

SUMMARY
BCR-ABL1 point mutation-mediated resistance to tyrosine kinase inhibitor (TKI) therapy in Philadelphia chromosome-positive (Ph+) leukemia is effectively managed with several approved drugs, including ponatinib for BCR-ABL1T315I-mutant disease. However, therapy options are limited for patients with leukemic clones bearing multiple BCR-ABL1 mutations. Asciminib, an allosteric inhibitor targeting the myristoyl-binding pocket of BCR-ABL1, is active against most single mutants but ineffective against all tested compound mutants. We demonstrate that combining asciminib with ATP-site TKIs enhances target inhibition and suppression of resistant outgrowth in Ph+ clinical isolates and cell lines. Inclusion of asciminib restores ponatinib’s effectiveness against currently untreatable compound mutants at clinically achievable concentrations. Our findings support combining asciminib with ponatinib as a treatment strategy for this molecularly defined group of patients.
Keywords: asciminib, ABL001, allosteric inhibitors, chronic myeloid leukemia, ponatinib, targeted therapy, compound mutation
INTRODUCTION
Outcomes for patients with chronic myeloid leukemia (CML) have been greatly improved by the development and implementation of small-molecule BCR-ABL1 tyrosine kinase inhibitors (TKIs) in the clinic, a translational paradigm initially paved by imatinib (Hochhaus et al., 2017). However, one in five patients with chronic phase (CP) CML will develop resistance to treatment, and responses are only transient in most patients with lymphoid or myeloid blast crisis CML or Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL) (Eide and O’Hare, 2015; O’Hare et al., 2012). Resistance is most often caused by acquisition of point mutations within the BCR-ABL1 kinase domain that impair drug binding, restoring the oncoprotein’s constitutively active tyrosine kinase activity (Gorre et al., 2001; O’Hare et al., 2012; Shah and Sawyers, 2003).
Second- and third-generation TKIs such as nilotinib (Kantarjian et al., 2006), dasatinib (Talpaz et al., 2006), bosutinib (Cortes et al., 2011), and ponatinib (Cortes et al., 2013) provide effective control of point mutation-mediated resistance. Ponatinib is the only U.S. Food and Drug Administration (FDA)-approved TKI with activity against all known BCR-ABL1 point mutations, including BCR-ABL1T315I. However, the emergence of compound mutations (two mutations within the same BCR-ABL1 allele) has been linked to resistance to all approved BCR-ABL1 TKIs, including ponatinib, posing a clinical challenge with limited treatment options (Shah et al., 2007; Zabriskie et al., 2014).
All BCR-ABL1 TKIs currently approved by the FDA target the ATP-binding site (Hantschel et al., 2012). The activity of native ABL1 is tightly regulated in part through the interaction of a myristoyl group near its N-terminus with a myristoyl-binding site located between the end of the ABL1 kinase domain and select helices of the ABL1 C-terminus, which stabilizes a catalytically inactive conformation of the kinase domain (Nagar et al., 2003). In the BCR-ABL1 fusion protein, this auto-inhibitory mechanism is compromised by loss of the N-terminal cap region including the myristoylated glycine residue at position 2. The retained myristoyl-binding pocket within the BCR-ABL1 kinase domain represents a site amenable to allosteric inhibition, and such inhibitors are predicted to be invulnerable to the set of point mutations that cause resistance to ATP-site TKIs (Qiang et al., 2017). The presence of a second inhibitory site also offers the potential for simultaneous targeting of both the myristoyl-binding and ATP-binding sites for enhanced kinase inhibition (Zhang et al., 2010).
Asciminib (formerly ABL001) is an allosteric inhibitor of BCR-ABL1 that binds to the myristoyl-binding site (Figure 1A), affording high selectivity for inhibition of BCR-ABL1 kinase and its downstream signaling (Wylie et al., 2017). Asciminib is in phase 1 trials for the treatment of refractory CML and Ph+ ALL. Herein, we profile asciminib for its efficacy against BCR-ABL1 single and compound mutants, alone and in combination with other ATP-site BCR-ABL1 TKIs.
Figure 1. The allosteric BCR-ABL1 inhibitor asciminib demonstrates selective activity against CML cell signaling, growth, and proliferation.

(A) Structural diagram of the ABL1 kinase domain highlighting the ATP-binding site (green) and myristoyl-binding pocket (blue). Nilotinib (purple) and asciminib (yellow) are shown bound to each site, respectively (PDB ID: 5MO4). (B and C) FACS (B) and immunoblot (C) analyses of CRKL phosphorylation in primary CML cells from a newly diagnosed patient treated with asciminib. (D) Myeloid colony formation assays using primary CML and healthy donor cells treated with asciminib. Colony numbers were normalized to untreated controls and are reported as the mean percent of untreated colonies ± SEM. (E) Dose-response curves for asciminib in human CML cell lines (K562 and LAMA84) and non-CML human leukemia cell lines (HL-60 and U937). Data points represent the mean percent of untreated ± SEM. See also Figure S1.
RESULTS
Asciminib Selectively Inhibits Signaling, Clonogenicity, and Proliferation of CML Cells
The activity of asciminib was first assessed by treating primary CML patient cells ex vivo. Fluorescence activated cell sorting (FACS) and immunoblot analyses showed a concentration-dependent reduction in phosphorylation of CRKL, a clinical biomarker of BCR-ABL1 signaling activity (Figure 1B & 1C). Consistent with specific inhibition of BCR-ABL1 signaling, asciminib selectively inhibited colony formation by primary CML cells but not by healthy donor controls (Figure 1D). Furthermore, the growth of human Ph+ leukemia cell lines (K562 and LAMA84) was potently inhibited by asciminib (IC50: 8.1 and 3.2 nM, respectively), whereas an IC50 for Ph− leukemia lines (HL-60 and U937) was not reached at 1 μM (Figures 1E and S1). Together, these findings confirm asciminib as a potent, selective inhibitor of BCR-ABL1-mediated signaling and cell growth.
Asciminib Exhibits Differential Activity Against BCR-ABL1 Point Mutants and Suppresses Emergence of Resistant Point Mutations When Combined With Nilotinib or Ponatinib
To determine the profile of asciminib against BCR-ABL1 point mutants that confer resistance to FDA-approved TKIs, proliferation assays were performed with Ba/F3 cells expressing native BCR-ABL1 or a BCR-ABL1 single mutant. Asciminib potently inhibited Ba/F3 cells harboring native BCR-ABL1 (IC50: 3.8 nM), but not Ba/F3 parental cells, and reduced native BCR-ABL1 autophosphorylation and tyrosine phosphorylation of downstream target STAT5 (Figure 2A and 2B).
Figure 2. Asciminib demonstrates differential sensitivity among clinically-relevant BCR-ABL1 single mutants associated with resistance to approved ATP-site TKIs.

(A) Cell proliferation curves for Ba/F3 MIG BCR-ABL1 cells and Ba/F3 parental cells treated with asciminib. Data points represent the mean percent of untreated ± SEM. (B) Immunoblot analysis of phosphorylation of BCR-ABL1 (Y393) and STAT5 (Y694) following treatment of Ba/F3 BCR-ABL1 cells with asciminib or imatinib. (C) Heat map summary of IC50 values for BCR-ABL1 TKIs against a panel of Ba/F3 cell lines expressing MIG BCR-ABL1 single mutants. Data for imatinib, nilotinib, dasatinib, and ponatinib are from (Zabriskie et al., 2014) and are included for comparison purposes. (D) Immunoblot analysis of BCR-ABL1 tyrosine autophosphorylation (Y393) for select Ba/F3 pSRα BCR-ABL1 single mutants following treatment with asciminib or imatinib. See also Figure S2 and Table S1.
Among ten BCR-ABL1 single mutants tested, asciminib potently inhibited five (G250E, Y253H, E255V, T315I, H396R) with IC50 values below 30 nM (Figure 2C and Table S1). By contrast, some variants (C/I/V) of F359 were insensitive to asciminib (IC50 >2500 nM). Further immunoblot analysis confirmed differences in sensitivity tracked with retention of BCR-ABL1 kinase activity (Figures 2D and S2).
Given asciminib being an allosteric inhibitor of BCR-ABL1, cell-based accelerated mutagenesis screens (Bradeen et al., 2006; Eide et al., 2011; O’Hare et al., 2009; Zabriskie et al., 2014) were used to identify mutations uniquely resistant to asciminib. To account for the possibility that mutations outside the ABL1 kinase domain may confer resistance to asciminib, sequencing of resistant clones was expanded to include the SH3, SH2, and C-terminal domains of BCR-ABL1. A concentration-dependent decrease in recovered resistant clones was observed, with 23% of wells demonstrating outgrowth at 1600 nM, the highest concentration of asciminib tested (Figure 3A and Table S2). Sequencing of resistant clones revealed multiple variants at positions located near or within the myristoyl-binding pocket (Figure S3), including A344P, P465S, and G671R, suggesting compromised asciminib binding. Similar findings were observed from independent experiments (Table S3). A fraction of resistant clones recovered with single-agent asciminib harbored no BCR-ABL1 point mutation within the surveyed region. It is conceivable these clones acquired resistance through overexpression of an ABC transporter, as recently reported (Qiang et al., 2017).
