

Abstract
Platinum-based chemotherapy is still widely applied for the treatment of advanced non-small cell lung cancer (NSCLC). However, acquired chemoresistance compromises the curative effect of this drug. In this study, we found that glucose-6-phosphate dehydrogenase (G6PD), a critical enzyme of the pentose phosphate pathway, contributed to cisplatin resistance in NSCLC. The experimental results showed that transforming growth factor beta 1 (TGFβ1) increased the expression of G6PD by activating the forkhead box protein M1-high mobility group AT-hook 1-G6PD (FOXM1-HMGA1-G6PD) transcriptional regulatory pathway, in which TGFβ1 inhibited the ubiquitination and degradation of FOXM1 protein. Additionally, HMGA1 induced TGFβ1 expression, and neutralized TGFβ1 in the culture medium downregulated HMGA1 levels, suggesting the existence of a TGFβ1-FOXM1-HMGA1-TGFβ1 positive feedback loop and its role in maintaining G6PD expression. Further investigations showed that exogenous TGFβ1 enhanced the cisplatin resistance of NSCLC cells, while disrupting the FOXM1-HMGA1-G6PD pathway, thereby sensitizing the cells to cisplatin. Consistently, the TGFβ1-FOXM1-HMGA1-G6PD axis was confirmed in NSCLC tissues, and overactivation of this axis predicted poor survival in NSCLC patients. Collectively, the results of this study demonstrate that the TGFβ1-FOXM1-HMGA1-TGFβ1 positive feedback loop plays a crucial role in the cisplatin resistance of NSCLC by upregulating the expression of G6PD, providing a potential therapeutic target to restore chemosensitivity in cisplatin-resistant NSCLC.
Keywords: Non-small-cell lung cancer (NSCLC), transforming growth factor β1 (TGFβ1), forkhead box protein M1 (FOXM1), high-mobility group A1 (HMGA1), glucose-6-phosphate dehydrogenase (G6PD), cisplatin resistance
Introduction
Lung cancer is the most common and fatal cancer worldwide, and non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancers. At present, platinum-based chemotherapy is the standard treatment for NSCLC [1]; however, the response rates range between 10% and 25% because of acquired resistance [2,3]. The mechanisms underlying the resistance to platinum are not fully understood.
Transforming growth factor beta 1 (TGFβ1) promotes the epithelial to mesenchymal transition (EMT), angiogenesis, and metastasis during the advanced stages of tumor development [4]. Recently, some studies have focused on the role of TGFβ1 in chemoresistance. In drug-resistant colorectal carcinoma cells, 5-fluorouracil (5FU)-induced TGFβ1 protected cells against the toxic action of the drug [5]. Another study showed that TGFβ1-dependent activation of SMAD/ERK signaling conferred temozolomide resistance in glioblastoma [6]. Additionally, it was found that TGFβ1 enhances the gemcitabine resistance of bladder cancer, and TGFβ1 stimulation induces leukemia cells into cell cycle arrest resistant to arabinosyl cytosine [7,8]. As major components of the tumor microenvironment, tumor-associated macrophages and fibroblasts also secrete TGFβ1 to facilitate chemoresistance [9,10]. Interestingly, Shen et al. [11] reported that the autocrine TGFβ1 decreases the susceptibility of cisplatin-resistant lung cancer cells to natural killer cell cytotoxicity by upregulating programmed death-ligand 1 levels [11].
Forkhead box M1 (FOXM1) is a transcription factor characterized as a regulator of cell cycle progression, which is overexpressed in a large variety of human tumors [12]. Except for proliferation, FOXM1 regulates many aspects of tumor progression including metastasis, angiogenesis, and chemoresistance [13,14]. In NSCLC, high FOXM1 expression has been detected in an invasive subgroup identified by poor prognosis, high incidence of metastases, and poor tumor differentiation [15]. In addition, the upregulation of FOXM1 also correlates with recurrence after NSCLC resection, resulting in shorter disease-free survival [16].
High-mobility group AT-hook 1 (HMGA1) is a chromatin architectural transcription factor that binds the minor groove of AT-rich DNA, thereby changing chromatin structure and facilitating the assembly of transcriptional complexes, consequently controlling the transcription of downstream effectors involved in several fundamental cellular processes such as differentiation, transformation, and apoptosis [17,18]. Compared with normal cells and tissues, HMGA1 is abundant in malignant carcinomas including NSCLC [19], and its overexpression correlates with metastatic potential, drug resistance, and reduced survival in NSCLC patients [20,21].
As the rate-limiting enzyme of the pentose phosphate pathway (PPP), glucose-6-phosphate dehydrogenase (G6PD) catalyzes the oxidation of G6P to 6-phosphogluconate leading to the production of nicotinamide adenine dinucleotide phosphate (NADPH). Elevated level and activity of G6PD usually appear during cancer development and progression, which are associated with resistance to therapy. Cancer cells overexpressing G6PD have shown high activity of the pentose phosphate pathway and increased resistance to multiple drugs including doxorubicin, cisplatin, oxaliplatin, and adriamycin [22-25]. Recently, Hong et al. [26] reported that inhibition of G6PD restored the cisplatin sensitivity of NSCLC cells by influencing redox homeostasis. Thus, understanding the dysregulation of G6PD may help overcome the resistance to current chemotherapy drugs.
In this study, it was demonstrated that TGFβ1 induced HMGA1 expression by increasing the stability of FOXM1 protein, thereby activating the transcription of G6PD, which was required for cisplatin resistance in NSCLC. In addition, the induction of TGFβ1 by HMGA1 suggested the existence of the TGFβ1-FOXM1-HMGA1-TGFβ1 positive feedback loop, which maintained G6PD levels and resistance to cisplatin. These findings reveal a novel regulatory pathway for G6PD as well as its role in chemoresistance.
Materials and methods
Cell culture and transfection
Human NSCLC cell lines, H1299, H226, Calu-3, H460 and A549, and the human lung epithelial cell line, BEAS-2B, were purchased from American Type Culture Collection (Manassas, VA, USA). All the cell lines were cultured in RPMI-1640 medium containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in 5% CO2 at 37°C. H1299 and H226 cells were treated with cisplatin (P4394; Sigma, St Louis, MO, USA) and recombinant human TGFβ1 (ab50036; Abcam, Cambridge, UK). Overexpression plasmids and small interfering RNAs (siRNAs) were transfected using LipofectamineTM 3000 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions.
Plasmids, siRNAs, and antibodies
FOXM1, HMGA1, and G6PD overexpression plasmids were constructed based on pcDNA3.1. The siRNAs targeting FOXM1, HMGA1, and G6PD were synthesized from GenePharma (Shanghai, China). Antibodies against TGFβ1 (ab92486), HMGA1 (ab168260), and G6PD (ab210702) were purchased from Abcam. Antibodies against FOXM1 (sc-376471), Flag (OctA, sc-166355), Ku80 (sc-5280), and gamma H2A histone family member X (γ-H2AX) (sc-517348) were purchased from Santa Cruz (Santa Cruz, CA, USA).
Western blot
Western blot analysis was conducted according to our previous study [27]. Whole cell lysate was obtained using RIPA lysis buffer (Millipore, CA, USA) and then examined using the indicated antibodies. IRDye 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA) were used for staining and proteins were detected using the Odyssey infrared imaging system (LI-COR) (Figure S1).
Quantitative real-time PCR
Total RNA was extracted using the RNAisoTM Plus reagent (Takara, Otsu, Japan) and reverse transcribed using a PrimeScriptTM RT reagent kit (Takara). Quantitative PCR (qPCR) was performed with SYBR Green Mix (Takara) according to the manufacturer’s instructions. β-actin served as the loading control. The sequences of the qPCR primers are listed in Table S1.
Immunofluorescence
Immunofluorescence was performed according to our previous study [27]. The slides were mounted and visualized by fluorescence microscope.
DNA pull-down assay
The DNA pull-down assay was performed according to our previous study [28]. Briefly, nuclear proteins were extracted from TGFβ1-treated or control H1299 cells. DNA probes of the HMGA1 promoter were obtained by PCR with the primers (one of which was labeled with biotin). The biotinylated DNA probes were pre-incubated with M-280 streptavidin Dynabeads (Invitrogen), and then the beads were added to the nuclear extracts and incubated. After washing, loading buffer was added to the precipitates, boiled, and separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins pulled down by the DNA probes were analyzed by silver stain, liquid chromatography-tandem mass spectrometry (LC-MS/MS), and immunoblotting.
