57BL/6 OT-II mice as above and then cultured with freshly isolated splenocytes from OT-II mice for 5 days. For splenocyte isolation, spleens were removed, trimmed of fat and connective tissue, and then ruptured, and cells were dispersed over a sterile 75-μm cell strainer. Strainers were flushed with 5 ml of RPMI 1640, and the resulting cells were pelleted and added to the cultures.
For ex vivo BMDC experiments using marrow from infected or uninfected control or TNFα-depleted mice, four femurs from each group were isolated and marrow was prepared as above at 7 dpi, taking care to keep each condition separate.
Method details
BMDC programming and challenge
For the programming phase, IFNγ was used at a concentration of 100 ng/ml, TNFα was used at a concentration of 20 ng/ml, and cells were incubated for 24 hours. For the challenge phase, programming phase medium was aspirated, wells were washed with sterile PBS, and challenge medium containing IL-4 was used at a concentration of 20 ng/ml, while IFNγ control-challenge medium was at the same concentration as above. For experiments using HKC, cultures of strain 52D were counted, diluted to 2 × 108 CFU/ml, and heat-killed by incubating at 65°C for 6 hours. Heat-killed cultures were added to the BMDCs at a multiplicity of infection of 10 during the programming phase for 24 hours and washed out before the challenge phase. For experiments using MM102 (Tocris, Bristol, UK), the inhibitor of MLL1, it was used at a concentration of 50 μM during the programming phase for 24 hours and washed out before challenge. For experiments using TNFRI and/or TNFRII blockade, sterile validated LEAF-purified blocking antibodies for TNFRI and TNFRII were purchased from BioLegend and used at a concentration of 10 μg/ml for 24 hours during the programming phase and the challenge phase. For medium- and TNFα-only control wells (using the same TNFα concentration as above), cells were incubated for 24 hours in their respective medium, washed identically to experimental wells, and further incubated for 24 hours in plain medium. Supernatants for enzyme-linked immunosorbent assay (ELISA) were harvested at this point, and the remaining cells were either lysed in TRIzol for quantitative reverse transcription (qRT)–PCR or detached for flow cytometry.
Experiments using BMDCs from infected and uninfected control and αTNFα mice were also performed as above.
OT-II cocultures
For OT-II DC–T cell cocultures, DCs were generated as described above and then treated with IFNγ at a concentration of 100 ng/ml in the presence or absence of TNFα at 20 ng/ml and C. neo at a multiplicity of infection of 10. After the programming phase, wells were washed and OT-II CD4 T cells were added at a ratio of four T cells to one DC. Cultures were incubated for 5 days, and then cells were removed and their surface markers and cytokine production were analyzed by flow cytometry.
Serum preparation
Mice were humanely euthanized by CO2 asphyxiation and then exsanguinated to confirm death. Whole blood was collected in Eppendorf tubes, spun down to consolidate blood, and allowed to rest at room temperature for 20 min. Blood was then incubated at 4°C for 20 min and then centrifuged at 5000 rpm to separate serum from erythrocytes. The top serum layer was collected and frozen at −80°C until ELISA or cytometric bead array for cytokine analysis.
Lung leukocyte isolation
At the time of data collection, lungs were perfused with 3 ml of sterile nonpyrogenic saline, removed, washed in RPMI 1640, and enzymatically dispersed as previously described (62, 63). Briefly, excised lungs from each mouse were minced with scissors and digested enzymatically at 37°C for 30 min in 5 ml/mouse digestion buffer [RPMI 1640, 5% fetal bovine serum, penicillin and streptomycin (Invitrogen, Grand Island, NY), collagenase A (1 mg/ml; Roche Diagnostics, Indianapolis, IN), and deoxyribonuclease (30 mg/ml; Sigma)]. The cell suspension and tissue fragments were further dispersed by repeated aspiration through the bore of a 10-ml syringe and centrifuged. Erythrocytes in the cell pellets were lysed by the addition of 3 ml of NH4Cl buffer [0.829% NH4Cl, 0.1% KHCO3, and 0.0372% Na2EDTA (pH 7.4)] for 3 min, followed by a 10-fold excess of RPMI 1640. Cells were resuspended, and a second cycle of syringe dispersion and filtration through a sterile 100-mm nylon screen (Nitex, Kansas City, MO) was performed. The filtrate was centrifuged for 25 min at 1500g in the presence of 40% Percoll (Sigma-Aldrich) in complete RPMI 1640 [RPMI 1640, 5% fetal bovine serum, penicillin and streptomycin (10 U/ml; Invitrogen, Grand Island, NY), 1× sodium pyruvate, 1× GlutaMAX, 1× nonessential amino acids, 2-mercaptoethanol] with no brake to separate leukocytes from cell debris and epithelial cells. Leukocyte pellets were resuspended in 5 ml of complete RPMI 1640 medium and enumerated on a hemocytometer after dilution in trypan blue (Sigma-Aldrich).
Lung CFU assay
For determination of fungal burden in the lungs, 100 μl was removed from the enzymatically dispersed lungs before centrifugation and 10-fold dilutions were plated in duplicate on Sabouraud dextrose agar plates. Colonies were counted after 48 hours of growth at room temperature, and CFU was calculated on a per-organ basis.