Figure 3. Combining asciminib with ATP-site TKIs suppresses emergence of BCR-ABL1 single mutant-based resistance, including asciminib-resistant myristoyl-binding site mutants.

(A) Ba/F3 pSRα BCR-ABL1 cells were treated with N-ethyl-N-nitrosourea (ENU) overnight, plated in fresh complete medium in the presence of graded concentrations of asciminib, and monitored for outgrowth for 28 days. BCR-ABL1 mutations identified from resistant clones are summarized in the box above the graph. (B and C) Results for assays similar to (A) carried out using the combination of asciminib with nilotinib (B) or ponatinib (C). BCR-ABL1 mutations identified from recovered combination treatment-resistant clones are shown above each graph. (D) Structural illustration of the myristoyl-binding pocket, highlighting the residues of asciminib-resistant mutations (orange spheres) relative to bound asciminib (blue). (E) Cell proliferation assays using TKIs against Ba/F3 cells expressing asciminib-resistant BCR-ABL1 myristoyl-binding site mutants. Bars represent mean fold-change in IC50 ± SEM. (F) Immunoblot analysis of BCR-ABL1 autophosphorylation in Ba/F3 cells expressing BCR-ABL1 myristoyl-binding site mutants treated as indicated. See also Figure S3 and Tables S1–S4.
Based on complementary profiles of asciminib and ATP-site inhibitors, resistance screens were performed combining asciminib with nilotinib or ponatinib in Ba/F3 BCR-ABL1 cells. In comparison to either single agent, the combination of asciminib and nilotinib reduced resistance outgrowth, with the lowest tested combination of 10 nM asciminib + 50 nM nilotinib yielding <20% outgrowth compared with outgrowth in nearly all wells for either inhibitor individually. Moreover, no clones that grew out of combined treatment harbored myristoyl-binding site mutations; all recovered mutations are known to confer clinical resistance to nilotinib, including multiple substitutions at position 359, consistent with results of cell proliferation studies (Figure 3B and Table S4). Combining asciminib and ponatinib also suppressed resistance outgrowth, with 10 nM asciminib + 2.5 nM ponatinib yielding <4% outgrowth (Figure 3C and Table S4) and no resistant clones observed at higher concentrations. These results suggest mutations of select residues in both the BCR-ABL1 kinase domain and myristoyl-binding site may confer resistance to single-agent asciminib, but this is largely overcome by combination treatment. As the greatest suppression of resistant outgrowth was observed with the combination of asciminib and ponatinib, and ponatinib is the only FDA approved TKI with activity against BCR-ABL1T315I, subsequent analyses of TKI combination efficacy focused on this pairing.
BCR-ABL1 Myristoyl-Binding Pocket Mutations Confer Resistance to Asciminib but Remain Sensitive to ATP-Site TKIs
Ba/F3 cells harboring BCR-ABL1 myristoyl-binding site mutations identified from our screens (A344P, P465S) or reported recently in asciminib resistance (A337V, P465S, V468F) (Wylie et al., 2017) (Figure 3D) were highly resistant to asciminib in vitro but remained sensitive to ATP-site BCR-ABL1 TKIs (Figure 3E and Table S1). Immunoblot analysis showed that phosphorylated BCR-ABL1 in myristoyl-binding site mutants was inhibited by nilotinib and ponatinib but persisted despite asciminib treatment (Figure 3F). Together, these data suggest mutations in or around the myristoyl-binding site may hinder the ability of asciminib to bind BCR-ABL1, representing a mechanism of resistance unique to this class of TKI.
BCR-ABL1 Kinase Domain Variants of Position 359 Selectively Expand or Persist in Patients Treated With Asciminib
To examine the dynamics of BCR-ABL1 mutations in patients on asciminib therapy, serial specimens from six patients enrolled in the phase 1 clinical trial who eventually discontinued asciminib monotherapy were obtained from participating sites (Table S5 and Figure S4). In each sample, BCR-ABL1 was amplified and subjected to next-generation sequencing to monitor mutational status over time. In three of the patients analyzed, there was evidence of selective expansion of variants of position 359 on asciminib therapy. Patient 1 had a diagnosis of CP CML and had failed nilotinib and dasatinib prior to starting asciminib (20 mg twice daily). T315I and F359V mutations were detected at baseline, and again in the day 29 sample (variant allele frequency (VAF): 21% T315I, 79% F359V). The T315I clone became undetectable by day 113 and never reemerged, tracking with a transient reduction in BCR-ABL1 transcripts. Meanwhile, expansion of the F359V clone, reaching a VAF of 92% in the day 533 sample, was accompanied by increasing molecular disease burden forcing subsequent dose escalations (Figure 4A), implicating this mutation in resistance.
Figure 4. Mutations of position 359 of the BCR-ABL1 kinase domain confer resistance to asciminib and are expanded in asciminib-treated CML patients.

(A-C) Graphical summaries of the BCR-ABL1 transcript level (on the International Scale) and variant allele frequency (VAF) of detected BCR-ABL1 variants of chronic phase CML patients 1 (A), 2 (B), and 3 (C) are displayed aligned to their clinical timeline and treatment dosing on asciminib. (D) Immunoblot analysis of BCR-ABL1 autophosphorylation in Ba/F3 cells expressing MIG BCR-ABL1F3591 or BCR-ABL1F359V treated with asciminib or imatinib. (E) Illustration of spatial position of residue F359 (orange sphere) relative to the binding of asciminib (blue) within the myristoyl pocket. See also Table S5 and Figures S4 and S5.
Patient 2 had CP CML at the time of starting asciminib, having previously failed radotinib, imatinib, and dasatinib. A T315I mutation was reported at baseline and confirmed in the day 29 sample (VAF: 29%; Figure 4B). Although this patient did not achieve reduction in BCR-ABL1 transcripts on asciminib treatment, multiple changes in mutations were evident. T315I became the dominant clone by day 57 (96%) and persisted at day 197 (100%), but by day 533 (13%) had been surpassed by F359I (65%). Several lower-level mutants also emerged at this time, including A337T (9%) and A433D (11%), both of which are located near the entrance to the myristoyl-binding pocket. This suggests that F359I along with less dominant myristoyl-binding site mutations may underlie the resistance in this patient on asciminib treatment.
Patient 3 was started on 120 mg asciminib once daily while in CP CML after failure of imatinib and bosutinib, and had a dominant T315I clone (VAF: 76%) at baseline, which temporally expanded by day 83 (96%) amid a slight reduction in BCR-ABL1 transcripts. On day 141, the asciminib dose was increased to 200 mg once daily, which was followed by emergence of an F359I clone coinciding with a reduction in T315I abundance (51% and 49%, respectively) in the day 251 sample. By day 308, F359I was the dominant clone (93%), while only a low level of T315I remained (7%) (Figure 4C). Throughout treatment, BCR-ABL1 transcripts remained ≥12%, even after an additional dose escalation to 200 mg twice daily on day 314. Ultimately, the patient discontinued treatment with single-agent asciminib at this dose on day 469.
Consistent with in vitro resistance profiling (Figure 2C and Table S1), we found that Ba/F3 cell lines expressing the F359V or F359I mutant demonstrated no inhibition of BCR-ABL1 autophosphorylation with asciminib, in contrast to concentration-dependent sensitivity to imatinib (Figure 4D). Furthermore, primary CML patient cells harboring BCR-ABL1F359I exhibited decreased CRKL phosphorylation following ex vivo treatment with ponatinib but not asciminib, which was slightly enhanced with the combination of both inhibitors (Figure S5). While the precise explanation behind the vulnerability of asciminib to mutations of this position (Figure 4E) requires more detailed investigation, these cases provide clinical evidence that variants of position 359 represent problematic sources of resistance to asciminib.
Combining Asciminib with Ponatinib or Nilotinib Improves Inhibition of BCR-ABL1 Signaling and CML Cell Growth Relative to Either TKI Alone
Lowering plasma TKI concentrations while maintaining efficacy may reduce dose-dependent toxicity such as cardiovascular toxicity in the case of ponatinib and nilotinib (Latifi et al., 2019). Given the capacity of asciminib combined with ponatinib or nilotinib to suppress emergence of BCR-ABL1 point mutation-based resistance, we sought to confirm that this extended to BCR-ABL1-driven signaling and clonogenicity of primary CML cells. Ponatinib alone decreased CRKL phosphorylation in primary CML patient cells, which was further reduced upon adding asciminib (Figure 5A & 5B). Consistently, ponatinib treatment inhibited colony formation of primary CP CML cells, which was augmented by asciminib (Figure 5C). Similar results were observed for the combination of asciminib with nilotinib. None of the combination treatments resulted in toxicity to healthy donor bone marrow cells. These results suggest combination treatment with asciminib and an ATP-site BCR-ABL1 TKI at concentrations that are non-toxic to normal hematopoietic progenitors provides an effective therapeutic strategy for eliminating primary CML cells.
