

Abstract
Although the mechanisms underlying prion propagation and infectivity are now well established, the processes accounting for prion toxicity and pathogenesis have remained mysterious. These processes are of enormous clinical relevance as they hold the key to identification of new molecular targets for therapeutic intervention. In this review, we will discuss two broad areas of investigation relevant to understanding prion neurotoxicity. The first is the use of in vitro experimental systems that model key events in prion pathogenesis. In this context, we will describe a hippocampal neuronal culture system we developed that reproduces the earliest pathological alterations in synaptic morphology and function in response to PrPSc. This system has allowed us to define a core synaptotoxic signaling pathway involving the activation of NMDA and AMPA receptors, stimulation of p38 MAPK phosphorylation and collapse of the actin cytoskeleton in dendritic spines. The second area concerns a striking and unexpected phenomenon in which certain structural manipulations of the PrPC molecule itself, including introduction of N‐terminal deletion mutations or binding of antibodies to C‐terminal epitopes, unleash powerful toxic effects in cultured cells and transgenic mice. We will describe our studies of this phenomenon, which led to the recognition that it is related to the induction of large, abnormal ionic currents by the structurally altered PrP molecules. Our results suggest a model in which the flexible N‐terminal domain of PrPC serves as a toxic effector which is regulated by intramolecular interactions with the globular C‐terminal domain. Taken together, these two areas of study have provided important clues to underlying cellular and molecular mechanisms of prion neurotoxicity. Nevertheless, much remains to be done on this next frontier of prion science.
Keywords: antibody, cell culture, dendrite, glutamate, ionic current, mitogen‐activated protein kinase (MAPK), neurotoxicity, neurodegeneration, prion, spine, synapse, transgenic mouse
Introduction
Prion diseases are a group of fatal, infectious neurodegenerative diseases affecting humans and animals. A defining feature of prion diseases is their association with a protein‐only infectious agent, PrPSc, which is self‐propagating and transmissible 87, 88. Once PrPSc is introduced into individuals from the environment or is generated endogenously, it converts the normal, cellular conformer, designated PrPC, into additional molecules of PrPSc. Consistent with this model, PrP knockout mice, in which PrPC expression is absent, are completely resistant to prion infection 14, 89. Probably the strongest evidence for the prion model is the fact that infectious molecules can be generated and amplified in vitro from purified or recombinant PrPC in reactions seeded by small amounts of PrPSc 18, 32, 125. A prion‐like mechanism may play a role in the spread of misfolded protein aggregates in other, more common neurodegenerative disorders 59, and in the maintenance of certain physiological states 81.
While a great deal is now known about the mechanisms of prion infectivity and propagation, we have a much more limited understanding of how misfolded PrP actually damages neurons and causes the neuropathological abnormalities characteristic of the disease. Previous studies have established several relevant points. First, there is considerable evidence that PrPC plays an essential role in mediating prion neurotoxicity, beyond its function as a required precursor to PrPSc 12, 75. In this regard, it has been hypothesized that PrPC may act as a cell surface receptor that binds PrPSc and transduces downstream neurotoxic signals, a process that could involve the subversion of a normal, physiological activity of PrPC 7, 93. Second, analogous to the case of Aβ in Alzheimer's disease 123, it is likely that oligomeric, possibly noninfectious forms of misfolded PrP, rather than large, self‐propagating polymers of PrPSc, are the most neurotoxic 23. Finally, there is strong evidence that, as in other neurodegenerative disorders, synapses are the earliest anatomical targets of prion neurotoxicity, and their degeneration and dysfunction are primary causes of the cognitive and motor symptoms of the disease 76.
Further investigation of the mechanisms of prion neurotoxicity has been hampered by two major obstacles. First is the lack of suitable in vitro model systems that can be interrogated at the cellular and molecular levels. There are a limited number of cell lines capable of propagating prions in culture 16, 92, and none of these exhibit signs of cytotoxicity as a result of chronic prion infection. Moreover, most of the cells used to propagate prions are transformed cell lines (e.g., N2a neuroblastoma cells), and there is very little published literature on prion infection of cultured primary neurons 30, 47. Beyond their importance for understanding the biology of prion neurodegeneration, in vitro systems are necessary to test potential therapeutic compounds in a systematic manner.
A second major obstacle to understanding prion neurotoxicity has been the limited information available about the normal function of PrPC. PrPC is a GPI‐anchored, cell surface glycoprotein that is widely expressed on neurons throughout the CNS beginning early in development 48, 77. A number of physiological functions have been proposed for PrPC 128, but the absence of a strong phenotype in PrP knockout animals has precluded the definitive identification of a normal molecular or cellular activity for PrPC 127, 132. Importantly, PrP knockout animals do not display symptoms of a prion disease 15, suggesting that the disease phenotype is due primarily to a gain‐of‐function attributable to PrPSc or a related toxic species, rather than to a loss of the normal function of PrPC. However, there are now a large number of studies demonstrating that certain manipulations of the PrPC molecule, including the introduction of particular mutations in the flexible N‐terminal domain or the binding of antibodies to specific epitopes in the globular, C‐terminal domain, can endow the protein with powerful neurotoxic activities 70, 104, 111. How these artificially induced toxic activities relate to the pathophysiology of prion diseases, and whether they are triggered by conversion of PrPC to PrPSc is uncertain. Taken together, however, these studies raise the possibility that prion neurotoxicity could involve the subversion of the normal physiological activity of PrPC as a result of disruption of structural interactions within the protein.
In this contribution, we will review work over the past several years from our laboratory and several others in two areas relevant to understanding the mechanisms of prion neurotoxicity. In the first section, we will discuss the use of simplified, in vitro experimental systems (brain slices, cultured neurons) that model key features of prion neurotoxicity. We will discuss in detail the results obtained from a novel neuronal culture system developed in our laboratory that we believe reproduces some of the earliest events in prion pathogenesis at the level of individual synapses. This system has allowed us to define what we think is a core signaling pathway activated by PrPSc. In the second section, we will discuss how artificial manipulations of the PrP molecule (deletion mutations, antibody binding) can, in the absence of PrPSc, trigger neurotoxic sequelae in mice, brain slices and cultured cells. We will review work from our own laboratory, involving a combination of electrophysiological, cellular and biophysical approaches, which leads to a new structural model to explain toxic as well as physiological activities of PrPC.
We will begin by first reviewing several topics that are conceptually relevant to understanding the mechanisms of prion neurotoxicity.
Infectivity vs. Neurotoxicity
There is evidence that infectivity (the ability to self‐propagate) and neurotoxicity (the ability to produce neuropathology) may be distinct properties attributable to different molecular forms of misfolded PrP 7. Misfolded forms of PrP purified from infected brain are known to be heterogeneous in terms of aggregation state, protease resistance and possibly protein conformation 63, 74, 96, 97, 134. Although, historically, proteinase K (PK) resistance has been used to define PrPSc in biochemical analyses, it has been estimated that a large fraction of the PrPSc present in the brain after prion infection is actually sensitive to PK digestion 63, 96, 97, 134. There is debate about the relative infectivity of the PK‐resistant and PK‐sensitive forms, and it has been suggested that the latter species may represent small aggregates that are particularly neurotoxic without being infectious 6, 7, 23. Our own previous work demonstrates that noninfectious, weakly PK‐resistant, oligomeric forms of PrP are responsible for neuropathology in a mouse transgenic model of a familial prion disease 6, 22, 23. Consistent with a distinction between infectivity and neurotoxicity, there is evidence that prion disease progression in mice is characterized by two, mechanistically discrete phases: rapid accumulation of PrPSc and infectivity, followed by slower development of neuropathology and clinical symptoms over a time course that is inversely related to the expression levels of PrPC 99, 100. It was postulated that the second phase occurred because of the accumulation of toxic, protease‐sensitive species, although these studies did not biochemically isolate or characterize these forms.
The Role of PrPC in Prion Neurotoxicity
An important clue to the mechanism underlying prion neurotoxicity is the observation that PrPC knockout neurons are relatively resistant to the toxic effects of PrPSc that is supplied exogenously by wild‐type astrocytes or by neighboring neurons 12, 75. This result suggests that a critical neurotoxic signal is generated as part of the process by which endogenous cell surface PrPC is converted into PrPSc, and in the absence of PrPC, this signal is not produced. As PrPC is normally attached to the cell surface by a glycolipid anchor, one might predict that a signal‐transducing function for PrPC would require its membrane anchoring. Consistent with this prediction, scrapie‐inoculated mice expressing an anchorless form of PrPC show an altered neuropathological profile, suggesting that the neurotoxic signaling processes normally mediated by PrPC require its attachment to the plasma membrane 20, 21. Although there are a number of studies suggesting signal‐transducing activities for cell surface PrPC 72, the pathways by which its interaction with PrPSc produces neurotoxic signals remain mysterious.
