

SUMMARY
Diphenylphosphonates (DPPs) have been used for 50 years to inactivate serine hydrolases, creating adducts representative of tetrahedral intermediates of this important class of enzymes. Failure to react at active site serine residues, however, can thwart their usefulness. Here we describe a facile route and allied mechanistic studies to highly reactive, structurally complex organofluorophosphonates (FPs) by direct fluorinative hydrolysis of DPPs. Advantages over current preparations of FPs are exemplified by the synthesis of a β-lactam containing peptide substrate analogue capable of modifying the C-terminal, dual-function thioesterase involved in nocardicin A biosynthesis. Although this serine hydrolase was found to resist modification by classical DPP inhibitors, active site selective phosphonylation by the corresponding FP occurs rapidly to form a stable adduct. This simple, late-stage method enables the ready preparation of FP probes that retain important structural motifs of native substrates, thus promoting efforts in mechanistic enzymology by accessing biologically relevant enzyme-inhibitor co-structures.
INTRODUCTION
Serine hydrolases (SHs) are among the largest enzyme superfamilies in phylogeny, utilizing catalytic triads or dyads to hydrolyze amide, ester, and thioester bonds in small molecules or proteins (Ekici et al., 2008). Mechanistically, a general base first mediates the formation of an acyl-enzyme intermediate with an active site serine, which is subsequently hydrolyzed to provide a free acid product and regenerated enzyme. During both acylation and hydrolysis, a tetrahedral transition state or intermediate is stabilized by an oxy-anion hole formed by the active site peptide backbone (Buller and Townsend, 2013). The involvement of SHs in a broad span of normal and pathological processes including metabolism (Long and Cravatt, 2011; Whitcomb and Lowe, 2007), blood clotting (Coughlin, 2000; Davie et al., 2006), neurotransmission (Colović et al., 2013), cancer (Shields et al., 2010), and immunity (Ortega et al., 2016; Skoreński and Sieńczyk, 2013) continues to foster interest in its members as pharmaceutical targets (Drag and Salvesen, 2010). Beyond their roles in human biology, the ubiquity of SHs in bioactive and therapeutic natural product biosynthesis poses challenging mechanistic questions important to basic enzymology and bioengineering efforts (Horsman et al., 2016; Newman et al., 2014).
Of particular interest is the chemistry performed by non-ribosomal peptide synthetases (NRPSs), multi-modular megaenzymes involved in the production of structurally and pharmacologically diverse natural products (Süssmuth and Mainz, 2017). Recently, investigations of the NRPS responsible for the synthesis of pro-nocardicin G (1), a key precursor of the paradigm monocyclic β-lactam antibiotic, nocardicin A (3), revealed an unprecedented bifunctional thioesterase domain (NocTE) performing C-terminal L→D epimerization prior to canonical hydrolytic product release (Figure 1, A) (Gaudelli and Townsend, 2014). Kinetic and biochemical experiments revealed NocTE to function as a “gatekeeper” in the NRPS assembly line because its strict substrate selectivity ensures prior β-lactam ring formation by the upstream condensation domain (Gaudelli et al., 2015). In order to study the epimerase/hydrolase activity of NocTE and its notably stringent preference for β-lactam containing substrates at the molecular level, an irreversible inhibitor bearing structural likeness to its native substrate was desired.
Figure 1.

(A). Abridged biosynthesis of nocardicin A. The pentamodular NRPS comprised of NocA and NocB performs amino acid activation, peptide elongation, epimerization, and beta-lactam formation to produce a terminal thioesterase (NocTE) bound intermediate. NocTE catalyzes C-terminal L→D epimerization prior to hydrolytic release of pro-nocardicin G (1). Production of nocardicin G (2) by post-NRPS trimming of pro-nocardicin G, followed by late-stage tailoring steps, results in nocardicin A (3). (B). Proposed DPP and FP NocTE substrate analogue inhibitors. Electrophilic warheads highlighted in red.
Previous work determining the substrate specificity of the excised NocTE domain served as an excellent starting point for inhibitor design (Gaudelli and Townsend, 2013). Although its native substrate is a carrier domain-bound pentapeptide bearing an embedded β-lactam ring, thioesterase activity was observed with simpler synthetic N-acetylcysteamine (NAC) thioesters of pro-nocardicin G, nocardicin G (2), and their C-terminal epimers (Gaudelli and Townsend, 2014). Proposed structural and mechanistic probes of NocTE, 4, 5, and 6 were therefore designed to conserve the azetidinone ring and appropriate amino acid sequence to maximize substrate fidelity while substituting the carboxyl terminus for a suitable active site seryl-reactive warhead (Figure 1, B).