Figure 5. Asciminib potentiates the efficacy of ATP-site TKIs to inhibit BCR-ABL1 signaling and clonogenicity in primary CML cells.

(A and B) FACS (A) and immunoblot (B) analyses of CRKL phosphorylation in primary newly diagnosed CML cells treated with ponatinib alone or in combination with asciminib. (C) Myeloid colony formation assay of primary CML and healthy donor cells treated with either nilotinib or ponatinib alone or in combination with asciminib. Colony numbers were normalized to those of untreated controls, and bars represent the mean ± SEM.
TKI-refractory BCR-ABL1 Compound Mutants Are Resistant to Single-Agent Asciminib or Ponatinib but Suppressed at Therapeutically Relevant Concentrations in Combination
To further define the resistance profile of asciminib, we used Ba/F3 cells expressing ten clinical BCR-ABL1 compound mutants (seven included T315I and three did not) (Table S6). Unlike native BCR-ABL1 and most kinase domain point mutants, BCR-ABL1 compound mutants uniformly conferred resistance to asciminib (IC50 >2500 nM) (Figure 6A & 6B). Ponatinib was also ineffective against T315I-inclusive BCR-ABL1 compound mutants but displayed varying degrees of activity against non-T315I compound mutants, as previously reported (Zabriskie et al., 2014). Addition of asciminib (50 or 250 nM) reduced the IC50 of ponatinib by 1.9- to 18.5-fold for T315I-inclusive compound mutants and 3.1- to 6.3-fold for non-T315I compound mutants (Figure 6A & 6B). For example, in the case of the T315I/H396R compound mutant, inclusion of 50 nM asciminib reduced the ponatinib IC50 by 12.6-fold. In contrast, Ba/F3 parental cells exhibited no toxicity to a matrix of ponatinib and asciminib concentrations (Figure S6A). Adding asciminib also enhanced dasatinib efficacy for non-T315I compound mutants, but T315I-inclusive compound mutants remained insensitive (Figure S6B), consistent with the incompatibility of the T315I substitution with binding of any of the approved ATP-site TKIs other than ponatinib.
Figure 6. Combining asciminib with ponatinib at clinically relevant concentrations restores efficacy against highly-resistant BCR-ABL1 compound mutations.

(A) Cellular proliferation IC50 values (mean ± SEM) of Ba/F3 cells expressing MIG BCR-ABL1 compound mutants treated with ponatinib alone or in combination with 50 nM or 250 nM asciminib. Dashed lines represent the clinically achievable steady-state plasma concentration for ponatinib (35, 84, and 101 nM for 15, 30, and 45 mg/day, respectively) (Cortes et al., 2012; Gozgit et al., 2013). (B) Heat map summary of TKI sensitivities in cellular proliferation assays for Ba/F3 cells expressing MIG BCR-ABL1 compound mutants. A color gradient from white (sensitive) to dark blue (insensitive) denotes the sensitivity to asciminib alone, ponatinib alone, or ponatinib in combination with either 50 or 250 nM asciminib. (C) Immunoblot analysis of BCR-ABL1 autophosphorylation in Ba/F3 cells expressing MIG BCR-ABL1T315M, BCR-ABL1Y253H/T315I, or BCR-ABL1E255V/T315I following treatment with ponatinib alone or in combination with 250 nM asciminib. (D) Ba/F3 pSRα BCR-ABL1T315I cells were treated with ENU overnight, plated in fresh complete medium in the presence of graded concentrations of asciminib alone, ponatinib alone, or the indicated matrix of combinations and monitored for outgrowth for 28 days. BCR-ABL1 compound mutations identified from resistant clones are summarized in the box above the graph. See also Tables S6–S8 and Figure S6.
The enhanced efficacy of the combination of asciminib and ponatinib included the three reported clinical compound mutants most resistant to ponatinib: T315M (a single amino acid change that requires two nucleotide changes [ACC to ATG]), Y253H/T315I, and E255V/T315I (Zabriskie et al., 2014). Asciminib alone had no inhibitory effect on Ba/F3 cells expressing BCR-ABL1T315M, BCR-ABL1Y253H/T315I, or BCR-ABL1E255V/T315I (IC50: >2,500 nM), and the IC50 values for ponatinib were 415, 316, and 661 nM, respectively, which are well above clinically achievable levels (reported at 101 nM for 45 mg once daily) (Cortes et al., 2012). Supplementing with 50 or 250 nM asciminib reduced the IC50 for ponatinib in each instance: T315M by 5.6-fold (74 nM) and 18.9-fold (22 nM), respectively; Y253H/T315I by 7.6-fold (47 nM) and 10.2-fold (35 nM), respectively; and E255V/T315I by 5.0-fold (132 nM) and 10.1-fold (66 nM), respectively (Figure 6A & 6B). Immunoblot analysis confirmed the enhanced efficacy of the combination tracked with reduced BCR-ABL1 phosphorylation and signaling compared to ponatinib alone (Figure 6C).
Cell-based resistance screens starting from Ba/F3 BCR-ABL1T315I cells revealed enhanced suppression of resistant clones for all asciminib-ponatinib combination treatments. For example, among the eight tested combinations involving 40 nM or lower ponatinib, three recovered no resistant clones, and the remaining five showed a maximum of 4.2% outgrowth (range: 1.0–4.2%) (Figure 6D and Table S7). The small number of resistant clones recovered from these combinations included clinically relevant compound mutants (Cortes et al., 2013; Zabriskie et al., 2014): Q252H/T315I, Y253H/T315I, and E255V/T315I. Similar findings were observed in independent experiments (Table S8). Together, these results suggest that while single-agent asciminib fails to control BCR-ABL1 compound mutants, the combination of asciminib and ponatinib may provide a treatment strategy for patients having CML with such mutations who currently have no effective TKI options. Additionally, combinations may provide an opportunity for upfront mitigation of the emergence of compound mutation-based resistance.
Combining Ponatinib and Asciminib Prolongs Survival and Inhibits Tumor Growth of Mice with Leukemia Induced by a T315I-Inclusive BCR-ABL1 Compound Mutant
We next evaluated efficacy of the asciminib-ponatinib combination in vivo in a mouse model of BCR-ABL1T315I-inclusive compound mutant-driven disease. We previously reported that a patient with CML harboring BCR-ABL1T315I/H396R at baseline experienced clinical progression on ponatinib without an intervening period of response (Zabriskie et al., 2014). This mutation also confers complete resistance to asciminib (Figure 6B). Nod-SCID mice were injected by tail vein with Ba/F3 cells expressing BCR-ABL1T315I/H396R and treated once daily with vehicle, asciminib, ponatinib, or the combination of asciminib and ponatinib (n=10 mice/group) for up to 19 days (Figure 7A). Consistent with our in vitro findings, combination treatment provided a significant, albeit modest, prolongation of survival (median: 25 days) compared to vehicle or either single agent (Figure 7B). Neither asciminib nor ponatinib alone provided a survival advantage over vehicle (p>0.6 for both), with median survival of 22, 23, and 23 days, respectively. Luciferase-based imaging showed decreased tumor burden in the combination-treated group at the last day of treatment (day 21) compared to all other groups (Figure 7C and 7D). Some mice experienced weight loss, most severely on ponatinib treatment. Animals treated with the combination were less prone to weight loss (Figure S7). These findings support the tolerability and efficacy of the combination of asciminib and ponatinib in targeting T315I-inclusive compound mutant tumor growth in vivo and provide evidence this combination should be further evaluated clinically.
Figure 7. Combined treatment with asciminib and ponatinib in vivo prolongs survival and inhibits T315I-inclusive compound mutant tumor growth in a xenograft mouse model.

(A) Design for in vivo mouse model to evaluate the combination of asciminib and ponatinib against the BCR-ABL1T315I/H396R mutant. (B) Survival curves for single-agent and combination treatments. Dosing period (from day 4 through 21) is highlighted in gray. Curves were compared by log-rank Mantel-Cox test. (C) IVIS imaging of luminescence signal in mice on treatment. Luminescence imaging was performed at days 14 and 21 for all mice in each treatment arm (n=10 per group at baseline). (D) Quantification of luminescence signal as a measure of tumor burden in (C) at end of treatment (day 21). Individual dots represent each animal, with the middle horizontal lines indicating the median of each group. Box-and-whiskers representation of the interquartile range and maximum/minimum of each group is included for reference. See also Figure S7.
