

Abstract
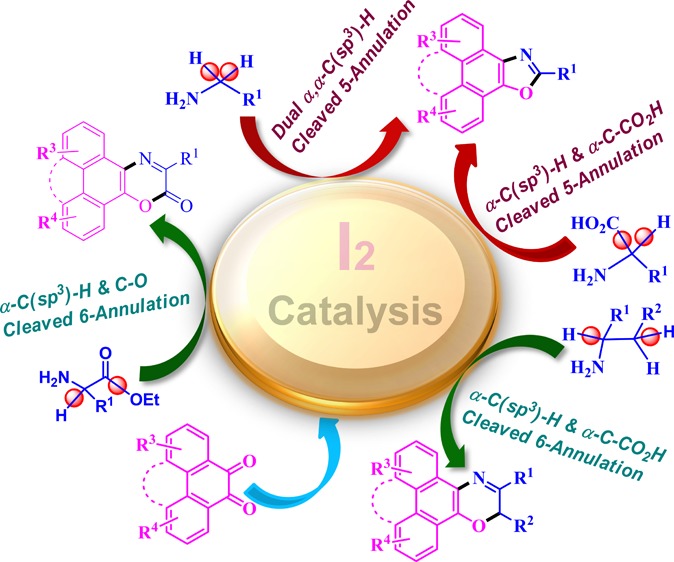
Unprecedented I2-catalyzed α,α-C(sp3)-H, decarboxylative α-C(sp3)-H, lactonized α-C(sp3)-H, and α,β-C(sp3)-H functionalized 5- and 6-annulation as well as α-C(sp3)-H activated 6-lactonization of primary aliphatic amines are devised under aerobic conditions. The metal-free sustainable strategy was employed for the diverse construction of valuable five-and six-membered polycyclic N,O-heteroaromatics such as oxazoles, 1,4-oxazines, and oxazin-2-one with a rapid reaction rate and high yield. The viability of this mild nonmetallic catalysis is successfully verified through syntheses of labile chiral heterocyclic analogues. In contrast to the common practice, this method is not limited to use of prefunctionalized amines, directing groups (DGs) and/or transient DGs, metal catalysts, and traditional oxidants. The possible mechanistic pathway of the annulation reaction is investigated by control experiments and ESI-MS data collected for a reaction mixture of the ongoing reaction. The synthesized new compounds are potent organic nanobuilding blocks to achieve valuable organic nanomaterials of different sizes, shapes, and dimensions, which are under investigation for the discovery of high-tech devices of innovative organic nanoelectronics and photophysical properties.
Introduction
Aliphatic primary amines are represented in a wide range of chemical feedstock and display great biological, pharmaceutical, agricultural, materials, and synthetic applications.1−6 Of late, functionalization of C(sp3)-H has emerged as a promising synthetic tool.7−10 The C(sp3)-H activated functionalization of secondary and tertiary aliphatic amines is well investigated employing transition-metal catalysts.11−14 However, site-selective functionalization of inactivated C(sp3)-H bonds of primary aliphatic amines is more challenging because of their strong nucleophilic, reducing, chelation, and deactivating properties, which may hamper or deactivate catalytic power of the possible metal catalysts. Thus, use of prefunctionalized amines, directing group (DGs), and/or transient DG, their removal after the desired transformation, and intramolecular annulation are frequently exercised in the metal catalysis processes.15−18 A limited number of approaches were devoted for the site-selective C(sp3)-H functionalization of primary aliphatic amines, such as ZrIV/NiII-catalyzed α-selective cyclization,19,20 PdII-tuned β-arylation,21 PdII-AgI guided γ-substitution,18,22−25 and PdII-activated δ-arylation.26,27 However, a metal-free C(sp3)-H functionalization is always a more appealing strategy to be developed for minimizing the harmful impact on nature.28−30
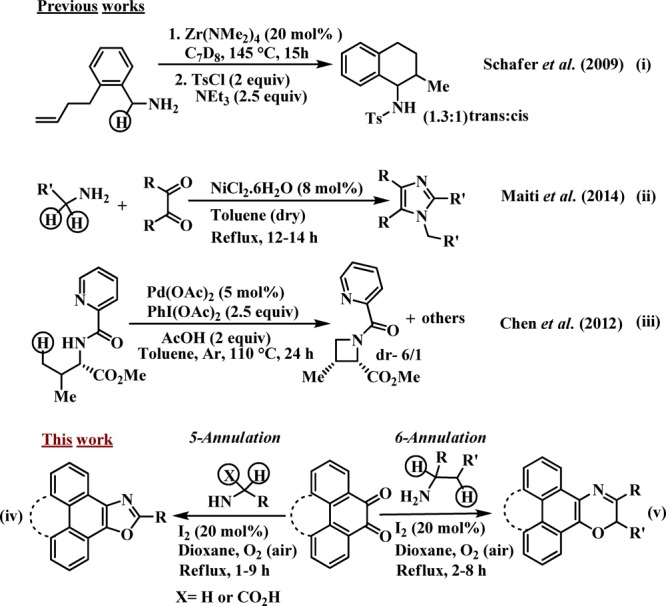
So far, few nonmetallic C(sp3)-H activation processes were developed utilizing pyridine-N-oxide, TBAI/TBHP, DTBP, hypervalent iodines, and iodine.31−37 Molecular iodine has emerged as an excellent catalyst38−43 because of its high solubility in the reaction media, easy handling, low cost, and environmentally friendly nature in comparison to heavy metals. The cyclization through C(sp3)-H functionalization has become an indispensable synthetic strategy to deliver invaluable heterocyclic molecules.44−50 Earlier, Schafer et al. introduced an α-C(sp3)-H activated intramolecular 6-annulation with the Zr(NM2)4 catalyst ((i), Scheme 1).19 We established NiX2·nH2O-catalyzed bimolecular 5-annulation ((ii), Scheme 1).20 Chen et al. reported Pd(OAc)2-PhI(OAc)2-mediated intramolecular 4-annulation of protected primary aliphatic amines ((iii), Scheme 1).26 The major limitations of these methods are the utilization of toxic heavy metal catalysts, intramolecular cyclization ((i) and (iii), Scheme 1), severe water susceptibility to catalysis ((ii), Scheme 1), high reaction temperature (145 °C), requirements of a stoichiometric oxidant [PhI(OAc)2] and base in excess (2.5 equiv.), and slow reaction rates. In a continuous effort to synthesize novel polycyclic heteroaromatics for design, synthesis, and fabrication of new organic nanomaterials to discover their innovative organic electronic properties51−54 for developing new generation devices,55,56 herein, we disclose a direct C–O bond forming 5-annulation ((iv), Scheme 1) and 6-annulation ((v), Scheme 1) through C(sp3)-H functionalization through nonmetallic catalysis to furnish polycyclic N,O-heteroaromatics.
Scheme 1. Annulation with Primary Aliphatic Amine-C(sp3)-H.
1,3-Oxazoles are a fundamental class of five-membered privileged heterocyclic motifs that have profound importance in natural products, pharmaceuticals, agrochemicals, and materials.57−62 For instance, the benzoxazole-based natural product nataxazoles (A, Figure 1) displayed anticancer, antibacterial, and cytotoxic bioactivities,61 and the heterocycle-grafted graphene oxide organic material was used as a valuable high-performance supercapacitor electrode (B, Figure 1).62 The widespread application of the heterocycles led to the development of several synthetic strategies such as CuII-catalyzed oxidative cyclization,63 [2 + 2 + 1] annulation of alkyne and nitrile,64 dehydrogenative annulation,65 PdII-catalyzed annulation of amides,66 and photocatalytic Ru catalysis.67 The importance of metal-free synthesis was also realized using organocatalysis,68 cyclization of aminoacids,69 PhIO-TfOH,70 and dehydrogenative I2-TBHP cyclization.71 1,4-Oxazines have shown immense importance in biological and material sciences.72−80 For example, oxazine derivatives (C, D, Figure 1) were employed in the treatment of neurodegenerative, inflammatory,78 autoimmune, and cardiovascular79 disorders, and the thin films of naphthoxazine-based materials (E, Figure 1) showed unique surface plasmon polarization with emission enhancement properties. Syntheses of oxazines were mainly achieved through PdII catalysis of bisvinylphosphate81 and intramolecular cyclization with the CuI-catalyst82 and triphenylphosphine.83
Figure 1.

Important benzoxazole and oxazine analogues.
Results and Discussion
Intending to synthesize the heterocyclic moiety through nonmetallic amine-C(sp3)-H functionalization, at first, we focused on α-C(sp3)-H functionalization, and for that, we have selected phenanthrenequinone (1a) and benzylamine (2a) as two reacting partners, which might yield the corresponding oxazole derivative (4a, Table 1). The feasibility of the 5-annulation catalysis was examined with 20 mol % triflic acid and camphor sulfonic acid (CSA) as possible catalysts (entries 1, 2, Table 1) under aerobic conditions in toluene at 100 °C (bath temperature) for 15 h. In these cases, the α,α-C(sp3)-H-amine derived oxazole was obtained successfully in moderate yields (48, and 57%, respectively). To our delight, on the use of I2, the reaction was rapidly completed (2 h), and the yield significantly improved (79%, entry 3). Keeping I2 as a promising catalyst (20 mol %), different solvents such as protic EtOH (entry 4), relatively low boiling point EDC (entry 5), and highly polar DMF and DME (entries 6, 7, respectively) were screened to achieve moderate to high yields (58–78%). Gratifyingly, dioxane proved to be the best reaction medium providing the desired product (4a) rapidly (1 h), and 4a was furnished almost in quantitative yield (98%, entry 8). The increase in reaction time (6 h) resulted in a decrease in yield of the desired product (95%, entry 9). In the absence of oxygen (argon atmosphere), the yield drastically dropped to 22% even after continuation of the reaction for 24 h (entry 10). Surprisingly, the change of catalyst loading (entries 11, 12) led to a considerable decrease in the yield of the desired product (4a). The 5-annulation was unsuccessful in the absence of the catalyst under the same reaction conditions (entry 13).
Table 1. Development of C(sp3)-H-Functionalized 5-Annulationa.

| entry | catalyst | solventb | time (h) | yield (%)c |
|---|---|---|---|---|
| 1 | triflic acid | toluene | 15 | 48 |
| 2 | CSA | toluene | 15 | 57 |
| 3 | I2 | toluene | 2 | 79 |
| 4d | I2 | EtOH | 1.5 | 58 |
| 5 | I2 | EDC | 1.5 | 70 |
| 6 | I2 | DMF | 1.5 | 75 |
| 7 | I2 | DME | 1.5 | 78 |
| 8 | I2 | dioxane | 1 | 98 |
| 9d | I2 | dioxane | 6 | 95 |
| 10e | I2 | dioxane | 24 | 22 |
| 11f | I2 | dioxane | 12 | 79 |
| 12g | I2 | dioxane | 1 | 94 |
| 13 | dioxane | 24 | NDh |
Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), catalyst (20 mol %).
Volume of solvent: 2 mL.
Yield of pure 4a after silica gel column chromatography using ethyl acetate in petroleum ether as an eluent.
Under reflux (∼80 °C for EtOH and ∼100 °C for dioxane).
Under argon.
Catalyst (15 mol %).
Catalyst (25 mol %).
Not detected.