Traditionally, covalent inhibitors of SHs exploit their specifically enhanced nucleophilicity to form stable adducts (Powers et al., 2002). First developed in the 1930s, the organofluorophosphonates (phosphonyl fluorides, phosphonofluoridates, FPs) wield highly electrophilic centers capable of phosphonylating active site seryl residues to produce oxyanion hole-stabilized tetrahedral adducts. The reactivity of simple FPs, derived from the strongly polarized phosphorus–fluorine bond, has found use in chemical nerve agents (sarin, soman) and insecticides (diisopropylfluorophosphate-DFP) as general SH inhibitors marked by unfettered enzyme promiscuity. Early investigations integrating the FP warhead into simple amino acid analogues, however, demonstrated that modest selectivity for chymotrypsin, elastase, or trypsin-like enzymes was achievable depending on side-chain identity (Bartlett and Lamden, 1986; Lamden and Bartlett, 1983; Ni and Powers, 1998). Indeed, even largely aliphatic FP probes resembling substrates of diacylglycerol lipases or ghrelin esterases were found to selectively inhibit specific SHs with minimal off-target reaction (Bisogno et al., 2013; Eubanks et al., 2011). The indiscriminate reactivity of the FP warhead toward individual SHs may therefore be focused by its combination with appropriate structural recognition elements. Present synthetic strategies, however, are incompatible with many functional groups or require chemical protection schemes that confound the synthesis of more structurally complex probes (Sampson and Bartlett, 1991). For example, the widespread use of diethylaminosulfur trifluoride (DAST) to prepare FPs by fluorination of monoalkylphosphonic acids limits warhead incorporation to functionally restricted systems. Among its repertoire of transformations, DAST’s efficient conversion of ketones and aldehydes to gem-difluorides, isolated alcohols to fluorides, and β-hydroxy amides—prevalent in seryl-or threonyl-containing peptides—to oxazolines compromises the synthesis of complex probes resembling many SH substrate classes (e.g. peptides and polyketides) (Hudlický, 1988; Singh and Shreeve, 2002). Bound by preparative constraints, peptide FPs remain greatly under investigated as a class of protease inhibitors (Joachimiak and Błażewska, 2018). An efficient method capable of installing the FP warhead on more chemically diverse substrate analogues would result in more useful inactivators and probes by better exploiting the nuanced structural specificities of SHs.
RESULTS AND DISCUSSION
In comparison to FPs, diarylphosphonate inhibitors historically exhibit a better balance of reactivity and functional group tolerance. First developed by Oleksyszyn and Powers, peptide α-aminoalkyl diphenylphosphonates owe their reactivity with SHs to electron-deficient aryl phosphonate diesters (Oleksyszyn and Powers, 1991). The compatibility of the DPP warhead with common synthetic transformations has permitted the systematic modification of probe structural elements to confer impressive differential inhibition and structural characterization of SHs across different classes (Grzywa and Sienczyk, 2013; Kasperkiewicz et al., 2015; Oleksyszyn and Powers, 1991; Serim et al., 2013). In addition to peptide-based protease inhibitors, polyketide DPP analogues have been used to structurally dissect the macrolactonization reaction catalyzed by PikTE, a PKS thioesterase involved in pikromycin biosynthesis, which shares its catalytic triad and α/β hydrolase fold with NocTE (Akey et al., 2006; Giraldes et al., 2006). While considerably less reactive than FPs, the greater stability, synthetic accessibility, and precedented success of peptide DPPs as selective SH inhibitors made DPPs 4 and 5 attractive first targets for irreversibly modifying NocTE.
Synthesis of Nocardicin G and Pro-Nocardicin G C-Terminal Diphenylphosphonates and Incubation with NocTE
Drawing on previously reported preparations of the nocardicins, the synthesis of nocG and pro-nocG DPP analogues [nocGP(OPh)2 (4), and pro-nocGP(OPh)2 (5), respectively] proceeded in a C- to N-terminal fashion (Scheme 1) (Salituro and Townsend, 1990; Gaudelli and Townsend, 2013). The O-benzyl protected DPP analogue of p-hydroxyphenylglycine (Hpg, 7) was obtained without chromatography by amidoalkylation of p-benzyloxybenzaldehyde with t-butyl carbamate and triphenylphosphite in acetic acid (Oleksyszyn and Subotkowska, 1979). followed by acid deprotection and precipitation with dilute HCl. PyBOP coupling of 7 with the dicyclohexylammonium salt of l-serine N-protected by Sheehan’s (Ox) group readily produced the dipeptide 8 as a C-terminal diastereomeric mixture. Exposure of 8 to modified Mitsunobu conditions promoted intramolecular cyclodehydration to the azetidinone 9. Pressurized hydrogenolysis of the benzyl and Ox protecting groups in the presence of HCl, yielded pure 3-aminonocardicinic acid DPP analogue 10 as the hydrochloride salt. Subsequent coupling of 10 with N-Boc-D-Hpg provided the protected β-lactam tripeptide, which after deprotection yielded C-terminal epimeric nocGP(OPh)2 analogues 4 separable by HPLC, but which interchange in aqueous solution. Convergent preparation of pro-nocGP(OPh)2 analogues 5 was achieved by coupling of 4 with the protected dipeptide 11 (Gaudelli and Townsend, 2013), followed by deprotection and HPLC purification.