RNA sequencing analysis
RNA sequencing (RNA-seq) was performed as previously described [27]. In brief, RNA from H1299 cells with or without HMGA1 knockdown was extracted and purified for quantification, RNA-seq library preparation, and sequencing. The libraries were sequenced on the Illumina HiSeq 2500 platform. The reads containing adapter or poly-N and reads of low quality were removed from raw data to generate clean reads for further analyses. Based on the clean reads, the Q20 (>90%), Q30 (>85%), and error rate (<0.1%) of the clean data were required. Then mapped reads were obtained by Tophat2 by aligning clean reads to the human genome reference (hg19). The number of mapped clean reads for each unigene was counted and normalized into reads/kb/million reads to calculate the expression level of the unigene.
G6PD activity, NADPH, and reactive oxygen species measurement
H1299 and H226 cells were seeded in six-well plates. Following transfection and treatment, G6PD activity, NADPH level, and reactive oxygen species (ROS) level were tested using the Glucose 6 Phosphate Dehydrogenase Assay Kit (ab102529; abcam), NADP/NADPH Assay Kit (ab65349; abcam), and ROS/Superoxide Detection Assay Kit (ab139476; abcam) according to each manufacturer’s specifications. The optical density 450/490 was measured using an automatic plate reader.
Luciferase assay
The promoter region of human G6PD was amplified by PCR and inserted into the pGL3 vector. The reporter constructs containing various lengths of G6PD promoter or mutated AT-hooks were generated by subsequent PCR-based cloning. H1299 cells were plated in 24-well plates and then co-transfected with pGL3 constructs, pRL-SV40 plasmid, and HMGA1 overexpression plasmid or siRNA. After 48 h, luciferase activity was measured using a dual-luciferase reporter assay system (Promega, Madison, WI, USA) and a luminometer (LB 9507; Berthold, BadWildbad, Germany).
Chromatin immunoprecipitation
The chromatin immunoprecipitation (ChIP) was performed according to our previous study [27]. Briefly, chromatin was crosslinked using 1% formaldehyde and sonicated to obtain DNA fragments of 200-500 base pairs. After centrifugation, the supernatants were subjected to immunoprecipitation overnight with antibodies against HMGA1 (ab168260). Protein A/G PLUS-Agarose (sc-2003; Santa Cruz) was used to isolate the chromatin-antibody complexes. The crosslinking was reversed, and the precipitated DNA fragments were purified and analyzed by qPCR and agarose gel with the primers (G6PD promoter) listed in Table S1.
Cell viability assay
H1299 and H226 cells in logarithmic growth were plated and then transfected or treated. After the indicated times of culture, cell viability was determined using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA), and the optical density at 450 nm was measured using an automatic plate reader.
Immunohistochemistry
In total, 94 NSCLC cases were from the National Human Genetic Resources Sharing Service Platform (HLugA180Su06; Tianjin, China); 58 NSCLC cases were from Chinese and Western Combined Hospital of Taizhou (Zhejiang Sheng, China) and 64 NSCLC cases were from Sanmen People’s Hospital of Zhejiang (Sanmen, China). Of the 216 cases, 205 had a survival time. Informed consent was obtained from all patients. The study was approved by the Ethics Committee of Chinese and Western Combined Hospital of Taizhou, the Ethics Committee of Sanmen People’s Hospital of Zhejiang, and Shanghai Outdo Biotech Company (Shanghai, China). Immunohistochemistry was performed and analyzed as previously described [27].
Statistical analysis
Statistical analysis was performed with SPSS (version 22.0) and GraphPad Prism (version 5.0). Analysis of differences was performed using the two-tailed Student’s t-test or analysis of variance (one-way with Tukey’s post hoc test; two-way with Sidak’s post hoc test). The results are presented as the mean ± standard deviation of three separate experiments. χ2 test or Fisher’s exact probability test was used to compare the clinicopathological features of patients with protein expression. The Spearman’s rank correlation test was used for analyzing the correlation. Kaplan-Meier plots and log-rank tests were used for survival analysis. P<0.05 was considered statistically significant.
Results
TGFβ1 indicates a poor prognosis and induces the expression of HMGA1 in NSCLC
The link between TGFβ1 and HMGA1 has been established in breast and thyroid cancer [29,30]. To verify the relationship in lung cancer, immunohistochemical analyses of NSCLC tissues from 216 cases was performed. The representative images in Figure 1A, 1B showed that cases with high TGFβ1 expression had stronger HMGA1 staining than those with low expression, and statistical analysis of IHC positively correlated the expression levels of TGFβ1 with the degree of HMGA1. In addition, for 205 cases with survival time, it was noticed that overall survival (OS) was worse in patients with high TGFβ1 staining than in those with low staining (Figure 1C). Furthermore, we analyzed the link between TGFβ1 and HMGA1 in NSCLC cells. The results showed that the expression levels of TGFβ1 paralleled those of HMGA1 in different NSCLC cell lines, and higher levels of TGFβ1 and HMGA1 were detected in the NSCLC cell lines compared with the lung epithelial cell line (Figure 1D). Exogenous TGFβ1 treatment increased the mRNA and protein levels of HMGA1 in H1299 and H226 cells, while neutralizing TGFβ1 in culture medium with a specific antibody inhibited the expression of HMGA1 (Figure 1E-G). These results support the TGFβ1-mediated regulation of HMGA1 in NSCLC cells.
Figure 1.
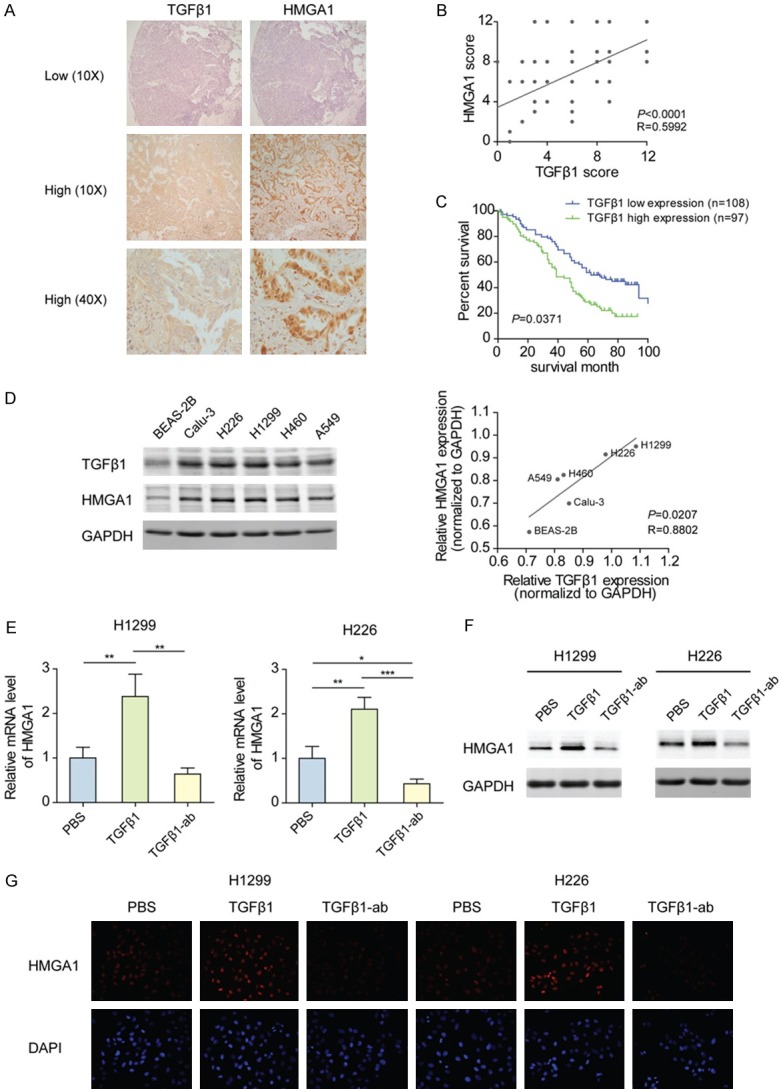
TGFβ1 indicates a poor prognosis and induces the expression of HMGA1 in NSCLC. (A) Representative image of TGFβ1 and HMGA1 immunostaining in NSCLC tissues from two cases, showing the low or high expression of TGFβ1 and HMGA1. (B) Correlation between concurrent immunostaining scores of TGFβ1 and HMGA1 in NSCLC tissues from 216 cases. (C) OS of 205 NSCLC patients with low and high expression of TGFβ1. (D) Left panel: Western blot analyses of TGFβ1 and HMGA1 in BEAS-2B, Calu-3, H226, H1299, H460, and A549 cells. Right panel: Band density in the left panel was calculated by Image J software. Based on the relative expression (densityTGFβ1 or HMGA1/densityGAPDH), the correlation between TGFβ1 and HMGA1 in human lung epithelial and NSCLC cell lines was analyzed. (E) The mRNA level of HMGA1 in H1299 and H226 cells treated with 10 ng/mL TGFβ1 or 100 ng/mL TGFβ1 antibody for 48 h was analyzed by qPCR. *P<0.05, **P<0.01, ***P <0.001. (F) The protein level of HMGA1 in H1299 and H226 cells treated as described in (E) was analyzed by western blotting. (G) Immunofluorescence analysis of HMGA1 protein in H1299 and H226 cells treated as described in (E).