The Importance of the Synapse
The terminal neuropathology of prion diseases encompasses a number of features, including spongiform change, amyloid deposition, astrogliosis and neuronal loss 71. However, some of the earliest and potentially most critical changes occur at the level of the synapse 76. Neuropathological and in vivo imaging studies in prion‐infected mice suggest that synaptic degeneration begins very early in the disease process, predating other pathological changes, and eventually contributing to the development of clinical symptoms 5, 17, 27, 31, 41, 58, 67, 76. Synaptic pathologies include morphological and functional abnormalities, leading eventually to complete elimination of synapses. Suggesting a causative role for PrPSc in synaptic pathology, PrPSc is often found in neuropil deposits that are referred to as “synaptic‐like” because they appear to surround synaptic sites 5, 17, 27, 31, 58, 67. Two‐photon imaging studies of living, prion‐infected animals demonstrate that the swelling of dendritic shafts and the retraction of dendritic spines occur early during the disease course, well before symptoms appear 41. Taken together, these studies pinpoint synapses, in particular dendrites and dendritic spines, as important initial targets of prion neurotoxicity. Dendritic spines are protuberances on dendrites at which synaptic contacts (usually excitatory) occur 83. Changes in their morphology are now believed to underlay synaptic plasticity associated with learning and memory, as well as degenerative events that occur during aging and neurological disease 51, 98. Given these considerations, any experimental system designed to study prion neurotoxicity must be capable of reproducing acute degenerative effects on synapses and dendritic spines.
In Vitro Systems for Studying Prion Neurotoxicity
A major roadblock in studying prion neurotoxicity has been the lack of experimentally tractable model systems in which pathological changes can be studied in vitro. As noted above, the transformed cell lines typically used for prion propagation show no cytopathology, and so are not useful for studying the toxic effects of prions 16, 92. In principle, one might model prion synaptotoxicity using cultures of dissociated neurons (primary or iPS‐derived), three‐dimensional neuronal cultures, acute or chronic brain slices or brain organoids. Studies of prion neurotoxicity have thus far been restricted to brain slices and dissociated neurons (Figure 1).
Figure 1.
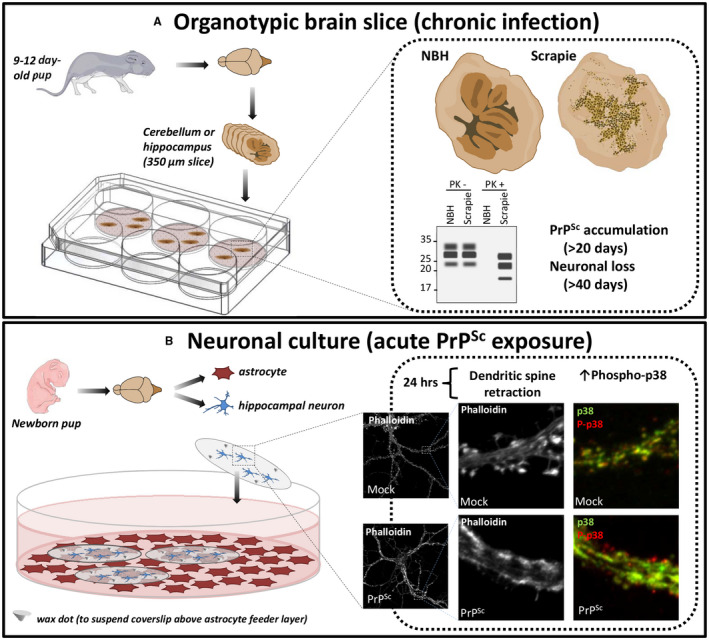
In vitro systems to study prion neurotoxicity. A. An organotypic brain slice system registers the effects of chronic prion infection, monitored by reductions in neuronal viability as PrPSc accumulates 34, 35, 36. Left: Cerebellar or hippocampal slices from WT or Tga20 (PrP‐over‐expressing) mice are prepared from neonatal pups and are treated with normal brain homogenate (NBH) or scrapie‐infected brain homogenate. Right: The slices become chronically infected, accumulating PrPSc (detectable by Western blotting for PK‐resistant PrP) and suffering neuronal loss over a matter of weeks. B. A hippocampal neuronal culture system allows analysis of the acute toxic effects of prions on synaptic structure and function 37, 38. Left: Neurons are isolated from hippocampi of neonatal pups and cultured at low density on coverslips that are suspended facedown, via wax dots, over a feeder layer of astrocytes. Right: Neurons in this system are susceptible to the synaptotoxic effects of prions within 24 hrs. Treatment with purified PrPSc, but not mock‐purified material, leads to retraction of dendritic spines, revealed by staining with fluorescent phalloidin, as well as local increases in p38 MAPK phosphorylation, visualized by staining with antibodies to phospho‐p38 (red) and total p38 (green). Electrophysiological abnormalities in synaptic transmission are also detectable using patch‐clamping techniques 38. Images of mice were taken from Servier Medical Art (http://smart.servier.com).
Aguzzi and colleagues recently described an organotypic brain slice system in which slices of cerebellum or hippocampus can be infected with scrapie prions 34, 35, 36 (Figure 1, top). The cytotoxic readout in this system is typically neuronal death, which is registered by staining the slices with a neuronal marker like NeuN or the DNA‐binding dye propidium iodide. This system has been used to demonstrate a role for calpains, reactive oxygen species, metabotropic glutamate receptors and the unfolded protein response in prion neurotoxicity, and to analyze the transcriptional response to prion infection 36, 43, 52. A drawback of this system is that it requires chronic infection of the slices with scrapie prions to generate sufficient PrPSc, which takes place over several weeks. This makes it difficult to disentangle the effects of prion propagation from prion neurotoxicity, which in vivo are thought to represent distinct phases of the disease process 99, 100. In addition, the synaptic abnormalities (dendritic spine loss) observed in these slices 17 occur in the context of more general pathological changes secondary to neuronal death, making it challenging to dissect the acute effects of PrPSc on synaptic structure and function.
There have been very few published studies of prion infection of neurons in culture. In one early study, Cronier et al 30 showed that cultured cerebellar granule neurons and astrocytes from transgenic mice over‐expressing ovine PrP could be infected by ovine prions, with a proportion of the infected neurons undergoing apoptosis after several weeks in culture as the amount of PrPSc increased. In a second study, Hannaoui et al 47 infected cultured cerebellar granule, striatal, or cortical neurons with three different strains of scrapie prions, and reported strain‐specific effects on neuronal viability that were related temporally to the accumulation of PrPSc over a time period of 10–14 days. These experiments demonstrate that chronic infection of cultured neurons results in the gradual accumulation of PrPSc, and this correlates with decreases in neuronal viability. Again, however, these systems do not allow the assessment of prion toxicity independent of propagation. In addition, the use of neuronal viability as a readout is likely to miss earlier, more subtle effects on synaptic structure and function.
A Novel Neuronal Culture System to Study Prion Neurotoxicity
We were interested in analyzing the acute synaptotoxic effects of prions over the course of 0–48 h, independent of the development of a chronic infected state. We therefore adopted the strategy of adding purified preparations of PrPSc directly to the extracellular medium bathing neuronal cultures. This allowed us to assess the earliest neuronal responses elicited by interaction of exogenous PrPSc with endogenous, cell surface PrPC. There is evidence that this interaction results in an extremely rapid conversion reaction, with generation of new molecules of PrPSc within minutes of the initial contact of the two conformers 44, 45. We anticipated that this system would allow us to monitor the acute effects of PrPSc formation without the necessity for the establishment of a chronic infected state, which is thought to involve additional steps related to intracellular trafficking of PrPSc 121.
We have adopted a specialized kind of neuronal culture system (Figure 1, bottom) that has several advantages over conventional methods for co‐culturing neurons and astrocytes on the same substrate 37, 60. In this system, neonatal mouse hippocampal neurons are cultured at low density on coverslips suspended face‐down over a feeder layer of astrocytes. A key feature of this system is that the neurons are physically separated from the astrocyte feeder layer. This means that visualization of neuronal morphology is greatly improved. In addition, the coverslip can be readily removed for extraction of proteins and RNA from the neurons, with minimal contamination from astrocytes, a crucial advantage for subsequent proteomic or genomic analyses. In this system, neurons can be maintained for as long as four weeks, during which time they elaborate highly differentiated axons and dendrites, and they develop functionally active synapses that can be analyzed by patch‐clamping techniques. The dendrites are studded with large numbers of mature, mushroom spines, which are the postsynaptic sites of excitatory transmission. The spines can be readily visualized using fluorescent phalloidin, which stains F‐actin in the spine cytoskeleton, or by filling them with cytoplasmic GFP. The cultures can be co‐stained for additional markers to reveal the overall morphology of axons and dendrites, as well as the location of presynaptic and postsynaptic elements. This system is ideal for studying the effects of PrPSc on synaptic structure and function, and it provides a facile platform for genetic and pharmacological manipulation of the underlying processes.