The substrate scope of the dual α-C(sp3)-H-functionalized 5-annulation was investigated using a wide range of primary aliphatic amines (2a–f, Scheme 2) and aromatic 1,2-diketones (1) under the developed optimized reaction conditions (entry 8, Table 1). The ester-substituted primary amine (2b, Scheme 2) and phenanthrenequinone (1a) as well as pyrene-based 1,2-diketones (1b) smoothly transformed into the corresponding desired products (4b, 4c, Scheme 2), which did not require any change in the developed reaction conditions (entry 8, Table 1).
Scheme 2. Dual α-C(sp3)-H-Functionalized 5-Annulation to Oxazoles.
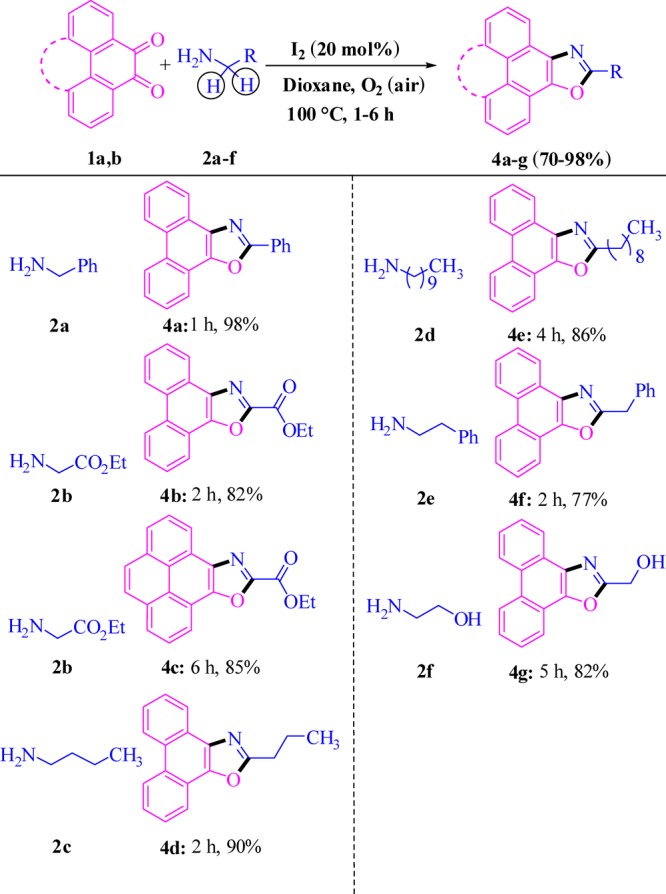
The presence of weak hydrophobic interactions through the installation of the hydrocarbon residue is frequently needed for the generation of organic nanomaterials.51−56 Herein, use of n-butyl-, decyl-, and phenethylamine (2c–e) was successful in achieving corresponding potent nanobuilding blocks (4d–f) with high yields (77–90%) in 2–4 h. Hydrogen bonding is one of the most common gluing interactions operating between the organic nanounits, and the synthesis of an oxazole derivative (4g) bearing the −OH group worked well with the coupling partner aminoethanol (2f).
After dual α-C(sp3)-H functionalization of various primary aliphatic amines, we turned our attention for making the strategy more diverse and general through replacement of one of the α-C(sp3)-H by −CO2H so that inexpensive and easily available amino acids may be employed as the key substrate for decarboxylative 5-annulation. To our delight, the attempted reaction between phenyl glycine (3a) and phenanthrenequinone (1a) rapidly (3 h) furnished the desired product (4a, Scheme 3) under similar reaction conditions (entry 8, Table 1) with high yield (80%). Many aliphatic and aromatic residues, phenolic −OH, alcoholic −OH, −SMe, and chirality were well tolerated to produce a variety of polycyclic oxazoles in high yield, and reaction rates were faster on use of a wide range of amino acids (3a–k). In a competitive experiment of cleaving C(sp3)-H versus −CO2H in glycine (3k) possessing two α-C(sp3)-H groups as well as one α-CO2H group furnished oxazole derivative 4p through a decarboxylation process exclusively, rather than formation of the −CO2H group tethered oxazole derivative (4q) by functionalization of consecutive two C(sp3)-H. Thus, C-CO2H breaking is more favorable under the catalytic conditions with respect to C(sp3)-H cleavage. The structure of compound 4j is established by single-crystal X-ray diffraction analysis.84 Further, 2,7-dibromo-phenanthrene-9,10-dione under optimized reaction conditions (entry 8, Table 1) led to exclusive construction of corresponding oxazole derivatives 4r and 4s through the decarboxylation process.
Scheme 3. Decarboxylative α-C(sp3)-H-Functionalized 5-Annulation.

Next, we envisioned functionalization of both α-C(sp3)-H and β-C(sp3)-H under the reaction conditions leading to the construction of valuable six-annulated polycyclic oxazine derivatives (Scheme 4). To verify that we have employed primary amines (5) possessing a β-C(sp3)-H, which was obtained by just replacing the α-H with an alkyl group of 2 (Scheme 4). To our surprise, six-annulated desired oxazine derivative 6a (Scheme 4) was rapidly (2 h) formed upon treatment of 1-phenylethylamine (5a) with 9,10-phenanthrenequinone (1a) under the catalytic conditions in high yield (80%). Herein, phenyl (6a, 6b, 6e, 6g, 6i) and activated aromatic residues such as naphthyl (6c), 4-tolyl (6d), 4-hydroxyphenyl (6h), and methyl [γ-C(sp3)-H, 6e) as well as the ester functionality (6g, 6h) are well tolerated to furnish selectively polyaromatic oxazine systems (6a–i) in 2–8 h with high yields (68–81%). In the presence of the cyclohexyl group, the desired product tautomerized to 6f through migration of a double bond. Probably, high steric and electronic repulsion appeared due to the presence of axial and equatorial C–H bonds (cyclohexyl residue) in the close vicinity of the C=N bond and lone pair in 6fi, which led to release of unwanted repulsive forces to obtain thermodynamically stable and fully aromatic 3-cyclohexyl-4H-phenanthro[9,10-b][1,4]oxazines (6f).
Scheme 4. α,β-C(sp3)-H-Functionalized 6-Annulation to Oxazines.
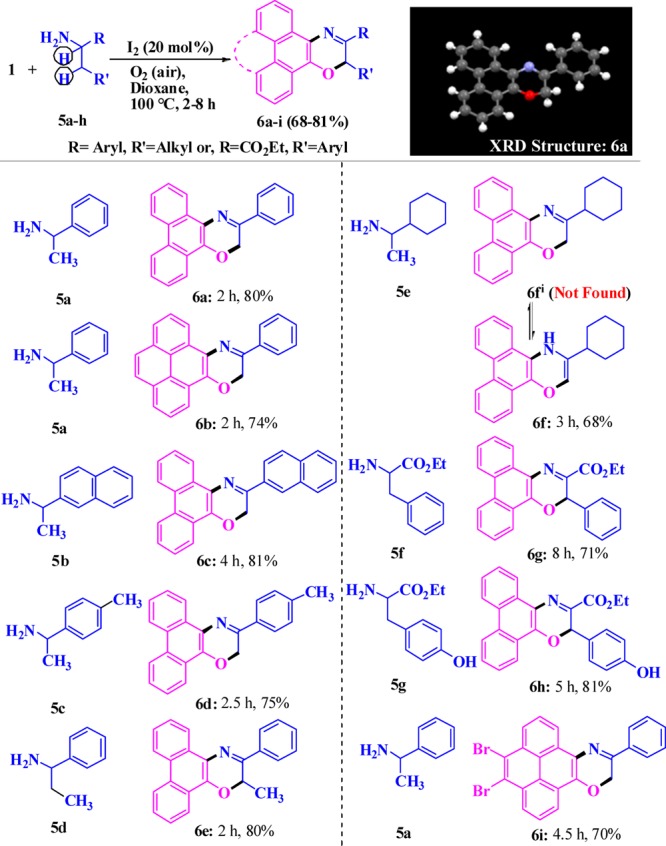
To understand the reactivity and versatility of the C(sp3)-H-functionalized annulation strategy, we have replaced the R group (5, Scheme 4) by a −CO2Et group in the aliphatic amines (7a–c, Scheme 5). To our surprise, a 6-annulation reaction occurred under the reaction conditions through α-C(sp3)-H functionalization of the primary aliphatic amines as well as an O–C coupling with the ester group involving the release of −OEt to afford valuable polynuclearoxazine-2-ones (8a–c). The molecular-iodine-catalyzed synthesis of 3-alkyl-2H-phenanthro[9,10-b][1,4]oxazin-2-one (8) was rapid (2–4.5 h) and high yielding (72–77%). Formation of all new oxazoles, oxazines, oxazine-2-ones, and analogues was confirmed by spectroscopic analyses, recorded melting points (Supporting Information), and also single-crystal X-ray diffraction analyses of 4j(84) and 6a.85 It is worthy to note that, although most of the reported metal catalysts for C–H-activated functionalization reactions are moisture-sensitive, herein, the catalyst I2 efficiently performed the diverse C–H-functionalized annulation catalysis even in the presence of water.
Scheme 5. α-C(sp3)-H-Functionalized Lactonization to Oxazine-2-ones.
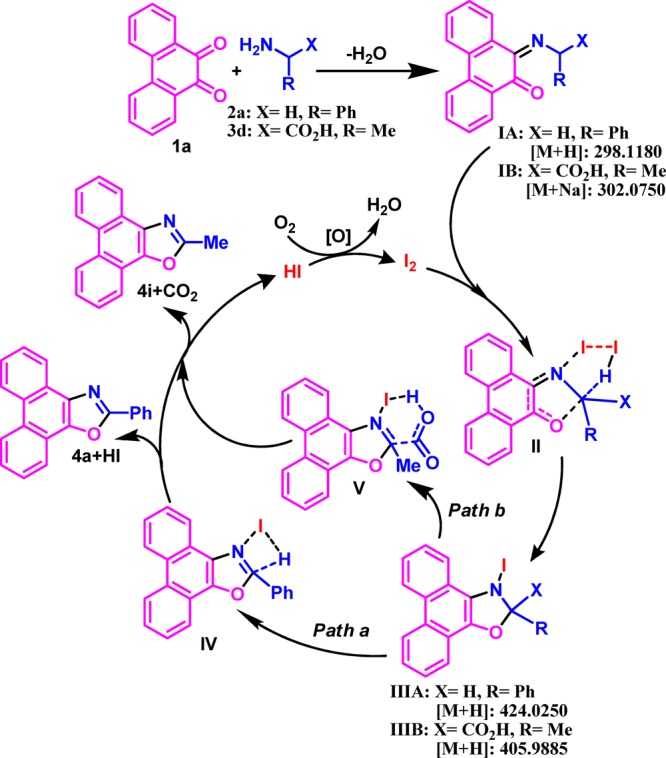
Out of several possibilities,86−91 the current catalysis is expected to pass through the initial formation of a monoimine IA and IB, which may proceed through activation of an α-C(sp3)-H subsequent formation of a five-membered transition state (II) with the catalyst (I2) to intermediates IIIA and IIIB (Scheme 6). The formation of intermediates IA and IB, as well as IIIA and IIIB, was detected in the mass spectral analyses of the ongoing reaction (Supporting Information). A second α-C(sp3)-H activation of IIIA with the close vicinity of the iodine substituent possessing lone pairs and larger size (IV) may release HI to furnish the dual α-C(sp3)-H-functionalized 5-annulated product (4a, path a). On the other hand, decarboxylative α-C(sp3)-H 5-annulation of IIIB is possibly passing through a six-membered transition state (V) to 4i (path b). In both cases, the generated HI is expected to oxidize immediately by aerial O2 to regenerate I2 for the next catalytic cycle. The role of inexpensive O2 as an oxidant was verified (entry 10, Table 1) by performing the reaction in the absence of O2 (argon atmosphere) where the yield of 4a drastically reduced (22%) in the presence of the catalyst. Herein, decarboxylation through a six-membered transition state (V, path b, Scheme 6) is energetically more favorable than the second α-C(sp3)-H activation by a four-membered one (IV, path a), which was reflected in the 5-annulation of glycine to produce exclusively 4p instead of 4q.