Preparation and enumeration of lung leukocytes
Following enzymatic dispersal of the lungs, 50,000 cells were cytospun onto glass slides, fixed, stained with Wright-Giemsa stain, and dried. Monocytes, eosinophils, neutrophils, and lymphocytes were counted as described previously (64).
Flow cytometry
Enzymatically dispersed lungs were counted and stained extracellularly and then fixed, permeabilized, and stained intracellularly. Briefly, cells were stained with fixable LIVE/DEAD stain (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions, washed twice in fluorescence-activated cell sorting (FACS) buffer (Difco Laboratories, Detroit, MI), resuspended in 100 μl of staining buffer (FACS buffer with Fc block; BioLegend, San Diego, CA), and incubated for 30 min at 4°C in the dark with labeled antibodies diluted in 100 μl of staining buffer. Final antibody concentrations were 1 to 2 μg/106 cells. Intracellular staining was performed using the FoxP3/Transcription Factor Staining Buffer Kit from eBioscience (San Diego, CA) according to the manufacturer’s instructions. Cells were then run on an LSR II flow cytometer using FACSDiva software (BD Biosciences, San Jose, CA) and analyzed further using FlowJo software (Tree Star, San Carlos, CA).
Gating for lung myeloid cells proceeded as follows: The CD45+ cells were identified, lymphocytes were removed (CD19+/CD3+), neutrophils were removed (Ly6G+/CD11b+), eosinophils were excluded (SSChigh/CD11cint), and then CD11c+ (myeloid cells) or CD11b+/CD11c+ (DCs) cells `were analyzed for expression of activation markers. Gating for lung CD4 T cells proceeded as follows: The CD45+ cells were identified, cells were selected on CD3+, and CD19/CD11b/CD11c+ cells were excluded. T cells were then gated on CD4 versus CD8 expression, and the CD4 single-positive population was analyzed for surface activation marker expression and intracellular expression of TH polarization cytokines. Intracellular staining was performed using the FoxP3/Transcription Factor Staining Buffer Kit from eBioscience (San Diego, CA). For specific antibodies, see each respective Methods section.
For intranuclear flow cytometry of H3K4me3, purified H3K4me3 (catalog no. 61379) was obtained from Active Motif (Carlsbad, CA) and the PE/Cy7 Conjugation Kit (Abcam, Cambridge, United Kingdom) was used to attach the Phycoerythrin (PE)/Cy7 fluorophore to the antibody according to the manufacturer’s protocol. Intranuclear staining was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience, San Diego, CA) following the manufacturer’s protocol. Antibodies used are listed in the “Key resources” table.
Key resources table.
| Reagent or resource | Source | Identifier |
| Antibodies | (Clone) | |
| IFNγ | BioLegend | XGM1.2 |
| IL-17 | BioLegend | TC11-18H10.1 |
| IL-13 | eBioscience | eBio13A |
| TNFα | BioLegend | MP6-XT22 |
| MHCII | BD Biosciences | 11-5.2 |
| CD86 | BioLegend | GL-1 |
| CD206 | BioLegend | C068C2 |
| Gal3 | BioLegend | eBioM3/38 |
| H3K4me3 | Active Motif | MABI 0304 |
| CD45 | BioLegend | 30-F11 |
| CD3ε | BioLegend | 145-2C11 |
| CD19 | BioLegend | 6D5 |
| CD11b | BioLegend | M1/70 |
| CD11c | BioLegend | N418 |
| Ly6C | BioLegend | HK1.4 |
| Ly6G | BioLegend | 1A8 |
| Lin (CD3/Gr1/CD11b/B220/Ter119) | BioLegend | N/A |
| SCA-1 | BioLegend | D7 |
| Flt-3 | BioLegend | A2F10 |
| c-kit | BioLegend | 2B8 |
| CD115 | BioLegend | AFS98 |
| CD4 | BioLegend | GK1.5 |
| CD8 | BioLegend | 53-6.7 |
| Fizz1 | R&D Systems | 226033 |
| TNFRI | BioLegend | 55R-170 |
| TNFRII | BioLegend | TR75-32.4 |
| Anti-goat Alexa Fluor 488 | Abcam | Catalog no. ab150129 |
| Anti-rabbit Alexa Fluor 594 | Abcam | Catalog no. ab150076 |
| Fungal strain | ||
| C. neo strain 52D | American Type Culture Collection | 24067 |
| Chemicals, peptides, and recombinant proteins | ||
| TNFα recombinant antibody | Leinco | Clone TN3-19.12 |
| MM102 (MLL1 inhibitor) | Tocris | 5307 |
