

Abstract
STUDY QUESTION
Does the X chromosome inactivation (XCI) of Klinefelter syndrome (KS)-derived human induced pluripotent stem cells (hiPSCs) correspond to female human pluripotent stem cells (hPSCs) and reflect the KS genotype?
SUMMARY ANSWER
Our results demonstrate for the first time that KS-derived hiPSCs show similar XCI behavior to female hPSCs in culture and show biological relevance to KS genotype-related clinical features.
WHAT IS KNOWN ALREADY
So far, assessment of XCI of KS-derived hiPSCs was based on H3K27me3 staining and X-inactive specific transcript gene expression disregarding the at least three XCI states (XaXi with XIST coating, XaXi lacking XIST coating, and XaXe (partially eroded XCI)) that female hPSCs display in culture.
STUDY DESIGN, SIZE, DURATION
The study used hiPSC lines generated from two azoospermic patients with KS and included two healthy male (HM) and one healthy female donor.
PARTICIPANTS/MATERIALS, SETTING, METHODS
In this study, we derived hiPSCs by reprograming fibroblasts with episomal plasmids and applying laminin 521 as culture substrate. hiPSCs were characterized by karyotyping, immunocytochemistry, immunohistochemistry, quantitative PCR, teratoma formation, and embryoid body differentiation. XCI and KS hiPSC relevance were assessed by whole genome transcriptomics analysis and immunocytochemistry plus FISH of KS, HM and female fibroblast, and their hiPSC derivatives.
MAIN RESULTS AND THE ROLE OF CHANCE
Applying whole genome transcriptomics analysis, we could identify differentially expressed genes (DEGs) between KS and HM donors with enrichment in gene ontology terms associated with fertility, cardiovascular development, ossification, and brain development, all associated with KS genotype-related clinical features. Furthermore, XCI analysis based on transcriptomics data, RNA FISH, and H3K27me3 staining revealed variable XCI states of KS hiPSCs similar to female hiPSCs, showing either normal (XaXi) or eroded (XaXe) XCI. KS hiPSCs with normal XCI showed nevertheless upregulated X-linked genes involved in nervous system development as well as synaptic transmission, supporting the potential use of KS-derived hiPSCs as an in vitro model for KS.
LIMITATIONS, REASONS FOR CAUTION
Detailed clinical information for patients included in this study was not available. Although a correlation between DEGs and the KS genotype could be observed, the biological relevance of these cells has to be confirmed with further experiments. In addition, karyotype analysis for two hiPSC lines was performed at passage 12 but not repeated at a later passage. Nevertheless, since all XCI experiments for those lines were performed between passage 11 and 15 the authors expect no karyotypic changes for those experiments.
WIDER IMPLICATIONS OF THE FINDINGS
As KS patients have variable clinical phenotypes that are influenced by the grade of aneuploidy, mosaicism, origin of the X chromosome, and XCI ‘escapee’ genes, which vary not only among individuals but also among different tissues within the same individual, differentiated KS hiPSCs could be used for a better understanding of KS pathogenesis.
STUDY FUNDING/COMPETING INTEREST(S)
This study was supported by grants from the Knut and Alice Wallenberg Foundation (2016.0121 and 2015.0096), Ming Wai Lau Centre for Reparative Medicine (2-343/2016), Ragnar Söderberg Foundation (M67/13), Swedish Research Council (2013-32485-100360-69), the Centre for Innovative Medicine (2–388/2016–40), Kronprinsessan Lovisas Förening För Barnasjukvård/Stiftelsen Axel Tielmans Minnesfond, Samariten Foundation, Jonasson Center at the Royal Institute of Technology, Sweden, and Initial Training Network Marie Curie Program ‘Growsperm’ (EU-FP7-PEOPLE-2013-ITN 603568). The authors declare no conflicts of interest.
Keywords: integration-free, xeno-free, laminin 521, human induced pluripotent stem cells, Klinefelter syndrome, XXY condition, X chromosome inactivation
Introduction
Klinefelter syndrome (KS), with a 1:500 to 1:1000 incidence, is the most frequent sex chromosome abnormality in males. It is an X chromosome aneuploidy characterized in 80–90% of the cases by a nonmosaic 47, XXY karyotype, while higher-grade aneuploidies (such as 48, XXXY) and mosaic forms of KS (such as 46, XY/47, XXY) account for the remaining 10–20%. The supernumerary X chromosome can be of either maternal or paternal origin, caused by failures in chromosome disjunction in meiosis I (maternal or paternal), meiosis II (maternal), or, in rare cases, during the first mitotic cell divisions of zygotes (maternal) (Lanfranco et al., 2004; Bonomi et al., 2017).
The clinical picture of KS is commonly described as a tall stature, gynecomastia, sparse body hair, small and firm testes, and hypogonadism resulting in azoospermia with hyalinization and fibrosis of the seminiferous tubules (Bonomi et al., 2017). Although the majority of patients with KS have germ cells in their testes before puberty, azoospermia is predominant in adulthood, making infertility the most common cause for KS diagnosis (Lanfranco et al., 2004; Wikstrom and Dunkel, 2011; Rohayem et al., 2016; Bonomi et al., 2017; Kanakis and Nieschlag, 2018). However, a broad phenotypic spectrum has been described for KS, including higher risk for metabolic disorders, osteoporosis, breast cancer, and autoimmune diseases, as well as verbal processing and attention deficits (Bonomi et al., 2017; Kanakis and Nieschlag, 2018). The variable clinical phenotype of KS is at least partly influenced by the grade of aneuploidy, mosaicism, and origin of the X chromosome, whereas the relatively mild phenotype is thought to result from X chromosome inactivation (XCI), which is a mechanism by which one of the two X chromosomes normally in female somatic cells is transcriptionally silenced to equalize the dosage of X-linked genes between males and females, resulting in one active (Xa) and one inactive (Xi) X chromosome.
XCI in KS patients has been shown by the presence of the Barr body in buccal smears, which has also been used as a screening test for KS diagnosis (Kamischke et al., 2003). As further evidence of XCI, KS patients have upregulated expression of X-inactive specific transcript (XIST), a long noncoding RNA coating the Xi in cis, inducing nuclear reorganization and recruitment of histone-modification complexes, thereby helping the initiation and maintenance of XCI (Vallot et al., 2013; Reik and Surani, 2015; Belling et al., 2017). In addition, several studies have utilized the variable (CAG) n repeat of the X-linked androgen receptor gene and methylation-sensitive HpaII restriction site to show inactivation of one of the alleles in KS patients. This assay has also been used as evidence of skewed XCI (preferential XCI in one X chromosome over the other) in some of the KS cases (Tuttelmann and Gromoll, 2010). While random XCI leads to a normal genetic mosaic of cells, with half of the cells expressing either one of the X chromosomes, skewed patterns of XCI lead to phenotypic variability in a number of human diseases linked to the X chromosome.
XCI in female cells is incomplete, and at least 23% of X-linked genes ‘escape’ XCI and are expressed also from the Xi, although at lower levels, resulting in sex biases in gene expression (Tukiainen et al., 2017). On the other hand, pseudoautosomal regions of the X chromosome consistently escape XCI to achieve balanced expression dosage between females and males. Thus, the KS phenotype may be influenced by two active copies or, in the case of X-Y homologous genes, three active copies of XCI escaping genes. In addition, some of the escaping genes show variability between individuals and tissues (Tukiainen et al., 2017), which is likely to introduce phenotypic diversity in KS patients and the severity of impact for certain tissues over others. Most of the molecular studies involving KS patients have used samples from whole blood (Vawter et al., 2007; Huang et al., 2015; Zitzmann et al., 2015; Belling et al., 2017), while only a few studies have focused on brain (Viana et al., 2014) or testes (D’Aurora et al., 2015, 2017; Winge et al., 2018). For better understanding the pathogenesis of KS, multiple cell types should be analyzed, which are often inaccessible.
Human induced pluripotent stem cells (hiPSCs) can be differentiated into any cell type of the body and thus provide a useful model system to study condition-based effects in early development. In addition, human pluripotent stem cells (hPSCs) have clonal XCI with the same parental X-chromosome inactivated in all cells, which allows a more detailed analysis of XCI from transcriptomics data (Tchieu et al., 2010; Sahakyan et al., 2017). One study with a KS patient-derived hiPSC line suggested XCI based on H3K27me3 staining (histone tri-methylation of lysine 27 on histone H3) and XIST gene expression and, regardless, showed aberrant gene expression of X-linked genes (Ma et al., 2012). However, female hPSCs have at least three XCI states: XaXi with XIST coating, XaXi lacking XIST coating, and XaXe (partially eroded XCI) (Patel et al., 2017). The state varies not only between cell lines but also between cell passages and even within a cell population; thus, it is important to match the samples when studying the connection of XCI and gene expression.