Increased stabilization of FOXM1 by deubiquitination promotes the transcription of HMGA1 in response to TGFβ1 stimulation
To understand the mechanisms responsible for the regulation of HMGA1 expression by TGFβ1, we examined the transcriptional activities of deletion constructs from HMGA1 promoter after TGFβ1 treatment. It was shown that the HMGA1 promoter lacking the region between -3000 and -2000 lost the ability to respond to TGFβ1 (Figure 2A). Then, DNA pull-down assays with biotin-labeled HMGA1 promoter probe (-3000/-2000) were performed. The protein complexes were captured by the DNA probe from nuclear extracts of H1299 cells with TGFβ1 stimulation or not. The captured proteins were analyzed by silver staining, and the different bands between TGFβ1 treatment and control groups were cut off to identify the components by LC-MS/MS spectrometry (Figure 2B). The most significantly enriched proteins in the TGFβ1 treatment group are shown in Figure 2C. Next, we determined whether FOXM1 mediated the TGFβ1-induced expression of HMGA1. A putative binding site for FOXM1 (-2069/-2061) was predicted at the HMGA1 promoter, and DNA pull-down assays coupled with western blot analysis showed that the binding of FOXM1 to the wild-type HMGA1 promoter probe was markedly enhanced by TGFβ1 exposure, while FOXM1 did not interact with the probe containing the mutant FOXM1 binding site (Figure 2D). Furthermore, it was found that overexpression of FOXM1 upregulated the mRNA and protein levels of HMGA1 in H1299 cells and knockdown of FOXM1 downregulated expression (Figure 2E, 2F). Additionally, silencing FOXM1 impaired the induction of HMGA1 that arose from TGFβ1 treatment (Figure 2G, 2H). These results indicate the crucial role of FOXM1 in the regulation of HMGA1 by TGFβ1. Considering the increased level of FOXM1 protein after TGFβ1 treatment (Figure 2D), we next studied the effect of TGFβ1 on FOXM1 expression. However, no significant change of FOXM1 mRNA level was detected in both H1299 and H226 cells (Figure 2I). Further investigations showed that TGFβ1 stimulation dramatically increased the half-life of FOXM1, measured by a CHX-chase assay (Figure 2J). To determine whether the ubiquitin proteasome pathway is involved in TGFβ1-mediated FOXM1 stability, we analyzed the ubiquitination of FOXM1 protein and found that FOXM1 had a lower ubiquitination level in TGFβ1-treated cells than in control cells (Figure 2K). Together, these findings indicate that TGFβ1 induces the deubiquitination of FOXM1 protein and enhances its stability, thereby transactivating HMGA1.
Figure 2.
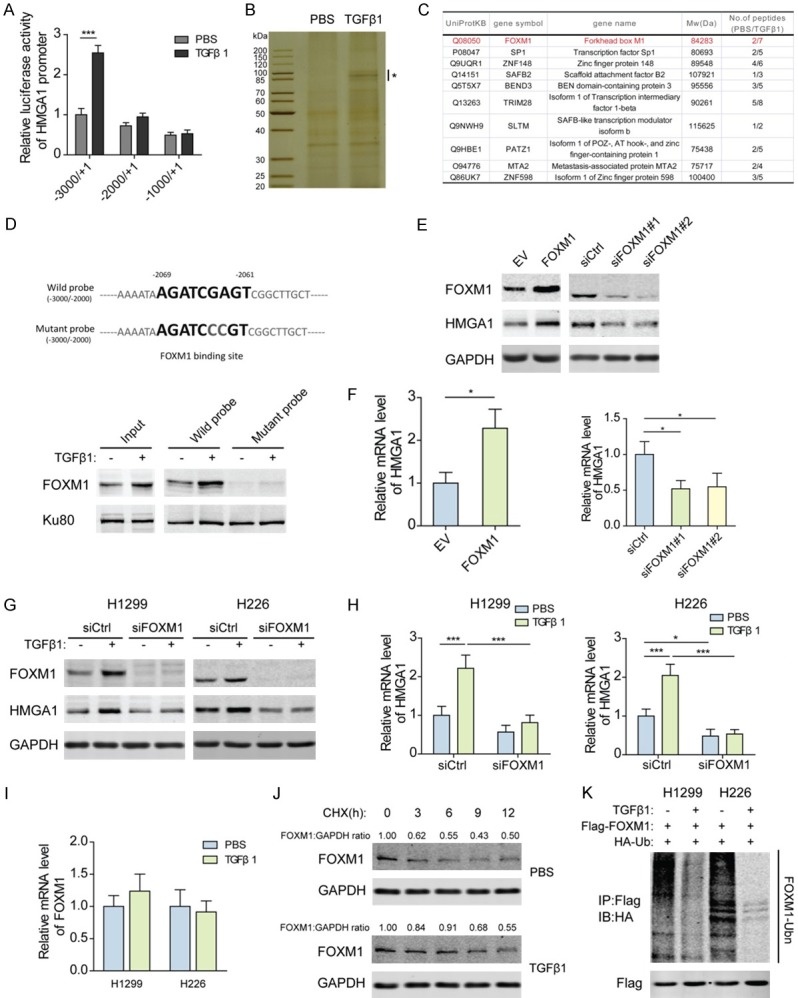
Increased stabilization of FOXM1 by deubiquitination promotes the transcription of HMGA1 in response to TGFβ1 stimulation. (A) Transcription activity of the truncated HMGA1 promoter sequences after 10 ng/mL TGFβ1 treatment for 48 h as measured by luciferase reporter assays in H1299 cells. ***P<0.001. (B) Nuclear proteins extracted from H1299 cells exposed to 10 ng/mL TGFβ1 for 48 h were pulled down by a biotin-labeled HMGA1 promoter (-3000/-2000) DNA probe, separated with 10% SDS-PAGE, and analyzed after silver staining. *Indicates the different bands between TGFβ1 treatment and control groups. (C) The different bands from (B) were cut off to identify the components by LC-MS/MS spectrometry. The most significantly enriched proteins in TGFβ1 treatment group are shown. (D) A putative binding site for FOXM1 was predicted at -2069/-2061 of HMGA1 promoter. H1299 cells were exposed to 10 ng/mL TGFβ1 for 48 h, and the binding of FOXM1 to the wild-type HMGA1 promoter probe (-3000/-2000) or the probe with mutant FOXM1 binding site was assessed by DNA pull-down assays coupled with western blot analysis. Ku80 served as a control. (E) Protein level of HMGA1 in H1299 cells transfected with FOXM1 overexpression plasmids or siRNAs for 48 h was analyzed by western blotting. (F) The mRNA level of HMGA1 in H1299 cells transfected as described in (E) was analyzed by qPCR. *P<0.05. (G) Protein levels of FOXM1 and HMGA1 in H1299 and H226 cells transfected with FOXM1 siRNAs and treated with 10 ng/mL TGFβ1 for 48 h were analyzed by western blotting. (H) The mRNA levels of FOXM1 and HMGA1 in H1299 and H226 cells transfected as described in (G) were analyzed by qPCR. *P<0.05, ***P<0.001. (I) The mRNA levels of FOXM1 in H1299 and H226 cells treated with 10 ng/mL TGFβ1 for 48 h were analyzed by qPCR. (J) Western blot analyses of FOXM1 in TGFβ1-treated or control H1299 cells pretreated for 15 min with 20 mmol/L cycloheximide. (K) HA-ubiquitin and Flag-tagged FoxM1 plasmids were transfected into H1299 cells. After exposure to 10 ng/mL TGFβ1 for 12 h, cells were treated with 25 nM MG132 for 6 h. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody, followed by immunoblotting with anti-Flag and anti-HA antibody.