| TNFα | PeproTech | 315-01A |
| IFNγ | Sigma-Aldrich | I4777-.1MG |
| IL-4 | PeproTech | 214-14 |
| Critical commercial assays | ||
| PE-Cy7 conjugation kit | Abcam | ab102903 |
| APC-Cy7 conjugation kit | Abcam | ab102859 |
| LEGENDplex mouse inflammation panel | BioLegend | 740150 |
| LEGENDplex mouse Th cytokine panel | BioLegend | 740005 |
| RT2 profiler PCR array mouse TH cell differentiation | Qiagen | PAMM-503Z |
| Foxp3 Transcription Factor Staining Buffer Kit | Thermo Fisher Scientific | A25866A |
| Experimental models: Organisms/strains | ||
| CBA/J mice | Jackson Laboratory | 000656 |
| OT-II mice | Jackson Laboratory | 004194 |
| Oligonucleotides | Sense primer | Antisense primer |
| 18S | GAGGCCCTGTAATTGGAATGAG | GCAGCAACTTTAATATCCGCTATTGG |
| iNOS | TTTGCTTCCATGCTAATGCGAAAG | GCTCTGTTGAGGTCTAAAGGCTCCG |
| Arginase | CAGAAGAATGGAAGAGTCAG | CAGATATGCAGGGAGTCACC |
| Fizz1 | GGTCCCAGTGCATATGGATGAGACCATAGA | CACCTCTTCACTCGAGGGACAGTTGGCAGC |
| MHCII | GCGACGTGGGCGAGTACC | CATTCCGGAACCAGCGCA |
| CD206 | CTCTGTTCAGCTATTGGACGC | CGGAATTTCTGGGATTCAGCTTC |
| IL-12b | GGAAGCACGGGGAGCAGAATA | AACTTGAGGGAGAAGTAGGAATGG |
| IL-13 | GCCAGCCCACAGTTCTACAGC | GAGATGTTGCTCAGCTCCTCA |
| iNOS (promoter) | TCCCTAGTGAGTCCCAGTTTTGA | CTGGTCGCCCGTCCAAGG |
| IL-12b (promoter) | TTCCCCCAGAATGTTTTGACA | TGATGGAAACCCAAAGTAGAAACTG |
| β-Actin (promoter) | AAGGACTCCTATGTGGGTGACGA | ATCTTCTCCATGTCGTCCCAGTTC |
| Fizz1 (promoter) | TGCAATTCTTTGATGCTGTGTCT | AGCACCCTCAACCCAAAGTG |
| KDM5c | GACCCATCGCCGAGAAGTC | TCGGGGAGTAAACCTGAAGTT |
| MLL1 | ATCCTCTCAGACCCATCTGTGT | GTAGGAGGTCTTCCTCTCTTC |
| Software and algorithms | ||
| Microsoft Excel | ||
| GraphPad Prism v6 | ||
| MetaMorph | ||
| FlowJo v7 | ||
| FACSDiva | ||
Multiplex ELISA
Cytokine levels were analyzed by LEGENDplex Th Cytokine and Inflammation panels. Serum from whole blood and standard curve was diluted in Matrix C reagent and stained as per the manufacturer’s instructions (BioLegend, San Diego, CA). Bead-antibody-protein complexes were fixed and run on an LSR II flow cytometer using FACSDiva acquisition software. Results were analyzed using software included with LEGENDplex kits.
Quantitative polymerase chain reaction
Cells were spun down and directly resuspended in 1 ml of TRIzol reagent (Life Technologies Inc., Gaithersburg, MD) in polypropylene tubes. Samples were allowed to incubate at room temperature, and 200 μl of chloroform per 1 ml of TRIzol was added to them. Samples were spun at 10,000 rpm for 15 min, the aqueous phase was transferred into fresh tubes, and equal volumes of isopropanol were added. Samples were then incubated at −20°C for 90 min to precipitate the RNA and centrifuged again as described above. Pellets were washed with 1 ml of 70% ethanol and centrifuged. RNA was resuspended in nuclease-free water (Ambion). The yield and purity of the RNA were determined spectrophotometrically at 260 and 280 nm. Complementary DNA (cDNA) was synthesized using QuantiTect Reverse Transcription Kit (Qiagen) using 1 μg of RNA according to the manufacturer’s instructions. For CD4 cells, TH differentiation qPCR arrays with 96 genes per sample were purchased from SABiosciences (Qiagen) and used initially, while individual qPCRs were performed later to confirm and expand analyzed genes. For CD11c cells, custom qPCR arrays with 48 genes per sample were used initially, while individual qPCRs were performed later to confirm and expand analyzed genes. For individual qRT-PCRs, cDNA was quantified with SYBR Green–based detection using an MX3000P system (Stratagene, La Jolla, CA) according to the manufacturer’s protocols. Forty cycles of PCR (94°C for 15 s, followed by 60°C for 30 s and 72°C for 30 s) were performed on a cDNA template. Primer sequences can be found in table S1. The mRNA levels were normalized to 18S mRNA levels. Primer sequences can be found in the “Key resources” table.