Dual Binding of Ponatinib and Asciminib Stabilizes the Pan-TKI-Resistant BCR-ABL1Y253H/T315I Compound Mutant in the Catalytically Inactive Conformation
One of the benefits afforded by combined treatment with asciminib and ponatinib was efficacy against the BCR-ABL1Y253H/T315I compound mutant (Figure 8A). To better understand the mechanism by which combining asciminib with ponatinib is effective against BCR-ABL1 compound mutants that are highly resistant to either alone, molecular dynamics and docking simulations of the ABL1 kinase domain were performed. Both ponatinib (O’Hare et al., 2009) and asciminib (Wylie et al., 2017) preferentially bind to catalytically inactive conformations of the ABL1 kinase domain. In the native ABL1 kinase domain, the inactive conformation features interaction of the SH2 and SH3 domains with the C-lobe, promoting breakage of the alpha-I-helix, formation of the alpha-I’ helix, and assembly of the myristoyl-binding pocket. Modeled structural ensembles of the uninhibited apo-form suggest that the Y253H/T315I mutant exists predominantly in the catalytically active, DFG-in conformation wherein the alpha-I helix of the ABL1 kinase domain is straight rather than bent, precluding the SH2/SH3-C-lobe interaction critical for the formation of the myristoyl-binding pocket (Figure 8B). Furthermore, in comparison to the native ABL1 kinase domain, the Y253H/T315I mutant exhibits substantially more mobility in select structural motifs, most notably involving residues of the C-helix and the activation loop of the kinase domain. This additional movement, along with that of residues near the end of the kinase domain, is mitigated upon binding of ponatinib, stabilizing an asciminib-sensitive conformation (Figure 8C). Although BCR-ABL1Y253H/T315I is resistant to ponatinib, ponatinib may be able to transiently occupy the active site and induce a conformational change from active to inactive state. This conformational change greatly reduces the flexibility of the myristoyl-binding pocket, enabling its assembly and the pursuant binding of asciminib. Binding of asciminib in turn is predicted to result in further, sustained stabilization of ponatinib binding and the inactive conformation of the BCR-ABL1 kinase domain, culminating in potent kinase inhibition (Figure 8D and 8E).
Figure 8. Ponatinib-induced shift of BCR-ABL1Y253H/T315I to inactive conformation is required to enable asciminib binding.
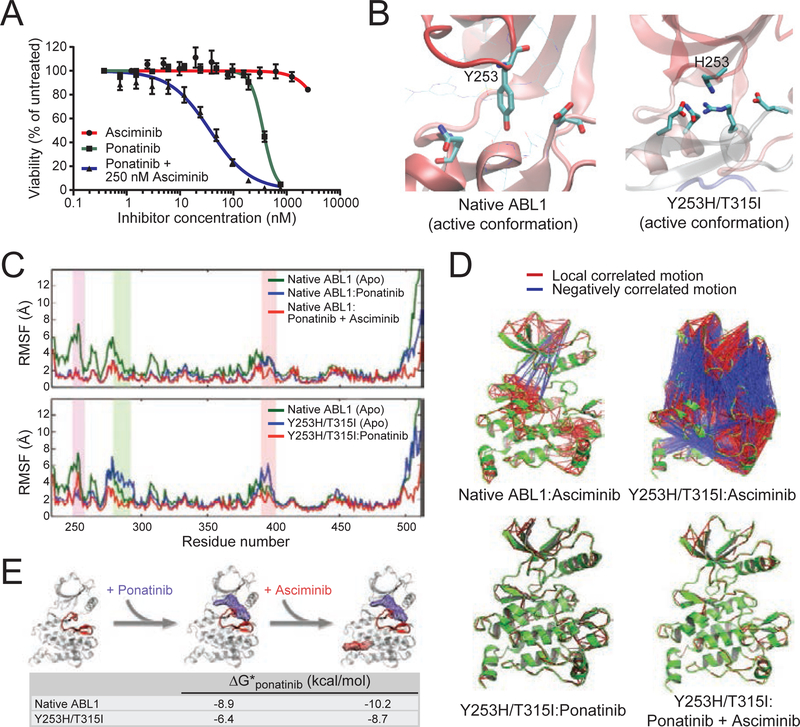
(A) Cell proliferation curves for Ba/F3 MIG BCR-ABL1Y253H/T315I cells treated with asciminib, ponatinib, or the combination of both TKIs. Data points represent the mean ± SEM. (B) Molecular dynamics-based modeling of the kinase domain of native ABL1 and the ABL1Y253H/T315I: mutant in the DFG-in, catalytically active conformation. The relevant sidechains at position 253 are highlighted, as H253 in the context of the mutant engages a stabilizing network of interactions including formation of a salt-bridge/hydrogen-bond. (C) Root mean square fluctuation (RMSF) profiles for the native and Y253H/T315I-mutant ABL1 kinase domains alone (apo) and in complex with ponatinib or with ponatinib and asciminib. Increased RMSF values for a given residue-numbered region indicate higher levels of flexibility during simulation. The P-loop, C-helix, and A-loop are highlighted in purple, green, and pink, respectively. (D) Cross-correlation analysis of forces within ABL1 kinase domain residues before and after TKI binding. Ribbon diagrams for each of the indicated kinase:TKI complexes (all featuring the inactive conformation) are shown in green, with local, positively correlated motion and negatively correlated motion denoted by red and blue lines, respectively, between residues. (E) Proposed model of cooperative binding of ponatinib and asciminib against the ABL1Y253H/T315I mutant. Gibbs free energy (ΔG) values for modeled ponatinib binding before and after asciminib co-binding are provided for the inactive conformation of both native ABL1 and the ABL1Y253H/T315I mutant. Increasingly negative values reflect more favorable and stable ponatinib binding; the A-loop of the ribbon structure is highlighted in red.
DISCUSSION
As all current clinically available BCR-ABL1 TKIs target the ATP-binding site, allosteric inhibitors may offer the opportunity for effectively inhibiting mutants resistant to ATP-site drugs (Gray and Fabbro, 2014; Hantschel et al., 2012). In the ABL1b isoform, autoinhibition is enforced at several levels, one of which involves delivery and binding of the myristoylated glycine residue at position 2 of the N-terminus to a distant hydrophobic pocket within the Clobe of the kinase to exert an allosteric effect that stabilizes the autoinhibited conformation. The extreme N-terminal region of ABLlb kinase is not retained in BCR-ABL1, resulting in loss of this auto-regulatory mechanism but preservation of the allosteric pocket. Efforts to design a small-molecule mimic of myristate to inhibit BCR-ABL1 identified GNF-2 and GNF-5, which lacked sufficient drug-like properties and potency to warrant clinical development (Zhang et al., 2010). Further refinement that incorporated aspects of the GNF scaffold led to asciminib (Wylie et al., 2017), an allosteric inhibitor of BCR-ABL1 that occupies the vestigial myristoyl-binding pocket. We verified that asciminib potently blocks BCR-ABL1-driven signaling and selectively inhibits proliferation and clonogenicity of BCR-ABL1-positive cells, albeit with higher concentrations of inhibitor required in the latter, consistent with experience using these assays in previous studies for currently approved ABL1 TKIs (Corbin et al., 2011; Hamilton et al., 2012; Zhang et al., 2012)..
Allosteric sites on kinases, while often difficult to identify and target, offer structurally unique binding pockets within the kinome compared with the largely conserved ATP-binding sites (Cox et al., 2011). This is exemplified by the narrow target profile (ABL1 and ABL2 only) reported for asciminib (Wylie et al., 2017). While most likely advantageous for CML-selective efficacy and limited toxicity, asciminib’s high selectivity may also limit its efficacy. For instance, inhibition of KIT signaling may contribute to the effects of imatinib (Corbin et al., 2013), suggesting that asciminib as a single agent may be subject to a broader scope of potential resistance mechanisms. Given a body of evidence suggesting that primitive CML CD34+CD38− cells are insensitive to highly potent inhibition of BCR-ABL1 kinase activity and rely on auxiliary pathways for survival (Corbin et al., 2011; Hamilton et al., 2012; Ma et al., 2014; Zhang et al., 2012), asciminib’s extremely narrow target selectivity also suggests that it alone or in combination with ATP-site ABL1 TKIs would be unlikely to eliminate leukemic stem cells. However, the observation of rapid and deep molecular responses to ponatinib in the EPIC trial of newly diagnosed CP CML (Lipton et al., 2016), which was stopped due to ponatinib cardiovascular toxicity, suggests that the near-complete, continuous suppression of BCR-ABL1 kinase activity achievable with a non-toxic ponatinib plus asciminib combination may translate into greater clinical efficacy and warrants additional studies.