Effect of PrPSc on synaptic morphology
Our initial validation of the system involved monitoring the effects of PrPSc on the morphology of dendritic spines 37. We found that the addition of purified PrPSc to the hippocampal cultures resulted in a rapid and dramatic retraction or collapse of dendritic spines on neurons throughout the culture, as monitored by staining with fluorescent phalloidin (Figure 1, bottom). This effect was dose‐dependent, and could be detected at PrPSc concentrations as low as 2.2 μg/mL. Retraction of spines was detectable within 24 h of PrPSc exposure, and occurred prior to other changes in overall dendritic morphology or neuronal cell death, and well before chronic prion infection was established. The effect on spines was specific to PrPSc‐containing samples, and was seen both with crude PrPSc‐containing brain homogenates, as well as with highly purified preparations of PrPSc. Importantly, spine collapse caused by exposure to PrPSc requires the expression of full‐length PrPC by the target neurons, consistent with an essential signal transduction role for cell surface PrPC 12, 75.
Taken together, these results demonstrated that our neuronal culture system was capable of reproducing the earliest changes in dendritic spine morphology that have been observed in the brains of living, scrapie‐infected mice by two‐photon imaging 41, as well as in postmortem brain sections 5, 27, 31, 58, 67. As detailed in the following sections, we have utilized this culture system to elucidate key cellular and molecular mechanisms underlying prion synaptotoxicity, to identify new, druggable therapeutic targets and to compare prion synaptotoxic pathways with the pathways operative in other neurodegenerative diseases.
Highly selective effects of PrPSc on synaptic function
We have found that PrPSc produces a strikingly selective synaptotoxic effect, specifically targeting the postsynaptic elements of excitatory synapses 38. Treatment of hippocampal neurons with purified PrPSc from infected brains caused a marked reduction in the frequency of miniature excitatory postsynaptic currents (mEPSCs), and a less pronounced but statistically significant decrease in mEPSC amplitude. These effects were not observed in neurons derived from PrP knockout mice, demonstrating that the functional as well as the morphological effects of PrPSc on synapses are entirely PrPC‐dependent. In contrast, PrPSc did not significantly affect the frequency or amplitude of miniature inhibitory postsynaptic currents (mIPSCs). In addition, PrPSc treatment did not change the number of inhibitory synapses, as determined by staining for gephyrin (a postsynaptic anchoring component for glycine and GABAA receptors). These results indicate that PrPSc targets primarily excitatory and not inhibitory synapses.
The reduction in mEPSC amplitude and frequency caused by PrPSc could be because of the effects on either presynaptic processes (e.g., synaptic release) or postsynaptic characteristics (e.g., number and distribution of active zones). When neurons were treated with purified PrPSc, there was a dramatic reduction in staining for GluR1, an AMPA receptor subunit, but not in staining for synaptophysin, a presynaptic marker. These data demonstrate that PrPSc exerts a highly selective effect on postsynaptic elements, with no detectable effect on presynaptic structures, even in the face of massive morphological changes in dendritic spines.
The requirement for PrPC
PrPSc‐induced synaptotoxicity in our neuronal culture system requires the expression of cell surface PrPC by the target neurons 37. In contrast to wild‐type neurons, PrP knockout neurons showed no significant change in spine number or area after treatment with PrPSc. Moreover, knockout neurons, unlike WT neurons, did not suffer any functional changes in synaptic transmission or alterations in intracellular calcium levels in response to PrPSc. These observations demonstrate that the toxicity of PrPSc is not attributable to nonspecific effects of the protein aggregates on neuronal integrity or viability, but rather require the mediation of a specific cell surface receptor (PrPC). Moreover, they are consistent with numerous reports that neuronal expression of membrane‐anchored PrPC is necessary for prion‐induced neurodegeneration in vivo 12, 20, 21, 75.
The PrPC requirement in our assay system has allowed us to dissect which parts of the PrPC molecule are essential for mediating synaptotoxic effects 37. Hippocampal neurons from two lines of transgenic mice expressing N‐terminally deleted forms of PrP (Δ23‐31 and Δ23‐111) were completely resistant to the toxic effects of purified PrPSc. This result indicates that a small, polybasic region of PrPC (residues 23‐31; KKRPKPGGW) expressed on target neurons is essential for spine loss induced by exogenously applied PrPSc. This result could be attributable to the previously documented role of residues 23‐31 in PrPC binding to PrPSc 78, 106, 117. Alternatively, the N‐terminal domain may play a direct role in the ability of PrPC or PrPSc to elicit downstream neurotoxic signals.
What is the precise mechanistic role of PrPC in mediating prion neurotoxicity? One possible scenario is that synaptic degeneration results directly from binding of PrPSc to PrPC on the cell surface. This idea is consistent with evidence that PrPC binds very selectively to PrPSc, and that this interaction represents the first step of the conversion process by which more molecules of PrPSc are generated 32, 54, 125. In this scenario, PrPC may be acting as a toxicity‐transducing receptor, akin to other cell surface receptors that generate intracellular signals upon binding of extracellular ligands. In fact, there is evidence that PrPC can participate in certain kinds of signal transduction phenomena 72. A related hypothesis is that PrPC serves to trap and concentrate PrPSc molecules at the cell surface, and that the captured PrPSc molecules then cause toxic effects by virtue of their interaction with the lipid bilayer or with other cell surface proteins. This mechanism would be akin to the function of some GPI‐anchored proteins that serve as non‐signaling co‐receptors for binding of extracellular ligands 119. A third possibility is that cell surface PrPC is first converted to PrPSc (or some other misfolded form) which then elicits a toxic signal. This hypothesis is consistent with recent reports 45 that cell surface PrPC is converted to PrPSc within minutes of contact with exogenously applied PrPSc. Future studies will be directed toward testing these three hypotheses. The astrocyte feeder layer in our culture system is required for trophic support of the adjacent neurons, but the contribution of astrocytes to PrPSc generation and synaptotoxicity remains to be investigated.
Assaying the neurotoxicity of different forms of PrP
What is the neurotoxic form of PrP? As outlined above, there are reasons to draw a distinction between infectious and neurotoxic forms of PrP. The neuronal culture system we have described allows us to assay neurotoxicity independent of infectivity. PrPSc purified from brain typically consists of both PK‐resistant and PK‐sensitive species. PK treatment results in the digestion of ~90% of the PrPSc, which represents PK‐sensitive PrPSc 96, 100. We have found that there is no significant difference between the toxicity of equivalent amounts of undigested and PK‐digested PrPSc, in terms of the ability of these preparations to induce dendritic spine retraction in our culture system 37. This result suggests that both PK‐sensitive and PK‐resistant forms of PrPSc contribute to dendritic spine loss. In addition, it demonstrates that residues 23 through ~90 of PrPSc are not essential for synaptotoxicity. In the future, our system can be used to assay the neurotoxicity of various PrPSc‐like forms generated by in vitro conversion reactions. As some of these species are infectious, while others are not 84, the results obtained will allow analysis of the structural relationship between the toxic and infectious properties of misfolded PrP. Thus far, all of our toxicity studies have been carried out with the RML strain of scrapie prions. In the future, we can use our system to investigate whether different prion strains, which are associated with characteristic PrPSc conformations, produce strain‐specific synaptotoxic effects.
The role of the neuronal cytoskeleton
Actin is abundant in dendritic spines and has been shown to regulate spine morphology 112. In addition, there is evidence that most signaling pathways linking synaptic activity to spine morphology influence local actin dynamics 53, 55. We found that PrPSc‐induced spine retraction was prevented by SiR‐actin 73, a fluorogenic, cell‐permeable peptide derived from jasplakinolide that both stabilizes and labels F‐actin 38. In contrast, the microtubule‐stabilizing agent, taxol did not have a protective effect. SiR‐actin also prevented the loss of the AMPA receptor subunit, GluR1, in response to PrPSc, as well as the decreases in mEPSC amplitude and frequency caused by PrPSc treatment. These data demonstrate that actin dynamics play an important role in the morphological changes in dendritic spines induced by PrPSc, and that stabilizing actin filaments can prevent these changes.
The role of glutamate receptors and calcium influx
We have found that NMDA receptor blockers, and to a lesser extent, AMPA receptor blockers, protect neurons from the synaptotoxic effects of PrPSc 38. In addition, we observed that purified PrPSc caused a rapid (within 30 minute) increase in intracellular Ca2+ as monitored with the calcium‐sensitive dye Fluo‐3 38. The effect of PrPSc on Ca2+ levels was absent in neurons derived from Prnp 0/0 mice, and was completely blocked by the NMDA receptor antagonist, memantine. These results imply that one of the first steps in the PrPSc synaptotoxic signaling cascade is the activation of NMDA‐ and AMPA‐type glutamate receptors.
This conclusion is consistent with several previous observations in the literature. First, there is evidence that PrPC modulates the NMDA receptor's function 9 and posttranslational modification 42, and also interacts physically with NMDA receptor subunits 62, processes that could be altered by binding of PrPSc to cell surface PrPC. Second, NMDA receptors have been shown to regulate the actin cytoskeleton in dendritic spines via Ca2+‐induced effects on cytoplasmic actin‐binding proteins 39. Finally, glutamate receptor‐dependent excitotoxicity contributes to the pathogenesis of many neurodegenerative diseases, and is known to cause morphological changes in dendrites similar to those observed here in response to PrPSc 2, 56, 64, 69, 103.