Scheme 6. Mechanistic Hypothesis for I2-Catalyzed 5-Annulation.
The 6-annulation is expected to proceed in a similar fashion through the initial formation of a monoimine VI (Scheme 7), which may proceed via an iodine-mediated activation of an α-C(sp3)-H through an eight-membered transition state (VII). Removal of HI leads to the generation of the putative intermediate VIII, which further tautomerizes to transition state IX. It undergoes O–C coupled cyclization, oxidative C=N formation (X) to desired product 6, and the regeneration of molecular iodine for the next catalytic cycle.
Scheme 7. Mechanistic Hypothesis for I2-Catalyzed 6-Annulation.

Next, we moved to investigate morphological characteristics of some final compounds through fabrication by the spin coating method. Thus, we choose three representative compounds, namely, 4n, 6b, and 6h, carrying strong aromatic electron clouds, polar functional groups, phenolic −OH, van der Waals interaction, and π–π stacking attractive forces, which might operate in between the small organic molecules (nanobuilding block) to fabricate the desired organic nanomaterials through the spin coating, deep coating, and Langmuir–Blodget techniques. The SEM images of the spin-coated materials displayed nanomorphologies such as the existence of the rod-like structure of compound 4n (Figure 2), the sheet-like architecture of compound 6b (Figure 3), and the flower-like nanostructure of compound 6h (Figure 4). We are now investigating the development of their innovative optical, nanoelectronics, and I–V characteristics for potential application in the valuable solar cell, supercapacitor, and nonvolatile memory devices.
Figure 2.

Rod-like nanostructures of 4n observed in SEM imaging.
Figure 3.

Sheet-like nanostructures of 6b observed in SEM imaging.
Figure 4.

Flower-like nanostructures of 6h observed in SEM imaging.
Conclusions
In conclusion, we have demonstrated a general nonmetallic synthetic strategy for diverse C(sp3)-H functionalization of unprotected primary aliphatic amines with an I2 catalyst to intermolecular 5- and 6-annulation. A variety of unsubstituted, substituted, acid, ester, alcohol, and thiol derivatives of primary amines and their chiral analogues were successfully coupled to 1,2-diketone analogues to obtain a series of new polyaromaticoxazoles, 1,4-oxazines, oxazin-2-one, and chiral heterocycles with rapid reaction rates and high yields. This newly established I2-catalyzed α,α-C(sp3)-H, decarboxylative α-C(sp3)-H, lactonized α-C(sp3)-H, and α,β-C(sp3)-H-functionalized 5- and 6-annulation as well as α-C(sp3)-H-activated 6-lactonization of primary aliphatic amines will open up another avenue for developing a metal-free sustainable strategy for simple, rapid, and diverse construction of functional-group-decorated heteroaromatics, which will find considerable application in organic synthesis, medicinal chemistry, materials science, and organic nanoelectronics for smart devices.
Experimental Section
General Methods
All reagents were purchased from commercial suppliers and used without further purification. Petroleum ether used in our experiments was in the boiling range of 60–80 °C. Column chromatography was performed on silica gel (100–200 and 230–400 mesh). Reported melting points are uncorrected. Prior to melting point determination, recrystallization was carried out; for compounds whose NMR spectra were taken in CDCl3, recrystallization was carried out in CDCl3, and for those whose NMR spectra were taken in DMSO-d6, recrystallization was carried out in an ethyl acetate/hexane mixture. 1H NMR and 13C NMR spectra were recorded at ambient temperature in CDCl3/DMSO-d6 solution. Chemical shifts are reported in ppm (δ) relative to internal reference tetramethylsilane. Coupling constants are quoted in Hz (J). Proton multiplicities are represented as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), and m (multiplet). Splitting patterns that could not be interpreted are designated as multiplet (m). Infrared spectra were recorded on an FT-IR spectrometer in thin films. HR-MS data were acquired by electron spray ionization on a Q-tof-micro quadruple mass spectrophotometer. X-ray crystallographic data were taken using an X-ray diffractometry instrument.
General Procedure for the Synthesis of Oxazoles 4a–s (GP-I)
To a mixture of phenanthrenequinone (1, 1.0 mmol) and amine (2a–f)/amino acid (3a–k, 1.1 mmol, 1.1 equiv.) in dioxane (2 mL), I2 (20 mol %, 50 mg) was added, and the solution was refluxed under air to complete the reaction, which was monitored by TLC. Dioxane was removed from the reaction mixture, and the residue was purified by silica gel column chromatography using a suitable eluent to afford the desired product.
2-Phenylphenanthro[9,10-d]oxazole (4a)92−94
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and benzylamine (1.1 mmol, 0.12 mL). Purification by column chromatography (8% EtOAc-pet ether) afforded the title compound as a white solid (289 mg, 0.98 mmol, 98% yield). 1H NMR (300 MHz, CDCl3): δ 7.54–7.59 (m, 3H), 7.61–7.75 (m, 4H), 8.27–8.37 (m, 3H), 8.59–8.66 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3): δ 120.7, 120.9, 122.8, 123.3, 123.6, 126.0, 126.1, 126.2, 127.0, 127.1, 127.3, 127.5, 128.8, 129.1, 130.8, 135.4, 144.7, 162.0; FT-IR (KBr, cm–1): 708.3, 755.3, 1058.9, 1235.1, 1450.9, 1480.0, 1548.4, 2853.2, 2923.7.
Ethyl Phenanthro[9,10-d]oxazole-2-carboxylate (4b)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and ethyl glycinate (1.1 mmol, 112 mg). Purification by column chromatography (8% EtOAc-pet ether) afforded the title compound as a yellow solid (239 mg, 0.82 mmol, 82% yield). mp 168–170 °C; 1H NMR (300 MHz, CDCl3): δ 1.50 (t, J = 7.2 Hz, 3H), 4.58 (q, J1 = 6.9 Hz, J2 = 15 Hz, 2H), 7.49–7.68 (m, 4H), 8.23–8.26 (m, 1H), 8.53–8.59 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3): δ 14.3, 29.7, 63.0, 120.3, 121.7, 123.1, 123.4, 123.7, 125.6, 126.9, 127.5, 127.8, 127.9, 129.2, 130.6, 134.7, 146.3, 151.8, 156.3; FT-IR (KBr, cm–1): 722.9, 755.4, 1148.0, 1278.8, 1451.1, 1534.4, 1733.4, 2857.4, 2925.2; HRMS (ESI-TOF) m/z calcd for C18H14NO3 [M + H]+: 292.0974, found 292.0971.
Ethyl Pyreno[4,5-d]oxazole-10-carboxylate (4c)
The compound was prepared following GP-I employing pyrene-4,5-dione (1.0 mmol, 232 mg) and ethyl glycinate (1.1 mmol, 112 mg). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a reddish orange solid (268 mg, 0.85 mmol, 85% yield). mp 146–148 °C; 1H NMR (300 MHz, CDCl3): δ 11.54 (t, J = 7.2 Hz, 3H), 4.63 (q, J1 = 12 Hz, J2 = 7.2 Hz, 2H), 7.97–8.13 (m, 4H), 8.18 (d, J = 6.9 Hz, 2H), 8.53 (d, J = 7.5 Hz, 1H), 8.81 (d, J = 7.5 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 14.3, 63.1, 119.0, 119.5, 120.6, 123.5, 124.4, 124.7, 126.1, 126.4, 126.7, 126.8, 127.3, 128.2, 131.7, 131.8, 135.5, 147.1, 152.1, 156.3; FT-IR (KBr, cm–1): 668.9, 1215.8, 1732.5, 2927.6, 3019.7; HRMS (ESI-TOF) m/z calcd for C20H14NO3 [M + H]+: 316.0974, found 316.0977.
2-Propylphenanthro[9,10-d]oxazole (4d)92−94
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and butyl amine (1.1 mmol, 0.11 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (235 mg, 0.90 mmol, 90% yield). 1H NMR (300 MHz, CDCl3): δ 1.02 (t, J = 6.9 Hz, 3H), 1.86–1.99 (m, 2H), 2.97 (t, J = 7.5 Hz, 2H), 7.50–7.64 (m, 4H) 8.08–8.11 (m, 1H), 8.42 (d, J = 7.8 Hz, 1H), 8.58 (q, J1 = 4.5 Hz, J2 = 7.8 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 13.9, 21.0, 30.8, 120.6, 121.1, 122.7, 123.4, 123.7, 128.8, 126.06, 126.13, 127.1, 127.3, 128.7, 128.9, 134.2, 144.7, 166.1.
2-Nonylphenanthro[9,10-d]oxazole (4e)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and decyl amine (1.1 mmol, 0.22 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a brown solid (297 mg, 0.86 mmol, 86% yield). mp 52–54 °C; 1H NMR (300 MHz, CDCl3): δ 0.84–0.89 (m, 3H), 1.34–1.47 (m, 12H), 1.88–1.96 (m, 2H), 3.02 (t, J = 7.2 Hz, 2H), 7.53–7.70 (m, 4H), 7.13 (d, J = 7.8 Hz, 1H), 8.48 (d. J = 7.8 Hz, 1H), 8.58–8.61 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 14.0, 22.6, 27.3, 28.8, 29.2, 29.3, 31.8, 120.4, 120.9, 122.5, 123.2, 123.5, 125.6, 125.8, 126.0, 127.0, 127.1, 128.5, 128.7, 134.1, 144.5, 166.0; FT-IR (KBr, cm–1): 754.9, 1215.6, 1346.8, 1560.2, 1579.6, 2855.1, 2927.0; HRMS (ESI-TOF) m/z calcd for C24H28NO [M + H]+: 346.2171, found 346.2166.
2-Benzylphenanthro[9,10-d]oxazole (4f)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and phenethylamine (1.1 mmol, 0.14 mL). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a yellow solid (238 mg, 0.77 mmol, 77% yield). mp 98–100 °C; 1H NMR (300 MHz, CDCl3): δ 4.41 (s, 2H), 7.25–7.35 (m, 3H), 7.43 (d, J = 7.2 Hz, 2H), 7.61–7.71 (m, 4H), 8.14–8.17 (m, 1H), 8.49 (d, J = 7.8 Hz, 1H), 8.66–8.70 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 35.2, 120.6, 120.9, 122.6, 123.3, 123.5, 125.8, 126.0, 126.1, 127.1, 127.2, 128.6, 128.7, 128.8, 134.2, 135.3, 145.1, 163.7; FT-IR (KBr, cm–1): 711.3, 760.3, 1111.9, 1235.1, 1450.9, 1480.0, 1556.4, 2859.3, 2925.9; HRMS (ESI-TOF) m/z calcd for C22H16NO [M + H]+: 310.1232, found 310.1236.