Given the allosteric myristoyl-binding pocket being distant from the catalytic site in the kinase domain of BCR-ABL1, a reasonable expectation is that asciminib should be insulated from point mutations that impart resistance to ATP-site TKIs. However, we found that the potency exerted by asciminib for native BCR-ABL1 is not maintained for three different F359 mutants that impart resistance to one or more approved BCR-ABL1 TKIs. A recent update of the phase 1 clinical trial with asciminib (single-agent or combined with imatinib, nilotinib, or dasatinib) in patients with CML or Ph+ ALL with resistance to at least one TKI and no other options (clinicaltrials.gov; NCT02081378) described at least one patient failing single-agent asciminib therapy with evidence of an F359C mutation and multiple myristoyl-binding site mutations (Hughes et al., 2016). Similarly, we observed expansion of F359 variants at the time of failure in a small cohort of patient samples collected serially on asciminib treatment. The phenylalanine at position 359, located near the C-lobe of the kinase domain, provides a favorable binding interaction with both imatinib and nilotinib (Shah et al., 2002; Tse and Verkhivker, 2015), and mutations of this position confer varying degrees of resistance to these inhibitors. While the precise mechanism underlying resistance of F359 mutants to asciminib requires further investigation, our findings suggest the possibility that select BCR-ABL1 kinase domain mutations outside of the myristoyl-binding site may represent vulnerabilities for single-agent asciminib therapy. Furthermore, F359 mutants may remain a liability for combination treatment with asciminib and nilotinib, whereas combinations of asciminib and ponatinib may offer broader efficacy for preventing emergence of resistance.
An important consideration accompanying a drug entering the clinic is not only how it addresses resistance vulnerabilities of existing drugs but also whether other mechanisms of resistance will emerge in patients. Cell-based resistance screens identified multiple asciminib-resistant BCR-ABL1 mutations in or around the myristoyl-binding site. Consistent with the GNF scaffold-influenced development of asciminib, mutations involving residues A337, A344, and P465 were all previously identified as vulnerable residues for GNF-2 (Zhang et al., 2010). The degree to which these mutations will arise in patients on therapy may in part depend on achievable drug plasma concentrations. At the last update of the aforementioned asciminib phase 1 trial, a maximum tolerated dose had not yet been reached. A dose of 40 mg twice daily was recommended for CP CML patients, corresponding to a peak plasma level of ~1.2 μM (Hughes et al., 2016). This dose of asciminib is also being tested versus bosutinib in a phase 3 trial in CP CML patients without BCR-ABLT315I who failed at least two TKIs (clinicaltrials.gov; NCT03106779). Our in vitro asciminib IC50 values for myristoyl-binding pocket mutants suggest they would not be effectively controlled at such levels but should be effectively managed with approved ATP-site TKIs. Furthermore, the enhanced efficacy of combining asciminib with nilotinib or ponatinib we observed in screens of resistant outgrowth suggest an opportunity to leverage the distinct binding modes of these agents to induce upfront deeper and more durable remissions in patients and preempt resistance.
FDA-approved BCR-ABL1 TKIs effectively control point mutation-based resistance, but additional resistance concerns remain. Ponatinib is currently the only approved TKI option for patients with the T315I mutant, and dose-dependent cardiovascular toxicity can complicate its management in some patients (Breccia et al., 2017; Cortes et al., 2012; Hoy, 2014; Moslehi and Deininger, 2015), making the ability to minimize the required dose of ponatinib an appealing aspect of combining with asciminib. Importantly, a recent update from the asciminib phase I trial suggests a dose of 200 mg twice daily is highly active and well tolerated in patients with BCR-ABL1T315I (Rea et al., 2018). Thus, the efficacy of asciminib against this mutant or the potential for low-dose ponatinib combined with asciminib may offer viable clinical strategies for optimal management of these patients.
While the value of combining ATP-site and allosteric TKIs in the context of kinase inhibition has been investigated (Wylie et al., 2017; Zhang et al., 2010), our studies of combination treatment with asciminib and ponatinib demonstrate the capacity of such a combination to tackle previously out-of-reach, clinically relevant resistant compound mutants, as well as suppress their emergence preemptively. Importantly, we show that this specific combination, as compared with asciminib paired with other approved ATP-site ABL1 TKIs, is uniquely capable of addressing T315I-inclusive compound mutations, which are among the most common and clinically challenging compound mutations reported to date (Zabriskie et al., 2014). Previous end-of-treatment analysis of patients participating in the phase 2 PACE trial found BCR-ABL1 compound mutations to be a recurrent mechanism of ponatinib failure (Cortes et al., 2013; Zabriskie et al., 2014), more commonly detected in advanced CML and Ph+ ALL than in CP CML patients (Deininger et al., 2016). While further evaluations of the clinical efficacy of the combination of asciminib and ponatinib in patients with blast crisis will be required, we believe our data support a beneficial application of this approach to at least those patients with compound mutations that have virtually no other available targeted therapy options.
The current phase 1 trial design for asciminib also includes cohorts pairing this drug with imatinib, nilotinib, or dasatinib to assess whether simultaneous targeting of the catalytic and allosteric pockets will provide enhanced clinical therapeutic benefit over either TKI alone. Based on our data, we predict such combinations will be inactive against T315I-inclusive compound mutations. In contrast, the clinically unexplored possibility of combining asciminib with ponatinib could have additional important implications for maximum disease control in the setting of TKI failure, especially due to T315I-inclusive compound mutations. In support of this, the combination of ponatinib and asciminib reduced tumor growth and modestly improved survival in mice injected with a T315I-inclusive compound mutant. The combination was well tolerated at the physiologically achievable doses used. The reported steady state plasma levels of ponatinib in humans are: 35 nM (15 mg once-daily dosing), 84 nM (30 mg once-daily dosing) and 101 nM (45 mg once-daily dosing) (Cortes et al., 2012; Gozgit et al., 2013). This suggests that asciminib can be combined with very low-dose ponatinib to effectively control compound mutants, while simultaneously reducing ponatinib-associated toxicity.
Studies of the asciminib precursors GNF-2 and GNF-5 found evidence of distal movement of residues within the ATP site upon docking to the myristoyl-binding site (Zhang et al., 2010), and asciminib was recently reported to demonstrate additive properties for inhibiting native ABL1 kinase when combined with imatinib, nilotinib, or dasatinib (Wylie et al., 2017). Our findings are conceptually different and important on several levels, however, and suggest that an application of the asciminib/ponatinib combination may lie in targeting highly resistant BCR-ABL1 compound mutants. First, we leverage synergy between different TKIs (asciminib and ponatinib) at the level of the kinase target through a previously unrecognized interaction between allosteric regulatory and ATP binding sites. Second, whereas additivity between two TKIs with partially overlapping resistance profiles has been shown (Eide et al.. 2011; O’Hare et al., 2008; Wylie et al., 2017; Zhang et al., 2010), we demonstrate that the combination of two mechanistically distinct ABL1 TKIs with no individual efficacy can cooperatively inhibit even the most resistant BCR-ABL1 compound mutants. Importantly, we also provide a structural model that explains how this combination uniquely targets clinically challenging T315I-inclusive compound mutants. Previous computational modeling suggests that compound mutation-mediated resistance to ponatinib is due in part to constriction of the ponatinib-binding pocket, which compromises but does not entirely preclude drug binding compared to either constituent mutation in isolation (Zabriskie et al., 2014). Our present modeling work suggests that transient binding of ponatinib to the Y253H/T315I compound mutant facilitates binding of asciminib, which in turn further stabilizes ponatinib binding. Comprehensive crystallographic and biochemical analyses will be required to fully elucidate this process.
In total, our findings highlight the efficacy of the allosteric BCR-ABL1 inhibitor asciminib as a highly active addition to the set of clinically available agents for the treatment of Ph+ leukemia. The most important benefit of asciminib may lie in its ability to potentiate the efficacy of ATP-site TKIs, particularly ponatinib. Asciminib-based drug combinations may offer exciting opportunities for more rapid and deeper remissions, with potential implications for treatment-free remission, prevention of the emergence of BCR-ABL1 compound mutations in advanced Ph+ leukemias, and further improvements in long-term outcomes of patients with CML and Ph+ ALL.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Michael Deininger (Michael.Deininger@hci.utah.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Consents
All patients were consented in accordance with the Declaration of Helsinki and the Belmont Report. All studies with primary human specimens were approved by the University of Utah Institutional Review Board or the Oregon Health & Science University Institutional Review Board.
Animal Studies Protocol Approval
In vivo xenograft study procedures involving Nod-SCID mice injected with Ba/F3 BCR-ABL1 compound mutant cells and treated with TKIs, as detailed below, were conducted in compliance with and following approval by The University of Utah Institutional Animal Care and Use Committee (IACUC).
Cell Lines
Ba/F3 transfectants (expressing full-length, native BCR-ABL1 or BCR-ABL1 with a single or compound kinase domain mutation in the pSRα or pMIG vector) were maintained in RPMI 1640 supplemented with 10% FBS, 1 unit/mL penicillin G, and 1mg/mL streptomycin (complete medium) at 37°C and 5% CO2. Parental Ba/F3 cells were supplemented with 15% vol/vol WEHI-3B-conditioned media as a source of IL-Prior to cell proliferation assays, the BCR-ABL1 kinase domain was amplified from each Ba/F3 cell line, and kinase domain mutations were confirmed by RT-PCR followed Sanger sequencing analysis. In addition to the Ba/F3 lines tested, two human CML (K562 and LAMA) and two non-CML (HL-60 and U937) leukemia cell lines were obtained from ATCC and maintained in complete media at 37°C and 5% CO2. No cell line was cultured longer than 4 weeks continuously prior to use in an experiment.