HMGA1 is required for the expression of G6PD and TGFβ1 in NSCLC
To identify the target genes of HMGA1 in NSCLC, RNA-seq was performed to profile the transcriptome changes after knocking down HMGA1 in H1299 cells. In total, 1152 genes with significantly differential expression (P<0.05) were obtained following HMGA1 knockdown. Among these genes, 439 were upregulated and 713 were downregulated (Figure 3A; Table S2). The differentially expressed genes (DEGs) with >2-fold change were represented in the heatmap (Figure 3B), among which G6PD and TGFβ1 had a 0.37-fold and 0.46-fold decrease, respectively. To confirm these findings, we modified the expression of HMGA1 in H1299 and H226 cells. The results showed that ectopic expression of HMGA1 markedly increased TGFβ1 and G6PD at the mRNA and protein levels, while TGFβ1 and G6PD were downregulated following knockdown of HMGA1 (Figure 3C, 3D, 3F, 3G), and evident accumulation of G6PD around the nucleus was observed after overexpressing HMGA1 (Figure 3F). Importantly, the secretion of TGFβ1 was consequently induced by HMGA1, as indicated by the TGFβ1 concentration in culture medium, suggesting a positive feedback between HMGA1 and TGFβ1 (Figure 3E). Because G6PD functions as the central regulator of PPP, we also examined the enzymatic activity of G6PD and the level of NADPH, an important antioxidant produced by G6PD. Consistent with G6PD expression, overexpressed HMGA1 enhanced the enzymatic activity of G6PD and promoted the production of NADPH, and knockdown of HMGA1 suppressed them (Figure 3H, 3I). These results demonstrate that HMGA1 regulates the expression of G6PD and TGFβ1 in NSCLC cells.
Figure 3.
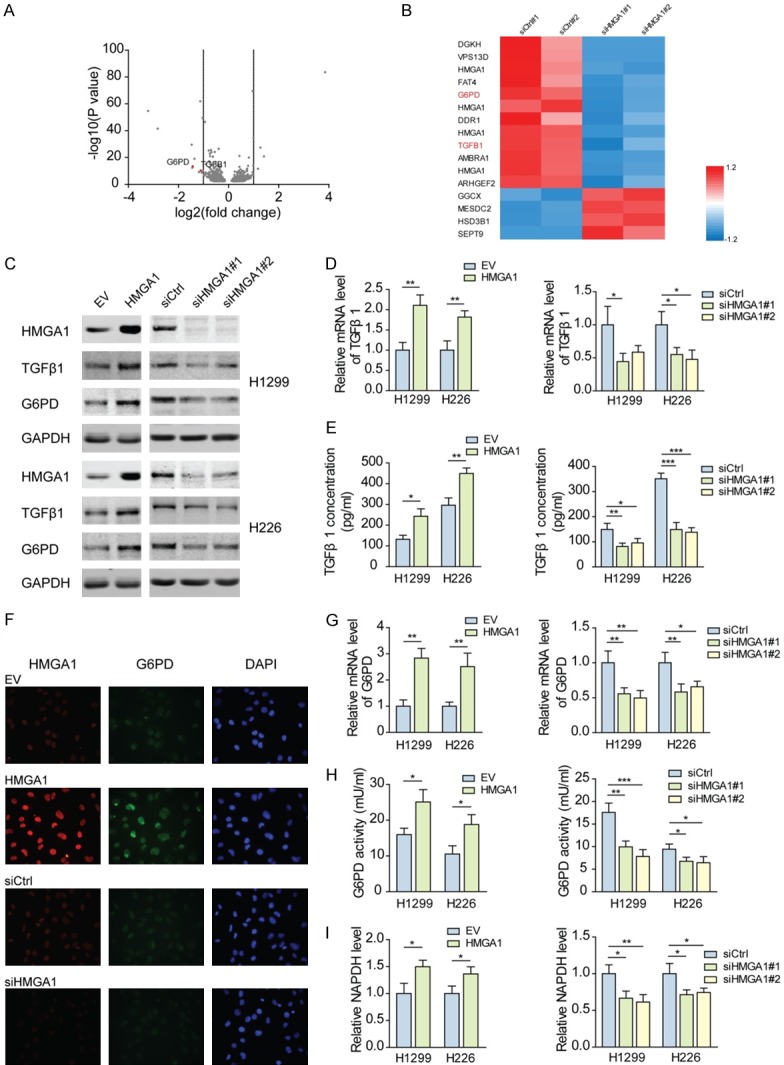
HMGA1 is required for the expression of G6PD and TGFβ1 in NSCLC. (A) Total number of genes with significant change in gene expression (P<0.05). 439 genes were upregulated and 713 genes were downregulated in H1299 cells transfected with HMGA1 siRNAs for 48 h, compared with the control. The differentially expressed genes were shown in volcano plots. (B) Heatmap of significantly differentially expressed genes (>2 fold) in HMGA1-silenced group versus control group. The fold change (fc) was normalized using (fc-meanrow)/SDrow. (C) Protein levels of TGFβ1 and G6PD in H1299 and H226 cells transfected with HMGA1 overexpression plasmids or siRNAs for 48 h were analyzed by western blotting. (D) The mRNA levels of TGFβ1 in H1299 and H226 cells transfected as described in (C) were analyzed by qPCR. *P<0.05, **P<0.01. (E) The concentrations of TGFβ1 secreted by H1299 and H226 cells transfected as described in (C) were analyzed by enzyme-linked immunoassay. *P<0.05, **P<0.01, ***P<0.001. (F) Immunofluorescence analysis of G6PD protein in H1299 cells transfected as described in (C). (G) The mRNA levels of G6PD in H1299 and H226 cells transfected as described in (C) were analyzed by qPCR. *P<0.05, **P<0.01. (H) The enzymatic activity of G6PD was measured in H1299 and H226 cells transfected as described in (C). *P<0.05, **P<0.01, ***P<0.001. (I) NADPH level was measured in H1299 and H226 cells transfected as described in (C). *P<0.05, **P<0.01.
TGFβ1-induced HMGA1 directly activates the transcription of G6PD
Based on the aforementioned results, it was determined whether HMGA1 directly activates the transcription of G6PD. The 2 kb region upstream of G6PD transcription start site was searched for putative HMGA1-binding sites. Serial deletion constructs of the promoter region were examined using luciferase reporter assays to identify the elements responsive to HMGA1 in NSCLC cells (Figure 4A). Activity of the full-length promoter was increased by HMGA1 overexpression and decreased by HMGA1 knockdown, but the promoter lacking the region between -1819 and -1345 was irresponsive to HMGA1 (Figure 4B). Around this region, three AT-rich sequences predicted to be bound by HMGA1 were identified (Figure 4C). Only the reporter activity of G6PD promoter with mutated #4 site was not affected by HMGA1 (Figure 4D). ChIP data also confirmed that HMGA1 directly bound to the G6PD promoter close to the #4 site. Notably, TGFβ1 stimulation strengthened the binding of HMGA1 to the G6PD promoter, while neutralizing TGFβ1 by specific antibody undermined the binding (Figure 4E, 4F). Because HMGA1 directly regulates the transcription of G6PD, it was necessary to confirm whether TGFβ1 induces the expression of G6PD via HMGA1. As shown in Figure 4G, 4H, exogenous TGFβ1 upregulated the mRNA and protein levels of G6PD, and as expected, the upregulation of G6PD was inhibited in HMGA1-silenced NSCLC cells. Similarly, TGFβ1 increased the enzymatic activity of G6PD and NADPH level in NSCLC cells, whereas knocking down HMGA1 attenuated the effect of TGFβ1 on G6PD and NADPH (Figure 4I, 4J). Thus it can be concluded that the induction of G6PD by TGFβ1 depends on HMGA1-mediated transcriptional regulation.