2-(Phenanthro[9,10-d]oxazol-2-yl)ethanol (4g)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and ethanolamine (1.1 mmol, 0.07 mL). Purification by column chromatography (20% EtOAc-pet ether) afforded the title compound as a yellow solid (204 mg, 0.82 mmol, 82% yield). mp 174–176 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.99 (s, 2H), 6.14 (brs, 1H), 7.76–7.98 (m, 4H), 8.22–8.28 (m, 1H), 8.63 (d, 1H, J = 10.8 Hz), 9.25 (t, 2H, J = 9 Hz); 13C{1H} NMR (75 MHz, DMSO-d6): δ 56.1, 119.8, 119.9, 121.6, 123.6, 123.8, 125.0, 126.0, 126.4, 127.4, 127.5, 127.8, 128.1, 133.0, 143.7, 164.5; FT-IR (KBr, cm–1): 669.1, 756.6, 1215.9, 1408.8, 1456.3, 1634.2, 1727.9, 2927.1, 3019.8; HRMS (ESI-TOF) m/z calcd for C16H12NO2 [M + H]+: 250.0868, found 250.0871.
4-(Phenanthro[9,10-d]oxazol-2-yl-methyl)phenol (4h)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and tyrosine (1.1 mmol, 200 mg). Purification by column chromatography (20% EtOAc-pet ether) afforded the title compound as a yellow solid (195 mg, 0.60 mmol, 60% yield). mp 200–202 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.29 (s, 2H), 6.72 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.60–7.75 (m, 4H), 8.03–8.06 (m, 1H), 8.28–8.31 (m, 1H), 8.79 (t, J = 6.6 Hz, 2H), 9.37 (s, 1H); 13C{1H} NMR (75 MHz, DMSO-d6): δ 33.0, 115.0, 119.7, 119.8, 121.7, 123.5, 123.7, 124.9, 125.1, 125.8, 126.1, 127.2, 127.3, 127.7, 127.8, 129.5, 133.2, 143.6, 156.0, 164.4; FT-IR (KBr, cm–1): 723.7, 756.2, 809.8, 1256.7, 1450.9, 1518.7, 1594.6, 2924.0, 3399.9; HRMS (ESI-TOF) m/z calcd for C22H16NO2 [M + H]+: 326.1181, found 326.1178.
2-Methylphenanthro[9,10-d]oxazole (4i)92−94
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and alanine (1.1 mmol, 98 mg). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a light green solid (198 mg, 0.85 mmol, 85% yield). mp 128–130 °C; 1H NMR (300 MHz, CDCl3): δ 2.72 (s, 3H), 7.48–7.68 (m, 4H), 8.07–8.09 (m, 1H), 8.41 (d, J = 7.8 Hz, 1H), 8.60 (d, J = 7.5 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 14.5, 120.4, 120.9, 122.4, 123.2, 123.5, 125.7, 125.85, 125.92, 127.0, 127.2, 128.5, 128.7, 134.2, 144.7, 162.3; FT-IR (KBr, cm–1): 723.1, 753.9, 1032.8, 1231.8, 1451.8, 1587.7, 1737.7, 2853.7, 2924.4.
2-Isopropylphenanthro[9,10-d]oxazole (4j)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and valine (1.1 mmol, 129 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (232 mg, 0.89 mmol, 89% yield). mp 76–78 °C; 1H NMR (300 MHz, CDCl3): δ 0.74 (s, J = 6.9 Hz, 6H), 2.54–2.63 (m, 1H), 6.73–6.89 (m, 4H), 7.36 (d, J = 7.5 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.80–7.83 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 20.7, 29.1, 120.5, 121.0, 122.6, 123.3, 123.5, 125.7, 125.9, 126.1, 127.0, 127.2, 128.5, 128.7, 134.0, 144.4; FT-IR (KBr, cm–1): 722.7, 730.7, 755.7, 1033.7, 1078.8, 1350.7, 1450.3, 1558.0, 1578.4, 2928.9, 2962.1; HRMS (ESI-TOF) m/z calcd for C18H16NO [M + H]+: 262.1232, found 262.1229.
10-Isopropylpyreno[4,5-d]oxazole (4k)
The compound was prepared following GP-I employing pyrene-4,5-dione (1.0 mmol, 232 mg) and valine (1.1 mmol, 129 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (256 mg, 0.90 mmol, 90% yield). mp 96–98 °C; 1H NMR (300 MHz, CDCl3): δ 1.61 (d, J = 6.9 Hz, 6H), 3.43–3.52 (m, 1H), 7.95–8.13 (m, 6H), 8.38 (d, J = 7.5 Hz, 1H); 8.72 (d, J = 7.8 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 20.8, 29.3, 117.4, 120.0, 120.3, 122.9, 124.8, 125.2, 126.0, 126.3, 127.3, 128.0, 131.7, 131.8, 145.2, 170.4; FT-IR (KBr, cm–1): 716.1, 826.2, 1178.6, 1303.8, 1564.2, 1603.8, 1726.9, 2925.6, 2969.4; HRMS (ESI-TOF) m/z calcd For C21H16NO [M + H]+: 286.1232, found 286.1227.
2-Isobutylphenanthro[9,10-d]oxazole (4l)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and leucine (1.1 mmol, 144 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a light brown solid (253 mg, 0.92 mmol, 92% yield). mp 68–70 °C; 1H NMR (300 MHz, CDCl3): δ 1.03 (d, J = 6.6 Hz, 6H), 2.29–2.38 (m, 1H), 2.90 (d, J = 7.2 Hz, 2H), 7.54–7.67 (m, 4H), 8.13–8.16 (m, 1H), 8.46 (d, J = 7.8 Hz, 1H), 8.60–8.65 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 22.5, 27.9, 37.8, 120.6, 121.1, 122.7, 123.4, 123.6, 125.8, 126.05, 126.14, 127.1, 127.3, 128.6, 128.9, 134.3, 144.7, 165.5; FT-IR (KBr, cm–1): 724.3, 751.8, 1051.7, 1323.7, 1450.5, 1556.5, 1578.9, 2870.9, 2926.6, 2959.9; HRMS (ESI-TOF) m/z calcd for C19H18NO [M + H]+: 276.1388, found 276.1393.
(R)-2-sec-Butylphenanthro[9,10-d]oxazole (4m)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and isoleucine (1.1 mmol, 144 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as yellow oil (250 mg, 0.91 mmol, 91% yield). [α]D20: +1.46° (c 2.667, CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.95 (t, J = 7.5 Hz, 3H), 1.47 (d, J = 6.9 Hz, 3H), 1.74–1.83(m, 1H), 1.94–2.10 (m, 1H), 3.13–3.20 (m, 1H), 7.56–7.67 (m, 4H), 8.16–8.19 (m, 1H), 8.45–8.48 (m, 1H), 8.64–8.69 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 11.8, 18.3, 28.4, 36.1, 120.6, 121.1, 122.7, 123.3, 123.6, 125.7, 125.9, 126.2, 126.5, 127.0, 127.2, 128.5, 128.8, 134.1, 144.5, 169.4; FT-IR (KBr, cm–1): 724.1, 754.9, 1053.2, 1324.3, 1452.1, 1521.4, 1578.5, 1618.7, 2932.3, 2967.9; HRMS (ESI-TOF) m/z calcd for C19H18NO [M + H]+: 276.1388, found 276.1383.
2-(2-(Methylthio)ethyl)phenanthro[9,10-d]oxazole (4n)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and methionine (1.1 mmol, 164 mg). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a deep yellow solid (270 mg, 0.92 mmol, 92% yield). mp 82–84 °C; 1H NMR (300 MHz, CDCl3): δ 2.17 (s, 3H), 3.07 (t, J = 7.5 Hz, 2H), 3.33 (s, J = 7.5 Hz, 2H), 7.57–7.69 (m, 4H), 8.11 (d, J = 7.5 Hz, 1H), 8.44 (d, J = 7.8 Hz, 1H), 8.60 (t, J = 4.1 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 15.6, 29.3, 31.3, 120.6, 120.9, 122.6, 123.4, 123.6, 125.9, 126.0, 126.2, 127.2, 127.3, 128.7, 128.9, 134.2, 144.8, 163.9; FT-IR (KBr, cm–1): 726.6, 763.0, 1054.3, 1321.7, 1432.1, 1558.7, 1576.7, 2853.3, 2922.5; HRMS (ESI-TOF) m/z calcd for C18H16NOS [M + H]+: 294.0953, found 294.0957.
1-(Phenanthro[9,10-d]oxazol-2-yl)ethanol (4o)
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and threonine (1.1 mmol, 131 mg). Purification by column chromatography (10% EtOAc-pet ether) afforded the title compound as a yellow solid (218 mg, 0.83 mmol, 83% yield). mp 178–180 °C; 1H NMR (300 MHz, CDCl3): δ 1.61 (dd, J1 = 1.8 Hz, J2 = 1.8 Hz, 3H), 5.04–5.10 (m, 1H), 6.00 (brs, 1H), 7.66–7.78 (m, 4H), 8.19 (d, J = 7.5 Hz, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.89 (t, J = 8.1 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 21.0, 62.1, 119.8, 121.6, 123.6, 123.8, 125.0, 125.8, 126.3, 127.3, 127.4, 127.8, 128.0, 132.8, 143.4, 167.1; FT-IR (KBr, cm–1): 715.8, 1245.9, 1426.8, 1501.3, 1654.2, 1729.5, 2928.7, 3087.5, 3325.6; HRMS (ESI-TOF) m/z calcd for C17H14NO2 [M + H]+: 264.1025, found 264.1030.
Phenanthro[9,10-d]oxazole (4p)92−94
The compound was prepared following GP-I employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and glycine (1.1 mmol, 83 mg). Purification by column chromatography (1% EtOAc-pet ether) afforded the title compound as a yellow solid (193 mg, 0.88 mmol, 88% yield). 1H NMR (300 MHz, CDCl3): δ 7.59–7.70 (m, 4H), 8.12–8.18 (m, 2H), 8.47 (d, J = 7.5 Hz, 1H), 8.59 (d, J = 6.3 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 120.85, 120.93, 122.7, 123.4, 123.6, 126.0, 126.2, 127.2, 128.9, 129.4, 133.4, 144.5, 151.2.
5,10-Dibromo-2-methylphenanthro[9,10-d]oxazole (4r)
The compound was prepared following GP-I employing 1,6-dibromo-9,10-phenanthrenequinone (1.0 mmol, 366 mg) and alanine (1.1 mmol, 98 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a light yellow solid (320 mg, 0.82 mmol, 82% yield). mp 150–152 °C; 1H NMR (300 MHz, CDCl3): δ 2.62 (s, 3H), 6.89 (d, 1H, J = 2.4 Hz), 6.95–6.98 (m, 1H), 7.17–7.28 (m, 1H), 7.27 (d, 1H, J = 8.7 Hz), 7.46–7.53 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 15.1, 119.8, 119.9, 121.4, 121.8, 122.8, 128.9, 130.8, 131.4, 131.8, 135.0, 136.5, 137.6, 139.6, 140.2, 141.1, 164.0; FT-IR (KBr, cm–1): 744.7, 756.8, 1046.7, 1228.8, 1399.8, 1601.7, 1775.6, 2826.8, 2970.5; HRMS (ESI-TOF) m/z calcd for C16H10Br2NO [M + H]+: 389.9129, found 389.9133 (one of the major peaks).