The present study focuses on the characterization of KS hiPSCs from two azoospermic donors in regards to their XCI state as well as their potential applicability as an in vitro KS disease model.
Materials and Methods
Fibroblast derivation and culture
Skin punch (4 mm punch) biopsies from the upper lateral quadrant of the gluteal region of healthy male (HM) donors (HM1, 31 years; HM2, 34 years) and azoospermia patients diagnosed with KS (KS1, 31 years; KS2, 34 years) at the Division of Reproductive Medicine at the Karolinska Hospital Huddinge, were obtained with a written informed consent and with the approval of the Stockholm Regional Ethics Board (Dnr: 2013/1132-32). After removing the dermis, tissue was cut into c.1 mm3 pieces and plated on fetal bovine serum (FBS; Life Technologies, USA) coated plates with human dermal fibroblast (HDF) medium consisting of Dulbecco’s Modified Eagle Medium (Life Technologies), 10% FBS, and 1× penicillin–streptomycin (Life Technologies). The resulting fibroblast cultures were passaged with 0.05% Trypsin (Life Technologies). Additional fibroblasts used were human female dermal fibroblasts (f-HF1; 31 years; Lonza, Switzerland).
Reprogramming and culture of hiPSCs and human embryonic stem cells
Reprogramming of fibroblasts with episomal plasmids was performed as previously reported (Okita et al., 2011) with some modifications. Briefly, 6 × 105 cells were electroporated with 1 μg of each plasmid; pCXLE-hOCT3/4-shp53, pCXLE-hSK, and pCXLE-hUL (gift from Dr Shinya Yamanaka), using the 100 μl tip Neon System (Life Technologies) with 1650 V, 30 ms, and 1 pulse settings. Transfected cells were plated onto noncoated cell culture plates with HDF medium for 6 days in 5% CO2, at 37°C. Cells were collected using 0.25% Trypsin (Life Technologies) and plated onto 10 μg/ml laminin 521 (LN521; Biolamina, Sweden) coated plates or onto human embryonic stem cell (hESC)-qualified Matrigel (BD Biosciences, USA) coated plates at a density of 5 × 103 cells/cm2 and transferred to low oxygen conditions (5% O2 and 5% CO2) at 37°C. The following day, HDF medium was replaced by Nutristem hPSC XF Medium (Biological Industries, USA) with medium changes every 2–3 days. Cell colonies were manually picked onto LN521 coated plates 23–40 days after transfection and maintained in Nutristem hPSC XF Medium throughout culture. Upon confluency, cells were passaged as single cells using 1× TrypLE Select (Life Technologies). The hESC line HS980, 46,XX (Rodin et al., 2014) was used in this study with the approval of the Stockholm Regional Ethics Board (Dnr: 454/02).
Spontaneous in vitro differentiation
Confluent cell cultures were detached as small cell clumps and plated onto ultra-low adhesion plates (Corning, USA) with Nutristem hPSC XF Medium, GF-free (Biological Industries) containing 10 μM ROCK inhibitor Y-27632 (Millipore, USA) for the first 24 h with subsequent media changes every 2–3 days. After 2 weeks in ultra-low adhesion plates, the embryoid bodies were plated for an additional 2 weeks onto LN521 coated glass chamber slides (Corning). After a total culture of 4 weeks, the cells were fixed with 4% formaldehyde (Sigma-Aldrich, USA).
Teratoma assay
Confluent cell cultures were detached as small cell clumps and plated onto ultra-low adhesion plates with Nutristem hPSC XF Medium containing 10 μM Y-27632 (Millipore) for 24 h. Sphere suspension was then mixed with hESC-qualified Matrigel and injected s.c. into severely compromised immunodeficient/Beige mice (Taconic, USA) with ~1 × 106 cells/injection. Mice were sacrificed and tumors were collected at 3–8 weeks after injection. Tumors were fixed in 4% formaldehyde and paraffin embedded cross-sections stained for hematoxylin and eosin. Animal work was performed with the approval of Stockholm south ethical committee S14–15.
Quantitative PCR
For plasmid copy number analysis, genomic DNA (gDNA) was collected using the DNeasy Blood & Tissue kit (Qiagen, Netherlands) according to the manufacturer’s protocol. Quantitative PCR (qPCR) analysis was performed with the StepOnePlus Real-Time PCR System (Life Technologies) using SYBR Select Master Mix (Life Technologies). The pCXLE-EGFP plasmid and gDNA from hESCs (H9) were used to generate standard curves to determine the plasmid or gene copy numbers from the threshold cycle values.
Epstein–Barr nuclear antigen 1 (EBNA-1) primers (forward: ATC AGG GCC AAG ACA TAG AGA TG; reverse: GCC AAT GCA ACT TGG ACG TT) were used to detect plasmid copies and F-box protein 15 (FBXO15) primers (forward: GCC AGG AGG TCT TCG CTG TA; reverse: AAT GCA CGG CTA GGG TCA AA) were used as genomic copy number controls. The cell number in each reaction was determined by dividing the estimated FBXO15 copy number by two (two alleles in one genome). Approximately 1 × 104 cells were used in one PCR reaction, and results were determined based on at least three reactions.
For gene expression analysis, RNA from three consecutive passages was extracted using the RNeasy Mini Kit (Qiagen) with on-column DNaseI treatment (Qiagen) according to the manufacturer’s protocol. qPCR analysis was performed with StepOnePlus Real-Time PCR System (Life Technologies) using Taqman Universal PCR Master Mix (Life Technologies) with Taqman assays (all from Life Technologies) for glycerinaldehyd-3-phosphate-dehydrogenase (GAPDH, 4333764F), Nanog homeobox (NANOG, Hs02387400_g1), POU class 5 homeobox 1 (POU5F1, Hs03005111_g1), SRY-Box 2 (SOX2, Hs01053049_s1), growth differentiation factor 3 (GDF3, Hs00220998_m1), and XIST (Hs01079824_m1). Relative quantity was determined with 2-ΔΔCt, using GAPDH as an endogenous control and hESCs (HS980) or f-HF1 as control sample.
Alkaline phosphatase staining and reprogramming efficiency calculation
For hiPSC colony counting in reprogramming conditions, cells were fixed with 4% formaldehyde for 2 min and washed with 0.05% Tween-20 in Tris-buffered saline (TBS-T; Sigma-Aldrich). Cells were then stained for 15 min using an Alkaline Phosphatase Detection Kit (Millipore) and washed with TBS-T. Positive colonies were counted and imaged with an Olympus IX81. Reprogramming efficiency was calculated by the following formula: number of alkaline phosphatase (AP) positive colonies obtained multiplied by Day 6 plating ratio (total cell count at Day 6/number of cells plated at Day 6) divided by number of cells transfected at Day 0. Statistical significance was tested with unpaired Student’s t-test using SigmaPlot 13.0 (Systat Software, Inc., USA), significance was accepted at P < 0.05, n = 3.
Immunofluorescence staining
For hiPSC characterization, cells were fixed with 4% formaldehyde for 15 min and permeabilized for 10 min with 0.03% Triton X-100 (Sigma-Aldrich) in PBS (Life Technologies). Cells were blocked in PBS containing 10% donkey serum (Jackson ImmunoResearch) and 1% bovine serum albumin (BSA; Sigma-Aldrich) for 1 h and incubated with primary antibodies in blocking buffer at 4°C overnight. After washing, cells were incubated with secondary antibodies in blocking buffer for 1 h and counterstained with 1 μg/ml DAPI (Life Technologies). hiPSCs were imaged with a confocal microscope (LSM700; Zeiss, Germany). Primary antibodies and concentrations used were rabbit a-POU5F1 (0.005 mg/ml; ab19857; Abcam, UK), rabbit a-SOX2 (0.002 mg/ml; ab97959; Abcam), rabbit a-NANOG (0.004 mg/ml; ab21624; Abcam), mouse anti-stage-specific embryonic antigen-4 (a-SSEA4; 0.05 mg/ml; ab16287; Abcam), mouse anti-neuron-specific class III beta-tubulin (a-TUJ1; 0.002 mg/ml; 801201; BioLegend, USA), mouse anti-alpha-smooth muscle actin (a-SMA; ascites 1:500; a2547; Sigma-Aldrich), mouse anti-alpha-fetoprotein (a-AFP; 0.005 mg/ml; ab3980; Abcam), and rabbit a-H3K27me3 (0.0003 mg/ml; ABE44; Millipore). For negative controls mouse IgG (0.4 mg/ml; sc-2025; SantaCruz, USA) and rabbit IgG (1.775 mg/ml; ab172730; Abcam) were used. Secondary antibodies and concentrations used were Alexa Fluor 488 Donkey a-mouse IgG (0.003 mg/ml; 715–546-150; Jackson ImmunoResearch) and Cy3 Donkey a-rabbit IgG (0.0028 mg/ml; 711–166-152; Jackson ImmunoResearch).