Scheme 1.

Synthesis of nocGP(OPh)2 and pro-nocGP(OPh)2 C-Terminal Epimers. Reaction conditions: (a) (1) BocNH2, P(OPh)3, AcOH, (2) TFA, DCM, (3) NaHCO3, Et2O, (4) HCl, THF, 75% over four steps; (b) l-Ox-Ser-DCA, PyBOP, DIPEA, DMF, 68%; (c) P(OEt)3, DEAD, THF, 65%; (d) cat. 10% Pd/C, 50 psi H2, HCl, EtOAc/EtOH, 62%; (e) (1) L-Boc-Hpg, isobutylchloroformate, 2,6-lutidine, cat. N-methylmorpholine, acetone, (2), 10, 2,6-lutidine, DMF, 69% over two steps; (f) TFA, DCM, quant.; (g) (1) N-Boc-L-Hpg-NG-Pbf-L-Arg (11), PyBOP, DIPEA, DMF, (2) TFA, (3) HPLC, 25% over three steps.
To determine the potency of the DPP natural product mimics as covalent inhibitors of NocTE, purified recombinant thioesterase was incubated with excess 4 overnight (Figure S2, A). Directly assaying for the formation of covalent complexes by intact protein UPLC-HRMS avoided time-and material-consuming kinetics experiments. Against expectation, however, mass shifts corresponding to the formation of irreversible protein adducts were not observed. Possible regeneration of apo-NocTE by enzyme-adduct hydrolysis was ruled out due to the absence of hydrolyzed inhibitor in the reaction mixture. Notwithstanding being better NAC-substrates for NocTE, pro-nocGP(OPh)2 pentapeptide analogues 5 were similarly ineffective at covalently modifying the thioesterase (Figure S2, B)(Gaudelli and Townsend, 2014)(Gaudelli and Townsend, 2014). Despite their nearly absolute substrate mimicry, the disappointing failure of both the nocGP(OPh)2 and pro-nocGP(OPh)2 probes pointed to a lack of reactivity between the DPP warhead and TE catalytic seryl residue.
Two-Step Conversion of Peptide Diphenylphosphonates to O-Methyl-Fluorophosphonates
Although systematic amidoalkylation of p-benzyloxybenzaldehyde with triarylphosphites bearing electron-withdrawing groups would result in increasingly electrophilic and reactive α-aminoalkyl diarylphosphonates (Guarino et al., 2014), the linearity of the synthesis and unlikely survival of most electron deficient groups discouraged a de novo approach. Instead, late-stage conversion of the DPP warhead to a more reactive electrophile could efficiently provide two potential inhibitors from one synthesis. Such a method was fortuitously discovered when attempts to produce the free phosphonic acid of 4 by ammonium fluoride-mediated hydrolysis unexpectedly yielded the fluorophosphonate ammonium salt 12 (Figure 2, A) (Kim et al., 2009). Review of the literature revealed that fluorinative hydrolysis of simple diarylphosphates to corresponding fluorophosphates using tetrabutylammonium fluoride (TBAF) has been reported (Murai et al., 2008, 2011). However, the organic insolubility of fluorophosphonate ammonium salts relative to tetrabutylammonium salts allowed the direct removal of organic byproducts through trituration with diethyl ether, thus facilitating isolation of the product. The unanticipated production of 12 not only presented an opportunity to convert the DPP group under mild conditions to the FP warhead but also inspired experiments directed at the underlying mechanism of the transformation.
Figure 2.

(A). Synthesis of FP 6. Reaction conditions: (a) NH4F, ACN, 60 oC, quant.; (b) CH2N2, DMSO. (B). Proposed fluorinative hydrolysis mechanism. Intermediates 13, 14, 16, and 18 were directly observed by 31P-NMR and UPLC-HRMS. (C). HRMS of 18 supplemented (top) and unsupplemented (bottom) with 15NH4Cl indicating by isotopic mass shift the incorporation of 15N into the molecule.