ChIP and qPCR
ChIP was performed using the protocol from the laboratory of Y. Dou at the Department of Pathology, University of Michigan. Briefly, magnetically separated CD11c+ cells were isolated from the lungs of mice at 14 dpi and fixed in formaldehyde at a final concentration of 1%. The fixation was stopped with 5 M glycine, cells were pelleted, the supernatant was aspirated, and pellets were frozen at −80°C until 25 × 106 to 30 × 106 cells per treatment were accumulated. Cells were thawed and lysed in SDS lysis buffer [1% SDS, 10 mM EDTA, 50 mM tris (pH 8.0), 1× cOmplete protease inhibitor cocktail] and homogenized by aspiration through a 27-gauge needle twice. Cells were sonicated on ice in a Bioruptor using the highest setting for 15 min with 30 s on/30 s off cycles. Sonication efficiency and chromatin shearing were assessed by running a small aliquot of sample on 1% agarose gel before proceeding. Reactions were centrifuged to pellet debris, and supernatant was removed and diluted in ChIP dilution buffer [0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM tris-HCl (pH 7.5), 167 mM NaCl, 1× cOmplete protease inhibitor cocktail]. Input fraction (1%) was removed here, and then the diluted lysate was aliquoted to Eppendorf tubes and antibodies were added at a concentration of 2.5 μg. Reactions were incubated overnight rotating at 4°C. For immunoprecipitation, 30 μl of protein G Dynabeads (Life Technologies, Carlsbad, CA) was used for each reaction. Reactions were washed with increasingly stringent washes: low-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 7.5), 150 mM NaCl], high-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 7.5), 500 mM NaCl], LiCl wash buffer [0.25 M LiCl, 1% NP-40, 1% deoxycholic acid, 1 mM EDTA, 10 mM tris-HCl (pH 7.5)], and TE wash buffer [10 mM tris-HCl (pH 7.5) and 1 mM EDTA]. Last, antibody-protein-DNA complexes were eluted using elution buffer (1% SDS, 0.5 mM NaHCO3) and heated at 37°C for 1 hour. Cross-linking was reversed by the addition of 5 M NaCl and incubated overnight at 65°C. DNA was purified using phenol-chloroform extraction and isopropanol precipitation. qPCR was performed as described above using recovered genomic DNA as the template and primers specific to the promoter regions of β-actin, Fizz, iNOS, and IL-12b.
Cell sorting for ChIP-seq
DCs were sorted using a FACSAria III and antibodies against murine CD11c (clone N418) and CD11b (clone M1/70). CD11b+CD11chi cells were sorted into PBS from single-cell suspensions obtained from the lungs that were digested with collagenase A/deoxyribonuclease and centrifuged over a Percoll gradient to remove debris and red blood cells.
High-throughput ChIP-seq
ChIP-seq was performed on purified lung CD11b+CD11chi DCs from four experimental groups: naïve (control), naïve plus anti-TNFα treatment (control anti-TNF), C. neo day 14 after intratracheal inoculation with 104 CFUs (C. neo), and C. neo day 14 after intratracheal inoculation with 104 CFUs plus anti-TNFα treatment (C. neo anti-TNF). Each experimental group contained three biological replicates. Starting cell numbers and quality measures are detailed in the table. Given the low cell numbers in some samples, an ultralow-input ChIP-seq protocol was used (65). Briefly, after sorting, the cells were placed in 20 μl of nuclear isolation buffer and were frozen at −80°C. The samples then underwent a micrococcal nuclease digestion (7.5 min at 21°C for 10,000 to 20,000 cells and 5 min at 37°C for 21,000 to 200,000 cells), and 10% of the sample was then removed and processed as input. The remaining samples were incubated overnight with 0.5 to 1 μg of anti-H3K4me3 (Abcam, ab8580) in the presence of protein A:protein G 1:1 dynabeads. The samples were then washed and eluted. The DNA was extracted using phenol:chloroform:isoamyl alcohol and were precipitated overnight using linear polyacrylamide. The samples were washed in 70% ethanol, and library construction was performed as previously described (65). After library preparation, the samples were pooled in equimolar concentrations and sequenced on an Illumina HiSeq 2500 platform (single-end 50-nucleotide reads). Sample quality was assessed using ChIPQC (66). Adapter sequences were trimmed from the raw sequencing reads using Trimmomatic (67). Raw reads were aligned to the GRCm38 Mus musculus genome using HISAT2 version 2.1.0 using the single-end option (68). Peaks were called against input using MACS2 version 2.1.1.20160309 (69). DiffBind was used to compare peaks between different experimental groups (70). As expected, most of the H3K4me3 peaks (up to 70%) were localized in the strictly defined promoter region, and the H3K4me3 peak distribution plots yielded curves consistent with those previously reported (fig. S8) (71, 72).
Peaks were annotated, and gene ontology terms were identified using ChIPpeakAnno (73). Input background was subtracted from the H3K4me3 files with MACS2 using the -subtract option. The Integrative Genomics Viewer was used for data visualization (www.broadinstitute.org/igv). The datasets used in this manuscript are available in the Gene Expression Omnibus repository under GSE118543.
Fluorescence microscopy
CD11b+ pre-DCs were isolated from the BM of isotype or TNFα-depleted infected and control mice by magnetic bead separation as described above according to the manufacturer’s protocol. Cells were washed and counted, and then 20,000 cells were cytospun onto charged glass microscope slides, fixed, stained with primary antibodies against H3K4me3 (Active Motif, Carlsbad, CA) and Fizz1 (R&D Systems, Minneapolis, MN) or control immunoglobulin G (IgG) (R&D Systems), then stained using anti-rabbit Alexa Fluor 594 or anti-goat Alexa Fluor 488 secondary antibodies, and mounted with VECTASHIELD mounting medium plus 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Slides were visualized by confocal microscopy using a spinning disk confocal microscope (Olympus America Inc., Center Valley, PA) with a digital charge-coupled device camera (Hamamatsu Photonics, Hamamatsu, Japan) for image capture and an arc lamp illumination source providing excitation wavelengths of 350 to 700 nm and three-color emission analyses. The acquired digital images were processed and analyzed using Stereo Investigator software version 9 (MBF Bioscience, Williston, VT).