METHOD DETAILS
Experimental Design
Wherever possible, all experiments were performed at least twice with at least three technical replicates. Results are summarized as the mean of all replicate experimental values, and SEM is indicated in conjunction. Appropriate controls were included in all assays, such as Ba/F3 parental cells, healthy donor, and untreated controls.
Inhibitors
Novartis Pharmaceuticals provided asciminib, and nilotinib was either obtained from Novartis or purchased from Selleck. Imatinib and ponatinib were purchased from Selleck; dasatinib was purchased from either Selleck or LC Laboratories. Inhibitors were prepared as 10.0 mM stock solutions in DMSO and stored at - 20°C. Independent aliquots of stock solutions were thawed prior to use in each experiment.
Cellular Proliferation Assays
Ba/F3 BCR-ABL1-expressing cells were plated in 96-well plates (2×103 cells/well), or 384-well plates (1×103 cells/well) and incubated in 2-fold escalating concentrations of asciminib, imatinib, nilotinib, dasatinib, or ponatinib for 72 h. Proliferation was assessed by methanethiosulfonate (MTS)-based viability assay (CellTiter 96 AQueous One; Promega). IC50 values were calculated using Graphpad Prism software using non-linear regression curve fit analysis and are reported as the mean ± SEM of three independent experiments performed in quadruplicate.
Isolation of Primary Ph+ Leukemia Cells from Blood or Bone Marrow
Mononuclear cells (MNCs) were isolated from primary patient peripheral blood or bone marrow specimens by Ficoll-separation. Where specified, CD34+ cells were enriched by magnetic column separation using a CD34 human microbead kit and the POSSELDS program (AutoMACS; Miltenyi). Purity of the CD34+ fraction was determined to be >90% by fluorescence-activated cell sorting.
Sequencing of the BCR-ABL1 Kinase Domain
RNA obtained from primary Ph+ leukemia cell lysates (QIAGEN RNeasy Mini Kit) served as template for cDNA synthesis (BioRad iScript cDNA Synthesis Kit) as recommended by the manufacturer. Amplification of the BCR-ABL1 kinase domain was done by two-step PCR to exclude amplification of normal ABL1. PCR products were electrophoresed on a 2% agarose gel to confirm amplification, purified (QIAquick PCR Purification Kit; QIAGEN), and subjected to conventional Sanger sequencing in both directions using BigDye terminator chemistry on an ABI3730 instrument. Next-generation sequencing of BCR-ABL1 was performed by ARUP Laboratories (Salt Lake City, UT).
Immunoblot Analysis of BCR-ABL1 and STAT5 Tyrosine Phosphorylation
Ba/F3 cells expressing native or mutant BCR-ABL1 or primary CML patient cells were cultured for 4–6 hr in standard medium alone or with escalating concentrations of TKI(s), followed by lysis and boiling for 10 min in SDS-PAGE loading buffer. Lysates were separated on 4–15% Tris-glycine gels, transferred, and immunoblotted with antibodies for the BCR N-terminus (3902; Cell Signaling), phospho-ABLl (Y393 [1a numbering]; 2865; Cell Signaling), ABL1 (554148; BD Biosciences), phospho-CRKL (Tyr207) (3181S; Cell Signaling), phospho-STAT5 (Tyr694) (9351S; Cell Signaling), STAT5 (610192; BD Biosciences), β-actin (2691430; Millipore) and/or β-tubulin (05–66; Millipore).
Single-Agent Accelerated Mutagenesis Screens
Ba/F3 cells expressing native BCR-ABL1 in the pSRα vector at a density of 7.5×105 cells/mL were treated overnight with N-ethyl-N-nitrosourea (ENU; 50 μg/mL), pelleted, resuspended in fresh complete medium, and distributed into 96-well plates at a density of 1×105 cells/well in 200 μL complete medium supplemented with graded concentrations of asciminib (25, 50, 100, 200, 400, 800, and 1600 nM). Additional similar experiments but with different concentrations were carried out using Ba/F3 MIG BCR-ABL1 cells. ENU-containing waste media was safely inactivated overnight with a solution of sodium thiosulfate (20% w/v) and sodium hydroxide (100 mM) and disposed of according to established protocol (source: Jackson Labs). Wells were observed for cell growth by visual inspection under an inverted microscope and media color change every two days throughout the course of the 28-day experiment. The contents of wells in which cell outgrowth was observed were transferred to a 24-well plate containing 1 mL/well of complete medium supplemented with inhibitor at the same concentration as in the initial 96-well plate. If growth was simultaneously observed in all 96 wells of a given condition, a representative subset of wells was expanded for further analysis. At confluency, cells in 24-well plates were collected by centrifugation and stored at −80°C. For full details of results, see Tables S2 and S3.
Drug Combination Accelerated Mutagenesis Screens
To test the potential efficacy of the combination of asciminib with current clinical ABL1 inhibitors, in conjunction with the single-agent mutagenesis screen described above for asciminib, a small pilot screen of combination of asciminib (100 nM) with imatinib (2000 nM), nilotinib (500 nM), dasatinib (50 nM), or ponatinib (20 nM) was performed. Expanded drug combination screens similar to those described in the single-agent study above were performed using pairwise combinations of asciminib with either nilotinib or ponatinib. Specifically, for experiments starting from Ba/F3 cells expressing native BCR-ABL1, the matrix of combinations of the following concentrations was evaluated: asciminib (10, 25, 50, 100 nM) in combination with nilotinib (50, 100, 200 nM) or ponatinib (2.5, 5, 10 nM). For screens starting from Ba/F3 BCR-ABL1T3151 cells, the following matrix of combinations was evaluated: asciminib (200, 400, 800, 1600 nM) in combination with ponatinib (20, 40, 80, 160 nM). Concentrations of each drug alone (asciminib: 200, 400, 800, 1600, 3200, 6400 nM; ponatinib: 20, 40, 80, 160 nM) were also tested for comparison. Clones recovered were expanded and harvested as described above for single-agent screens. Additional similar combination screens were performed using Ba/F3 MIG BCR-ABL1 or BCR-ABLT3151 cells with different concentrations. For full details, see Tables S4, S6, and S7.
Colony Formation Assays
Primary mononuclear cells were isolated by Ficoll density gradient centrifugation from bone marrow samples from either newly diagnosed CML or healthy donor specimens. Cells were plated in triplicate wells (5×104 cells/well) in 6-well plates in a 9:1 mix of CFU-GM MethoCult medium (H4534; StemCell Technologies, Seattle, WA) and IMDM supplemented with the indicated concentrations of asciminib alone or in combination with nilotinib or ponatinib. Colonies were enumerated after 14 days using a StemVision Colony Counter instrument (StemCell Technologies, Seattle, WA), and counts were normalized to untreated control wells.
Molecular Dynamics Simulations
Mutant conformations of the ABL1 kinase were prepared using standard methods to generate ABL1Y253H/T315I. For each mutant, both the active (PDB entry 2GQG) and inactive (PDB entry 2HYY) conformations of ABL1 kinase were created. The NAMD simulation package was used for molecular dynamics simulation, and the Amber ff12SB force field was employed for standard protein parameters.
Docking Simulations
The Schrödinger suite of programs (Suite 2012: Maestro, version 9.3) was used for docking studies. In the final 50 ns of the simulation, 50 conformations were extracted as docking receptors. Selected conformations were prepared using Protein Preparation Wizard. Ligands (ponatinib and dasatinib) were prepared (Suite 2012: LigPrep, version 2.5) and initial docking simulation was performed using the GlideXP module (version 5.7) of the Schrödinger program. To enhance binding conformations and allow receptor flexibility, docked conformations were subjected to induced fit simulations. Docking scores were computed using the GlideXP module.
p-CRKL Flow Cytometry
Primary CML CD34+ cells were cultured overnight in complete medium in the presence of cytokines. Asciminib, ponatinib, or the combination was added to the cells for 4 h. Cells (1 × 106) were pelleted (1000×g, 5 min, 4°C) and washed once in D-PBS, at which point a sample was removed for viability analysis, resuspended in 1:1,000 LIVE/DEAD Fixable Aqua Stain (Invitrogen, Carlsbad, CA), and stored on ice for 15 min. Cells were pelleted, washed once in 1 mL of D-PBS, resuspended in 250 μL of D-PBS and supplemented with 250 μL of warm Fixation Buffer I (BD Bioscience, San Jose, CA), and placed in a 37°C water bath for 10 min. Cells were pelleted then resuspended in 1% fetal bovine serum and 0.09% sodium azide in D-PBS (wash block buffer, WBB) and pelleted again, removing as much liquid as possible. The pellet was vortexed briefly then 600 μL of Perm Buffer III (BD) was added, two drops at a time with a brief vortex in between sets of drops. The permeabilized cells were stored on ice for 30 min, at which time 500 μL WBB was added (Wash 1), and the cells were washed and pelleted a total of three times in 500 μL WBB, removing as much liquid as possible after the third wash. WBB (50 μL) containing 1:5 human Fc block (eBioscience, Waltham, MA) was added to the cell pellet and cells were vortexed and incubated on ice for 10 min. A prepared solution containing 20 μL anti-pCrkL-PE (Tyr-207; 560788; BD Biosciences) or isotype control (Clone MOPC-173; 558595; BD Biosciences), 10 μL human Fc block, and 20 μL WBB was added to each 50 μL cell suspension. Samples were incubated on ice for 45 min, then transferred to a 96-well plate. Cells were washed by the addition of 100 μL of WBB, washed again in 200 μL WBB, then resuspended in 200 μL WBB and analyzed on a BD Fortessa instrument. Gates were set on the FSC-A high, SSC-A high population then single cells (defined as the majority population from FSC-W × SSC-W and FSC-H × SSC-H), then Aqua negative (live cells), then the median fluorescence intensity (MFI) of PE was determined. Data was collected with FACS Diva and analyzed with FlowJo software (Ashland, OR).