5,10-Dibromo-2-isopropylphenanthro[9,10-d]oxazole (4s)
The compound was prepared following GP-I employing 1,6-dibromo-9,10-phenanthrenequinone (1.0 mmol, 366 mg) and valine (1.1 mmol, 129 mg). Purification by column chromatography (1% EtOAc-pet ether) afforded the title compound as a light yellow solid (335 mg, 0.80 mmol, 80% yield). mp 118–120 °C; 1H NMR (300 MHz, CDCl3): δ 1.25 (d, J = 6 Hz, 6H), 2.85–2.94 (m, 1H), 6.98–7.04 (m, 1H), 7.07–7.11 (m, 1H), 7.32–7.41 (m, 1H), 7.53–7.66 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3): δ 21.1, 30.2, 119.1, 119.8, 120.6, 122.3, 123.0, 125.5, 128.5, 130.3, 133.7, 138.7, 140.1, 140.6; FT-IR (KBr, cm–1): 701.7, 748.7, 801.7, 1051.7, 1101.9, 1299.7, 1508.7, 1602.0, 1659.4, 2889.7, 2971.2; HRMS (ESI-TOF) m/z calcd for C18H14Br2NO [M + H]+: 417.9442, found 417.9437 (one of the major peaks).
General Procedure for the Synthesis of Oxazines 6a-i (GP-II)
To a mixture of phenanthrenequinone (1, 1.0 mmol) and α-substituted amine (5a-e)/amino acid ester (5f, g, 1.1 mmol, 1.1 equiv.) in dioxane (2 mL), I2 (20 mol %, 50 mg) was added, and the solution was refluxed under air to complete the reaction, which was monitored by TLC. Dioxane was removed from the reaction mixture, and the residue was purified by silica gel column chromatography using a suitable eluent to afford the desired product.
3-Phenyl-2H-Phenanthro[9,10-b][1,4]oxazine (6a)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and α-methylbenzylamine (1.1 mmol, 0.14 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (247 mg, 0.80 mmol, 80% yield). mp 136–138 °C; 1H NMR (300 MHz, CDCl3): δ 5.22 (s, 2H), 7.49–7.68 (m, 7H), 8.10 (t, J = 3.6 Hz, 2H), 8.25–8.28 (m, 1H), 8.61 (t, J = 9.3 Hz, 2H), 8.74 (d, J = 8.1 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 62.8, 122.5, 122.7, 122.77, 122.82, 124.7, 124.8, 125.1, 126.7, 126.8, 127.0, 127.1, 127.2, 128.8, 130.2, 130.7, 130.9, 135.8, 139.0, 154.7; FT-IR (KBr, cm–1): 722.6, 752.0, 1127.5, 1324.6, 1449.1, 2853.6, 2924.5; HRMS (ESI-TOF) m/z calcd for C22H16NO [M + H]+: 310.1232, found 310.1237.
11-Phenyl-10H-pyreno[4,5-b][1,4]oxazine (6b)
The compound was prepared following GP-II employing pyrene-4,5-dione (1.0 mmol, 232 mg) and α-methylbenzylamine (1.1 mmol, 0.14 mL). Purification by column chromatography (20% DCM-pet ether) afforded the title compound as a yellow solid (246 mg, 0.74 mmol, 74% yield). mp 146–148 °C; 1H NMR (300 MHz, CDCl3): δ 5.26 (s, 2H), 7.457.49 (m, 3H), 7.90–8.11 (m, 8H), 8.43 (d, J = 7.8 Hz, 1H), 8.90 (dd, J1 = 1.5 Hz, J2 = 6 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 62.8, 119.8, 120.0, 121.5, 123.8, 124.3, 125.3, 125.6, 126.0, 126.3, 126.7, 127.7, 128.7, 129.2, 130.9, 131.0, 135.7, 140.0, 155.0; FT-IR (KBr, cm–1): 717.3, 825.1, 1051.7, 1384.6, 1455.5, 1567.5, 1646.3, 2857.4, 2932.2; HRMS (ESI-TOF) m/z calcd for C24H16NO [M + H]+: 334.1232, found 334.1227.
3-(Naphthalen-2-yl)-2H-phenanthro[9,10-b][1,4]oxazine (6c)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and 1-(naphthalen-2-yl)-ethylamine (1.1 mmol, 0.13 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow crystalline solid (291 mg, 0.81 mmol, 81% yield). mp 104–106 °C; 1H NMR (300 MHz, CDCl3): δ 5.13 (s, 2H), 7.19–7.74 (m, 8H), 7.80–7.99 (m, 3H), 8.28–8.34 (m, 1H), 8.58–8.78 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3): δ 65.6, 122.4, 122.67, 122.74, 122.9, 124.2, 124.6, 125.0, 125.2, 125.3, 125.9, 126.3, 126.6, 126.8, 127.0, 127.2, 127.3, 128.3, 128.6, 130.1, 130.7, 133.0, 134.1, 134.5, 139.0, 157.1; FT-IR (KBr, cm–1): 718.6, 749.3, 756.5, 1026.2, 1235.2, 1508.1, 1674.4, 2852.0, 2923.0; HRMS (ESI-TOF) m/z calcd for C26H18NO [M + H]+: 360.1388, found 360.1393.
3-p-Tolyl-2H-phenanthro[9,10-b][1,4]oxazine (6d)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and 1-(4-methylphenyl)-ethylamine (1.1 mmol, 0.16 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (242 mg, 0.75 mmol, 75% yield). mp 118–120 °C; 1H NMR (300 MHz, CDCl3): δ 2.41 (s, 3H), 5.19 (s, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.54–7.68 (m, 4H), 7.99 (d, J = 8.1 Hz, 2H), 8.24–8.27 (m, 1H), 8.58–8.64 (m, 2H), 8.73–8.76 (m, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 21.5, 62.6, 122.3, 122.5, 122.6, 124.6, 124.7, 124.9, 126.5, 126.6, 127.0, 129.4, 130.1, 130.4, 133.0, 138.8, 141.3, 154.7; FT-IR (KBr, cm–1): 721.9, 755.9, 1032.4, 1126.2, 1179.5, 1384.2, 1494.5, 1608.1, 2853.1, 2923.9; HRMS (ESI-TOF) m/z calcd for C23H18NO [M + H]+: 324.1388, found 324.1386.
2-Methyl-3-phenyl-2H-phenanthro[9,10-b][1,4]oxazine (6e)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and α-ethylbenzylamine (1.1 mmol, 0.16 mL). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a yellow solid (258 mg, 0.80 mmol, 80% yield). mp 172–174 °C; 1H NMR (300 MHz, CDCl3): δ 1.43 (d, J = 6.6 Hz, 3H), 5.80 (q, J = 6.9 Hz, 1H), 7.42–7.72 (m, 7H), 8.12–8.15 (m, 2H), 8.30–8.33 (m, 1H), 8.60–8.66 (m, 2H), 8.82 (dd, J1 = 0.6 Hz, J2 = 9 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 16.1, 67.6, 122.4, 122.5, 122.7, 123.4, 124.9, 125.5, 126.6, 126.7, 126.8, 127.0, 127.9, 128.4, 128.7, 130.0, 130.7, 132.8, 135.4, 136.3, 157.8; FT-IR (KBr, cm–1): 682.5, 760.5, 1057.9, 1427.8, 1564.1, 2853.9, 2925.5; HRMS (ESI-TOF) m/z calcd for C23H18NO [M + H]+: 324.1388, found 324.1383.
3-Cyclohexyl-4H-phenanthro[9,10-b][1,4]oxazine (6f)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and 1-cyclohexylethylamine (1.1 mmol, 0.16 mL). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a pale yellow low melting solid (215 mg, 0.68 mmol, 68% yield). 1H NMR (300 MHz, CDCl3): δ 1.51–1.63 (m, 4H), 1.73–1.77 (m, 2H), 1.84–1.88 (m, 2H), 2.13–2.23 (m, 2H), 2.84–2.92 (m, 1H),6.81 (s, 1H), 7.52–7.64 (m, 5H), 8.05–8.08 (m, 1H), 8.26–8.28 (m, 1H), 8.66 (d, J = 8.1 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3): δ 26.0, 29.7, 31.7, 37.8, 99.6, 120.3, 121.1, 122.6, 123.4, 123.6, 123.9, 124.8. 125.2, 126.75, 126.83, 127.6, 128.0, 128.4, 147.8, 163.1; FT-IR (KBr, cm–1): 754.3, 1215.7, 1450.9, 1632.5, 1728.5, 2854.6, 2927.0, 3019.2; HRMS (ESI-TOF) m/z calcd for C22H22NO [M + H]+: 316.1701, found 316.1697.
Ethyl 2-Phenyl-2H-phenanthro[9,10-b][1,4]oxazine-3-carboxylate (6g)95
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and phenylalanine ethyl ester (1.1 mmol, 193 mg). Purification by column chromatography (8% EtOAc-pet ether) afforded the title compound as a yellow solid (271 mg, 0.71 mmol, 71% yield). 1H NMR (300 MHz, CDCl3): δ 1.41 (t, J = 7.2 Hz, 3H), 4.41–4.46 (m, 2H), 6.60 (s, 1H), 7.17–7.22 (m, 3H), 7.33–7.37 (m, 2H), 7.54–7.68 (m, 4H), 8.32–8.35 (m, 1H), 8.54 (dd, J1 = 8.1 Hz, J2 = 15 Hz, 2H), 8.64 (dd, J1 = 1.2 Hz, J2 = 15 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 14.1, 29.6, 62.2, 122.3, 122.7, 122.8, 123.4, 123.5, 124.4, 125.4, 126.7, 126.9, 127.4, 128.5, 128.6, 129.0, 129.5, 132.1, 135.6, 139.5, 147.4, 163.3.
Ethyl 2-Phenyl-2H-phenanthro[9,10-b][1,4]oxazine-3-carboxylate (6h)
The compound was prepared following GP-II employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and tyrosine ethyl ester (1.1 mmol, 209 mg). Purification by column chromatography (15% EtOAc-pet ether) afforded the title compound as a deep yellow solid (322 mg, 0.81 mmol, 81% yield). mp 192–194 °C; 1H NMR (300 MHz, CDCl3): δ 1.39 (t, J = 7.2 Hz, 3H), 4.40 (q, J = 6.9 Hz, 2H), 6.49 (s, 1H), 6.60 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 10.5 Hz, 2H), 7.51–7.66 (m, 5H), 8.28 (d, J = 7.8 Hz, 1H), 8.46–8.57 (m, 2H), 8.65 (d, J = 8.1 Hz, 1H); 13C{1H} NMR (75 MHz, DMSO-d6): δ 13.5, 61.2, 70.8, 115.1, 121.5, 122.2, 122.47, 122.54, 122.8, 123.5, 124.7, 125.2, 125.8, 126.9, 127.3, 128.2, 128.3, 128.5, 130.8, 138.2, 148.1, 158.0, 161.9; FT-IR (KBr, cm–1): 724.4, 755.0, 1170.0, 1229.5, 1514.2, 1613.1, 1715.7, 2853.4, 2924.7, 3370.8; HRMS (ESI-TOF) m/z calcd For C25H20NO4 [M + H]+: 398.1392, found 398.1388.