Quantification and statistical analysis
All values are reported as means ± SEM. Continuous ratio scale data were evaluated by unpaired Student’s t test (for comparison between two samples) or analysis of variance (ANOVA) (for multiple comparisons) with post hoc analysis using Student’s t test with Bonferroni adjustments. Nonparametric analysis was performed using the Kruskal-Wallis, Kolmogorov-Smirnov, or Mann-Whitney tests. Statistical calculations were performed using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA). Statistical difference was accepted at P < 0.05, symbols are explained in the figure legend, and descriptions of the samples compared are included in the figure legend. For each experiment, n is indicated in the figure legend and refers to biological replicates, often either mice or individual wells of an experiment. Where applicable, the number of repeated experiments is indicated in the figure legend.
Supplementary Material
Acknowledgments
Funding: This study was supported by the U.S. Department of Veterans Affairs (M.A.O.: 1I01BX000656 AND 1IK6BX003615; J.J.O.: BX002120-01), the National Institutes of Health (A.J.E. T32 AI007413, L.M.N. T32 HL007749), the University of Michigan (A.J.E.: University of Michigan Rackham Fellowship, the Herman & Dorothy Miller Award for Innovative Immunology Research), and the American Association of Immunologists (A.J.E. and M.A.O.: Careers in Immunology Fellowship). Author contributions: A.J.E., J.X., J.B., N.P., L.M.N., G.Z., A.M., and M.S. performed the experiments. J.B., A.d.D., L.M.N., M.S., and S.K. provided key experimental support and methods. A.J.E., J.X., J.B., J.J.O., I.K., and M.A.O. performed writing and revision or provided serious assistance to writing and revision. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaaw9051/DC1
Fig. S1. TNFα depletion in CBA/J mice results in fungal persistence and dysregulated immunity during C. neo infection.
Fig. S2. Schematic of BMDC experiments and testing model.
Fig. S3. TNFRI and TNFRII are both required for optimal DC1 programming.
Fig. S4. TNFα results in sustained DC1 profile at the protein level in pulmonary DCs during C. neo infection.
Fig. S5. MLL1 is uniquely induced in TNFα-programmed DC1.
Fig. S6. DC1 from control C. neo–infected mice epigenetically resemble TNFα-programmed DC1.
Fig. S7. Assessment of total and myeloid precursor populations in the BM during C. neo infection.
Supplemental file 1 C.neo H3K4me3 Peaks.xlsx
Supplemental file S2 52D Infected Unique and Shared Peaks.xlsx
Supplemental file S3 52D Infected Gene Ontology.xlsx
REFERENCES AND NOTES
- 1.Di Pietro A., Good-Jacobson K. L., Disrupting the code: Epigenetic dysregulation of lymphocyte function during infectious disease and lymphoma development. J. Immunol. 201, 1109–1118 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Carson W. F., Cavassani K. A., Soares E. M., Hirai S., Kittan N. A., Schaller M. A., Scola M. M., Joshi A., Matsukawa A., Aronoff D. M., Johnson C. N., Dou Y., Gallagher K. A., Kunkel S. L., The STAT4/MLL1 epigenetic axis regulates the antimicrobial functions of murine macrophages. J. Immunol 199, 1865–1874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh V., Prakhar P., Rajmani R. S., Mahadik K., Borbora S. M., Balaji K. N., Histone methyltransferase SET8 epigenetically reprograms host immune responses to assist mycobacterial survival. J Infect. Dis. 216, 477–488 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Ahmed R., Bevan M. J., Reiner S. L., Fearon D. T., The precursors of memory: Models and controversies. Nat. Rev. Immunol. 9, 662–668 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Netea M. G., Joosten L. A., Latz E., Mills K. H., Natoli G., Stunnenberg H. G., O’Neill L. A., Xavier R. J., Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netea M. G., Quintin J., van der Meer J. W., Trained immunity: A memory for innate host defense. Cell Host Microbe 9, 355–361 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Xu J., Eastman A. J., Flaczyk A., Neal L. M., Zhao G., Carolan J., Malachowski A. N., Stolberg V. R., Yosri M., Chensue S. W., Curtis J. L., Osterholzer J. J., Olszewski M. A., Disruption of early tumor necrosis factor alpha signaling prevents classical activation of dendritic cells in lung-associated lymph nodes and development of protective immunity against cryptococcal infection. MBio 7, e00510–e00516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida K., Maekawa T., Zhu Y., Renard-Guillet C., Chatton B., Inoue K., Uchiyama T., Ishibashi K.-i., Yamada T., Ohno N., Shirahige K., Okada-Hatakeyama M., Ishii S., The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat. Immunol. 16, 1034–1043 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Quintin J., Saeed S., Martens J. H., Giamarellos-Bourboulis E. J., Ifrim D. C., Logie C., Jacobs L., Jansen T., Kullberg B. J., Wijmenga C., Joosten L. A., Xavier R. J., van der Meer J. W., Stunnenberg H. G., Netea M. G., Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng S. C., Quintin J., Cramer R. A., Shepardson K. M., Saeed S., Kumar V., Giamarellos-Bourboulis E. J., Martens J. H., Rao N. A., Aghajanirefah A., Manjeri G. R., Li Y., Ifrim D. C., Arts R. J., van der Meer B. M., Deen P. M., Logie C., O'Neill L. A., Willems P., van de Veerdonk F. L., van der Meer J. W., Ng A., Joosten L. A., Wijmenga C., Stunnenberg H. G., Xavier R. J., Netea M. G., mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis J. L., Cell-mediated adaptive immune defense of the lungs. Proc. Am. Thorac. Soc. 2, 412–416 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwasaki A., Medzhitov R., Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Ginderachter J. A., Movahedi K., Hassanzadeh Ghassabeh G., Meerschaut S., Beschin A., Raes G., De Baetselier P., Classical and alternative activation of mononuclear phagocytes: Picking the best of both worlds for tumor promotion. Immunobiology 211, 487–501 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Osterholzer J. J., Chen G.