In Vivo Murine Model Studies
Female Nod-SCID mice were purchased from Jackson Labs at 9 weeks of age. At 12 weeks of age, mice were injected by tail vein with 1×106 Ba/F3 cells transduced with pMIG-BCR-ABL1T315I/H396R and pMMP-luciferase-hygromycin (Ridges et al., 2012) (kindly provided by Dr. Michael Engel, The University of Utah). Oral drug delivery was initiated 72 hr later. Mice received 50 μL of drug in solvent SCIE (66.25 mM sucrose, 13.25 mM citric acid, 40% isopropanol, and 6.7% ethanol; see below). Animals were dosed with asciminib (30 mg/kg), ponatinib (25 mg/kg), or the combination once daily. Luciferase was imaged on days 14 and 21 post-injection on a PerkinElmer (Waltham, MA) IVIS instrument. Dosing was held if mice lost >10% of body weight and maintained this loss on two consecutive days or if a mouse lost 4 grams in 24 hours. Mice were re-enrolled if they gained two grams or more. Mice were euthanized upon exhibiting signs of suffering, generally including lack of response to stimuli and/or lethargy.
Preparation of Vehicle and Drug Stocks for In Vivo Studies
SCIE vehicle (stock prepared once per week)
-
1)
Prepare 25 mM citric acid: 1 mL of 1 M citric acid in 39 mL water (stock prepared once at the beginning of study)
-
2)
Prepare sucrose (125 mM) in citric acid: 344 mg sucrose in 8 mL of 25 mM citric acid
-
3)
To 8 mL of sucrose:citric acid solution, add 6 mL isopropanol and 1 mL ethanol (final sucrose concentration = 67 mM in 15 mL)
NOTE: For drug stock preparation, use only barrier 1000 μL Denville tips (drugs stick to other tips).
Asciminib
-
1)
Add 650 μL of SCIE to 17.16 mg asciminib - do NOT mix up and down
-
2)
Vortex, shaker for 5 min
-
3)
Vortex several times, shaker for another 10 min
-
4)
Vortex until almost all chunks/grains disappear, leave on shaker as needed
-
5)
Transfer 315 μL to a new tube for combination solution - set aside
-
6)
Transfer 315 μL to a new tube, add 315 μL SCIE - this is single agent asciminib
Ponatinib
-
1)
Add 650 μL of SCIE to 14.3 mg ponatinib powder - do NOT mix up and down
-
2)
Vortex, shaker for 5 min
-
3)
Vortex several times, shaker for another 5 min
-
4)
Vortex until almost all chunks/grains disappear
-
5)
Transfer 315 μL to a new tube, add 315 μL SCIE - this is single agent ponatinib
Asciminib + Ponatinib Combination
-
1)
Add 315 μL ponatinib and 315 μL asciminib to the tube from step 4, vortex
-
2)
Leave on shaker as needed to fully mix
QUANTIFICATION AND STATISTICAL ANALYSIS
Cellular IC50 values were determined by non-linear regression curve-fit analysis across a broad concentrations range using Prism 6 statistical software (GraphPad). Median survival was compared between treatment arms for in vivo mouse studies using Kaplan-Meier curves and the Mantel-Cox log-rank method in Prism software. Molecular dynamics-based computational modeling was performed using the Schrödinger software suite.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BCR N-terminus | Cell Signaling | #3902 |
| Phospho-ABLI (Y393 [1a numbering]) | Cell Signaling | #2865 |
| ABL1 | BD Biosciences | #554148 |
| Phospho-CRKL (Tyr 207) | Cell Signaling | #3181S |
| Phospho-STAT5 (Tyr 694) | Cell Signaling | #9351S |
| STAT5 | BD Biosciences | #610192 |
| β-Actin | Millipore | #2691430 |
| β-Tubulin | Millipore | #05–66 |
| Anti-pCRKL-PE (Tyr 207) | BD Biosciences | #560788 |
| Isotype, Clone MOPC-173 | BD Biosciences | #558595 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Methanethiosulfonate (MTS), CellTiter 96 Aqueous One Solution Reagent | Promega | #G358X |
| ENU, N-Ethyl-N-nitrosourea | Sigma-Aldrich | #N3385 |
| Imatinib (STI571) | Selleck Chemicals | #S2475 |
| Nilotinib | Selleck Chemicals | #S1033 |
| Dasatinib | LC Laboratories | #D-3307 |
| Ponatinib (AP24534) | Selleck Chemicals | #S1490 |
| Asciminib (ABL001) | Novartis | N/A |
| CFU-GM MethoCult medium | StemCell Technologies | #H4534 |
| Critical Commercial Assays | ||
| CD34 MicroBead Kit, human | Miltenyi | #130-046-703 |
| Experimental Models: Cell Lines | ||
| Ba/F3 parental | ATCC | |
| Ba/F3 MIG BCR-ABL1 | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 G250E | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 Y253H | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 E255V | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 F311I | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 T315I | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 F317L | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 F359C | This paper | N/A |
| Ba/F3 MIG BCR-ABL1 F359I | This paper | N/A |
| Ba/F3 MIG BCR-ABL1 F359V | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 H396R | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 G250E/T315I | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 Y253H/T315I | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 E255V/T315I | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 T315I/M351T | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 T315I/H396R | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 T315I/E453K | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 T315L | This paper | N/A |
| Ba/F3 MIG BCR-ABL1 T315M | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 E255V/V299L | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 V299L/F317L | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 F317L/F359V | Huntsman Cancer Institute | N/A |
| Ba/F3 MIG BCR-ABL1 A337V | This paper | N/A |
| Ba/F3 MIG BCR-ABL1 P465S | This paper | N/A |
| Ba/F3 MIG BCR-ABL1 V468F | This paper | N/A |
| Ba/F3 pSRα BCR-ABL1 | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 G250E | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 Q252H | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 Y253F | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 Y253H | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 E255K | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 E255V | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 T315A | Gift from N. Shah, UCSF | N/A |
| Ba/F3 pSRα BCR-ABL1 T315I | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 F317L | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 F317V | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 M351T | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 F359V | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 G250E/T315I | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 E255K/T315I | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 E255V/T315I | OHSU Knight Cancer Institute | N/A |
| Ba/F3 pSRα BCR-ABL1 A344P | This paper | N/A |
| Ba/F3 pSRα BCR-ABL1 P465S | This paper | N/A |
| Experimental Models: Animal Studies | ||
| NOD.CB17-Prkdcscid/J (Nod-SCID) mice, female, 9 weeks old | The Jackson Laboratory | #001303 |
| Oligonucleotides | ||
| ABL1_4315F forward primer w/M13F tag: gtaaaacgacggccagtTCCAGTATCTCAGACGAAG | This paper | N/A |
| ABL1_e11R reverse primer w/M13R tag:caggaaacagctatgaccGGTGCTGACTCACTAGTCCC | This paper | N/A |
| ABL1_3984R reverse primer:TGTCATCAACCTGCTCAGGC | This paper | N/A |
| BCR-F4 forward primer:ACAGCATTCCGCTGACCATCAATA | OHSU Knight Cancer Institute | N/A |
| BCR-ABL_B2A forward primer:TTCAGAAGCTTCTCCCTGACAT | OHSU Knight Cancer Institute | N/A |
| ABL1_4317R reverse primer: AGCTCTCCTGGAGGTCCTC | OHSU Knight Cancer Institute | N/A |
| ABL1_3335F forward primer: ACCACGCTCCATT | OHSU Knight Cancer Institute | N/A |
| ABL1_4275R reverse primer: CCT GCAGCAAGGTAGTCA | OHSU Knight Cancer Institute | N/A |
| Software and Algorithms | ||
| GraphPad Prism software Non-linear regression curve fit analysis for calculating IC50 values and survival curve analysis for in vivo animal studies | Prism ver. 6.0, 1994–2014 | www.graphpad.com |
| Schrodinger Suite software package Molecular dynamics modeling simulations | Schrödinger Suite 2012: Maestro ver. 9.3 LigPrep ver 2.5 GlideXP ver. 5.7 |
www.schrodinger.com |
SIGNIFICANCE.