A p38 MAPK synaptotoxic signaling pathway
Mitogen‐activated protein kinases (MAPKs) are important signal transducers downstream of many kinds of intracellular and extracellular stimuli 79, including stressful stimuli like excitotoxicity 28. In mammals, the MAPKs are grouped into three main families, ERKs (extracellular signal‐regulated kinases), JNKs (Jun amino‐terminal kinases) and p38/SAPKs (stress‐activated protein kinases) 79. In the nervous system, p38 MAPK has been found to play a role in synaptic plasticity as well as in neuronal damage and survival, and it has been linked to a number of neurodegenerative diseases 28, 116. In this regard, there is evidence that p38 MAPK and its downstream substrates regulate AMPA receptor trafficking, dendritic spine morphology, cytoskeletal dynamics and synaptic transmission.
Several pieces of evidence demonstrate that p38 MAPK plays an essential role in mediating the toxic effects of PrPSc on synaptic structure and function in our neuronal culture system 38. First, pharmacological inhibitors of p38 MAPK, including those selective for the α isoform, blocked and even reversed dendritic spine collapse and defects in synaptic transmission induced by PrPSc. In contrast, pan‐isoform inhibitors of ERK and JNK, the two other major classes of MAPK, were without effect. Second, genetic suppression of p38 MAPK signaling by a dominant‐negative form of p38 MAPK prevented PrPSc‐induced synaptic abnormalities. Finally, the amount of phosphorylated p38 in dendritic spines increased after 1 h of PrPSc treatment, and remained elevated after 24 h in the region of collapsed spines. Using selective pharmacological inhibitors, we have shown that the likely downstream targets phosphorylated by p38 MAPK include MAPK‐activated protein kinase (MAPKAP or MK) isoforms 2 and 3.
Taken together, our results have allowed us to define a glutamate receptor/p38 MAPK‐dependent signaling pathway underlying prion synaptotoxicity 38 (Figure 2). Importantly, this work identifies a new, druggable target that could be used to block synaptotoxic sequelae in prion diseases: p38 MAPK. Inhibitors of this kinase are already in clinical use for the treatment of inflammatory and neurodegenerative disorders (including Alzheimer's disease) 1, 10, 29, 80, 101, 133, and some of these compounds might be easily repurposed for therapy of prion diseases. The neuronal culture system described here could be readily adapted to screen additional therapeutic agents, as we have already done for some novel anti‐PrPSc compounds 57.
Figure 2.
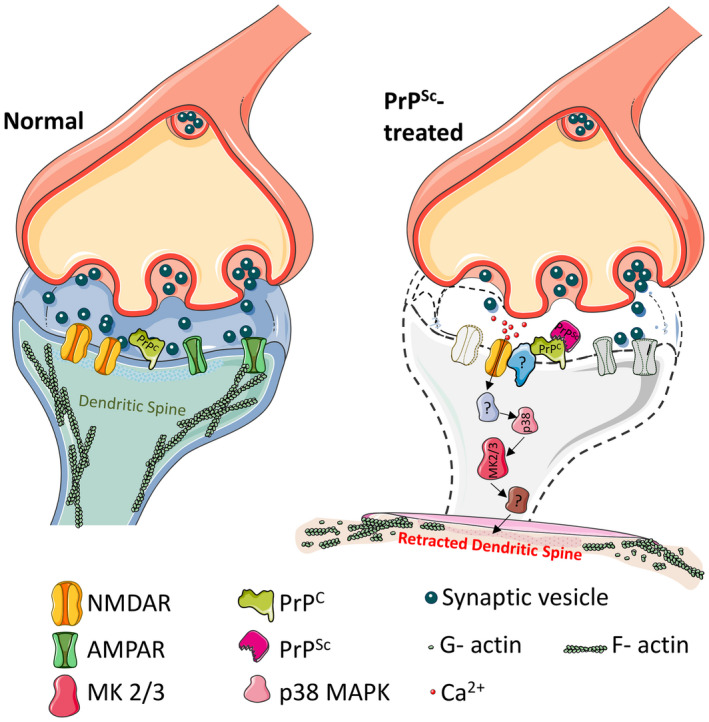
Model for a prion synaptotoxic pathway. The pathway is initiated by binding of PrPSc to endogenous PrPC on the cell surface. This binding event itself, or the subsequent conversion of PrPC to PrPSc, results in the activation of NMDA and AMPA receptors with influx of calcium ions. Calcium influx leads to subsequent activation of p38 MAPK and MK2/3, collapse of the dendritic spine cytoskeleton, spine retraction and decreases in synaptic transmission. Question marks indicate unknown components of the pathway. Figure was produced using Servier Medical Art (http://smart.servier.com).
PrPSc and Aβ active distinct synaptotoxic signaling pathways
Our neuronal culture system allows direct comparisons between pathogenic mechanisms involved in prion diseases and other neurodegenerative disorders. It has been proposed that PrPC is a cell surface receptor for Alzheimer's Aβ oligomers, and that it mediates some of the neurotoxic effects of these assemblies 11, 19, 40, 68, 82. Binding of Aβ oligomers to PrPC is thought to trigger a signaling pathway involving mGluR5 and Fyn kinase, and pharmacologically inhibiting this pathway has been shown to prevent Aβ neurotoxicity and ameliorate neurological symptoms in mice 46, 61, 118, 120.
We used specific pharmacological inhibitors to compare the synaptotoxic signaling pathways activated by PrPSc and Aβ oligomers 38. We found, consistent with a previous report 118, that the mGluR5 inhibitor, MPEP, completely blocked the ability of Aβ oligomers to cause dendritic spine retraction. In contrast, MPEP had no influence on PrPSc‐induced retraction of dendritic spines. Moreover, a p38 MAPK inhibitor, which completely blocked PrPSc synaptotoxicity, had no significant effect on Aβ oligomer‐induced dendritic spine loss. These data suggest that Aβ oligomers and PrPSc trigger different neurotoxic signaling pathways downstream of a common cell surface receptor, PrPC.
Manipiulations of the PrPC Molecule Produce Neurotoxic Effects
In this section of the review, we will discuss a body of work that began 20 years ago with the publication of a paper by Weissmann, Aguzzi and colleagues 104, in which the authors described a curious and striking phenomenon in some lines of transgenic mice expressing PrP molecules with deletions in the flexible, N‐terminal domain. These mice displayed an ataxic neurodegenerative disorder with specific cerebellar lesions, a phenotype that could be rescued by co‐expression of wild‐type PrP in the same animals. This phenomenon immediately attracted great interest because of the possibility that it could be a clue both to the normal physiological function of PrPC, as well as to how PrPC might generate neurotoxic signals during prion diseases. Our own laboratory began working on this subject approximately 10 years ago, and we continue to believe that it offers important insights about structural interactions within the PrP molecule, and how these influence the biological activities of the protein. The original work has now been extended to include a related phenomenon in which antibody binding to specific epitopes in the globular, C‐terminal domain mimics some of the toxic effects of the deletions within the N‐terminal domain 111, 131.
N‐terminal deletion mutants of PrP cause neurodegeneration
As described in the original publication 104, transgenic mice expressing PrP molecules with deletions of residues 32‐121 or 32‐134 within the flexible, N‐terminal domain developed ataxia and degeneration of the granular layer of the cerebellum at 2–3 months after birth. It was subsequently shown that smaller deletions within this region also produced a neurodegenerative phenotype 4, 13. Amazingly, we found that the smallest deletion to be examined (referred to as ΔCR), which removes only 21 amino acids from the Central Region of PrP (residues 105‐125) produced the most severe phenotype, causing death of the mice within a week of birth 70. In all cases, co‐expression of WT PrP in the same animals dose‐dependently suppressed the neurodegenerative phenotype, with larger amounts of WT PrP required for rescue of ΔCR mice than Δ32‐121/134 mice. This striking genetic interaction between mutant and WT PrP immediately suggested a molecular model in which the mutant and WT proteins physically interact with each as part of a molecular complex, in a dominant‐positive fashion, or else they compete with each other for binding to a common molecular target. Consideration of these models suggests the concept that deletions in the PrP molecule somehow alter the normal biological activity of PrPC in a way that produces neurotoxic effects.