-H., Olszewski M. A., Curtis J. L., Huffnagle G. B., Toews G. B., Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J. Immunol. 183, 8044–8053 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wozniak K. L., Vyas J. M., Levitz S. M., In vivo role of dendritic cells in a murine model of pulmonary cryptococcosis. Infect. Immun. 74, 3817–3824 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serbina N. V., Salazar-Mather T. P., Biron C. A., Kuziel W. A., Pamer E. G., TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19, 59–70 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Davis M. J., Eastman A. J., Qiu Y., Gregorka B., Kozel T. R., Osterholzer J. J., Curtis J. L., Swanson J. A., Olszewski M. A., Cryptococcus neoformans-induced macrophage lysosome damage crucially contributes to fungal virulence. J. Immunol. 194, 2219–2231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii M., Wen H., Corsa C. A., Liu T., Coelho A. L., Allen R. M., Carson W. F. IV, Cavassani K. A., Li X., Lukacs N. W., Hogaboam C. M., Dou Y., Kunkel S. L., Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arora S., Olszewski M. A., Tsang T. M., McDonald R. A., Toews G. B., Huffnagle G. B., Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect. Immun. 79, 1915–1926 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G. H., McNamara D. A., Hernandez Y., Huffnagle G. B., Toews G. B., Olszewski M. A., Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infect. Immun. 76, 2379–2391 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen G. H., Osterholzer J. J., Choe M. Y., McDonald R. A., Olszewski M. A., Huffnagle G. B., Toews G. B., Dual roles of CD40 on microbial containment and the development of immunopathology in response to persistent fungal infection in the lung. Am. J. Pathol. 177, 2459–2471 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pilmis B., Puel A., Lortholary O., Lanternier F., New clinical phenotypes of fungal infections in special hosts. Clin. Microbiol. Infect. 22, 681–687 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Gardam M. A., Keystone E. C., Menzies R., Manners S., Skamene E., Long R., Vinh D. C., Anti-tumour necrosis factor agents and tuberculosis risk: Mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Hage C. A., Wood K. L., Winer-Muram H. T., Wilson S. J., Sarosi G., Knox K. S., Pulmonary cryptococcosis after initiation of anti-tumor necrosis factor-alpha therapy. Chest 124, 2395–2397 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Herring A. C., Falkowski N. R., Chen G. H., McDonald R. A., Toews G. B., Huffnagle G. B., Transient neutralization of tumor necrosis factor alpha can produce a chronic fungal infection in an immunocompetent host: Potential role of immature dendritic cells. Infect. Immun. 73, 39–49 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herring A. C., Lee J., McDonald R. A., Toews G. B., Huffnagle G. B., Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect. Immun. 70, 2959–2964 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hijdra D., Vorselaars A. D., Grutters J. C., Claessen A. M., Rijkers G. T., Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J. Inflamm. 9, 38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maney N. J., Reynolds G., Krippner-Heidenreich A., Hilkens C. M., Dendritic cell maturation and survival are differentially regulated by TNFR1 and TNFR2. J. Immunol. 193, 4914–4923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sundquist M., Wick M. J., TNF-alpha-dependent and -independent maturation of dendritic cells and recruited CD11c(int)CD11b+ cells during oral Salmonella infection. J. Immunol. 175, 3287–3298 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Vecchiarelli A., Pietrella D., Lupo P., Bistoni F., McFadden D. C., Casadevall A., The polysaccharide capsule of Cryptococcus neoformans interferes with human dendritic cell maturation and activation. J. Leukoc. Biol. 74, 370–378 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Pietrella D., Corbucci C., Perito S., Bistoni G., Vecchiarelli A., Mannoproteins from Cryptococcus neoformans promote dendritic cell maturation and activation. Infect. Immun. 73, 820–827 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupo P., Chang Y. C., Kelsall B. L., Farber J. M., Pietrella D., Vecchiarelli A., Leon F., Kwon-Chung K. J., The presence of capsule in Cryptococcus neoformans influences the gene expression profile in dendritic cells during interaction with the fungus. Infect. Immun. 