Karyotyping
Cells were treated with 10 μg/ml colcemid (Life Technologies) for 5 h, collected with TrypLE Select and centrifuged. The cell pellet was resuspended in 0.4% potassium chloride solution and incubated for 40 min before fixation with methanol:acetic acid (3:1). The chromosomal G-band analysis was performed at the Genetics Clinic at Skåne University Hospital, Sweden, or at Karolinska University Hospital, Sweden, and at least 20 metaphases were counted.
RNA sequencing sample preparation and data analysis
RNA was extracted using the RNeasy Mini Kit (Qiagen) with on-column DNase I treatment (Qiagen) according to the manufacturer’s protocol from three different passages of f-HM1 (p8–10), f-HM2 (p7, p10–11), f-KS1 (p5, p8–9), f-KS2 (p5, p7–8), f-HF1 (p12–14), ips-HM1 (p13–15), ips-HM2 (p11–13), ips-KS1 (p13–15), ips-KS2 (p11–13), ips-HF1 (p15–17), and HS980 (p23–25). Library preparation and sequencing was performed at the Science for Life Laboratory, Sweden. Briefly, libraries were made using Illumina TruSeq® stranded mRNA with poly-A selection. Total two pools were made from the 33 samples, and each sample pool was sequenced on two lanes using Illumina HiSeq 2500 with 2 × 125 setup.
Initial quality control of generated paired-end reads was performed using the FastQC software. The reference index-based algorithm HISAT 2.1.0 (Kim et al., 2015) was used to align reads with the human reference genome (GRCh37) (Supplementary Table SI). The resulting BAM files were examined by MultiQC (version 1.0) (Ewels et al., 2016) and then used to estimate transcript abundance (FPKM; fragments per kilobase of exon per million fragments mapped) values using StringTie v1.3.3 (Pertea et al., 2015). Pearson product-moment correlation coefficient was calculated to examine closeness among replicates and one replicate (f-HM1 p8) was excluded due to low correlation. Quantified expression values (FPKM) were imported into R (version 3.4.3; R Project), and principal component analysis (PCA) was conducted with the top 10 000 highly expressed genes. Statistical significance for the sum of total X chromosome and autosome expression was tested with one-way ANOVA using SigmaPlot 13.0; significance was accepted at P < 0.05, n = 3.
Cuffdiff v2.2.1 (Trapnell et al., 2013) was used to identify differentially expressed genes (DEGs). The transcripts with a P-value <0.05, fold change ≥1.5, and average FPKM value ≥1 in higher expressing samples were considered to be significantly expressed. The Panther (Mi et al., 2013, 2017) online suite of tools was used for overrepresentation test of gene ontology (GO) biological processes with Fisher’s exact false discovery rate (FDR) multiple test correction. An FDR < 0.01 was considered significant.
Sample-specific single nucleotide polymorphisms (SNPs) for chromosome X were identified using the ‘mpileup2snp’ subcommand of VarScan2 (Koboldt et al., 2009) with a P-value 0.5. Only uniquely mapped reads were considered (k = 1). SNP locations with ≥30 reads were considered in downstream analysis. Informative SNPs for each donor were obtained by combining SNPs with ≥5 reads on minor allele in fibroblast replicates and in corresponding hiPSC replicates (f-HM1/ips-HM1: 436; f-HM2/ips-HM2: 368; f-KS1/ips-KS1: 880; f-KS2/ips-KS2: 725; f-HF1/ips-HF1: 920; HS980: 783). SNPs were considered bi-allelic/with variation when 20–80% of reads corresponded to the reference allele, and mono-allelic/no variation when <20% or >80% of reads corresponded to the reference allele. Further, only transcripts with all SNPs showing either bi-allelic or mono-allelic status in all replicates were considered informative.
Accession numbers
The RNA sequencing data from this publication have been submitted to the Array Express database under the accession number E-MTAB-734.
Single-molecule RNA fluorescence in situ hybridization
Single-molecule RNA FISH was performed as previously reported (La Manno et al., 2016) with minor modifications. Briefly, hiPSCs and fibroblasts cultured on chamber slides (Sarstedt, Germany) were fixed with 4% formaldehyde for 15 min at room temperature and permeabilized with pre-chilled methanol at −20°C for 10 min. After air drying for 30 min, cells were incubated for 10 min at 70°C in Tris–EDTA buffer at pH 8.0 (Ambion, USA), and washed with 2× saline–sodium citrate (SSC) buffer (Ambion). Cells were hybridized with 125 nM Stellaris FISH probes for human XIST with Quasar 570 Dye and human ATRX with Quasar 670 dye (Biosearch Technologies, UK) for 5 h at 38.5°C in hybridization buffer containing 10% deionized formamide (Ambion), 10% dextran sulfate (Sigma), 2 mg/ml tRNA from Escherichia coli (Sigma-Aldrich), 2 mM ribonucleoside vanadyl complex (New England Biolabs, USA), and 0.2 mg/ml BSA in 2× SSC (Ambion). Cells were washed three times with wash buffer (20% deionized formamide in 2× SSC buffer) for 15 min at 38.5°C and once with wash buffer containing 1 μg/ml Hoechst 33342 (Molecular Probes, USA) for 15 min at 38.5°C. Cells were mounted with ProLong Diamond Antifade Mountant (Molecular Probes). Image stacks (0.3 μm distance) were acquired using a Nikon Ti-E motorized inverted microscope with a plan Apo λ 60× oil immersion objective (NA = 1.4) and using SpectraX light source (Lumencor, USA) and Andor Zyla 4.2+ sCMOS camera.
Analysis was performed in Fiji ImageJ (ImageJ, USA) on three consecutive passages, counting all cells of four representative images per passage.
DNA fluorescence in situ hybridization
DNA FISH was performed on the same slides as used for RNA FISH after submerging the slides in PBS for 15 min at 37°C and removing the coverslips. Cells were re-fixed with 4% formaldehyde for 10 min at room temperature and dehydrated in 70%, 90%, and 100% ethanol for 2 min each followed by brief air drying. Cells were treated with 100 μg/ml RNaseA (Qiagen) in PBS for 30 min at 37°C followed by incubation with 10% pepsin (Sigma-Aldrich) in 0.01 M HCl for 5–8 min at 37°C. Cells were then re-fixed for 10 min with 1% formaldehyde and dehydrated in 70%, 90%, and 100% ethanol for 2 min each and air dried. A probe mixture consisting of Vysis CEP X (DXZ1) SpectrumGreen probe, Vysis CEP Y (DYZ1) SpectrumOrange probe and CEP hybridization buffer (all from Abbott Molecular, USA) was applied onto the slide with a coverslip and sealed with Fastik (Staples, USA). The sample plus probes were then co-denaturated for 3 min at 85°C in a heat block followed by overnight hybridization at 41°C in a humidity chamber. Slides were washed with 0.3% Tween-20 in 0.4× SSC for 2 min at 73°C and with 0.1% Tween-20 in 2× SSC for 30 s at room temperature. Cells were counterstained with 1 μg/ml Hoechst 33342 for 10 min and mounted with ProLong Diamond Antifade Mountant. Image stacks (2 μm distance) were acquired using a Nikon Ti-E motorized inverted microscope with a plan Apo λ 60× oil immersion objective (NA = 1.4), a SpectraX light source (Lumencor) and Andor Zyla 4.2+ sCMOS camera. The images were processed and analyzed using ImageJ.