N‐terminally deleted forms of PrP induce abnormal ionic currents
In an attempt to understand why ΔCR and related deletion mutants are so toxic, we used whole‐cell patch clamping techniques to record electrical activity from cells expressing these mutants. To our surprise, we observed large, spontaneous inward currents in a variety of transfected cell lines as well as primary neurons expressing the highly pathogenic ΔCR deletion mutant 8, 108, 109, 110. These currents, which were inward at negative holding potentials, were highly irregular, fluctuating randomly over a time course of seconds to minutes and reaching amplitudes of several thousand picoamps. They were present only at hyperpolarized holding potential (<−30 mV) 131. Ion substitution experiments demonstrated that the currents resulted from relatively nonselective, cation‐permeable pathways in the membrane. The unusual kinetic properties of the currents suggested that they reflect the transient formation of membrane pores or localized disruptions of the lipid bilayer, rather than the presence of conventional ion channels. Several different deletion mutants spanning the central region, including Δ94‐134, Δ94‐110, Δ111‐134 and Δ114‐121, all of which produce neurodegeneration in transgenic mice, as well as three disease‐associated point mutations, including P101L, G130V and G113V, also induced spontaneous currents 110. Importantly, WT PrP potently suppressed the currents induced by all of these mutants, in parallel with the ability of WT PrP to reverse the neurodegenerative phenotype of Tg(ΔCR) and Tg(Δ32‐121/134) mice 70, 104. The antagonistic effects of WT and mutant PrP in both settings argue that the current‐inducing activity of mutant PrPs observed in vitro is mechanistically related to their neurotoxicity in vivo.
The N‐terminal domain of PrP possesses a toxic effector function
We have carried out a series of experiments designed to identify which parts of the PrP molecule are responsible for the toxic effects of the deletion mutants. These studies revealed that the flexible, N‐terminal domain of PrPC possesses a powerful, toxic effector activity, which, when unregulated, produces neurodegenerative changes in mice and abnormal ion currents in cells. In support of this conclusion, we found that the spontaneous currents induced by ΔCR and related mutants are inhibited by treatment with ligands that bind to the N‐terminal domain of PrPC, including sulfated glycosaminoglycans (pentosan polysulfate), Cu2+ ions and antibodies 131. We interpret these results to mean that ligands bound to the N‐terminal domain prevent its interaction with the lipid bilayer and/or with other membrane‐associated targets that are essential for ion channel activity and other toxic effects.
We also found that ion channel activity depends on the presence of a polybasic amino acid segment at the very beginning of the N‐terminal domain of PrP (KKRPKPGGW, residues 23‐31) 110, 131. ΔCR PrP carrying a deletion of the KKRPKPGGW sequence, or mutations that reverse the positive in this region, completely lacked channel activity. A requirement for the KKRPKPGGW sequence is also seen in vivo: removal of this segment abolishes the neurotoxicity of Δ32‐134 PrP when expressed in transgenic mice 129.
The KKRPKPGGW region is similar in sequence to those of positively charged “cell penetrating peptides” or “protein transduction domains,” originally derived from the HIV Tat protein, which can transiently permeabilize the lipid bilayer by interacting with negatively charged membrane phospholipids 50, 122. An analogous mechanism may account for the ability of ΔCR PrP to create transient pores in the membrane, by acting as a “tethered protein transduction domain.” Support for this model is provided by our demonstration that a C‐terminally lipidated, recombinant form of ΔCR PrP incorporated into synthetic liposomes created membrane pores that were permeable to fluorescent dyes and ionic quenchers 25. An alternative model is that the N‐terminal polybasic domain interacts with other membrane proteins, for example endogenous ion channels or channel‐modulating proteins, to induce current activity.
In order to determine whether N‐terminal domain can function as an autonomous effector, we constructed a series of chimeric proteins (collectively designated PrP(N)‐EGFP‐GPI) consisting of various lengths of the N‐terminal domain of PrPC (residues 23‐109) fused to an EGFP molecule that was equipped with the GPI addition signal from PrPC 131. We found that the PrP(N)‐EGFP‐GPI constructs induced spontaneous currents similar to those produced by ΔCR PrP, with the amount of current activity increasing as the length of the N‐terminal segment became longer. These currents, like those associated with ΔCR PrP, were silenced by application of pentosan polysulfate, Cu2+ ions and N‐terminal antibodies, and by the removal of residues 1‐31.
Taken together, these results indicate that certain structural alterations (deletion of residues in the central region; absence of the C‐terminal domain) allow the N‐terminal domain of PrPC to acquire a powerful, toxic effector function, which is manifested by abnormal ionic currents in cultured cells and neurodegeneration in transgenic mice.
Anti‐prion antibodies cause neuronal toxicity
Several groups, including our own, have found that another kind of structural perturbation, caused by binding of antibodies to the C‐terminal domain of PrPC, also unmasks latent toxic activities. We have reported that three different antibodies (POM1, D18 and ICSM‐18) targeting overlapping epitopes in helix 1 induce ionic current activity in cells expressing WT PrPC 131. The properties of these currents are identical to those of spontaneous currents associated with ΔCR PrP, in terms of their sporadic nature, their blockage by N‐terminal ligands (pentosan sulfate and antibodies targeting the octapeptide repeat domain) and their absolute dependence on the presence of the 23‐31 region.
In addition to acutely inducing ionic currents, helix 1 antibodies (POM1, D18 and ICSM18) cause major degenerative changes in the dendrites of cultured hippocampal neurons when applied for 48 h 131. This antibody‐induced dendritic degeneration is, like the currents induced by the same antibodies, entirely dependent on the N‐terminal domain of PrPC, and is blocked by N‐terminal ligands and deletions of residues 23‐31. The parallel characteristics of the ionic currents and the dendritic changes induced by C‐terminal antibodies suggest a mechanistic connection between the two phenomena, perhaps via the activation of excitotoxic pathways. Dendritic varicosities similar to those caused by anti‐PrP antibodies are a characteristic feature of glutamate excitotoxicity 49, and glutamate excitotoxicity has been implicated in neuronal degeneration induced by ΔCR PrP 8, 24.
Studies from two other laboratories have also reported that antibodies targeting specific epitopes in the structured domain of PrPC cause neuronal death when administered in vivo or in brain slices 43, 95, 107, 111. Investigation of the underlying mechanisms revealed the involvement of several different pathways, including the generation of reactive oxygen species, calpain activation and stimulation of the PERK arm of the unfolded protein response 52, 111. It remains to be determined whether these pathways are operative in our neuronal culture system, in which we observe acute changes in dendritic morphology without loss of neuronal viability.
Aside from their relevance to understanding the mechanisms of PrP toxicity, these studies have important clinical implications, as anti‐PrP antibodies have been proposed as therapeutic agents for the treatment of both prion and Alzheimer's diseases 26, 66, 130. However, if the antibodies themselves have significant neurotoxic potential, this could constitute an unacceptable side effect precluding their use. Unfortunately, there is now considerable controversy surrounding this subject. Although several studies, including our own 131, have reported neurotoxic effects of anti‐PrP antibodies 52, 95, 107, 111, another study found that the same antibodies were nontoxic 65. It has been claimed that the neurotoxic effects observed are nonspecific, and related to the use of high antibody concentrations 90, 91, despite the fact that mice lacking PrP expression, or expressing N‐terminally deleted forms of PrP (Δ23‐31 or Δ23‐111), are resistant to antibody toxicity 94, 131. In light of these uncertainties, it would seem prudent to exercise caution in administering anti‐PrP antibodies to patients for therapeutic purposes.
Structural mechanisms regulating the toxic activity of PrPC
The findings described thus far suggest a structural model in which the flexible, N‐terminal domain of PrPC functions as a powerful toxicity‐transducing effector whose activity is tightly regulated in cis by the globular C‐terminal domain (Figure 3). Deletions of the hinge region connecting the N‐ and C‐terminal domains (as in ΔCR PrP), or complete elimination of the C‐terminal domain (as in the PrP(N)‐EGFP‐GPI constructs), would be predicted to interfere with the regulatory interaction between these two domains, thereby freeing the N‐terminal domain to produce spontaneous ionic currents and neurodegenerative changes. Antibodies targeting helix 1 in the C‐terminal domain would have a similar effect by disrupting the regulatory action of the C‐terminal domain on the N‐terminal domain. Ligands targeting the N‐terminal domain, as well as deletion of residues 23‐31, would abrogate these toxic effects. Aguzzi and colleagues have proposed a similar model to explain the effect of anti‐PrP antibodies 111.
Figure 3.