76, 1581–1589 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karatas H., Townsend E. C., Cao F., Chen Y., Bernard D., Liu L., Lei M., Dou Y., Wang S., High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J. Am. Chem. Soc. 135, 669–682 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salvana E. M., Salata R. A., Infectious complications associated with monoclonal antibodies and related small molecules. Clin. Microbiol. Rev. 22, 274–290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milam J. E., Herring-Palmer A. C., Pandrangi R., McDonald R. A., Huffnagle G. B., Toews G. B., Modulation of the pulmonary type 2 T-cell response to Cryptococcus neoformans by intratracheal delivery of a tumor necrosis factor alpha-expressing adenoviral vector. Infect. Immun. 75, 4951–4958 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis M. J., Tsang T. M., Qiu Y., Dayrit J. K., Freij J. B., Huffnagle G. B., Olszewski M. A., Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 4, e00264–e00213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faith A., McDonald J., Peek E., Richards D., Caulfield J., Chevretton E., Roberts D., Lee T., Corrigan C., Hawrylowicz C., Functional plasticity of human respiratory tract dendritic cells: GM-CSF enhances T(H)2 development. J. Allergy Clin. Immunol. 116, 1136–1143 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Clarke D. L., Clifford R. L., Jindarat S., Proud D., Pang L., Belvisi M., Knox A. J., TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J. Biol. Chem. 285, 29101–29110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohmori Y., Schreiber R. D., Hamilton T. A., Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem. 272, 14899–14907 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Robinson C. M., Hale P. T., Carlin J. M., NF-κB activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-γ and tumor necrosis factor-α. Cytokine 35, 53–61 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Carson W. F., Cavassani K. A., Dou Y., Kunkel S. L., Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics 6, 273–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyn-Kew K., Rich E., Zeng X., Wen H., Kunkel S. L., Newstead M. W., Bhan U., Standiford T. J., IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLOS ONE 5, e11145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen H., Dou Y., Hogaboam C. M., Kunkel S. L., Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 111, 1797–1804 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arts R. J., Blok B. A., Aaby P., Joosten L. A., de Jong D., van der Meer J. W., Benn C. S., van Crevel R., Netea M. G., Long-term in vitro and in vivo effects of gamma-irradiated BCG on innate and adaptive immunity. J. Leukoc. Biol. 98, 995–1001 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Arts R. J. W., Carvalho A., La Rocca C., Palma C., Rodrigues F., Silvestre R., Kleinnijenhuis J., Lachmandas E., Goncalves L. G., Belinha A., Cunha C., Oosting M., Joosten L. A. B., Matarese G., van Crevel R., Netea M. G., Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 17, 2562–2571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arts R. J. W., Moorlag S., Novakovic B., Li Y., Wang S. Y., Oosting M., Kumar V., Xavier R. J., Wijmenga C., Joosten L. A. B., Reusken C., Benn C. S., Aaby P., Koopmans M. P., Stunnenberg H. G., van Crevel R., Netea M. G., BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23, 89–100.e105 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Kaufmann E., Sanz J., Dunn J. L., Khan N., Mendonca L. E., Pacis A., Tzelepis F., Pernet E., Dumaine A., Grenier J. C., Mailhot-Leonard F., Ahmed E., Belle J., Besla R., Mazer B., King I. L., Nijnik A., Robbins C. S., Barreiro L. B., Divangahi M., BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176–190.e19 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Kleinnijenhuis J., Quintin J., Preijers F., Joosten L. A., Ifrim D. C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H. G., Xavier R. J., van der Meer J. W., van Crevel R., Netea M. G., Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U.S.A. 109, 17537–17542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Meer J. W., Joosten L. A., Riksen N., Netea M. G., Trained immunity: A smart way to enhance innate immune defence. Mol. Immunol. 68, 40–44 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Ifrim D. C., Quintin J., Meerstein-Kessel L., Plantinga T. S., Joosten L. A., van der Meer J. W., van de Veerdonk F. L., Netea M. G., Defective trained immunity in patients with STAT-1-dependent chronic mucocutaneaous candidiasis. Clin. Exp. Immunol. 181, 434–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leopold Wager C. M., Hole C. R., Campuzano A., Castro-Lopez N., Cai H., Caballero Van Dyke M. C., Wozniak K. L., Wang Y., Wormley F. L. Jr., IFN-gamma immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans. PLOS Pathog. 14, e1007358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ordonez M. E., Farraye F. A., Di Palma J. A., Endemic fungal infections in inflammatory bowel disease associated with anti-tnf antibody therapy. Inflamm. Bowel Dis. (2013). [DOI] [PubMed] [Google Scholar]