Results
Reprogramming efficiency of HDFs on LN521 and characterization of KS hiPSCs cultured on LN521
For the generation of patient-specific hiPSC lines, dermal fibroblast cultures were established from skin punch biopsies from two KS patients (f-KS1 and f-KS2), two HMs (f-HM1 and f-HM2) and commercially available healthy female fibroblasts (HF; f-HF1). Using human recombinant LN521 as a coating matrix and episomal plasmids pCXLE-hOCT3/4-shp53, pCXLE-hSK, and pCXLE-hUL (Okita et al., 2011) for reprogramming (Fig. 1A) we could observe several small AP positive colonies by Day 15 after transfection. The AP positive colonies were counted and the reprogramming efficiency was calculated to be 0.52% for LN521, which was significantly higher than for Matrigel (0.36%; P < 0.05; Supplementary Fig. S1A), which was used as a control.
Figure 1.
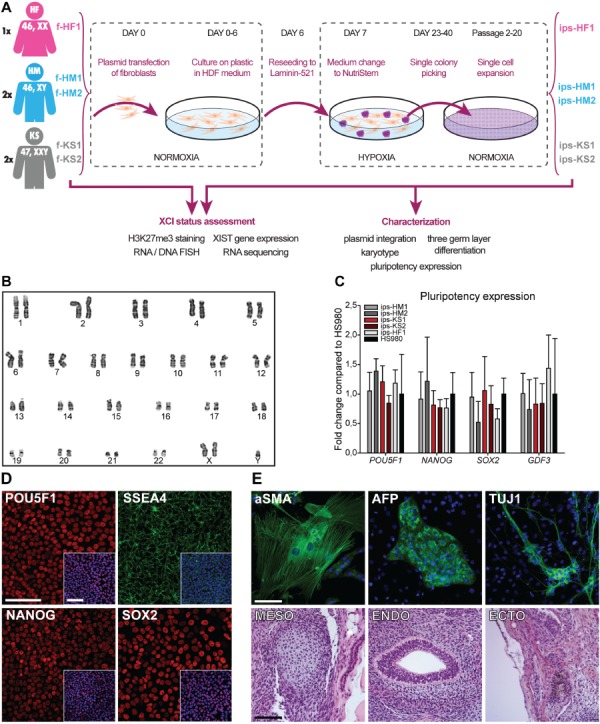
Feeder-free reprogramming conditions with nonintegrating episomal plasmids and characterization of hiPSCs. (A) Overview of reprogramming and characterization schema. (B) Chromosomal G-band analysis of at least 25 mitoses confirmed KS karyotype 47,XXY for ips-KS2 cells at passage 20. A representative image is shown. (C) Gene expression of pluripotency markers POU Class 5 Homeobox 1 (POU5F1), Nanog Homeobox (NANOG), SRY-Box 2 (SOX2), and Growth Differentiation Factor 3 (GDF3) for all hiPSC lines was similar to a hESC line (HS980) cultured on laminin 521 (LN521). Mean expression of three biological replicates with ±SD. (D) Expression of NANOG, POU5F1, SOX2, and stage-specific embryonic antigen-4 (SSEA4) was confirmed at protein level; ips-KS2 is shown as a representative line. Nuclei were counterstained with DAPI (blue) as shown in the small merged image in the lower right corner; scale bar is equal to 100 μm. (E) Three germ-layer formation from ips-KS2. Upper panel shows representative images with cytoskeletal staining of alpha-smooth muscle actin (aSMA, mesoderm) and neuron-specific class III beta-tubulin (TUJ1, ectoderm) as well as alpha fetoprotein (AFP, endoderm) in green with DAPI (blue) as counterstaining. Lower panel shows meso-, endo-, and ectodermal structures of paraffin-embedded teratomas stained with hematoxylin and eosin. Scale bar is equal to 100 μm. See also Supplementary Figure S1.
The reprogrammed cell colonies were manually picked between 23 and 40 days after transfection and further expanded on LN521 (Fig. 1A). We generated two KS (ips-KS1 and ips-KS2) and two HM (ips-HM1 and ips-HM2), as well as one HF (ips-HF1) hiPSC lines, with each monoclonal derived from a separate donor.
The loss of episomal plasmids for all five hiPSC lines was confirmed at passage 5 by genomic qPCR analysis (Supplementary Fig. S1B) and a karyotype of 47,XXY (non-mosaic), 46,XY, or 46,XX confirmed at passage 12 or passage 20 for all five hiPSCs (Fig. 1B and Supplementary Fig. S1C; Supplementary Table SI). All five hiPSC lines expressed the pluripotency genes NANOG, POU5F1, SOX2, and GDF3 at similar levels relative to a female hESC line (HS980) cultured on LN521 (Fig. 1C) and showed presence of NANOG, POU5F1, SOX2 (Fig. 1D and Supplementary Fig. S1D), and SSEA4 (Fig. 1D) at protein level.
The spontaneous in vitro differentiation and teratoma formation assay showed presence of markers representing all three germ layers (AFP for endoderm, aSMA for mesoderm, and TUJ1 for ectoderm) (Fig. 1E and Supplementary Fig. S1D) and structures representing all three germ layers (Fig. 1E and Supplementary Fig. S1D) in all five hiPSC lines that were reprogrammed and expanded for 20 passages on LN521. Negative controls are shown in Supplementary Figure S2.
GO term enrichment shows correlation of KS fibroblasts and hiPSCs with genotype-related clinical features of KS
To study KS-specific molecular features, we performed RNA-sequencing for the KS, HM, and HF fibroblasts and hiPSC, as well as for HS980 (Supplementary Table SII). PCA showed a separation of fibroblast and hPSC lines along principal component 1, while principal component 2 seemed to separate hPSCs based on karyotype with the exception of ips-HM2 (Fig. 2A). Comparing KS and HM fibroblasts, 311 DEGs could be observed (with fold change of ≥1.5 and P < 0.05), 219 upregulated and 92 downregulated in KS, while only 8 upregulated and 3 downregulated genes were X-linked genes (Fig. 2B; Supplementary Table SIII). On the contrary, comparison of KS and HM hiPSC lines showed 920 DEGs, 324 upregulated and 596 downregulated in KS, while 116 upregulated and 2 downregulated genes were X-linked genes (Fig. 2B; Supplementary Table SIV).
Figure 2.
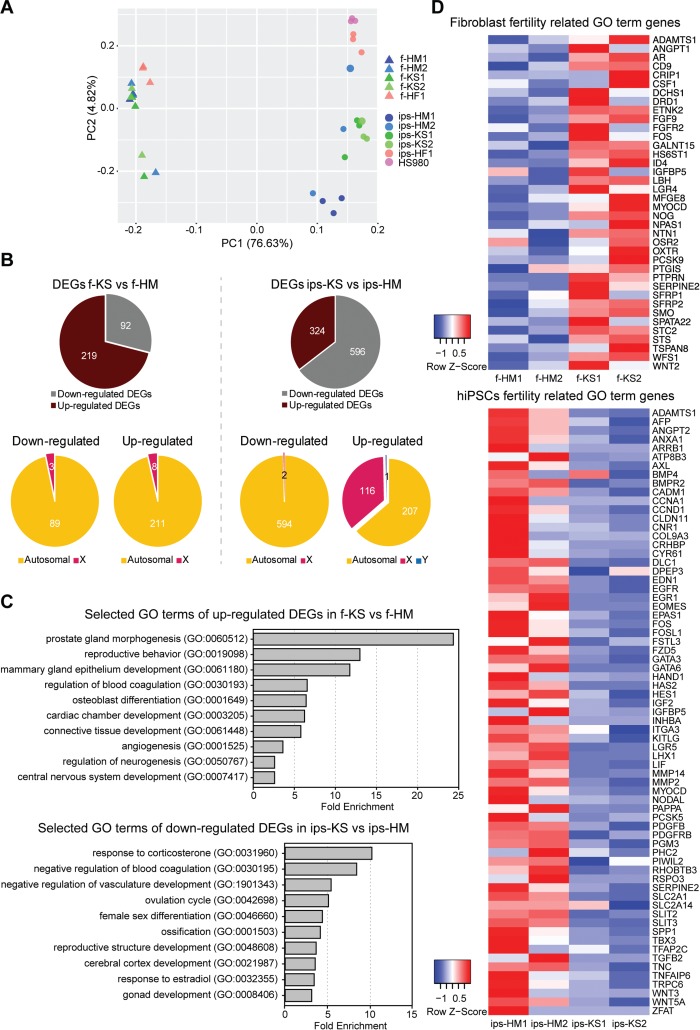
KS fibroblasts and hiPSCs show differentially expressed genes (DEGs) related to the clinical genotype of the KS disorder. (A) Principal component (PC) analysis with top 10 000 expressing genes of KS, HM and HF fibroblasts (triangle) and corresponding hiPSC as well as female hESC line HS980 (circle). See also Supplementary Tables SII. (B) Downregulated and upregulated DEGs comparing KS with HM fibroblast and KS with HM hiPSCs. P < 0.05 and fold change ≥1.5. See also Supplementary Tables SII and SIII. (C) Fold change of enriched GO terms of KS disorder-related terms for upregulated DEGs of KS fibroblasts, and downregulated DEGs of KS iPSCs, FDR <0.01. See also Supplementary Tables SIV–SVI. (D) Heatmap of gene expression levels associated with fertility-related GO terms in HM and KS fibroblasts and MH and KS hiPSCs. See also Supplementary Tables SV and SVII.