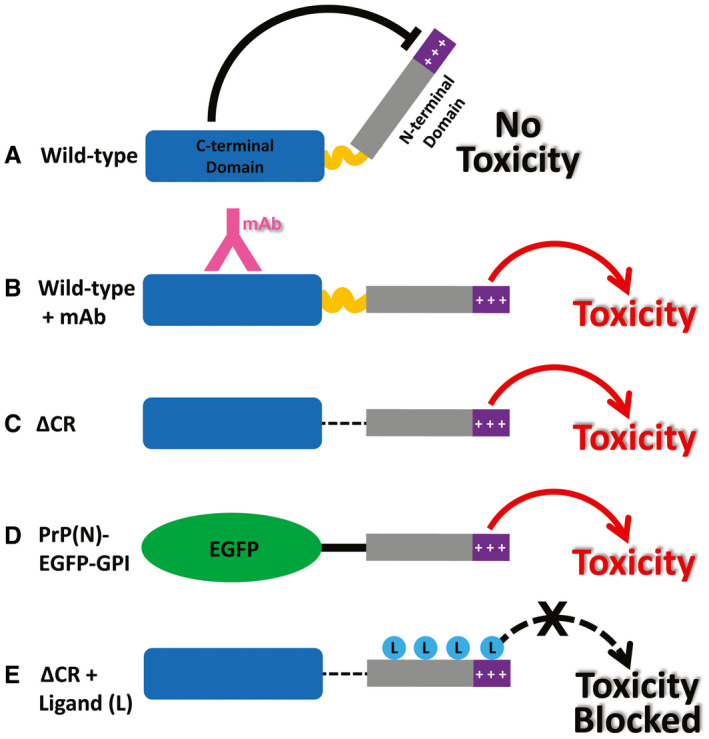
Models for the neurotoxic effects of PrP. A. The C‐terminal domain of PrPC negatively regulates the toxic effector function of the N‐terminal domain. +++, basic residues within the 23‐31 region at the extreme N‐terminus, which are essential for the toxic action of PrP. B. Binding of monoclonal antibodies to the C‐terminal domain disrupts this regulatory interaction, releasing the N‐terminal domain to produce toxic effects. C. Deletion of the central region, as in ΔCR PrP, produces a similar loss of regulation, with toxic consequences. D. When EGFP is substituted for the C‐terminal domain of PrPC, regulation is also lost. E. Binding of ligands (PPS, antibodies, Cu2+) to the N‐terminal domain of ΔCR PrP blocks its ability to exert toxic effects. Reprinted from 131.
NMR studies we have carried out in collaboration with the laboratory of Glenn Millhauser provide direct support for this model. In these experiments, we performed 1H‐15N HSQC NMR analysis of PrP in the presence and absence of Cu2+ ions. Cu2+ ions are physiological ligands that bind to histidine residues in the octapeptide repeat region within the N‐terminal domain 124. Broadening of specific NMR peaks in the structured C‐terminal domain as a result of Cu2+‐induced paramagnetic relaxation enhancement is then used as an indicator of the proximity of the corresponding residues to Cu2+ ions bound to the octapeptide repeats. Previous studies had shown that that both Cu2+ and Zn2+ ions drive the N‐terminus of wild‐type PrPC to associate with the C‐terminal domain 33, 113. We found that, compared to WT PrP, ΔCR PrP showed many fewer residues in the C‐terminal domain that were affected by the presence of Cu2+ ions bound to the N‐terminal domain. This indicated diminished interaction between the two domains in this mutant, consistent with the proposed release of regulatory inhibition 131. In subsequent experiments, we have used chemical cross‐linking in combination with mass spectrometry to pinpoint specific interacting residues in the N‐ and C‐terminal domains, and how these are affected by toxic mutations (manuscript submitted).
Implications for pathology and physiology
The toxic effects we have described above are all produced by artificial manipulations of the PrPC molecule, including the introduction of specific deletion mutations, or binding of antibodies. How do these effects relate to neurotoxic processes that occur during the course of a naturally occurring prion disease, or other neurodegenerative disorders where PrPC is thought to play a role? One hypothesis is that pathologic ligands, including PrPSc or Aβ, which bind to PrPC, produce neurotoxic effects by disrupting the normal regulatory cis‐interaction between the N‐ and C‐terminal domains, thereby unleashing the toxic effector activity of the N‐terminal domain. Consistent with this model, Aguzzi and colleagues have reported that prion infection and anti‐PrP antibodies activate similar downstream neurotoxic pathways in brain slices 52. On the other hand, mice expressing N‐terminally truncated PrPC remain susceptible to prion diseases 114, 117, arguing against a primary neurotoxic role for the N‐terminal domain. However, the disease course is considerably prolonged in these animals, which may reflect the recruitment of additional, less efficient toxic pathways, or else an effect of the deletions on the PrPC‐PrPSc conversion process. One test of the toxic effector hypothesis would be to determine whether the exposure of PrPC‐expressing cells to PrPSc or Aβ induces abnormal ionic currents similar to those produced by antibodies and deletion mutations. Thus far, we have not observed such currents (unpublished data). At this point, the disease relevance of the deletion‐ and antibody‐induced toxic activities of PrP requires further investigation.
It is also possible that the effects of deletion mutations and antibodies reflect aberrations in the normal physiological activity of PrPC. For example, natural ligands, including proteins, small molecules or metal ions, may exist, whose binding to PrPC regulates a physiologically relevant effector activity of the N‐terminal domain. Previous studies have implicated the N‐terminal domain in several physiological activities of PrPC 85, 86, 102, 105, 115, 126, some of which may be regulated by the interaction with the C‐terminal domain. Copper ions are examples of natural ligands, binding of which to the octapeptide repeats promotes docking of the N‐ and C‐terminal domains 33. This phenomenon may be important if PrPC functions as a sensor or transporter of these divalent metal ions. Endogenous ligands for the globular domain may also exist, which either enhance or disrupt the N‐C interaction.
SUMMARY AND PERSPECTIVE
Although the mechanisms underlying prion propagation and infectivity are now well established, the processes accounting for prion toxicity and pathogenesis have remained less clear. Beyond its role as a precursor to infectious PrPSc, the cellular form of the prion protein, PrPC, functions as an essential mediator or transducer of PrPSc‐initiated neurotoxicity. Progress in understanding the nature of prion neurotoxicity has been hampered by the dearth of suitable in vitro experimental systems that model the earliest neurotoxic events at the level of synapses, which are key pathological targets of prions. A hippocampal neuronal culture system we have developed has allowed us to define a core prion synaptotoxic pathway involving the activation of NMDA and AMPA receptors, and the stimulation of p38 MAPK phosphorylation. Another line of investigation into prion toxicity is based on the striking observation that certain structural manipulations of the PrPC molecule itself (deletion mutations and antibody binding) endow the protein with powerful toxic activities, including induction of neurodegeneration in transgenic mice and abnormal ionic currents in cultured cells. These effects are caused because of altered intramolecular interactions between the globular C‐terminal and flexible N‐terminal domains of PrPC, resulting in disinhibition of a toxic effector activity of the latter.
Much remains to be learned about prion neurotoxicity. We need to identify additional, missing steps in the synaptotoxic cascade shown in Figure 2, and understand the role of PrPC‐PrPSc conversion in this process. Discovery‐based genomic and proteomic approaches may help in this regard. We also need to validate findings from neuronal cell culture in other systems, including brain slices or organoids, and mouse models. We will also want to exploit the therapeutic potential of our neuronal culture system, in terms of identifying novel drugs and drug targets that inhibit prion neurotoxicity. Such compounds could be combined in a dual therapy with drugs that block prion replication. The artificially induced toxic effects of N‐terminal deletion mutants like ΔCR and anti‐C‐terminal PrP antibodies remain an intriguing puzzle requiring further investigation. We need to determine whether these effects relate to processes occurring during the disease state, and whether they reflect aberrations of normal, physiological states of the PrPC molecule. Finally, we will want to investigate how the neurotoxic mechanisms uncovered in prion diseases relate to other neurodegenerative disorders, many which may display features of prion‐like propagation 59, and some of which may even involve PrPC as a cell surface receptor for protein oligomers 3, 68. Investigations of prion neurotoxicity hold great promise from both a biological and medical standpoint, and they constitute the next major frontier of prion research.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
Work in the Harris laboratory is supported by grants from the N.I.H. (NS065244, NS101659, NS107755) and the Creutzfeldt‐Jakob Disease Foundation.