- 53.Wissmann G., Morilla R., Martin-Garrido I., Friaza V., Respaldiza N., Povedano J., Praena-Fernandez J. M., Montes-Cano M. A., Medrano F. J., Goldani L. Z., de la Horra C., Varela J. M., Calderon E. J., Pneumocystis jirovecii colonization in patients treated with infliximab. Eur. J. Clin. Invest. 41, 343–348 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Wood K. L., Hage C. A., Knox K. S., Kleiman M. B., Sannuti A., Day R. B., Wheat L. J., Twigg H. L. III, Histoplasmosis after treatment with anti-tumor necrosis factor-α therapy. Am. J. Respir. Crit. Care Med. 167, 1279–1282 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Athie-Morales V., Smits H. H., Cantrell D. A., Hilkens C. M., Sustained IL-12 signaling is required for Th1 development. J. Immunol. 172, 61–69 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Bekkering S., Arts R. J. W., Novakovic B., Kourtzelis I., van der Heijden C. D. C. C., Li Y., Popa C. D., ter Horst R., van Tuijl J., Netea-Maier R. T., van de Veerdonk F. L., Chavakis T., Joosten L. A. B., van der Meer J. W. M., Stunnenberg H., Riksen N. P., Netea M. G., Metabolic induction of trained immunity through the mevalonate pathway. Cell 172, 135–146.e139 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Mitroulis I., Ruppova K., Wang B., Chen L.-S., Grzybek M., Grinenko T., Eugster A., Troullinaki M., Palladini A., Kourtzelis I., Chatzigeorgiou A., Schlitzer A., Beyer M., Joosten L. A. B., Isermann B., Lesche M., Petzold A., Simons K., Henry I., Dahl A., Schultze J. L., Wielockx B., Zamboni N., Mirtschink P., Coskun Ü., Hajishengallis G., Netea M. G., Chavakis T., Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Mutairi N., Nour T., Al-Rqobah D., Onychomycosis in patients of nail psoriasis on biologic therapy: A randomized, prospective open label study comparing Etanercept, Infliximab and Adalimumab. Expert Opin. Biol. Ther. 13, 625–629 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Osawa R., Singh N., Colitis as a manifestation of infliximab-associated disseminated cryptococcosis. Int. J. Infect. Dis. 14, e436–e440 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Leentjens J., Kox M., Koch R. M., Preijers F., Joosten L. A., van der Hoeven J. G., Netea M. G., Pickkers P., Reversal of immunoparalysis in humans in vivo: A double-blind, placebo-controlled, randomized pilot study. Am. J. Respir. Crit. Care Med. 186, 838–845 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Peters van Ton A. M., Kox M., Abdo W. F., Pickkers P., Precision immunotherapy for sepsis. Front. Immunol. 9, 1926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olszewski M. A., Huffnagle G. B., McDonald R. A., Lindell D. M., Moore B. B., Cook D. N., Toews G. B., The role of macrophage inflammatory protein-1 alpha/CCL3 in regulation of T cell-mediated immunity to Cryptococcus neoformans infection. J. Immunol. 165, 6429–6436 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Olszewski M. A., Huffnagle G. B., Traynor T. R., McDonald R. A., Cook D. N., Toews G. B., Regulatory effects of macrophage inflammatory protein 1alpha/CCL3 on the development of immunity to Cryptococcus neoformans depend on expression of early inflammatory cytokines. Infect. Immun. 69, 6256–6263 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jain A. V., Zhang Y., Fields W. B., McNamara D. A., Choe M. Y., Chen G. H., Erb-Downward J., Osterholzer J. J., Toews G. B., Huffnagle G. B., Olszewski M. A.,. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect. Immun. 77, 5389–5399 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brind'Amour J., Liu S., Hudson M., Chen C., Karimi M. M., Lorincz M. C., An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 6, 6033 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Carroll T. S., Liang Z., Salama R., Stark R., de Santiago I., Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 5, 75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu L., Zinkgraf M., Petzold H. E., Beers E. P., Filkov V., Groover A., The Populus ARBORKNOX1 homeodomain transcription factor regulates woody growth through binding to evolutionarily conserved target genes of diverse function. New Phytol. 205, 682–694 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Ross-Innes C. S., Stark R., Teschendorff A. E., Holmes K. A., Ali H. R., Dunning M. J., Brown G. D., Gojis O., Ellis I. O., Green A. R., Ali S., Chin S. F., Palmieri C., Caldas C., Carroll J. S., Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang Y., Min S., Lui Y., Sun J., Su X., Liu Y., Zhang Y., Han D., Che Y., Zhao C., Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun. 13, 311–320 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Messier T. L., Gordon J. A., Boyd J. R., Tye C. E., Browne G., Stein J. L., Lian J. B., Stein G. S., Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 7, 5094 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu L. J., Gazin C., Lawson N. D., Pages H., Lin S. M., Lapointe D. S., Green M. R., ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaaw9051/DC1
Fig. S1. TNFα depletion in CBA/J mice results in fungal persistence and dysregulated immunity during C. neo infection.
Fig. S2. Schematic of BMDC experiments and testing model.
Fig. S3. TNFRI and TNFRII are both required for optimal DC1 programming.
Fig. S4. TNFα results in sustained DC1 profile at the protein level in pulmonary DCs during C. neo infection.
Fig. S5. MLL1 is uniquely induced in TNFα-programmed DC1.
Fig. S6. DC1 from control C. neo–infected mice epigenetically resemble TNFα-programmed DC1.
Fig. S7. Assessment of total and myeloid precursor populations in the BM during C. neo infection.
Supplemental file 1 C.neo H3K4me3 Peaks.xlsx
Supplemental file S2 52D Infected Unique and Shared Peaks.xlsx
Supplemental file S3 52D Infected Gene Ontology.xlsx