Looking at GO term enrichment of the 219 upregulated KS fibroblasts genes, multiple KS disorder-associated terms stood out (Fig. 2C; Supplementary Table SV), whereas for 92 downregulated KS fibroblasts genes, only a few GO terms were enriched and mostly associated with vasculature development (Supplementary Table SVI). Interestingly, the 324 upregulated KS hiPSCs genes showed no GO term enrichment, whereas the 596 downregulated genes showed similar KS disorder-associated terms to the KS fibroblasts upregulated genes (Fig. 2C; Supplementary Table SVII).
Enriched GO terms for DEGs of KS fibroblasts and hiPSCs included fertility-related terms, such as reproductive behavior, female sex differentiation, reproductive structure, ovulation cycle, and gonad development. The expression heatmap of genes associated with fertility-related GO terms for KS and HM fibroblasts as well as their hiPSC derivatives further demonstrates a dysregulation in gene expression levels, with KS fibroblasts showing upregulation and KS hiPSCs downregulation of fertility-related genes (Fig. 2D; Supplementary Tables SIV and SVI). Comparison of DEGs between KS fibroblast and their respective hiPSC line revealed only fibroblast or hiPSC specific genes with no KS-specific relevance in GO terms of upregulated or downregulated genes (data not shown).
H3K27me3 staining and RNA FISH indicate different XCI states for the KS hiPSC lines
The expression level of XIST in KS fibroblasts (f-KS1 and f-KS2), KS, and female hiPSCs (ips-KS1, ips-KS2, and ips-HF1) was similar to HF fibroblasts (f-HF1) (Fig. 3A), while no expression was observed for female hESCs (HS980). In alignment with a normal in vivo situation, HM fibroblasts (f-HM1 and f-HM2) and their hiPSCs derivatives (ips-HM1 and ips-HM2) showed no expression of XIST. Immunofluorescence staining for H3K27me3, a repressive chromatin marker that accumulates on the Xi, as well as RNA FISH for XIST, which coats the Xi, and ATRX, an X-linked gene that has one active transcription site for XaXi cells and two for XaXa cells, showed the presence of one XIST cloud in >90% of cells for KS fibroblasts and HF fibroblasts (Fig. 3B and C; Supplementary Table SVIII). No clouds were observed for HM fibroblasts. A small percentage of HF and KS fibroblasts showed expression of two XIST clouds (Fig. 3B and C; Supplementary Table SVIII), which were all confirmed to be tetraploid cells upon DNA FISH analysis of the same cells (Fig. 3C).
Figure 3.
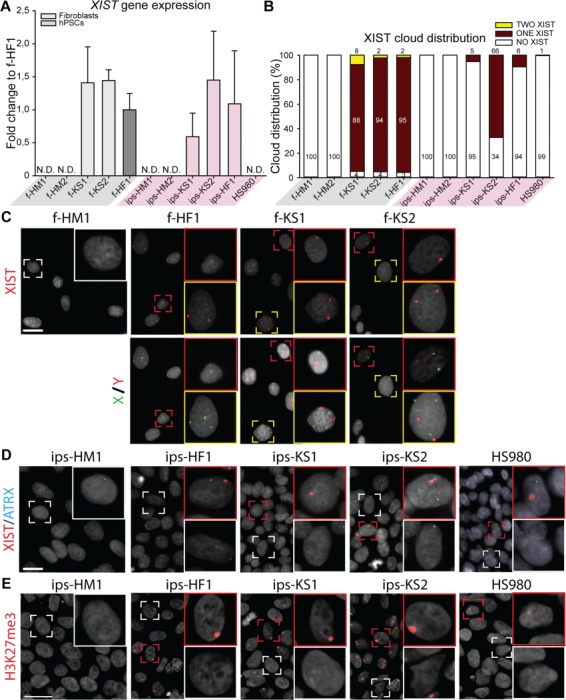
X chromosome inactivation (XCI) analysis of KS fibroblasts and hiPSCs indicates different XCI states. (A) Gene expression level of XCI marker, XIST, relative to female fibroblasts (f-HF1). Mean expression of three biological replicates with ±SD; N.D. means no detection. (B) Distribution of XIST cloud expression in each cell line based on RNA FISH: one cloud (red), no cloud (white), or two clouds (yellow). Total count from three independent experiments, with >193 cells counted per cell line. (C) Upper panel shows representative images of RNA fluorescence in situ hybridization (FISH) with one (red square) or two (yellow square) XIST clouds in HF (f-HF1) and KS (f-KS1 and f-KS2) fibroblasts. HM fibroblasts (f-HD1) were used as negative control showing no clouds (white square). Lower panel shows DNA FISH for X chromosome (green) and Y chromosome (red) for upper panel cells. DAPI (gray) was used as counterstaining. Scale bar is equal to 50 μm. (D) Representative images of RNA FISH with one (red square) or no (white square) XIST (red) cloud in HF (ips-HF1 and HS980) and KS (ips-KS1 and ips-KS2) hPSCs. HM hiPSCs (ips-HM1) were used as negative control showing no XIST (red) clouds. Active transcription site of ATRX is shown in blue. DAPI (gray) was used as counterstaining. Scale bar is equal to 50 μm. (E) Immunostaining of hPSCs for silencing methylation mark, H3K27me3 (red), with positive accumulation mark (red square) or no staining (white square). DAPI (gray) was used as counterstaining. Scale bar is equal to 50 μm.
The XCI analysis of hPSCs showed more variability; KS hiPSC lines (ips-KS1 and ips-KS2) and female hPSC lines (ips-HF1 and HS980) consisted of two cell populations, showing either one or no H3K27me3 accumulation (Fig. 3E) as well as one or no XIST cloud (Fig. 3B and D; Supplementary Table SVIII). However, all hPSC lines displayed only mono-allelic ATRX transcription site (Fig. 3D; Supplementary Table SVIII). Both HM hiPSC derivatives showed no H3K27me3 accumulation (Fig. 3E) as well as no XIST cloud (Fig. 3B and D; Supplementary Table SVIII) and one ATRX transcription site (Fig. 3D; Supplementary Table SVIII).
Transcriptomics analysis shows erosion of XCI in one of the KS hiPSC lines
Transcriptomics data showed similar levels of the sum of total X chromosome expression in KS and HF fibroblasts and ips-KS2 compared to HM fibroblasts and hiPSCs (Fig. 4A). On the contrary, the sum of total X chromosome expression was significantly increased for ips-KS1, ips-HF1, and HS980 (Fig. 4A). As a control, the sum of autosomal expression showed similar levels among all fibroblasts as well as similar levels amongst all hPSCs (Fig. 4B).
Figure 4.