4,5-Dibromo-11-phenyl-10H-pyreno[4,5-b][1,4]oxazine (6i)
The compound was prepared following GP-II employing 4,5-dibromopyrene-4,5-dione (1.0 mmol, 390 mg) and α-methylbenzylamine (1.1 mmol, 0.14 mL). Purification by column chromatography (15% DCM-pet ether) afforded the title compound as a bright yellow solid (344 mg, 0.70 mmol, 70% yield). mp 156–158 °C; 1H NMR (300 MHz, CDCl3): δ 4.93 (s, 2H), 7.64–7.68 (m, 3H), 8.10–8.30 (m, 8H); 13C{1H} NMR (75 MHz, CDCl3): δ 60.7, 118.8, 119.0, 120.5, 122.8, 123.3, 124.3, 124.6, 124.7, 125.3, 125.7, 126.7, 127.7, 128.2, 129.9, 130.0, 135.1, 139.1, 155.0; FT-IR (KBr, cm–1): 720.2, 900.1, 1021.5, 1121.5, 1314.9, 1422.7, 1601.7, 1678.6, 2823.2, 2954.7; HRMS (ESI-TOF) m/z calcd for C24H14Br2NO [M + H]+: 489.9442, found 489.9447 (one of the major peaks).
General Procedure for the Synthesis of Oxazine-2-ones 8a–c (GP-III)
To a mixture of phenanthrenequinone (1, 1.0 mmol) and amino acid ester (5i–k, 1.1 mmol, 1.1 equiv.) in dioxane (2 mL), I2 (20 mol %, 50 mg) was added, and the solution was refluxed under air to complete the reaction, which was monitored by TLC. Dioxane was removed from the reaction mixture, and the residue was purified by silica gel column chromatography using a suitable eluent to afford the desired product.
Ethyl 2-Phenyl-2H-phenanthro[9,10-b][1,4]oxazine-3-carboxylate (8a)
The compound was prepared following GP-III employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and alanine ethyl ester (1.1 mmol, 117 mg). Purification by column chromatography (8% EtOAc-pet ether) afforded the title compound as a yellow solid (201 mg, 0.77 mmol, 77% yield). mp 198–200 °C; 1H NMR (300 MHz, CDCl3): δ 2.66 (s, 3H), 7.63–7.75 (m, 4H), 8.42 (d, J = 8.1 Hz, 1H), 8.56–8.62 (m, 2H), 8.77–8.79 (m, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 21.3, 122.4, 122.5, 122.7, 123.4, 126.9, 127.4, 127.7, 128.0, 128.3, 129.0, 130.8, 131.3, 141.1, 153.6; FT-IR (KBr, cm–1): 723.7, 763.3, 1086.3, 1396.5, 1731.5, 1741.8, 2853.0, 2923.3. HRMS (ESI-TOF) m/z calcd For C17H12NO2 [M + H]+: 262.0868, found 262.0864.
3-Isopropyl-2H-phenanthro[9,10-b][1,4]oxazin-2-one (8b)
The compound was prepared following GP-III employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and valine ethyl ester (1.1 mmol, 145 mg). Purification by column chromatography (3% EtOAc-pet ether) afforded the title compound as a yellow solid (201 mg, 0.77 mmol, 77% yield). mp 148–150 °C; 1H NMR (300 MHz, CDCl3): δ 1.40–1.43 (m, 6H), 3.48–3.56 (m, 1H), 7.61–7.70 (m, 4H), 8.39 (d, J = 7.8 Hz, 1H), 8.50–8.56 (m, 2H), 8.80 (d, J = 7.8 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3): δ 20.0, 32.1, 122.4, 122.6, 122.7, 123.4, 123.5, 126.9, 127.4, 127.6, 128.0, 128.6, 128.9, 131.2, 153.0, 160.5; FT-IR (KBr, cm–1): 723.1, 751.1, 1036.3, 1451.7, 1623.9, 1731.3, 2853.6, 2925.8. HRMS (ESI-TOF) m/z calcd for C19H16NO2 [M + H]+: 290.1181, found 290.1176.
3-Isopropyl-2H-phenanthro[9,10-b][1,4]oxazin-2-one (8c)
The compound was prepared following GP-III employing 9,10-phenanthrenequinone (1.0 mmol, 208 mg) and leucine ethyl ester (1.1 mmol, 159 mg). Purification by column chromatography (2% EtOAc-pet ether) afforded the title compound as a pale yellow solid (218 mg, 0.72 mmol, 72% yield). mp 106–108 °C; 1H NMR (300 MHz, CDCl3): δ 1.01 (d, J = 6.6 Hz, 6H), 2.39–2.43 (m, 1H), 2.83 (d, J = 6.9 Hz, 2H), 7.54–7.66 (m, 4H), 8.25 (d, J = 7.8 Hz, 1H), 8.40–8.44 (m, 2H), 8.65–8.68 (m, 1H); 13C{1H} NMR (75 MHz, CDCl3): δ 22.6, 26.3, 42.5, 122.3, 122.4, 122.6, 123.2, 123.3, 126.8, 127.2, 127.5, 127.8, 128.3, 128.8, 131.0, 140.6, 153.4, 155.7. FT-IR (KBr, cm–1): 725.2, 764.7, 1193.4, 1293.8, 1397.2, 1451.1, 1495.8, 1554.6, 1734.2, 2870.4, 2926.6, 2955.6. HRMS (ESI-TOF) m/z calcd for C20H18NO2 [M + H]+: 304.1338, found 304.1333.
Acknowledgments
The research facility by CAS-SAP, DST-FIST, financial support (CSIR project no. 02(0250)/15/EMR-II), and research fellowship (SRF to S.D.) from CSIR and D. S. Kothari fellowship (T.D.) from UGC, India, are gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b03501.
The authors declare no competing financial interest.
Supplementary Material
References
- Jadhav M. S.; Righi P.; Marcantoni E.; Bencivenni G. Enantioselective α-Benzoyloxylation of Ketones Promoted by Primary Amine Catalyst. J. Org. Chem. 2012, 77, 2667–2674. 10.1021/jo2024976. [DOI] [PubMed] [Google Scholar]
- Solovyov A.; Amundsen T. J.; Daniels J. J.; Kim Y.-G.; Katz A. Primary Amine Confinement at the Interface of Grafted Calixarenes and Silica. Chem. Mater. 2008, 20, 6316–6318. 10.1021/cm801302g. [DOI] [Google Scholar]
- Alesi W. R. Jr.; Kitchin J. R. Evaluation of a Primary Amine-Functionalized Ion-Exchange Resin for CO2 Capture. Ind. Eng. Chem. Res. 2012, 51, 6907–6915. 10.1021/ie300452c. [DOI] [Google Scholar]
- Martin N. I.; Beeson W. T.; Woodward J. J.; Marletta M. A. NG-Aminoguanidines from Primary Amines and the Preparation of Nitric Oxide Synthase Inhibitors. J. Med. Chem. 2008, 51, 924–931. 10.1021/jm701119v. [DOI] [PubMed] [Google Scholar]
- Sarciaux M.; Pantel L.; Midrier C.; Serri M.; Gerber C.; de Figueiredo R. M.; Campagne J.-M.; Villain-Guillot P.; Gualtieri M.; Racine E. Total Synthesis and Structure–Activity Relationships Study of Odilorhabdins, a New Class of Peptides Showing Potent Antibacterial Activity. J. Med. Chem. 2018, 61, 7814–7826. 10.1021/acs.jmedchem.8b00790. [DOI] [PubMed] [Google Scholar]
- Chikhale R. V.; Barmade M. A.; Murumkar P. R.; Yadav M. R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. 10.1021/acs.jmedchem.8b00281. [DOI] [PubMed] [Google Scholar]
- White M. C. Adding Aliphatic C–H Bond Oxidations to Synthesis. Science 2012, 335, 807–809. 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- Dey A.; Pimparkar S.; Deb A.; Guin S.; Maiti D. Chelation-Assisted Palladium-Catalyzed γ-Arylation of Aliphatic Carboxylic Acid Derivatives. Adv. Synth. Catal. 2017, 359, 1301–1307. 10.1002/adsc.201601121. [DOI] [Google Scholar]
- Pati T. K.; Debnath S.; Kundu M.; Khamrai U.; Maiti D. K. 3-Amino-1-methyl-1H-pyridin-2-one-Directed PdII Catalysis: C(sp3)–H Activated Diverse Arylation Reaction. Org. Lett. 2018, 20, 4062–4066. 10.1021/acs.orglett.8b01618. [DOI] [PubMed] [Google Scholar]
- Clark J. R.; Feng K.; Sookezian A.; White M. C. Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization. Nat. Chem. 2018, 10, 583–591. 10.1038/s41557-018-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally A.; Haffemayer B.; Collins B. S. L.; Gaunt M. J. Palladium-catalysed C–H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 2014, 510, 129–133. 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]
- He C.; Gaunt M. J. Ligand-Enabled Catalytic C-H Arylation of Aliphatic Amines by a Four-Membered-Ring Cyclopalladation Pathway. Angew. Chem., Int. Ed. 2015, 54, 15840–15844. 10.1002/anie.201508912. [DOI] [PubMed] [Google Scholar]
- Willcox D.; Chappell B. G. N.; Hogg K. F.; Calleja J.; Smalley A. P.; Gaunt M. J. A general catalytic β-C-H carbonylation of aliphatic amines to β-lactams. Science 2016, 354, 851–857. 10.1126/science.aaf9621. [DOI] [PubMed] [Google Scholar]
- Verma S.; Baig R. B. N.; Nadagouda M. N.; Varma R. S. Oxidative C-H activation of amines using protuberant lychee-like goethite. Sci. Rep. 2018, 8, 2024–2030. 10.1038/s41598-018-20246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev V. G.; Shabashov D.; Daugulis O. Highly Regioselective Arylation of sp3 C–H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- Zhang S.-Y.; He G.; Nack W. A.; Zhao Y.; Li Q.; Chen G. Palladium-Catalyzed Picolinamide-Directed Alkylation of Unactivated C(sp3)–H Bonds with Alkyl Iodides. J. Am. Chem. Soc. 2013, 135, 2124–2127. 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]
- Chan K. S. L.; Wasa M.; Chu L.; Laforteza B. N.; Miura M.; Yu J.-Q. Ligand-enabled cross-coupling of C(sp3)–H bonds with arylboron reagents via Pd(II)/Pd(0) catalysis. Nat. Chem. 2014, 6, 146–150. 10.1038/nchem.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Ge H. Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 2017, 9, 26–32. 10.1038/nchem.2606. [DOI] [Google Scholar]
- Bexrud J. A.; Eisenberger P.; Leitch D. C.; Payne P. R.; Schafer L. L. Selective C–H Activation α to Primary Amines. Bridging Metallaaziridines for Catalytic, Intramolecular α-Alkylation. J. Am. Chem. Soc. 2009, 131, 2116–2118. 10.1021/ja808862w. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Roy D.; Khamarui S.; Maiti D. K. Ni(II)–salt catalyzed activation of primary amine-sp3Cα–H and cyclization with 1,2-diketone to tetrasubstitutedimidazoles. Chem. Commun. 2014, 50, 2477–2480. 10.1039/C3CC48437H. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Wang C.; Dong G. A Hydrazone-Based exo-Directing-Group Strategy for β-C-H Oxidation of Aliphatic Amines. Angew. Chem., Int. Ed. 2016, 55, 5299–5303. 10.1002/anie.201600912. [DOI] [PubMed] [Google Scholar]