Figure 4.
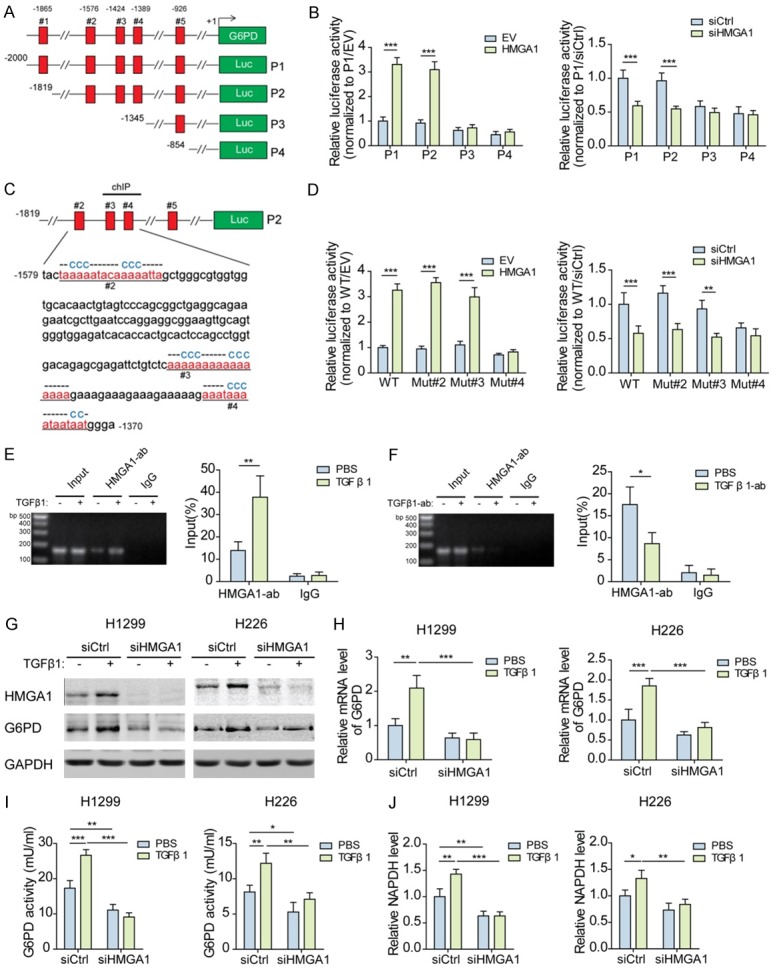
TGFβ1-induced HMGA1 directly activates the transcription of G6PD. (A) Schematic of the truncated G6PD promoter sequence with potential HMGA1 binding sites indicated by red rectangle. (B) Transcription activity of the truncated G6PD promoter sequences was measured by luciferase reporter assays in H1299 cells transfected with HMGA1 overexpression plasmids or siRNAs for 48 h. ***P<0.001. (C) The potential AT-hook sequences for HMGA1 binding were mutated as indicated. (D) Transcription activity of the wild-type or mutated G6PD promoter sequences was measured by luciferase reporter assays in H1299 cells transfected as described in (B). **P<0.01, ***P<0.001. (E) The binding of HMGA1 protein to G6PD promoter was detected by ChIP-PCR in H1299 cells treated with 10 ng/ml TGFβ1 for 48 h. **P<0.01. (F) The binding of HMGA1 protein to G6PD promoter was detected by ChIP-PCR in H1299 cells treated with 100 ng/mL TGFβ1 antibody for 48 h. *P<0.05. (G) The protein levels of HMGA1 and G6PD in H1299 and H226 cells transfected with HMGA1 siRNAs and treated with 10 ng/mL TGFβ1 for 48 h were analyzed by western blotting. (H) The mRNA levels of G6PD in H1299 and H226 cells transfected and treated as described in (G) were analyzed by qPCR. **P<0.01, ***P<0.001. (I) The enzymatic activity of G6PD was measured in H1299 and H226 cells transfected and treated as described in (G). *P<0.01, **P<0.01, ***P<0.001. (J) NADPH level was measured in H1299 and H226 cells transfected and treated as described in (G). *P<0.01, **P<0.01, ***P<0.001.
The TGFβ1-FOXM1-HMGA1-G6PD axis enhances the cisplatin resistance of NSCLC cells
Considering the crucial role of G6PD in chemoresistance, we analyzed the cisplatin resistance of NSCLC cells after activating or blocking the TGFβ1-FOXM1-HMGA1-G6PD axis. Cell viability assays showed that, compared with parental cells, TGFβ1-treated H1299 and H226 cells exhibited increased resistance to cisplatin, with resistance indexes of 1.56 or 1.76, respectively (Figure 5A). However, blocking the TGFβ1-FOXM1-HMGA1-G6PD axis by knocking down FOXM1, HMGA1, or G6PD individually impaired TGFβ1-induced cisplatin resistance in both H1299 and H226 cells (Figure 5B). Highly expressed G6PD enhances the supplies of NADPH and ribose for detoxifying ROS and repairing DNA damage that is elevated by chemotherapy agents. Hence, we measured the levels of ROS and γ-H2AX, an indicator for DNA damage, in NSCLC cells. The results showed that cisplatin increased ROS levels in NSCLC cells, while cisplatin-induced ROS decreased when exogenous TGFβ1 was added (Figure 5C). Additionally, silencing FOXM1, HMGA1, or G6PD prevented TGFβ1 from eliminating ROS in cisplatin-treated cells (Figure 5D). In line with the above findings, γ-H2AX formation caused by cisplatin was reversed by exogenous TGFβ1, meanwhile silencing of FOXM1, HMGA1, or G6PD resulted in the significant accumulation of γ-H2AX (Figure 5E). These data indicate that G6PD expression upregulated by the TGFβ1-FOXM1-HMGA1 pathway may lead to cisplatin resistance in NSCLC cells.
Figure 5.
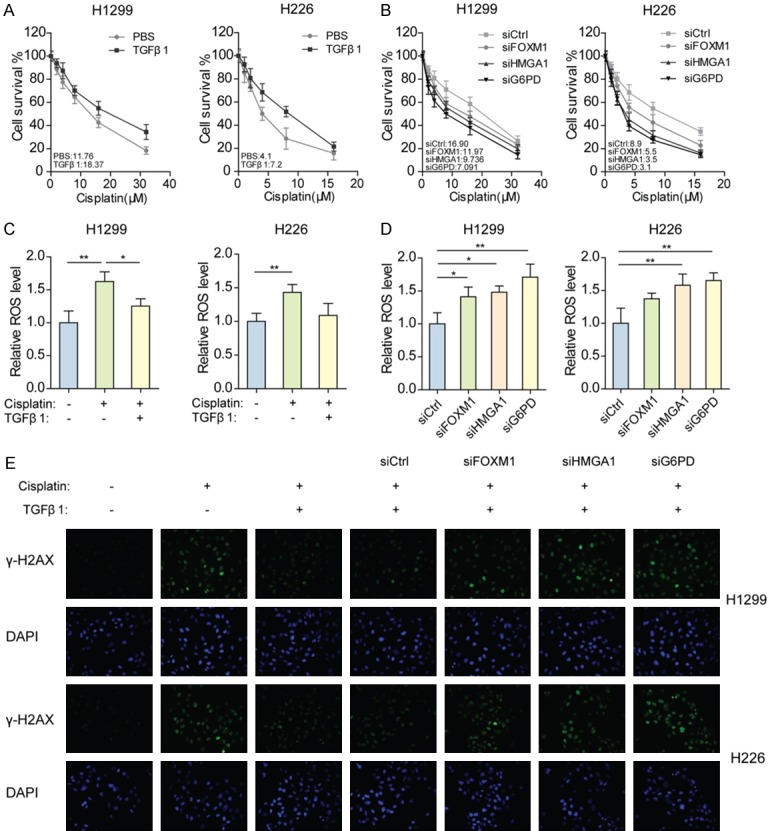
The TGFβ1-FOXM1-HMGA1-G6PD axis enhances the cisplatin resistance of NSCLC cells. A. Cell viability was measured in H1299 and H226 cells pre-exposed to 10 ng/mL TGFβ1 for 24 h, when treated with various concentrations of cisplatin for 48 h. B. Cell viability was measured in H1299 and H226 cells pre-exposed to 10 ng/mL TGFβ1 and pre-transfected with FOXM1, HMGA1, or G6PD siRNAs for 24 h, when treated with various concentrations of cisplatin for 48 h. C. ROS level was measured in H1299 and H226 cells pre-exposed to 10 ng/mL TGFβ1 for 24 h, when treated with 3 μM (H1299) or 1.5 μM (H226) cisplatin for 48 h. *P<0.01, **P<0.01. D. ROS levels were measured in H1299 and H226 cells pre-exposed to 10 ng/mL TGFβ1 and pre-transfected with FOXM1, HMGA1, or G6PD siRNAs for 24 h, when treated with 3 μM (H1299) or 1.5 μM (H226) cisplatin for 48 h. *P<0.01, **P<0.01. E. Immunofluorescence analysis of γ-H2AX protein in H1299 and H226 cells pre-exposed to 10 ng/mL TGFβ1 or pre-transfected with FOXM1, HMGA1, or G6PD siRNAs for 24 h, when treated with 3 μM (H1299) or 1.5 μM (H226) cisplatin for 48 h.