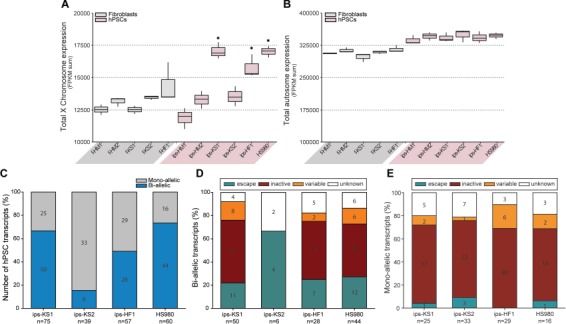
Transcriptomics analysis of XCI state of KS fibroblasts and hiPSC confirms the XaXe and XaXi state of KS hiPSCs. (A) Total X chromosome expression (sum of FPKM values for X-linked genes) for HM, KS and HF fibroblasts and hiPSCs, and HS980. Significant compared to HM fibroblasts (f-HM-1 and f-HM2), HM hiPSCs (ips-HM1 and ips-HM2), KS fibroblasts (f-KS1 and f-KS2), and ips-KS2. Box-and-whisker plots representing the median value with 50% of all data falling within the box. The `whiskers' extend to the fifth and 95th percentiles, P < 0.05. (B) Total autosomal expression (sum of FPKM values for autosomal genes) for HM, KS and HF fibroblasts and hiPSCs, and HS980. Box-and-whisker plots representing the median value with 50% of all data falling within the box. The `whiskers' extend to the fifth and 95th percentiles. (C) Number of transcripts showing mono-allelic or bi-allelic expression in KS and HF hPSCs. Results combined from three biological replicates. (D) Reported XCI status of bi-allelic transcripts (E) and mono-allelic transcripts in KS and HF hPSCs. See also Supplementary Figure SII and Supplementary Table SVIII.
To investigate whether the X chromosome linked genes had mono- or bi-allelic expression in hPSCs, SNP variation from the RNA sequencing data against a reference genome were analyzed. In order to exclude false positive transcripts, we analyzed the HM fibroblasts and their hiPSC derivatives and observed a total of 12 unique transcripts that appeared as bi-allelic in one or more HM cell line (Supplementary Fig. S3A; Supplementary Table SIX), from which seven have a known Y homologue (Supplementary Fig. S3B; Supplementary Table SIX). We found that the majority of the X-linked transcripts in ips-KS2 had mono-allelic expression (Fig. 4C; Supplementary Table SIX), and most of the bi-allelic transcripts were known XCI escapees (Fig. 4D; Supplementary Table SIX). On the contrary, a high percentage of bi-allelic transcripts was observed for ips-KS1, ips-HF1, and HS980 (Fig. 4C; Supplementary Table SIX), including many known XCI inactive genes (Fig. 4D; Supplementary Table SIX). As expected, the majority of mono-allelic transcripts of ips-KS1, ips-KS2, ips-HF1, and HS980 belong to known XCI inactive genes (Fig. 4E; Supplementary Table SIX).
Unlike hPSCs, fibroblasts are not clonally derived and therefore in the case of random XCI, both X chromosomes should be equally expressed in a cell population analysis. f-KS2 and f-HF1 cell lines showed SNP variation in >82% of transcripts, indicating random XCI (Fig. S2C; Supplementary Table SIX). However, the f-KS1 cell line showed SNP variation for only 24% of transcripts, suggesting a skewed XCI (Fig. S2C; Supplementary Table SIX). In addition, the majority of the transcripts that did show SNP variation are known XCI escaping genes, further supporting skewed XCI in f-KS1 (Supplementary Table SIX).
The XCI analysis showed that ips-KS1 had a XaXe state, a known abnormal XCI state commonly observed in female hPSCs, whereas ips-KS2 had normal XCI supporting its potential relevance in KS disease, especially for X-linked genes. Therefore, we performed DEG analysis by comparing ips-KS1 and ips-KS2 individually with HM hiPSCs. We observed 111 upregulated genes for ips-KS2 of which 24 were X-linked genes (Fig. 5A; Supplementary Table SX). In contrast, we observed 123 upregulated X-linked genes for ips-KS1, but a similar number of upregulated autosomal genes (68 ips-KS1 and 87 ips-KS2) (Fig. 5A). GO analysis for downregulated genes in ips-KS2 (265) showed enrichment for terms that were mostly shared with an analysis using both ips-KS1 and ips-KS2 lines (Supplementary Table SVII and SX), suggesting that XCI status affects mostly the X-linked genes. From the 24 upregulated X-linked genes in ips-KS2, 3 were also showing bi-allelic expression: A-kinase anchoring protein 17A (AKAP17A), glycogenin 2 (GYG2), haloacid dehalogenase-like hydrolase domain-containing protein 1 (HDHD1) (Fig. 5B; Supplementary Table SIX), and a substantial number of the genes are involved in nervous system development (doublecortin (DCX), neuroligin 3 (NLGN3), neuroligin 4 X-Linked (NLGN4X), proteolipid protein 1 (PLP1), and nucleosome assembly protein 1 like 3 (NAP1L3)) as well as synaptic transmission (gamma-aminobutyric acid type a receptor alpha3 subunit (GABRA3)) (Fig. 5B).
Figure 5.
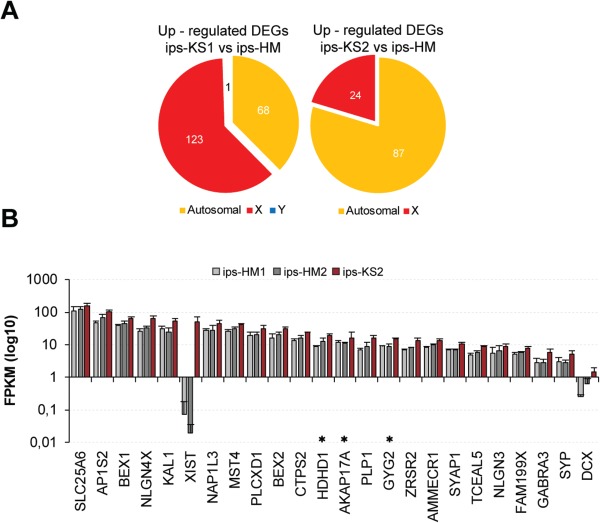
Upregulation of X-linked genes in ips-KS2 related to nervous system development, synaptic transmission, and metabolic processes. (A) Chromosomal distribution of upregulated DEGs comparing either ips-KS1 or ips-KS2 with HM hiPSCs. See also Supplementary Table SIX. (B) Expression of X-linked genes showing up-regulated expression in ips-KS2 compared to HM hiPSC. Mean expression of three biological replicates with ±SD. Asterisk indicates upregulated genes that showed also bi-allelic expression in ips-KS2. See also Supplementary Tables SVIII and SIX.
Discussion
The infrequency of KS diagnosis is related to the diverse phenotypes associated with the condition that, at least in part, are caused by the number of supernumerary X chromosomes, the skewedness of XCI, as well as the genes escaping XCI. Since XCI escapee genes vary between individuals and even within different tissue of the same individual, the analysis of a variety of tissue types is needed for a better understanding of KS pathogenesis. Nevertheless, most molecular studies involving KS patients use whole blood, being easily accessible samples, for analysis (Huang et al., 2015; Zitzmann et al., 2015; Belling et al., 2017). However, in view of the fact that KS patients suffer from infertility, cognitive dysfunction, metabolic disruption, disruption in bone density, and cardiovascular complications (Bonomi et al., 2017; Kanakis and Nieschlag, 2018), KS-derived hiPSCs provide a useful alternative to study early developmental defects of the KS disorder, as they can be differentiated into any cell type of the body.
Despite previous efforts in the generation of KS-derived hiPSCs from single patients, XCI analysis was either not performed (Shimizu et al., 2016) or based solely on the H3K27me3 staining pattern and XIST expression levels, yet aberrant gene expression of X-linked genes was reported (Ma et al., 2012) without consideration of the various XCI states that hPSC exhibit in culture (Patel et al., 2017). In the present study, we focused on the derivation of hiPSCs from two azoospermic KS patients combined with detailed analysis of their XCI state. As LN521 has been shown to homogenize the stemness expression of hPSCs during culture (Albalushi et al., 2018), LN521 was utilized as substrate to minimize variation in stemness states between the derived hiPSC lines while further allowing rapid expansion as monolayer cultures using an easy single-cell passaging method without the need of Rho kinase inhibitor (Rodin et al., 2014).
Using integration-free episomal plasmids (Okita et al., 2011) together with NutriStem medium and LN521 as a substrate we were able to efficiently derive two KS, two HM and one HF hiPSC lines, each from different donors, with a high reprograming efficiency (0.52%) compared to previous studies using the same episomal plasmids with laminin-511 E8 fragments (0.077%) (Nakagawa et al., 2014) or LN521 in combination with polycistronic lentiviral vectors (0.3%) (Lu et al., 2014).