Asciminib effectively inhibits native BCR-ABL1 by binding to the allosteric myristoyl-binding pocket. However, most BCR-ABL1 compound mutants are insensitive to asciminib as well as all approved ATP-site TKIs at clinically relevant concentrations. Dose escalation with a potentially effective TKI such as ponatinib is not an option to address such mutants due to dose-limiting toxicities. Asciminib engages BCR-ABL1 in an allosteric pocket distantly located from the ATP-binding site, providing the opportunity to use asciminib in conjunction with an ATP-site TKI. Here we demonstrate that addition of asciminib greatly reduces the concentration of ponatinib needed to inhibit compound mutants and control refractory Ph+ leukemias.
Acknowledgments
We thank Novartis Pharmaceuticals for providing asciminib and nilotinib under Material Transfer Agreements. We thank all members of the Deininger, O’Hare, and Druker laboratories for technical assistance and/or valuable discussions. We acknowledge support in conjunction with grant P30 CA042014 awarded to the Huntsman Cancer Institute (T.O.; M.W.D.). M.C.H. is supported by a VA Merit Review Grant (2I01BX000338-05). D.Y. is supported by a Special Fellow Award from the Leukemia & Lymphoma Society. T.O. is supported by the NIH/NCI (R01 CA178397). M.W.D. is supported by the NIH (HL082978-01, 5 P01 CA049639-23 and R01 CA178397). B.J.D. is an investigator for the Howard Hughes Medical Institute and is supported by the NIH/NCI (R01 CA065823-21).
Footnotes
Disclosure of Potential Conflicts of Interest
M.W.D. served on advisory boards and as a consultant for Bristol-Myers Squibb, ARIAD, and Novartis and receives research funding from Bristol-Myers Squibb, Celgene, Novartis and Gilead. B.J.D. potential competing interests -- SAB: Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene & Baxalta (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, Beta Cat, GRAIL, Third Coast Therapeutics, CTI BioPharma (inactive); Scientific Founder & Stock: MolecularMD; Board of Directors & Stock: Amgen; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (one Merck exclusive license).
REFERENCES
- Bradeen HA, Eide CA, O’Hare T, Johnson KJ, Willis SG, Lee FY, Druker BJ, and Deininger MW (2006). Comparison of imatinib, dasatinib (BMS-354825), and nilotinib (AMN107) in an n-ethyl-n-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood 108, 2332–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breccia M, Pregno P, Spallarossa P, Arboscello E, Ciceri F, Giorgi M, Grossi A, Mallardo M, Nodari S, Ottolini S, et al. (2017). Identification, prevention and management of cardiovascular risk in chronic myeloid leukaemia patients candidate to ponatinib: an expert opinion. Annals of hematology 96, 549–558. [DOI] [PubMed] [Google Scholar]
- Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, and Druker BJ (2011). Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. The Journal of clinical investigation 121, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin AS, O’Hare T, Gu Z, Kraft IL, Eiring AM, Khorashad JS, Pomicter AD, Zhang TY, Eide CA, Manley PW, et al. (2013). KIT signaling governs differential sensitivity of mature and primitive CML progenitors to tyrosine kinase inhibitors. Cancer research 73, 5775–5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, O’Hare T, Hu S, Narasimhan NI, Rivera VM, et al. (2012). Ponatinib in refractory Philadelphia chromosome-positive leukemias. The New England journal of medicine 367, 2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes JE, Kantarjian HM, Brummendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, et al. (2011). Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 118, 4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, et al. (2013). A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. The New England journal of medicine 369, 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KJ, Shomin CD, and Ghosh I. (2011). Tinkering outside the kinase ATP box: allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future medicinal chemistry 3, 29–43. [DOI] [PubMed] [Google Scholar]
- Deininger MW, Hodgson JG, Shah NP, Cortes JE, Kim DW, Nicolini FE, Talpaz M, Baccarani M, Muller MC, Li J, et al. (2016). Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood 127, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide CA, Adrian LT, Tyner JW, Mac Partlin M, Anderson DJ, Wise SC, Smith BD, Petillo PA, Flynn DL, Deininger MW, et al. (2011). The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer research 71, 3189–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide CA, and O’Hare T. (2015). Chronic myeloid leukemia: advances in understanding disease biology and mechanisms of resistance to tyrosine kinase inhibitors. Current hematologic malignancy reports 10, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, and Sawyers CL (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Gozgit JM, Schrock A, Chen TH, Clackson T, and Rivera VM (2013). Comprehensive Analysis Of The In Vitro Potency Of Ponatinib, and All Other Approved BCR-ABL Tyrosine Kinase Inhibitors (TKIs), Against a Panel Of Single and Compound BCR-ABL Mutants. Blood 122, 3992. [Google Scholar]
- Gray NS, and Fabbro D. (2014). Discovery of allosteric BCR-ABL inhibitors from phenotypic screen to clinical candidate. Methods in enzymology 548, 173–188. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Muller-Tidow C, Bhatia R, Brunton VG, et al. (2012). Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 119, 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O, Grebien F, and Superti-Furga G. (2012). The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer research 72, 4890–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S, et al. (2017). Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. The New England journal of medicine 376, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy SM (2014). Ponatinib: a review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs 74, 793–806. [DOI] [PubMed] [Google Scholar]
- Hughes TP, Goh YT, Ottmann OG, Minami H, Rea D, Lang F, Mauro MJ, DeAngelo DJ, Talpaz M, Hochhaus A, et al. (2016). Expanded Phase 1 Study of ABL001, a Potent, Allosteric Inhibitor of BCR-ABL, Reveals Significant and Durable Responses in Patients with CML-Chronic Phase with Failure of Prior TKI Therapy. Blood 128, 625.27297793 [Google Scholar]
- Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, et al. (2006). Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. The New England journal of medicine 354, 2542–2551. [DOI] [PubMed] [Google Scholar]
- Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, Ozawa K, Shentu W, Brown E, Shirai T, et al. (2019). Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 133, 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, Etienne G, Nicolini FE, le Coutre P, Clark RE, et al. (2016). Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol 17, 612–621. [DOI] [PubMed] [Google Scholar]
- Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, Zhu LJ, Hutchinson L, Cerny J, Khoury HJ, et al. (2014). A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med 6, 252ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslehi JJ, and Deininger M. (2015). Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol 33, 4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, and Kuriyan J. (2003). Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871. [DOI] [PubMed] [Google Scholar]
- O’Hare T, Eide CA, Tyner JW, Corbin AS, Wong MJ, Buchanan S, Holme K, Jessen KA, Tang C, Lewis HA, et al. (2008). SGX393 inhibits the CML mutant Bcr-AblT315I and preempts in vitro resistance when combined with nilotinib or dasatinib. Proceedings of the National Academy of Sciences of the United States of America 105, 5507–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al. (2009). AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer cell 16, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T, Zabriskie MS, Eiring AM, and Deininger MW (2012). Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nature reviews Cancer 12, 513–526. [DOI] [PubMed] [Google Scholar]
- Qiang W, Antelope O, Zabriskie MS, Pomicter AD, Vellore NA, Szankasi P, Rea D, Cayuela JM, Kelley TW, Deininger MW, and O’Hare T. (2017). Mechanisms of resistance to the BCR-ABL1 allosteric inhibitor asciminib. Leukemia 31, 2844–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea D, Lang F, Kim DW, Cortes JE, Hughes TP, Minami H, Breccia M, DeAngelo DJ, Hochhaus A, Talpaz M, et al. (2018). Asciminib, a Specific Allosteric BCR-ABL1 Inhibitor, in Patients with Chronic Myeloid Leukemia Carrying the T315I Mutation in a Phase 1 Trial. Blood 132, 792. [Google Scholar]
- Ridges S, Heaton WL, Joshi D, Choi H, Eiring A, Batchelor L, Choudhry P, Manos EJ, Sofla H, Sanati A, et al. (2012). Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood 119, 5621–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, and Sawyers CL (2002). Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer cell 2, 117–125. [DOI] [PubMed] [Google Scholar]
- Shah NP, and Sawyers CL (2003). Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene 22, 7389–7395. [DOI] [PubMed] [Google Scholar]
- Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, and Sawyers CL (2007). Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. The Journal of clinical investigation 117, 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, et al. (2006). Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. The New England journal of medicine 354, 2531–2541. [DOI] [PubMed] [Google Scholar]
- Tse A, and Verkhivker GM (2015). Molecular Determinants Underlying Binding Specificities of the ABL Kinase Inhibitors: Combining Alanine Scanning of Binding Hot Spots with Network Analysis of Residue Interactions and Coevolution. PloS one 10, e0130203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, et al. (2017). The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. [DOI] [PubMed] [Google Scholar]
- Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva M, Cortes JE, et al. (2014). BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer cell 26, 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, and Bhatia R. (2012). Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer cell 21, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, et al. (2010). Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.