- Rodríguez N.; Romero-Revilla J. A.; Fernández-Ibáñez M. Á.; Carretero J. C. Palladium-catalyzed N-(2-pyridyl)sulfonyl-directed C(sp3)–H γ-arylation of amino acid derivatives. Chem. Sci. 2013, 4, 175–179. 10.1039/C2SC21162A. [DOI] [Google Scholar]
- Chen K.; Wang D.; Li Z.-W.; Liu Z.; Pan F.; Zhang Y.-F.; Shi Z.-J. Palladium catalyzed C(sp3)–H acetoxylation of aliphatic primary amines to γ-amino alcohol derivatives. Org. Chem. Front. 2017, 4, 2097–2101. 10.1039/C7QO00432J. [DOI] [Google Scholar]
- Hu X.-X.; Liu J.-B.; Wang L.-L.; Huang F.; Sun C.-Z.; Chen D.-Z. The stabilizing effect of the transient imine directing group in the Pd(II)-catalyzed C(sp3)–H arylation of free primary amines. Org. Chem. Front. 2018, 5, 1670–1678. 10.1039/C8QO00094H. [DOI] [Google Scholar]
- Kapoor M.; Liu D.; Young M. C. Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. 2018, 140, 6818–6822. 10.1021/jacs.8b05061. [DOI] [PubMed] [Google Scholar]
- He G.; Zhao Y.; Zhang S.; Lu C.; Chen G. Highly Efficient Syntheses of Azetidines, Pyrrolidines, and Indolines via Palladium Catalyzed IntramolecularAmination of C(sp3)–H and C(sp2)–H Bonds at γ and δ Positions. J. Am. Chem. Soc. 2012, 134, 3–6. 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Young M. C.; Wang C.; Magness D. M.; Dong G. Catalytic C(sp(3))-H Arylation of Free Primary Amines with an exo Directing Group Generated In Situ. Angew. Chem., Int. Ed. 2016, 55, 9084–9087. 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]
- Anastas P. T.; Warner J. C.. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998. [Google Scholar]
- Li C.-J.; Trost B. M. Green chemistry for chemical synthesis. Proc. Natl. Acad. Sci. 2008, 105, 13197–13202. 10.1073/pnas.0804348105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn P. J. The importance of Green Chemistry in Process Research and Development. Chem. Soc. Rev. 2012, 41, 1452–1461. 10.1039/C1CS15041C. [DOI] [PubMed] [Google Scholar]
- Chen D.-F.; Han Z.-Y.; He Y.-P.; Yu J.; Gong L.-Z. Metal-free oxidation/C(sp3)-H Functionalization of UnactivatedAlkynes using Pyridine-N-oxide as the External Oxidant. Angew. Chem., Int. Ed. 2012, 51, 12307–12310. 10.1002/anie.201205062. [DOI] [PubMed] [Google Scholar]
- Gogoi A.; Modi A.; Guin S.; Rout S. K.; Das D.; Patel B. K. A metal free domino synthesis of 3-aroylindoles via two sp3 C–H activation. Chem. Commun. 2014, 50, 10445–10447. 10.1039/C4CC04407J. [DOI] [PubMed] [Google Scholar]
- Moteki S. A.; Usui A.; Selvakumar S.; Zhang T.; Maruoka K. Metal-free C-H bond activation of branched aldehydes with a hypervalent iodine(III) catalyst under visible-light photolysis: successful trapping with electron-deficient olefins. Angew. Chem., Int. Ed. 2014, 53, 11060–11064. 10.1002/anie.201406513. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Fang H.; Qian P.; Han J.; Pan Y. Metal-Free Oxidative C(sp3)–H Bond Functionalization of Alkanes and Conjugate Addition to Chromones. Org. Lett. 2014, 16, 5342–5345. 10.1021/ol502524d. [DOI] [PubMed] [Google Scholar]
- Osorio-Nieto U.; Chamorro-Arenas D.; Quintero L.; Höpfl H.; Sartillo-Piscil F. Transition Metal-Free Selective Double sp3 C–H Oxidation of Cyclic Amines to 3-Alkoxyamine Lactams. J. Org. Chem. 2016, 81, 8625–8632. 10.1021/acs.joc.6b01566. [DOI] [PubMed] [Google Scholar]
- Evoniuk C. J.; Gomes G. D. P.; Hill S. P.; Fujita S.; Hanson K.; Alabugin I. V. Coupling N–H Deprotonation, C–H Activation, and Oxidation: Metal-Free C(sp3)–H Aminations with Unprotected Anilines. J. Am. Chem. Soc. 2017, 139, 16210–16221. 10.1021/jacs.7b07519. [DOI] [PubMed] [Google Scholar]
- Gao B.; Chen K.; Bi X.; Wang J. Intramolecular functionalization of C(sp3)-H bonds adjacent to an amide nitrogen atom: Metal-free synthesis of 2-hydroxy-benzoxazinone derivatives. Tetrahedron 2017, 73, 7005–7010. 10.1016/j.tet.2017.09.001. [DOI] [Google Scholar]
- Wu X.; Zhao P.; Geng X.; Zhang J.; Gong X.; Wu Y.-d.; Wu A.-x. Direct Oxidative Cleavage of Multiple Csp3–H Bonds and a C–C Bond in 2-(Pyridin-2-yl)acetate Derivatives: Formal [3 + 1 + 1] Synthesis of 3-(Pyridin-2-yl)indolizine Skeletons. Org. Lett. 2017, 19, 3319–3322. 10.1021/acs.orglett.7b01492. [DOI] [PubMed] [Google Scholar]
- Duhamel T.; Stein C. J.; Martínez C.; Reiher M.; Muñiz K. Engineering Molecular Iodine Catalysis for Alkyl–Nitrogen Bond Formation. ACS Catal. 2018, 8, 3918–3925. 10.1021/acscatal.8b00286. [DOI] [Google Scholar]
- Ambethkar S.; Kalaiselvi M.; Ramamoorthy J.; Padmini V. I2-Catalyzed Oxidative Cross-Coupling Reaction of Methyl Ketones and 2-(2-Aminophenyl) Benzimidazole: Facile Access to Benzimidazo[1,2-c]quinazoline. ACS Omega 2018, 3, 5021–5028. 10.1021/acsomega.8b00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Zhou Y.; Yang Z.; Li Q.; Zhao L.; Liu P. Iodine-Catalyzed C–H Amidation and Imination at the 2α-Position of 2,3-Disubstituted Indoles with Chloramine Salts. J. Org. Chem. 2018, 83, 4665–4673. 10.1021/acs.joc.8b00286. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Hou J.; Liu J.; Yu W.; Chang J. Synthesis of imidazo[1,5-a]pyridinesviaI2-mediated sp3C–H amination. Org. Biomol. Chem. 2018, 16, 5653–5660. 10.1039/C8OB01501E. [DOI] [PubMed] [Google Scholar]
- Deshmukh D. S.; Bhanage B. M. Ruthenium-Catalyzed Annulation of N-Cbz Hydrazones via C–H/N–N Bond Activation for the Rapid Synthesis of Isoquinolines. Synthesis 2019, 51, 2506–2514. 10.1055/s-0037-1611795. [DOI] [Google Scholar]
- Colby D. A.; Bergman R. G.; Ellman J. A. Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation. Chem. Rev. 2010, 110, 624–655. 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung C. S.; Dong V. M. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon–Carbon Bonds by Oxidizing Two Carbon–Hydrogen Bonds. Chem. Rev. 2011, 111, 1215–1292. 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]
- Zhang S.-Y.; Zhang F.-M.; Tu Y.-Q. Direct sp3 α-C–H activation and functionalization of alcohol and ether. Chem. Soc. Rev. 2011, 40, 1937–1949. 10.1039/c0cs00063a. [DOI] [PubMed] [Google Scholar]
- Rouquet G.; Chatani N. Catalytic Functionalization of C(sp2)-H and C(sp3)-H Bonds by Using Bidentate Directing Groups. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Recent Advances in Radical C-H Activation/Radical Cross Coupling. Chem. Rev. 2017, 117, 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- Nairoukh Z.; Cormier M.; Marek I. Merging C–H and C–C bond cleavage in organic synthesis. Nat. Rev. Chem. 2017, 1, 1–17. 10.1038/s41570-017-0035. [DOI] [Google Scholar]
- Pandit P.; Chatterjee N.; Halder S.; Hota S. K.; Patra A.; Maiti D. K. PhIO as a Powerful Cyclizing Reagent: Regiospecific [3+2]-Tandem Oxidative Cyclization of Imine toward Cofacially Self-Aggregated Low Molecular Mass Organic Materials. J. Org. Chem. 2009, 74, 2581–2584. 10.1021/jo8028136. [DOI] [PubMed] [Google Scholar]
- Maiti D. K.; Halder S.; Pandit P.; Chatterjee N.; De Joarder D.; Pramanik N.; Saima Y.; Patra A.; Maiti P. K. Synthesis of Glycal-Based Chiral Benzimidazoles by VO(acac)2–CeCl3 Combo Catalyst and Their Self-Aggregated Nanostructured Materials. J. Org. Chem. 2009, 74, 8086–8097. 10.1021/jo901458k. [DOI] [PubMed] [Google Scholar]
- Maiti D. K.; Debnath S.; Nawaz S. M.; Dey B.; Dinda E.; Roy D.; Ray S.; Mallik A.; Hussain S. A. Composition-dependent nanoelectronics of amido-phenazines: non-volatile RRAM and WORM memory devices. Sci. Rep. 2017, 7, 13308. 10.1038/s41598-017-13754-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda T.; Maiti D. K.; Panda M. K. Inkless Writing and Self-Erasing Security Feature of (Z)-1,2-Diarylacrylonitrile-Based Materials: A Confidential Data Communication. ACS Appl. Mater. Interfaces 2018, 10, 29100–29106. 10.1021/acsami.8b08279. [DOI] [PubMed] [Google Scholar]
- Stępień M.; Gońka E.; Żyła M.; Sprutta N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]
- Vespa M.; Cann J. R.; Dayneko S. V.; Melville O. A.; Hendsbee A. D.; Zou Y.; Lessard B. H.; Welch G. C. Synthesis of a Perylene Diimide Dimer with Pyrrolic N–H Bonds and N-Functionalized Derivatives for Organic Field-Effect Transistors and Organic Solar Cells. Eur. J. Org. Chem. 2018, 4592–4599. 10.1002/ejoc.201801055. [DOI] [Google Scholar]
- Wipf P. Synthetic Studies of Biologically Active Marine Cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. 10.1021/cr00038a013. [DOI] [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- Yeh V. S. C. Recent advances in the total syntheses of oxazole-containing natural products. Tetrahedron 2004, 60, 11995–12042. 10.1016/j.tet.2004.10.001. [DOI] [Google Scholar]