Recent studies exploring the relation of KS transcription patterns to the clinical picture of KS have demonstrated a high association of DEGs between KS and HM controls toward phenotypic features such as waist circumference, metabolic syndrome, insulin resistance, verbal cognition, cardiac complications, and infertility (Huang et al., 2015; Zitzmann et al., 2015; Belling et al., 2017). Similarly, DEGs between KS and HM controls in our study showed disorder-related dysregulation in KS fibroblast and their hiPSC derivatives. As infertility is strongly associated with KS patients, we could observe dysregulation in fertility-related genes in KS fibroblasts and even more in KS hiPSC. Although it is possible to obtain spermatozoa from testicular sperm extractions for a few KS patients, the majority have suffered from germ cell loss upon diagnosis in adulthood. Therefore, approaches such as in vitro testicular somatic or germ cell differentiation from patient-derived hiPSCs could facilitate a better understanding of the germ cell loss mechanisms and potentially facilitate parenthood for those Sertoli-cell-only patients. Nevertheless, successful differentiation of hiPSCs into gametes and subsequently resulting in live birth have only been achieved in animal models to date, while differentiation to primordial germ cell up to spermatogonia stage has shown low efficiency in humans (Botman and Wyns, 2014; Irie et al., 2015; Sasaki et al., 2015).
In addition, the clinical picture of KS patients having cardiovascular problems, decreased bone density, and cognitive dysfunctions was reflected in enriched GO terms related to cardiovascular development, ossification, and brain development for both KS fibroblast and hiPSCs. Interestingly, the genes linked to these GO terms were upregulated in KS fibroblasts but downregulated in KS hiPSCs, reflecting a cell type-specific response. Furthermore, we observed additional enriched GO terms that were specific for either KS fibroblasts or hiPSCs. KS fibroblasts showed enrichment in GO terms for mammary gland epithelium development and prostate gland morphogenesis, possibly correlating with female phenotypic features such as gynecomastia as well as fertility related issues that KS patients suffer from. KS hiPSCs, however, showed enrichment in GO terms for response to corticosterone and estradiol, which are involved in metabolism and fertility and possibly impacting KS as early as during fetal development. Nevertheless, comparing KS fibroblasts to their respective hiPSC derivate revealed mostly fibroblast or hiPSC specific DEGs. Upon removal of genes common to comparisons of HM fibroblast with their hiPSC derivates, no KS specific GO terms could be observed, suggesting that a KS genotype may not have an effect on hiPSCs reprogramming.
Exploring the XCI status of KS fibroblasts we were able to observe similar expression levels of XIST and XIST cloud distribution relative to normal female fibroblasts, confirming XCI in KS fibroblasts. Although we could observe a small percentage of KS and HF fibroblasts showing two XIST clouds, we could confirm those cells as being tetraploid, tending to spontaneously occur as an artifact in fibroblast cultures (Mittwoch et al., 1965; Schwarzacher and Schnedl, 1965). In addition, transcriptional analysis revealed evidence for skewed XCI of one KS fibroblast line (f-KS1) as only 24% of transcripts showed SNP variation (compared to >82% in f-KS2 and f-HF1), and the majority of the transcripts were known XCI escaping genes. Skewed XCI has high prevalence in KS patients compared to females and is speculated to contribute to the range of mental deficiencies and phenotypic variation (Iitsuka et al., 2001).
Recent studies have shown that although female hPSC lines initially express one Xa and one Xi, the XCI maintenance during culture can be unstable resulting in cells with XCI (XaXi) with or without XIST coating or XCI with erosion of silencing (XaXe) (Vallot et al., 2015). In agreement, the female hPSCs in our study, ips-HF1 and HS980, had mostly XaXe status displayed by the loss of H3K27me3 marks and XIST clouds, while still showing only one active transcription site for ATRX, which is known to resist XCI erosion (Vallot et al., 2015). In addition, these cell lines displayed high X chromosomal expression and a high degree of bi-allelic transcripts, mostly classified as common XCI inactive genes. One KS hiPSC line, ips-KS1, showed similar evidence for XaXe, while the other line, ips-KS2, displayed normal XaXi status with H3K27me3 accumulation, XIST clouds, mono-allelic ATRX, a lower level of X chromosomal expression, and a low level of bi-allelic transcripts (being the XCI escaping genes AKAP17A, HDHD1, GYG2, and taxilin gamma (TXLNG)), or non-coding transcripts with unknown status (PIR-FIGF readthrough (PIR-FIGF) and CD99 molecule pseudogene 1 (CD99P1)).
Since X chromosomal expression is thought to directly influence the KS phenotype, we looked specifically for X-linked DEGs and analyzed ips-KS1 and ips-KS2 separately due their different XCI states. Confirming the XaXe state of ips-KS1, we found 123 upregulated X-linked genes, whereas ips-KS2 with the XaXi state had only 24 upregulated X-linked genes. Since XaXe is an aberrant XCI state common to hPSCs and may not represent KS correctly, we focused on the ips-KS2 line. From the 24 upregulated X-linked genes, 6 were found to be upregulated for KS in other studies: XIST (Vawter et al., 2007; Ma et al., 2012; Belling et al., 2017; Winge et al., 2018), AKAP17A (Belling et al., 2017; Winge et al., 2018), SLC25A6 (Zitzmann et al., 2015; Belling et al., 2017), HDHD1 (Zitzmann et al., 2015), NLGN4X (Winge et al., 2018), PLCXD1 (Belling et al., 2017), and XIST and HDHD1 were also upregulated in KS fibroblasts, highlighting the importance of these genes for future studies. In addition, many of the upregulated genes were related to nervous system development (DCX (Reiner, 2013; Ayanlaja et al., 2017), NLGN3, NLGN4X (Bottos et al., 2011), PLP1, GABRA3, and NAP1L3 (Carpanini et al., 2017)) underlining the biological relevance of KS hiPSCs with normal XCI, even in the undifferentiated state. Nevertheless, further studies with in vitro differentiation of KS hiPSCs will be needed for a more detailed analysis of KS-related defects, for example for neuronal or germ cell development. In addition, conversion of KS hiPSCs into a naive pluripotency state could provide new insights for early developmental defects caused by supernumerary X chromosomes, as female naive hPSCs have the XaXa status similar to early human pre-implantation embryos.
Conclusion
In conclusion, our results demonstrate that KS-derived hiPSCs show similar XCI behavior to female hPSCs in culture and show biological relevance to KS genotype-related clinical features associated with gonadal function and fertility. In addition, our results highlight the importance of XCI analysis for KS hiPSCs when studying dysregulation of X-linked genes associated with KS owing to the possibility of various XCI states in culture.
Supplementary Material
Acknowledgements
The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure. The authors would like to thank Dr Kenny Rodriguez-Wallberg, the nurses, midwifes, and the staff of Reproduction Medicine Unit at Karolinska University Hospital, Huddinge, for patient consenting and tissue collection; the staff of the Department of Laboratory Medicine at Karolinska University Hospital, Huddinge, for tissue processing; and Georgia Kokaraki for teratoma confirmation. We would like to thank all men who volunteered to donate tissue for this study.
Authors’ roles
S.P.: conception and design, data collection, analysis and interpretation, manuscript writing, and final approval; M.K.: data collection, analysis and interpretation, manuscript writing, and final approval; P.K.: data analysis and final approval of manuscript; H.A., S.P.S., and P.D.: data collection and final approval of manuscript; J.I.O.: provision of patients and final approval of manuscript; O.H.: conception and design and final approval of manuscript; F.L. and J.-B.S.: conception and design, data interpretation, financial support, and final approval of manuscript.
Funding
This study was performed at the Live Cell Imaging facility, Karolinska Institutet, Sweden, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Centre for Innovative Medicine, and the Jonasson Center at the Royal Institute of Technology, Sweden. This study was supported by grants from the Knut and Alice Wallenberg Foundation (2016.0121 and 2015.0096), Ming Wai Lau Centre for Reparative Medicine (2-343/2016), Ragnar Söderberg Foundation (M67/13), Swedish Research Council (2013-32485-100360-69), Centre for Innovative Medicine (2-388/2016-40), Kronprinsessan Lovisas Förening För Barnasjukvård/Stiftelsen Axel Tielmans Minnesfond, Samariten Foundation, and Jonasson Center at the Royal Institute of Technology, Sweden. M.K. was supported by the ITN Marie Curie program ‘Growsperm’ (EU-FP7-PEOPLE-2013-ITN 603568).
Conflict of interest
The authors declare no conflicts of interest.