Ectopic activation of the FOXM1-HMGA1-G6PD regulatory axis indicates a poor prognosis in NSCLC patients
To characterize the expression of FOXM1, HMGA1, and G6PD in NSCLC, immunohistochemical analysis in 216 NSCLC patients was performed. As shown in Figure 6A, different levels of FOXM1, HMGA1, and G6PD were detected in the NSCLC tissues from different patients. FOXM1 and HMGA1 immunostaining was detected in the nucleus, while G6PD was found to be distributed in the cytoplasm. The representative images showed a case with low expression of FOXM1, HMGA1 and G6PD as well as another case with high expression of the three factors, which were the most common in the immunohistochemical analysis. Statistical results indicated positive correlations among the expression levels of FOXM1, HMGA1, and G6PD in NSCLC tissues (Figure 6B-D). Clinical relevance analysis showed that all factors of the TGFβ1-FOXM1-HMGA1-G6PD axis were positively correlated with tumor-node-metastasis (TNM) stage (Table 1). Moreover, only TGFβ1 was positively associated with Lymph node metastasis and G6PD positively correlated with tumor size. The potential associations between immunostaining and OS were retrospectively evaluated in 205 NSCLC patients. Kaplan-Meier analysis showed that OS was worse among patients with high FOXM1, HMGA1, or G6PD staining than among those with low staining (Figure 6E-G). These results suggest that the TGFβ1-activated FOXM1-HMGA1-G6PD pathway is involved in NSCLC progression and metastasis.
Figure 6.

Ectopic activation of the FOXM1-HMGA1-G6PD regulatory axis indicates a poor prognosis in NSCLC patients. A. Representative image of FOXM1, HMGA1, and G6PD immunostaining in the NSCLC tissues from two cases, showing the low or high expression of FOXM1, HMGA1, and G6PD. B-D. The correlation among concurrent immunostaining scores of FOXM1, HMGA1, and G6PD in NSCLC tissues from 216 cases. E-G. The OS of 205 NSCLC patients with low and high expression of FOXM1, HMGA1, or G6PD.
Table 1.
Correlation of the expression of TGFβ1, HMGA1, FOXM1, and G6PD with clinicopathological features in NSCLC
| Cases | TGFB1 expression | HMGA1 expression | FOXM1 expression | G6PD expression | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||||
| Low | High | P Value | Low | High | P Value | Low | High | P Value | Low | High | P Value | ||
|
|
|
|
|
||||||||||
| Cases | Cases | Cases | Cases | Cases | Cases | Cases | Cases | ||||||
| Gender | 216 | 116 | 100 | 0.9728 | 99 | 117 | 0.6228 | 105 | 111 | 0.7566 | 114 | 102 | 0.1251 |
| Male | 137 | 73 | 64 | 63 | 74 | 66 | 71 | 68 | 69 | ||||
| Female | 79 | 43 | 36 | 36 | 43 | 39 | 40 | 46 | 33 | ||||
| Age | 0.3655 | 0.9277 | 0.1216 | 0.0528 | |||||||||
| <57 | 55 | 33 | 22 | 31 | 25 | 36 | 20 | 36 | 20 | ||||
| ≥57 | 161 | 83 | 78 | 68 | 92 | 69 | 91 | 78 | 82 | ||||
| Tumor size | 0.3614 | 0.3841 | 0.2096 | 0.019* | |||||||||
| ≤3 cm | 85 | 49 | 36 | 43 | 42 | 49 | 36 | 53 | 32 | ||||
| >3 cm | 131 | 67 | 64 | 56 | 75 | 56 | 75 | 61 | 70 | ||||
| Histological type | 0.0533 | 0.0726 | 0.4402 | 0.1207 | |||||||||
| Squamous cell carcinoma | 74 | 41 | 33 | 34 | 40 | 34 | 40 | 31 | 43 | ||||
| Adenocarcinoma | 113 | 66 | 47 | 57 | 56 | 61 | 52 | 68 | 45 | ||||
| Mixed/other | 29 | 9 | 20 | 8 | 21 | 10 | 19 | 15 | 14 | ||||
| Lymph node metastasis | 0.0141* | 0.0571 | 0.0563 | 0.3218 | |||||||||
| No | 80 | 53 | 27 | 46 | 34 | 46 | 34 | 48 | 32 | ||||
| Yes | 136 | 63 | 73 | 53 | 83 | 59 | 77 | 66 | 70 | ||||
| Differentiation status | 0.0979 | 0.5694 | 0.3117 | 0.6339 | |||||||||
| Well | 70 | 41 | 29 | 36 | 34 | 38 | 32 | 39 | 31 | ||||
| Moderate | 74 | 32 | 42 | 30 | 44 | 37 | 37 | 39 | 35 | ||||
| Poor | 72 | 43 | 29 | 33 | 39 | 30 | 42 | 36 | 36 | ||||
| TNM stage | 0.0187* | 0.0119* | 0.0200* | 0.0015* | |||||||||
| I/II | 130 | 79 | 51 | 67 | 63 | 72 | 58 | 81 | 49 | ||||
| III/IV | 86 | 37 | 49 | 32 | 54 | 33 | 53 | 33 | 53 | ||||
P<0.05.
Discussion
TGFβ1 is a cytokine with a paradoxical role in that it behaves as a tumor suppressor in pre-cancerous or early cancerous tissue, while leading to tumor invasiveness and metastasis during the late stages of cancer progression when TGFβ1 growth inhibitory signals are lost. As a constituent of resistance progression in the principal non-surgical interventions in lung cancer, TGFβ1 has been highlighted in different mechanisms that mediate resistance to cytotoxic chemotherapy. Induction of the EMT, promotion of drug efflux receptor expression, protection from cytotoxic therapy-induced apoptosis, and recruitment of a pro-tumor stromal niche leads to TGFβ1-drived chemoresistance in lung cancer [31]. In this study, we found that TGFβ1 promoted resistance to cytotoxic chemotherapy by altering metabolism in NSCLC cells, and the PPP was activated through the TGFβ1-FOXM1-HMGA1-G6PD regulatory axis. Thus, the link between microenvironment and abnormal metabolism in cancer progression may display a crucial role in chemoresistance.
Recent evidence has implicated that FOXM1 is dispensable in many aspects of the DNA damage response. Accordingly, FOXM1 drives the transcription of DNA damage sensors, mediators, signal transducers and effectors, playing an integral part in chemoresistance by maintaining genome integrity [14]. Knockdown of FOXM1 enhanced the cytotoxic and pro-apoptotic effects of docetaxel in NSCLC cells by inducing activation of the c-Jun N-terminal kinases/mitochondrial signaling pathway, and this mechanism was also found in cisplatin-induced NSCLC cell apoptosis when downregulating FOXM1 [32,33]. Consistently, clinical analysis has shown that NSCLC patients with high FOXM1 expression have a significantly lower response rate for cisplatin-based combination chemotherapy [34]. In this study, we found that FOXM1 mediated TGFβ1-induced cisplatin resistance in NSCLC cells by activating the downstream pathway HMGA1/G6PD. Recent evidence has indicated FOXM1 deregulation in the development of chemoresistance, showing that the imbalance between FOXO3 and FOXM1 plays a key role [35]. Here, we found that TGFβ1 enhanced the stability of FOXM1 protein by inhibiting its ubiquitination. Similarly, ubiquitination and degradation of FOXM1 were also suppressed in breast cancer with epirubicin resistance [36]. Although TGFβ1 was confirmed to repress degradation of FOXM1 in NSCLC, the regulation of FOXM1 protein by the TGFβ1 receptor was not studied in the present work.