References
- 1. Alam J, Blackburn K, Patrick D (2017) Neflamapimod: clinical phase 2b‐ready oral small molecule inhibitor of p38α to reverse synaptic dysfunction in early Alzheimer's disease. J Prev Alzheimers Dis 4:273–278. [DOI] [PubMed] [Google Scholar]
- 2. Ambrosi G, Cerri S, Blandini F (2014) A further update on the role of excitotoxicity in the pathogenesis of Parkinson's disease. J Neural Transm (Vienna) 121:849–859. [DOI] [PubMed] [Google Scholar]
- 3. Aulic S, Masperone L, Narkiewicz J, Isopi E, Bistaffa E, Ambrosetti E et al (2017) α‐Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell‐to‐cell spreading and block prion replication. Sci Rep 7:10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH et al (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 26:538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belichenko PV, Brown D, Jeffrey M, Fraser JR (2000) Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie‐infected mice. Neuropathol Appl Neurobiol 26:143–149. [DOI] [PubMed] [Google Scholar]
- 6. Biasini E, Medrano AZ, Thellung S, Chiesa R, Harris DA (2008) Multiple biochemical similarities between infectious and non‐infectious aggregates of a prion protein carrying an octapeptide insertion. J Neurochem 104:1293–1308. [DOI] [PubMed] [Google Scholar]
- 7. Biasini E, Turnbaugh JA, Unterberger U, Harris DA (2012) Prion protein at the crossroads of physiology and disease. Trends Neurosci 35:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biasini E, Unterberger U, Solomon IH, Massignan T, Senatore A, Bian H et al (2013) A mutant prion protein sensitizes neurons to glutamate‐induced excitotoxicity. J Neurosci 33:2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Black SA, Stys PK, Zamponi GW, Tsutsui S (2014) Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Front Cell Dev Biol 2:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borders AS, de Almeida L, Van Eldik LJ, Watterson DM (2008) The p38α mitogen‐activated protein kinase as a central nervous system drug discovery target. BMC Neurosci 9(Suppl. 2):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bove‐Fenderson E, Urano R, Straub JE, Harris DA (2017) Cellular prion protein targets amyloid‐beta fibril ends via its C‐terminal domain to prevent elongation. J Biol Chem 292:16858–16871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y et al (1996) Normal host prion protein necessary for scrapie‐induced neurotoxicity. Nature 379:339–343. [DOI] [PubMed] [Google Scholar]
- 13. Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P et al (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13:310–318. [DOI] [PubMed] [Google Scholar]
- 14. Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C (1993) Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. [DOI] [PubMed] [Google Scholar]
- 15. Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ et al (1992) Normal development and behaviour of mice lacking the neuronal cell‐surface PrP protein. Nature 356:577–582. [DOI] [PubMed] [Google Scholar]
- 16. Butler DA, Scott MR, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK et al (1988) Scrapie‐infected murine neuroblastoma cells produce protease‐resistant prion proteins. J Virol 62:1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Campeau JL, Wu G, Bell JR, Rasmussen J, Sim VL (2013) Early increase and late decrease of purkinje cell dendritic spine density in prion‐infected organotypic mouse cerebellar cultures. PLoS One 8:e81776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121:195–206. [DOI] [PubMed] [Google Scholar]
- 19. Chen S, Yadav SP, Surewicz WK (2010) Interaction between human prion protein and amyloid‐beta (Abeta) oligomers: role OF N‐terminal residues. J Biol Chem 285:26377–26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chesebro B, Race B, Meade‐White K, Lacasse R, Race R, Klingeborn M et al (2010) Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog 6:e1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chesebro B, Trifilo M, Race R, Meade‐White K, Teng C, LaCasse R et al (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439. [DOI] [PubMed] [Google Scholar]
- 22. Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21:1339–1351. [DOI] [PubMed] [Google Scholar]
- 23. Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si‐Hoe SL, Takao M et al (2003) Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 77:7611–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christensen HM, Dikranian K, Li A, Baysac KC, Walls KC, Olney JW et al (2010) A highly toxic cellular prion protein induces a novel, nonapoptotic form of neuronal death. Am J Pathol 176:2695–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chu NK, Shabbir W, Bove‐Fenderson E, Araman C, Lemmens‐Gruber R, Harris DA, Becker CF (2014) A C‐terminal membrane anchor affects the interactions of prion proteins with lipid membranes. J Biol Chem 289:30144–30160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD et al (2010) Anti‐PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci 11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clinton J, Forsyth C, Royston MC, Roberts GW (1993) Synaptic degeneration is the primary neuropathological feature in prion disease: a preliminary study. NeuroReport 4:65–68. [DOI] [PubMed] [Google Scholar]
- 28. Correa SA, Eales KL (2012) The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct 2012:649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA (2009) p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cronier S, Laude H, Peyrin JM (2004) Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc Natl Acad Sci U S A 101:12271–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP et al (2003) Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17:2147–2155. [DOI] [PubMed] [Google Scholar]
- 32. Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro . Proc Natl Acad Sci USA 104:9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Evans EG, Pushie MJ, Markham KA, Lee HW, Millhauser GL (2016) Interaction between prion protein's copper‐bound octarepeat domain and a charged C‐terminal pocket suggests a mechanism for N‐terminal regulation. Structure 24:1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Falsig J, Aguzzi A (2008) The prion organotypic slice culture assay–POSCA. Nat Protoc 3:555–562. [DOI] [PubMed] [Google Scholar]
- 35. Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A (2008) A versatile prion replication assay in organotypic brain slices. Nat Neurosci 11:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Falsig J, Sonati T, Herrmann US, Saban D, Li B, Arroyo K et al (2012) Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog 8:e1002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fang C, Imberdis T, Garza MC, Wille H, Harris DA (2016) A neuronal culture system to detect prion synaptotoxicity. PLoS Pathog 12:e1005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fang C, Wu B, Le NTT, Imberdis T, Mercer RCC, Harris DA (2018) Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog 14:e1007283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A (2000) Glutamate receptors regulate actin‐based plasticity in dendritic spines. Nat Neurosci 3:887–894. [DOI] [PubMed] [Google Scholar]
- 40. Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C et al (2013) An N‐terminal fragment of the prion protein binds to amyloid‐beta oligomers and inhibits their neurotoxicity in vivo . J Biol Chem 288:7857–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J (2007) Dendritic pathology in prion disease starts at the synaptic spine. J Neurosci 27:6224–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S‐nitrosylation. Antioxid Redox Signal 22:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goniotaki D, Lakkaraju AKK, Shrivastava AN, Bakirci P, Sorce S, Senatore A et al (2017) Inhibition of group‐I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog 13:e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ (2013) Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci 126:3552–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J et al (2011) Rapid cell‐surface prion protein conversion revealed using a novel cell system. Nat Commun 2:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS et al (2017) Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer's mouse phenotypes. Cell Rep 20:76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hannaoui S, Maatouk L, Privat N, Levavasseur E, Faucheux BA, Haik S (2013) Prion propagation and toxicity occur in vitro with two‐phase kinetics specific to strain and neuronal type. J Virol 87:2535–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harris DA, Lele P, Snider WD (1993) Localization of the mRNA for a chicken prion protein by in situ hybridization. Proc Natl Acad Sci USA 90:4309–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP (2001) Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci 21:2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herce HD, Garcia AE (2007) Molecular dynamics simulations suggest a mechanism for translocation of the HIV‐1 TAT peptide across lipid membranes. Proc Natl Acad Sci USA 104:20805–20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herms J, Dorostkar MM (2016) Dendritic spine pathology in neurodegenerative diseases. Annu Rev Pathol 11:221–250. [DOI] [PubMed] [Google Scholar]
- 52. Herrmann US, Sonati T, Falsig J, Reimann RR, Dametto P, O'Connor T et al (2015) Prion infections and anti‐PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 11:e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hlushchenko I, Koskinen M, Hotulainen P (2016) Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton (Hoboken) 73:435–441. [DOI] [PubMed] [Google Scholar]
- 54. Horiuchi M, Caughey B (1999) Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease‐resistant state. EMBO J 18:3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hotulainen P, Hoogenraad CC (2010) Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hynd MR, Scott HL, Dodd PR (2004) Glutamate‐mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int 45:583–595. [DOI] [PubMed] [Google Scholar]
- 57. Imberdis T, Heeres JT, Yueh H, Fang C, Zhen J, Rich CB et al (2016) Identification of anti‐prion compounds using a novel cellular assay. J Biol Chem 291:26164–26176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C et al (2000) Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie‐infected murine hippocampus. Neuropathol Appl Neurobiol 26:41–54. [DOI] [PubMed] [Google Scholar]
- 59. Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21:1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1:2406–2415. [DOI] [PubMed] [Google Scholar]
- 61. Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT et al (2015) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 77:953–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J et al (2008) Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol 181:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim C, Haldiman T, Cohen Y, Chen W, Blevins J, Sy MS et al (2011) Protease‐sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt‐Jakob disease are indicator of progression rate. PLoS Pathog 7:e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2016) Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol 275(Pt 1):162–171. [DOI] [PubMed] [Google Scholar]
- 65. Klohn PC, Farmer M, Linehan JM, O'Malley C, Fernandez de Marco M, Taylor W et al (2012) PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 335:52. [DOI] [PubMed] [Google Scholar]