- Farmanzadeh D.; Najafi M. Theoretical study of anticancer properties of indolyl-oxazole drugs and their interactions with DNA base pairs in gas phase and solvent. Struct. Chem. 2015, 26, 831–844. 10.1007/s11224-014-0546-8. [DOI] [Google Scholar]
- Ward D. N.; Talley D. C.; Tavag M.; Menji S.; Schaughency P.; Baier A.; Smith P. J. UK-1 and structural analogs are potent inhibitors of hepatitis C virus replication. Bioorg. Med. Chem. Lett. 2014, 24, 609–612. 10.1016/j.bmcl.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai W.; Zhou W.; Du Z.; Du Y.; Zhang H.; Jia X.; Xie L. M.; Yu T.; Huang W. Benzoxazole and benzimidazole heterocycle-grafted graphene for high-performance supercapacitor electrodes. J. Mater. Chem. 2012, 22, 23439–23446. 10.1039/c2jm35234f. [DOI] [Google Scholar]
- Wan C.; Zhang J.; Wang S.; Fan J.; Wang Z. Facile Synthesis of Polysubstituted Oxazoles via A Copper-Catalyzed Tandem Oxidative Cyclization. Org. Lett. 2010, 12, 2338–2341. 10.1021/ol100688c. [DOI] [PubMed] [Google Scholar]
- He W.; Li C.; Zhang L. An Efficient [2 + 2 + 1] Synthesis of 2,5-Disubstituted Oxazoles via Gold-Catalyzed Intermolecular Alkyne Oxidation. J. Am. Chem. Soc. 2011, 133, 8482–8485. 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Zhang C.; Jiao N. Synthesis of oxazoles through copper-mediated aerobic oxidative dehydrogenative annulation and oxygenation of aldehydes and amines. Angew. Chem., Int. Ed. 2012, 51, 11367–11370. 10.1002/anie.201206382. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Huang L.; Huang H.; Li H.; Wu W.; Jiang H. Palladium-Catalyzed Sequential C–N/C–O Bond Formations: Synthesis of Oxazole Derivatives from Amides and Ketones. Org. Lett. 2014, 16, 5906–5909. 10.1021/ol502916a. [DOI] [PubMed] [Google Scholar]
- Chatterjee T.; Cho J. Y.; Cho E. J. Synthesis of Substituted Oxazoles by Visible-Light Photocatalysis. J. Org. Chem. 2016, 81, 6995–7000. 10.1021/acs.joc.6b00989. [DOI] [PubMed] [Google Scholar]
- Xie J.; Jiang H.; Chenga Y.; Zhu C. Metal-free, organocatalytic cascade formation of C–N and C–O bonds through dual sp3 C–H activation: oxidative synthesis of oxazole derivatives. Chem. Commun. 2012, 48, 979–981. 10.1039/C2CC15813B. [DOI] [PubMed] [Google Scholar]
- Xu W.; Kloeckner U.; Nachtsheim B. J. Direct Synthesis of 2,5-Disubstituted Oxazoles through an Iodine-Catalyzed Decarboxylative Domino Reaction. J. Org. Chem. 2013, 78, 6065–6074. 10.1021/jo400753n. [DOI] [PubMed] [Google Scholar]
- Saito A.; Taniguchi A.; Kambara Y.; Hanzawa Y. Metal-Free [2 + 2 + 1] Annulation of Alkynes, Nitriles, and Oxygen Atoms: Iodine(III)-Mediated Synthesis of Highly Substituted Oxazoles. Org. Lett. 2013, 15, 2672–2675. 10.1021/ol4009816. [DOI] [PubMed] [Google Scholar]
- Gao W.-C.; Hu F.; Huo Y.-M.; Chang H.-H.; Li X.; Wei W.-L. I2-Catalyzed C–O Bond Formation and Dehydrogenation: Facile Synthesis of Oxazolines and Oxazoles Controlled by Bases. Org. Lett. 2015, 17, 3914–3917. 10.1021/acs.orglett.5b01933. [DOI] [PubMed] [Google Scholar]
- Nair M. G.; Salter O. C.; Kisliuk R. L.; Gaumont Y.; North G. Folate analogs. 22. Synthesis and biological evaluation of two analogs of dihydrofolic acid possessing a 7,8-dihydro-8-oxapterin ring system. J. Med. Chem. 1983, 26, 1164–1168. 10.1021/jm00362a015. [DOI] [PubMed] [Google Scholar]
- Buckman B. O.; Mohan R.; Koovakkat S.; Liang A.; Trinh L.; Morrissey M. M. Design, synthesis, and biological activity of novel purine and bicyclic pyrimidine factor Xa inhibitors. Bio. Org. Med. Chem. Lett. 1998, 8, 2235–2240. 10.1016/S0960-894X(98)00386-2. [DOI] [PubMed] [Google Scholar]
- Fringuelli R.; Pietrella D.; Schiaffella F.; Guarraci A.; Perito S.; Bistoni F.; Vecchiarelli A. Anti-Candida albicans properties of novel benzoxazine analogues. Bio. Org. Med. Chem. 2002, 10, 1681–1686. 10.1016/S0968-0896(02)00038-X. [DOI] [PubMed] [Google Scholar]
- Ilaš J.; Anderluh P. Š.; Dolenc M. S.; Kikelj D. Recent advances in the synthesis of 2H-1,4-benzoxazin-3-(4H)-ones and 3,4-dihydro-2H-1,4-benzoxazines. Tetrahedron 2005, 61, 7325–7348. 10.1016/j.tet.2005.05.037. [DOI] [Google Scholar]
- Vogelsang J.; Cordes T.; Forthmann C.; Steinhauer C.; Tinnefeld P. Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc. Natl. Acad. Sci. 2009, 106, 8107–8112. 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindhu T. J.; Arikkatt S. D.; Vincent G.; Chandran M.; Bhat A. R.; Krishnakumar K. Biological Activities of Oxazine and its Derivatives: a Review. Int. J. Pharm. Sci. Res. 2013, 4, 134–143. [Google Scholar]
- Höelscher P.; Jautelat R.; Rehwinkel H.; Jaroch S.; Süzle D.; Hillmann M.; Burton G. A.; McDonald F. M.. Benzoxazine derivatives and benzothiazine derivatives having nos-inhibitory and antioxidant properties Patent Appl. WO0181324, February 5, 2001.
- Burton G. A.; Rehwinkel H.; Jaroch S.; Hoelscher P.; Suelzle D.; Hillmann M.; McDonald F. M.. Benzoxazine and benzothiazine derivatives and their use in medicines Patent Appl. WO0017173, April 25, 2000.
- Wismanto W. Y.; Hidayat R.; Tjia M. O.; Fujiwara Y.; Murata K.; Ogawa Y.; Yoshida H.; Fujii A.; Ozaki M. Emission Enhancement Characteristics Of Oxazine In Pmma Matrix Influenced By Surface Plasmon Polariton Induced On Sinusoidal Silver Grating. J. Nonlinear Opt. Phys. Mater. 2012, 21, 1250013. 10.1142/S0218863512500130. [DOI] [Google Scholar]
- Claveau E.; Gillaizeau I.; Blu J.; Bruel A.; Coudert G. Easy Access to New Heterocyclic Systems: 1,4-Oxazine and Substituted1,4-Oxazines. J. Org. Chem. 2007, 72, 4832–4836. 10.1021/jo070528n. [DOI] [PubMed] [Google Scholar]
- Jangili P.; Kashanna J.; Das B. Synthesis of dihydrobenzo[1,4]oxazines using copper catalyzed intramolecular ring closure reaction. Tetrahedron Lett. 2013, 54, 3453–3456. 10.1016/j.tetlet.2013.04.090. [DOI] [Google Scholar]
- Mohebat R.; Abadi A. Y. E.; Soltani A.; Saghafi M. New and efficient synthesis of 1,4-oxazines through the reaction of acetylenic esters and nitrosonaphthols in the presence of phosphine derivatives. ARKIVOC 2016, 1. 10.3998/ark.5550190.p009.543. [DOI] [Google Scholar]
- CCDC no. 4j: 1551936.
- CCDC no. 6a: 1551938.
- Ishikawa T.; Kimura M.; Kumoi T.; Iida H. Coupled Flavin-Iodine Redox Organocatalysts: Aerobic Oxidative Transformation from N-Tosylhydrazones to 1,2,3-Thiadiazoles. ACS Catal. 2017, 7, 4986–4989. 10.1021/acscatal.7b01535. [DOI] [Google Scholar]
- Kano T.; Ueda M.; Maruoka K. Direct Asymmetric Iodination of Aldehydes Using an Axially Chiral Bifunctional Amino Alcohol Catalyst. J. Am. Chem. Soc. 2008, 130, 3728–3729. 10.1021/ja074003o. [DOI] [PubMed] [Google Scholar]
- Martín R.; Cuenca A.; Buchwald S. L. Sequential Copper-Catalyzed Vinylation/Cyclization: An Efficient Synthesis of Functionalized Oxazoles. Org. Lett. 2007, 9, 5521–5524. 10.1021/ol7024718. [DOI] [PubMed] [Google Scholar]
- Wan X.; Meng Q.; Zhang H.; Sun Y.; Fan W.; Zhang Z. An Efficient Synthesis of Chiral β-Hydroxy Sulfones via Ru-Catalyzed Enantioselective Hydrogenation in the Presence of Iodine. Org. Lett. 2007, 9, 5613–5616. 10.1021/ol702565x. [DOI] [PubMed] [Google Scholar]
- Takeda Y.; Kajihara R.; Kobayashi N.; Noguchi K.; Saito A. Molecular-Iodine-Catalyzed Cyclization of 2-Alkynylanilines via Iodocyclization–Protodeiodination Sequence. Org. Lett. 2017, 19, 6744–6747. 10.1021/acs.orglett.7b03497. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Peddinti R. K. Iodine-Catalyzed Regioselective Synthesis of Multisubstiuted Pyrrole Polyheterocycles Free from Rotamers and Keto–Enol Tautomers. J. Org. Chem. 2017, 82, 9360–9366. 10.1021/acs.joc.7b01538. [DOI] [PubMed] [Google Scholar]
- Sarkar R.; Mukhopadhyay C. A convenient strategy to 2,4,5-triaryl and 2-alkyl-4,5-diaryl oxazole derivatives through silver-mediated oxidative C-O cross coupling/cyclization. Tetrahedron Lett. 2015, 56, 3872–3876. 10.1016/j.tetlet.2015.04.103. [DOI] [Google Scholar]
- Jaffe G. M.; Day A. R. Reactions of phenanthraquinone and retenequinone with amines under pressure. J. Org. Chem. 1943, 08, 43–51. 10.1021/jo01189a008. [DOI] [Google Scholar]
- McCoy G.; Day A. R. The Reaction of ortho-Quinones and ortho-Quinonimines with Primary Amines. J. Am. Chem. Soc. 1943, 65, 1956–1959. 10.1021/ja01250a041. [DOI] [Google Scholar]
- Nicolaides D. N.; Gautam D. R.; Litinas K. E.; Manouras C.; Fylaktakidou K. C. Reaction of 2-(methoxyimino)benzene-1-ones with a-alkylethoxcarbonylmethylene(triphenyl)phosphoranes. Tetrahedron 2001, 57, 9469–9474. 10.1016/S0040-4020(01)00942-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.