HMGA1 overexpression is often associated with antineoplastic drug resistance. In this study, TGFβ1-induced FOXM1 was found to promote the expression of HMGA1 by directly activating its transcription. Knockdown of HMGA1 restored TGFβ1-impaired sensitivity to cisplatin in NSCLC cells, meanwhile ROS levels were upregulated and DNA damage was aggravated. Consistently, aberrant expression of HMGA1 facilitated cisplatin resistance in bladder cancer [37]. Interestingly, HMGA1 also induced TGFβ1 expression at the mRNA, protein, and secretion levels. Although whether HMGA1 directly activates the transcription of TGFβ1 was not determined in the present work, secreted TGFβ1 in turn drove FOXM1-HMGA1-G6PD axis to establish a positive feedback loop, thus maintaining the resistance to cisplatin. Addition of antibody against TGFβ1 into culture medium blocked the positive feedback loop, as indicated by the evident decrease in HMGA1.
With the exception of TGFβ1, G6PD was identified as another target of HMGA1, which functioned as an effector during chemoresistance progression. Silencing G6PD obviously promoted the accumulation of ROS and DNA damage caused by cisplatin. Consistently, Hong et al. [26] reported that cisplatin-resistant A549 cells exhibited increased levels of G6PD protein, G6PD enzymatic activity, NADPH, and glutathione. G6PD inhibition effectively induced apoptosis and more ROS accumulation after cisplatin exposure. In this study, the enzymatic activity and protein level of G6PD changed coordinately when the TGFβ1-FOXM1-HMGA1 pathway was activated or disrupted, suggesting that upregulation of enzymatic activity may result from the increased expression of G6PD. Previous studies have demonstrated that G6PD is highly regulated at multiple levels including transcription, translation, posttranslation, and intracellular location [38]. Here, localization of G6PD protein around the nucleus was observed especially after HMGA1 overexpression, as shown by immunofluorescence, which may facilitate the function of G6PD. In addition, whether posttranslational modification of G6PD is involved in TGFβ1-induced chemoresistance of NSCLC should be validated in the future. Interestingly, cisplatin-resistant cells exhibit increased glucose uptake and consumption, which further supports activation of the PPP [39].
Clinical analysis has shown that the high expression of TGFβ1, FOXM1, HMGA1, or G6PD indicates the short survival of NSCLC patients and is associated with TNM stage, revealing that the TGFβ1-FOXM1-HMGA1-G6PD axis accelerated NSCLC progression. However, only TGFβ1 was positively correlated with lymph node metastasis, and this was likely due to TGFβ1-induced EMT. Additionally, G6PD but not other factors was positively correlated with tumor size, most likely due to G6PD promotion of cell proliferation. Therefore, the TGFβ1-FOXM1-HMGA1-G6PD axis not only enhances chemoresistance but may affect other malignant phenotypes of NSCLC.
In summary, our findings reveal the existence of the TGFβ1-FOXM1-HMGA1-G6PD axis and its role in the cisplatin resistance of NSCLC. Deubiquitination-induced TGFβ1 signaling increased the stability of FOXM1 protein, which transactivated HMGA1. Furthermore, highly expressed HMGA1 activated the transcription of G6PD and then accelerated the PPP to provide NADPH and dNTP for antagonizing cisplatin-induced ROS and DNA damage. Additionally, HMGA1 promoted the production and secretion of TGFβ1, thereby establishing a positive feedback loop for continuous G6PD expression and resistance to cisplatin (Figure 7). Collectively, understanding the mechanism may contribute to a more accurate prognosis and more effective treatment for NSCLC patients with cisplatin resistance.
Figure 7.
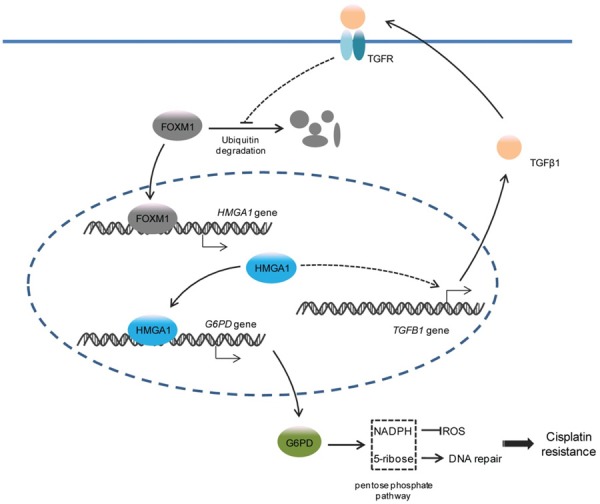
Schema indicating the role of TGFβ1-FOXM1-HMGA1-G6PD axis in cisplatin resistance of NSCLC. TGFβ1 stimulation prevents the ubiquitination and degradation of FOXM1 protein, a transcription factor for HMGA1 gene, thereby activating the transcription of HMGA1. Upregulated HMGA1 further activates the transcription of G6PD to promote the PPP, which supplies NADPH and dNTP against the ROS and DNA damage caused by cisplatin. Moreover, HMGA1 induces the production and secretion of TGFβ1, which in turn enhances TGFβ1 signaling to maintain G6PD expression and chemoresistance.
Acknowledgements
This work was supported by Science and Technology Program of Sanmen County Public Technology Social Development Project (16303).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Aggarwal C, Borghaei H. Treatment paradigms for advanced non-small cell lung cancer at academic medical centers: involvement in clinical trial endpoint design. Oncologist. 2017;22:700–708. doi: 10.1634/theoncologist.2016-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ettinger DS, Akerley W, Bepler G, Blum MG, Chang A, Cheney RT, Chirieac LR, D’Amico TA, Demmy TL, Ganti AK, Govindan R, Grannis FW Jr, Jahan T, Jahanzeb M, Johnson DH, Kessinger A, Komaki R, Kong FM, Kris MG, Krug LM, Le QT, Lennes IT, Martins R, O’Malley J, Osarogiagbon RU, Otterson GA, Patel JD, Pisters KM, Reckamp K, Riely GJ, Rohren E, Simon GR, Swanson SJ, Wood DE, Yang SC NCCN Non-Small Cell Lung Cancer Panel Members. Non-small cell lung cancer. J Natl Compr Canc Netw. 2010;8:740–801. doi: 10.6004/jnccn.2010.0056. [DOI] [PubMed] [Google Scholar]
- 3.Johnson DH, Schiller JH, Bunn PA Jr. Recent clinical advances in lung cancer management. J. Clin. Oncol. 2014;32:973–982. doi: 10.1200/JCO.2013.53.1228. [DOI] [PubMed] [Google Scholar]
- 4.Drabsch Y, ten Dijke P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012;31:553–568. doi: 10.1007/s10555-012-9375-7. [DOI] [PubMed] [Google Scholar]
- 5.Romano G, Santi L, Bianco MR, Giuffre MR, Pettinato M, Bugarin C, Garanzini C, Savarese L, Leoni S, Cerrito MG, Leone BE, Gaipa G, Grassilli E, Papa M, Lavitrano M, Giovannoni R. The TGF-beta pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget. 2016;7:22077–22091. doi: 10.18632/oncotarget.7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng H, Yang Z, Xu N, Liu B, Fu Z, Lian C, Guo H. Connective tissue growth factor promotes temozolomide resistance in glioblastoma through TGF-beta1-dependent activation of Smad/ERK signaling. Cell Death Dis. 2017;8:e2885. doi: 10.1038/cddis.2017.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhuang J, Shen L, Yang L, Huang X, Lu Q, Cui Y, Zheng X, Zhao X, Zhang D, Huang R, Guo H, Yan J. TGFbeta1 promotes gemcitabine resistance through regulating the LncRNA-LET/NF90/miR-145 signaling axis in bladder cancer. Theranostics. 2017;7:3053–3067. doi: 10.7150/thno.19542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamai M, Furuichi Y, Kasai S, Ando N, Harama D, Goi K, Inukai T, Kagami K, Abe M, Ichikawa H, Sugita K. TGFbeta1 synergizes with FLT3 ligand to induce chemoresistant quiescence in acute lymphoblastic leukemia with MLL gene rearrangements. Leuk Res. 2017;61:68–76. doi: 10.1016/j.leukres.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Xian G, Zhao J, Qin C, Zhang Z, Lin Y, Su Z. Simvastatin attenuates macrophage-mediated gemcitabine resistance of pancreatic ductal adenocarcinoma by regulating the TGF-beta1/Gfi-1 axis. Cancer Lett. 2017;385:65–74. doi: 10.1016/j.canlet.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Xie C, Yue J, Jiang Z, Zhou R, Xie R, Wang Y, Wu S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFbeta1 signaling loop in esophageal squamous cell carcinoma. Mol Carcinog. 2017;56:1150–1163. doi: 10.1002/mc.22581. [DOI] [PubMed] [Google Scholar]