- 66. Klyubin I, Nicoll AJ, Khalili‐Shirazi A, Farmer M, Canning S, Mably A et al (2014) Peripheral administration of a humanized anti‐PrP antibody blocks Alzheimer's disease Aβ synaptotoxicity. J Neurosci 34:6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kovacs GG, Preusser M, Strohschneider M, Budka H (2005) Subcellular localization of disease‐associated prion protein in the human brain. Am J Pathol 166:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature 457:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases‐what is the evidence? Front Neurosci 9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105‐125. EMBO J 26:548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liberski PP, Ironside JW (2004) An outline of the neuropathology of transmissible spongiform encephalopathies (prion diseases). Folia Neuropathol 42(Suppl. B):39–58. [PubMed] [Google Scholar]
- 72. Linden R (2017) The biological function of the prion protein: a cell surface scaffold of signaling modules. Front Mol Neurosci 10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lukinavicius G, Reymond L, D'Este E, Masharina A, Gottfert F, Ta H et al (2014) Fluorogenic probes for live‐cell imaging of the cytoskeleton. Nat Methods 11:731–733. [DOI] [PubMed] [Google Scholar]
- 74. Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Ostapchenko VG, Budka H et al (2012) A new mechanism for transmissible prion diseases. J Neurosci 32:7345–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302:871–874. [DOI] [PubMed] [Google Scholar]
- 76. Mallucci GR (2009) Prion neurodegeneration: starts and stops at the synapse. Prion 3:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J (1992) The prion protein gene: a role in mouse embryogenesis? Development 115:117–122. [DOI] [PubMed] [Google Scholar]
- 78. Miller MB, Geoghegan JC, Supattapone S (2011) Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog 7:e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morrison DK (2012) MAP kinase pathways. Cold Spring Harb Perspect Biol 4:a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Munoz L, Ammit AJ (2010) Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology 58:561–568. [DOI] [PubMed] [Google Scholar]
- 81. Newby GA, Lindquist S (2013) Blessings in disguise: biological benefits of prion‐like mechanisms. Trends Cell Biol 23:251–259. [DOI] [PubMed] [Google Scholar]
- 82. Nicoll AJ, Panico S, Freir DB, Wright D, Terry C, Risse E et al (2013) Amyloid‐beta nanotubes are associated with prion protein‐dependent synaptotoxicity. Nat Commun 4:2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. [DOI] [PubMed] [Google Scholar]
- 84. Noble GP, Supattapone S (2015) Dissociation of recombinant prion autocatalysis from infectivity. Prion 9:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC et al (2007) Cellular prion protein regulates beta‐secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A 104:11062–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pauly PC, Harris DA (1998) Copper stimulates endocytosis of the prion protein. J Biol Chem 273:33107–33110. [DOI] [PubMed] [Google Scholar]
- 87. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. [DOI] [PubMed] [Google Scholar]
- 88. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M et al (1993) Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti‐PrP antibodies. Proc Natl Acad Sci U S A 90:10608–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Purro SA, Mead S, Khalili‐Shirazi A, Nicoll AJ, Collinge J (2018) Reply to: intrinsic toxicity of antibodies to the globular domain of the prion protein. Biol Psychiatry 84:e53–e54. [DOI] [PubMed] [Google Scholar]
- 91. Purro SA, Nicoll AJ, Collinge J (2018) Prion protein as a toxic acceptor of amyloid‐beta oligomers. Biol Psychiatry 83:358–368. [DOI] [PubMed] [Google Scholar]
- 92. Race RE, Caughey B, Graham K, Ernst D, Chesebro B (1988) Analyses of frequency of infection, specific infectivity, and prion protein biosynthesis in scrapie‐infected neuroblastoma cell clones. J Virol 62:2845–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rambold AS, Muller V, Ron U, Ben‐Tal N, Winklhofer KF, Tatzelt J (2008) Stress‐protective signalling of prion protein is corrupted by scrapie prions. EMBO J 27:1974–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reimann RR, Aguzzi A (2018) Intrinsic toxicity of antibodies to the globular domain of the prion protein. Biol Psychiatry 84:e51–e52. [DOI] [PubMed] [Google Scholar]
- 95. Reimann RR, Sonati T, Hornemann S, Herrmann US, Arand M, Hawke S, Aguzzi A (2016) Differential toxicity of antibodies to the prion protein. PLoS Pathog 12:e1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H et al (2005) Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102:3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sajnani G, Silva CJ, Ramos A, Pastrana MA, Onisko BC, Erickson ML et al (2012) PK‐sensitive PrP is infectious and shares basic structural features with PK‐resistant PrP. PLoS Pathog 8:e1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sala C, Segal M (2014) Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94:141–188. [DOI] [PubMed] [Google Scholar]
- 99. Sandberg MK, Al‐Doujaily H, Sharps B, Clarke AR, Collinge J (2011) Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470:540–542. [DOI] [PubMed] [Google Scholar]
- 100. Sandberg MK, Al‐Doujaily H, Sharps B, De Oliveira MW, Schmidt C, Richard‐Londt A et al (2014) Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat Commun 5:4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Scheltens P, Prins N, Lammertsma A, Yaqub M, Gouw A, Wink AM et al (2018) An exploratory clinical study of p38α kinase inhibition in Alzheimer's disease. Ann Clin Transl Neurol 5:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sempou E, Biasini E, Pinzon‐Olejua A, Harris DA, Malaga‐Trillo E (2016) Activation of zebrafish Src family kinases by the prion protein is an amyloid‐beta‐sensitive signal that prevents the endocytosis and degradation of E‐cadherin/beta‐catenin complexes in vivo . Mol Neurodegener 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sepers MD, Raymond LA (2014) Mechanisms of synaptic dysfunction and excitotoxicity in Huntington's disease. Drug Discov Today 19:990–996. [DOI] [PubMed] [Google Scholar]
- 104. Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J et al (1998) Expression of amino‐terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93:203–214. [DOI] [PubMed] [Google Scholar]
- 105. Shyng SL, Moulder KL, Lesko A, Harris DA (1995) The N‐terminal domain of a glycolipid‐anchored prion protein is essential for its endocytosis via clathrin‐coated pits. J Biol Chem 270:14793–14800. [DOI] [PubMed] [Google Scholar]
- 106. Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA (2007) Toward molecular dissection of PrPC‐PrPSc interactions. J Biol Chem 282:7465–7471. [DOI] [PubMed] [Google Scholar]
- 107. Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez‐Alavez M, Sugama S et al (2004) Cross‐linking cellular prion protein triggers neuronal apoptosis in vivo . Science 303:1514–1516. [DOI] [PubMed] [Google Scholar]
- 108. Solomon IH, Biasini E, Harris DA (2012) Ion channels induced by the prion protein: mediators of neurotoxicity. Prion 6:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Solomon IH, Huettner JE, Harris DA (2010) Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem 285:26719–26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA (2011) An N‐terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem 286:14724–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S et al (2013) The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501:102–106. [DOI] [PubMed] [Google Scholar]
- 112. Spence EF, Soderling SH (2015) Actin out: regulation of the synaptic cytoskeleton. J Biol Chem 290:28613–28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Spevacek AR, Evans EG, Miller JL, Meyer HC, Pelton JG, Millhauser GL (2013) Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 21:236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ et al (2001) Identification of two prion protein regions that modify scrapie incubation time. J Virol 75:1408–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Taylor DR, Watt NT, Perera WS, Hooper NM (2005) Assigning functions to distinct regions of the N‐terminus of the prion protein that are involved in its copper‐stimulated, clathrin‐dependent endocytosis. J Cell Sci 118:5141–5153. [DOI] [PubMed] [Google Scholar]
- 116. Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. [DOI] [PubMed] [Google Scholar]
- 117. Turnbaugh JA, Unterberger U, Saa P, Massignan T, Fluharty BR, Bowman FP et al (2012) The N‐terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J Neurosci 32:8817–8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H et al (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79:887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Um JW, Ko J (2017) Neural gcosylphosphatidylinositol‐achored poteins in synaptic specification. Trends Cell Biol 27:931–945. [DOI] [PubMed] [Google Scholar]
- 120. Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A et al (2012) Alzheimer amyloid‐beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Vorberg I, Raines A, Priola SA (2004) Acute formation of protease‐resistant prion protein does not always lead to persistent scrapie infection in vitro . J Biol Chem 279:29218–29225. [DOI] [PubMed] [Google Scholar]
- 122. Wadia JS, Schaller M, Williamson RA, Dowdy SF (2008) Pathologic prion protein infects cells by lipid‐raft dependent macropinocytosis. PLoS One 3:e3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Walsh DM, Selkoe DJ (2007) Aβ oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- 124. Walter ED, Chattopadhyay M, Millhauser GL (2006) The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry 45:13083–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang F, Wang X, Yuan CG, Ma J (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Watt NT, Taylor DR, Kerrigan TL, Griffiths HH, Rushworth JV, Whitehouse IJ, Hooper NM (2012) Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun 3:1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Watts JC, Bourkas MEC, Arshad H (2018) The function of the cellular prion protein in health and disease. Acta Neuropathol 135:159–178. [DOI] [PubMed] [Google Scholar]
- 128. Westergard L, Christensen HM, Harris DA (2007) The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 1772:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Westergard L, Turnbaugh JA, Harris DA (2011) A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci 31:14005–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S et al (2003) Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422:80–83. [DOI] [PubMed] [Google Scholar]
- 131. Wu B, McDonald AJ, Markham K, Rich CB, McHugh KP, Tatzelt J et al (2017) The N‐terminus of the prion protein is a toxic effector regulated by the C‐terminus. Elife 6:e23473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wulf MA, Senatore A, Aguzzi A (2017) The biological function of the cellular prion protein: an update. BMC Biol 15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yasuda S, Sugiura H, Tanaka H, Takigami S, Yamagata K (2011) p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent Nerv Syst Agents Med Chem 11:45–59. [DOI] [PubMed] [Google Scholar]
- 134. Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I et al (2010) Variably protease‐sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]