References
- Albalushi H, Kurek M, Karlsson L, Landreh L, Kjartansdóttir KR, Söder O, Hovatta O, Stukenborg JB. Laminin 521 stabilizes the pluripotency expression pattern of human embryonic stem cells initially derived on feeder cells. Stem Cells Int 2018;2018:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayanlaja AA, Xiong Y, Gao Y, Ji G, Tang C, Abdikani Abdullah Z, Gao D. Distinct features of Doublecortin as a marker of neuronal migration and its implications in cancer cell mobility. Front Mol Neurosci 2017;10:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belling K, Russo F, Jensen AB, Dalgaard MD, Westergaard D, Rajpert-De Meyts E, Skakkebaek NE, Juul A, Brunak S. Klinefelter syndrome comorbidities linked to increased X chromosome gene dosage and altered protein interactome activity. Hum Mol Genet 2017;26:1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi M, Rochira V, Pasquali D, Balercia G, Jannini EA, Ferlin A, Klinefelter Italia NG. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. J Endocrinol Invest 2017;40:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botman O, Wyns C. Induced pluripotent stem cell potential in medicine, specifically focused on reproductive medicine. Front Surg 2014;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottos A, Rissone A, Bussolino F, Arese M. Neurexins and neuroligins: synapses look out of the nervous system. Cell Mol Life Sci 2011;68:2655–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpanini SM, Wishart TM, Gillingwater TH, Manson JC, Summers KM. Analysis of gene expression in the nervous system identifies key genes and novel candidates for health and disease. Neurogenetics 2017;18:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aurora M, Ferlin A, Di Nicola M, Garolla A, De Toni L, Franchi S, Palka G, Foresta C, Stuppia L, Gatta V. Deregulation of sertoli and leydig cells function in patients with Klinefelter syndrome as evidenced by testis transcriptome analysis. BMC Genomics 2015;16:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aurora M, Ferlin A, Garolla A, Franchi S, D’Onofrio L, Trubiani O, Palka G, Foresta C, Stuppia L, Gatta V. Testis transcriptome modulation in Klinefelter patients with hypospermatogenesis. Sci Rep 2017;7:45729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang L, Deng H, Chang L, Liu Q, Liu P. Global transcriptome analysis of peripheral blood identifies the most significant down-regulated genes associated with metabolism regulation in Klinefelter syndrome. Mol Reprod Dev 2015;82:17–25. [DOI] [PubMed] [Google Scholar]
- Iitsuka Y, Bock A, Nguyen DD, Samango-Sprouse CA, Simpson JL, Bischoff FZ. Evidence of skewed X-chromosome inactivation in 47,XXY and 48,XXYY Klinefelter patients. Am J Med Genet 2001;98:25–31. [PubMed] [Google Scholar]
- Irie N, Weinberger L, Tang WWC, Kobayashi T, Viukov S, Manor YS, Dietmann S, Hanna JH, Surani MA. SOX17 is a critical specifier of human primordial germ cell fate. Cell 2015;160:253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamischke A, Baumgardt A, Horst J, Nieschlag E. Clinical and diagnostic features of patients with suspected Klinefelter syndrome. J Androl 2003;24:41–48. [PubMed] [Google Scholar]
- Kanakis GA, Nieschlag E. Klinefelter syndrome: more than hypogonadism. Metabolism 2018;86:135–144. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009;25:2283–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, Borm LE, Stott SRW, Toledo EM, Villaescusa JC et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 2016;167:566–580e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet 2004;364:273–283. [DOI] [PubMed] [Google Scholar]
- Lu HF, Chai C, Lim TC, Leong MF, Lim JK, Gao S, Lim KL, Wan AC. A defined xeno-free and feeder-free culture system for the derivation, expansion and direct differentiation of transgene-free patient-specific induced pluripotent stem cells. Biomaterials 2014;35:2816–2826. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li C, Gu J, Tang F, Li C, Li P, Ping P, Yang S, Li Z, Jin Y. Aberrant gene expression profiles in pluripotent stem cells induced from fibroblasts of a Klinefelter syndrome patient. J Biol Chem 2012;287:38970–38979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45:D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013;8:1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittwoch U, Lele KP, Webster WS. Deoxyribonucleic acid synthesis in cultured human cells and its bearing on the concepts of endoreduplication and polyploidy. Nature 1965;208:204–208. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa M et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep 2014;4:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011;8:409–412. [DOI] [PubMed] [Google Scholar]
- Patel S, Bonora G, Sahakyan A, Kim R, Chronis C, Langerman J, Fitz-Gibbon S, Rubbi L, Skelton RJP, Ardehali R et al. Human embryonic stem cells do not change their X inactivation status during differentiation. Cell Rep 2017;18:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Surani MA. Germline and pluripotent stem cells. Cold Spring Harb Perspect Biol 2015;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O. LIS1 and DCX: implications for brain development and human disease in relation to microtubules. Scientifica (Cairo) 2013;2013:393975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin S, Antonsson L, Niaudet C, Simonson OE, Salmela E, Hansson EM, Domogatskaya A, Xiao Z, Damdimopoulou P, Sheikhi M et al. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nat Commun 2014;5:3195. [DOI] [PubMed] [Google Scholar]
- Rohayem J, Nieschlag E, Zitzmann M, Kliesch S. Testicular function during puberty and young adulthood in patients with Klinefelter’s syndrome with and without spermatozoa in seminal flui. Andrology 2016;4:1178–1186. [DOI] [PubMed] [Google Scholar]
- Sahakyan A, Kim R, Chronis C, Sabri S, Bonora G, Theunissen TW, Kuoy E, Langerman J, Clark AT, Jaenisch R et al. Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell 2017;20:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzacher HG, Schnedl W. Endoreduplication in human fibroblast cultures. Cytogenetics 1965;4:1–18. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Yokobayashi S, Nakamura T, Okamoto I, Yabuta Y, Kurimoto K, Ohta H, Moritoki Y, Iwatani C, Tsuchiya H et al. Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell 2015;17:178–194. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Shiohara M, Tai T, Nagao K, Nakajima K, Kobayashi H. Derivation of integration-free iPSCs from a Klinefelter syndrome patient. Reprod Med Biol 2016;15:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 2010;7:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A et al. Landscape of X chromosome inactivation across human tissues. Nature 2017;550:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttelmann F, Gromoll J. Novel genetic aspects of Klinefelter’s syndrome. Mol Hum Reprod 2010;16:386–395. [DOI] [PubMed] [Google Scholar]
- Vallot C, Huret C, Lesecque Y, Resch A, Oudrhiri N, Bennaceur-Griscelli A, Duret L, Rougeulle C. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat Genet 2013;45:239–241. [DOI] [PubMed] [Google Scholar]
- Vallot C, Ouimette JF, Makhlouf M, Feraud O, Pontis J, Come J, Martinat C, Bennaceur-Griscelli A, Lalande M, Rougeulle C. Erosion of X chromosome inactivation in human pluripotent cells initiates with XACT coating and depends on a specific heterochromatin landscape. Cell Stem Cell 2015;16:533–546. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Harvey PD, DeLisi LE. Dysregulation of X-linked gene expression in Klinefelter’s syndrome and association with verbal cognition. Am J Med Genet B Neuropsychiatr Genet 2007;144B:728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana J, Pidsley R, Troakes C, Spiers H, Wong CC, Al-Sarraj S, Craig I, Schalkwyk L, Mill J. Epigenomic and transcriptomic signatures of a Klinefelter syndrome (47,XXY) karyotype in the brain. Epigenetics 2014;9:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom AM, Dunkel L. Klinefelter syndrome. Best Pract Res Clin Endocrinol Metab 2011;25:239–250. [DOI] [PubMed] [Google Scholar]
- Winge SB, Dalgaard MD, Jensen JM, Graem N, Schierup MH, Juul A, Rajpert-De Meyts E, Almstrup K. Transcriptome profiling of fetal Klinefelter testis tissue reveals a possible involvement of long non-coding RNAs in gonocyte maturation. Hum Mol Genet 2018;27:430–439. [DOI] [PubMed] [Google Scholar]
- Zitzmann M, Bongers R, Werler S, Bogdanova N, Wistuba J, Kliesch S, Gromoll J, Tuttelmann F. Gene expression patterns in relation to the clinical phenotype in Klinefelter syndrome. J Clin Endocrinol Metab 2015;100:E518–E523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.