- 11.Shen M, Tsai Y, Zhu R, Keng PC, Chen Y, Chen Y, Lee SO. FASN-TGF-beta1-PD-L1 axis contributes to the development of resistance to NK cell cytotoxicity of cisplatin-resistant lung cancer cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:313–322. doi: 10.1016/j.bbalip.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Regan RM, Nahta R. Targeting forkhead box M1 transcription factor in breast cancer. Biochem Pharmacol. 2018;154:407–413. doi: 10.1016/j.bcp.2018.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zona S, Bella L, Burton MJ, Nestal de Moraes G, Lam EW. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim Biophys Acta. 2014;1839:1316–1322. doi: 10.1016/j.bbagrm.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu N, Wu SD, Wang H, Wang Q, Bai CX. Involvement of FoxM1 in non-small cell lung cancer recurrence. Asian Pac J Cancer Prev. 2012;13:4739–4743. doi: 10.7314/apjcp.2012.13.9.4739. [DOI] [PubMed] [Google Scholar]
- 17.Sgarra R, Zammitti S, Lo Sardo A, Maurizio E, Arnoldo L, Pegoraro S, Giancotti V, Manfioletti G. HMGA molecular network: from transcriptional regulation to chromatin remodeling. Biochim Biophys Acta. 2010;1799:37–47. doi: 10.1016/j.bbagrm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Sumter TF, Xian L, Huso T, Koo M, Chang YT, Almasri TN, Chia L, Inglis C, Reid D, Resar LM. The high mobility group A1 (HMGA1) transcriptome in cancer and development. Curr Mol Med. 2016;16:353–393. doi: 10.2174/1566524016666160316152147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 20.Wang YT, Pan SH, Tsai CF, Kuo TC, Hsu YL, Yen HY, Choong WK, Wu HY, Liao YC, Hong TM, Sung TY, Yang PC, Chen YJ. Phosphoproteomics reveals HMGA1, a CK2 substrate, as a drug-resistant target in non-small cell lung cancer. Sci Rep. 2017;7:44021. doi: 10.1038/srep44021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Wang Q, Chen F, Liu J. Elevated expression of HMGA1 correlates with the malignant status and prognosis of non-small cell lung cancer. Tumour Biol. 2015;36:1213–1219. doi: 10.1007/s13277-014-2749-4. [DOI] [PubMed] [Google Scholar]
- 22.Polimeni M, Voena C, Kopecka J, Riganti C, Pescarmona G, Bosia A, Ghigo D. Modulation of doxorubicin resistance by the glucose-6-phosphate dehydrogenase activity. Biochem J. 2011;439:141–149. doi: 10.1042/BJ20102016. [DOI] [PubMed] [Google Scholar]
- 23.Huang Y, Bell LN, Okamura J, Kim MS, Mohney RP, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α/SREBF1 protein interactions: bridging cell metabolism and cisplatin chemoresistance. Cell Cycle. 2012;11:3810–3827. doi: 10.4161/cc.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Liang S, Lian X, Liu L, Zhao S, Xuan Q, Guo L, Liu H, Yang Y, Dong T, Liu Y, Liu Z, Zhang Q. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci Rep. 2015;5:9301. doi: 10.1038/srep09301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin X, Tang B, Li JH, Wang Y, Zhang L, Xie XY, Zhang BH, Qiu SJ, Wu WZ, Ren ZG. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J Exp Clin Cancer Res. 2017;36:166. doi: 10.1186/s13046-017-0637-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong W, Cai P, Xu C, Cao D, Yu W, Zhao Z, Huang M, Jin J. Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front Pharmacol. 2018;9:43. doi: 10.3389/fphar.2018.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang Z, Gong C, Yu S, Zhou W, Hassan W, Li H, Wang X, Hu Y, Gu K, Chen X, Hong B, Bao Y, Chen X, Zhang X, Liu H. NFYB-induced high expression of E2F1 contributes to oxaliplatin resistance in colorectal cancer via the enhancement of CHK1 signaling. Cancer Lett. 2018;415:58–72. doi: 10.1016/j.canlet.2017.11.040. [DOI] [PubMed] [Google Scholar]
- 28.Gong C, Liu H, Song R, Zhong T, Lou M, Wang T, Qi H, Shen J, Zhu L, Shao J. ATR-CHK1-E2F3 signaling transactivates human ribonucleotide reductase small subunit M2 for DNA repair induced by the chemical carcinogen MNNG. Biochim Biophys Acta. 2016;1859:612–626. doi: 10.1016/j.bbagrm.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Zu X, Zhong J, Tan J, Tan L, Yang D, Zhang Q, Ding W, Liu W, Wen G, Liu J, Cao R, Jiang Y. TGF-beta1 induces HMGA1 expression in human breast cancer cells: implications of the involvement of HMGA1 in TGF-beta signaling. Int J Mol Med. 2015;35:693–701. doi: 10.3892/ijmm.2015.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong J, Liu C, Zhang QH, Chen L, Shen YY, Chen YJ, Zeng X, Zu XY, Cao RX. TGF-beta1 induces HMGA1 expression: the role of HMGA1 in thyroid cancer proliferation and invasion. Int J Oncol. 2017;50:1567–1578. doi: 10.3892/ijo.2017.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eser PO, Janne PA. TGFbeta pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol Ther. 2018;184:112–130. doi: 10.1016/j.pharmthera.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Zhu X, Zhang K, Zhu L, Zhou F. FoxM1 inhibition enhances chemosensitivity of docetaxel-resistant A549 cells to docetaxel via activation of JNK/mitochondrial pathway. Acta Biochim Biophys Sin (Shanghai) 2016;48:804–809. doi: 10.1093/abbs/gmw072. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Chen X, Gu Y, Zhu L, Qian Y, Pei D, Zhang W, Shu Y. FOXM1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and mediates sensitivity to cisplatin in A549 cells via the JNK/mitochondrial pathway. Neoplasma. 2015;62:61–71. doi: 10.4149/neo_2015_008. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Wen L, Zhao SH, Ai ZH, Guo JZ, Liu WC. FoxM1 expression is significantly associated with cisplatin-based chemotherapy resistance and poor prognosis in advanced non-small cell lung cancer patients. Lung Cancer. 2013;79:173–179. doi: 10.1016/j.lungcan.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Yao S, Fan LY, Lam EW. The FOXO3-FOXM1 axis: a key cancer drug target and a modulator of cancer drug resistance. Semin Cancer Biol. 2018;50:77–89. doi: 10.1016/j.semcancer.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karunarathna U, Kongsema M, Zona S, Gong C, Cabrera E, Gomes AR, Man EP, Khongkow P, Tsang JW, Khoo US, Medema RH, Freire R, Lam EW. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene. 2016;35:1433–1444. doi: 10.1038/onc.2015.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Liu M, Meng F, Sun B, Jin X, Jia C. The long noncoding RNA HIF1A-AS2 facilitates cisplatin resistance in bladder cancer. J Cell Biochem. 2019;120:243–252. doi: 10.1002/jcb.27327. [DOI] [PubMed] [Google Scholar]
- 38.Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64:362–369. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X, Xie L, Yang J, Yu H, Su J, Sun L. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int J Oncol. 2018;53:1055–1068. doi: 10.3892/ijo.2018.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.