Mechanistic Studies of Fluorinative Hydrolysis
Generally, the fluorinative hydrolysis of DPPs involves the substitution of both aryl esters with an equivalent of fluoride and water. The absence of chemical analogy to carbon-based esters, indeed behavior contrary to expectation, makes insights into the timing and formation of intermediates in the reaction particularly valuable. Two routes may be envisioned to account for the conversion of 13 to 16 (Figure 2, B). The first proposal reported suggests fluoride nucleophilic substitution at phosphorus produces monofluorinated intermediate 14, which undergoes direct hydrolysis to 16 with loss of phenol (Murai et al., 2011). Given the documented susceptibility of alkyl-or aryl-fluorophosphonates to fluoride-displacing hydrolysis, however, any reaction sequence invoking the hydrolytic substitution of phenol over hydrogen fluoride must be cast into doubt (Ni and Powers, 1998; Waters and de Worms, 1949). Alternatively, an equilibrium process providing a low concentration of highly reactive difluoride intermediate 15 would be capable of producing 16 by hydrolysis. If formed, the thermodynamically favorable reaction of 15 with water would selectively stop at the fluorophosphonate product 16 due to decreased phosphorus electrophilicity from anion resonance and is consistent with the observed hydrolysis of difluorophosphonates to monofluorophosphonates (Wagner et al., 2017).
To test our proposed mechanism, racemic, protected DPP Hpg analogue 13 was heated in an NMR tube containing excess NH4F and dry (ampoule) d6-DMSO (Figure S3 and S4). Monitoring of the reaction by 31P-NMR spectroscopy revealed the formation of monofluorinated intermediate 14, appearing as two sets of doublets (1JP-F = 1135 Hz) (Ni and Powers, 1998) and consistent with the now stereogenic phosphorus center, followed by accumulation of product 16 as a single doublet (1JP-F = 983 Hz) and an unknown species comparatively downfield present as two sets of doublets (1JP-F = 1036 Hz). Overnight reaction exclusively produced 16 and the unknown coproduct without detection of difluoride 15. UPLC-HRMS analysis of the crude reaction mixture revealed the mass of the unknown coproduct to be consistent with the P-N-P-bridged dimer 18. Reminiscent of imidophosphate nucleotide analogues (Reynolds et al., 1983), the structural assignment of 18 was corroborated by the addition of 15NH4Cl to the reaction mixture. Additional splitting of the 31P-NMR signals due to 31P-15N coupling, measurable upfield changes in chemical shift due to the heavy atom effect, as well as an isotopic mass shift in the mass-spectrum (Figure 2, C) support 15N-incorporation into the bridging position (Figure S4, D). Given that formation of 18 was not observed using KF or TBAF and is, therefore, unessential for the production of 16, we propose that the appearance of 18 is the result of an alternative reaction pathway particular to the NH4F reaction in DMSO. Furthermore, we conclude the formation of 18 implicates the intermediacy of difluoride 15 in the hydrolysis pathway. Despite the electrophilicity of 14, it is unlikely that 15 is formed in concentrations high enough for direct detection using current methods. Rather, reaction of 15 with high levels of ammonium transiently results in 17, capable of combining with another reactive equivalent of 15 to provide the P-N-P dimerization product. It follows that accumulation of the reaction-stable intermediate 18 is indirect proof of the pathway proceeding through difluoride 15. It is important to note that when NH4F-mediated fluorinative hydrolysis is performed in ACN, 18 is not produced in appreciable amounts and the desired fluorophosphonate 16 is readily obtained, in keeping with the important role of DMSO in solubilizing reactants and intermediates.
Whereas the free fluorophosphonate ammonium salt 12 was found to be stable in buffer but inert towards NocTE (Figure S2, C), the more reactive FP nocGP(F)(OMe) analogue 6 could be produced by fluorophosphonate O-alkylation with ethereal diazomethane (Figure 2, A). The selectivity of diazomethane for acidic functional groups allowed methylation to occur rapidly and in the presence of the more nucleophilic N-terminal amine. Although use of excess reagent resulted in some overmethylation of less acidic Hpg phenols (pKa ~ 10), subjecting 12 to limiting quantities of diazomethane constrained reaction to the fluorophosphonic acid moiety (pKa ~ 0.85) (Benschop et al., 1970).
Covalent Adduct Formation Between Nocardicin G O-Methyl-Fluorophosphonate 6 and NocTE
With the straightforward conversion of the DPP group to the FP warhead in hand, the capacity of 6 to act as an inactivator of NocTE was examined. Gratifyingly, UPLC-MS analysis of NocTE incubated with excess 6 revealed an adduct corresponding to the mass of inhibitor 6 with loss of hydrogen fluoride (Figure 3). Complete modification of NocTE was found to occur within 15 min at room temperature, with no observed regeneration of unliganded enzyme by hydrolysis after a week in buffer (Figure S2, D). The regioselectivity of the inactivator for the enzyme active site was verified by incubating 6 with a NocTE site-specific mutant substituting the catalytic serine with alanine (NocTE*S1179A) (Gaudelli and Townsend, 2014). The lack of change in the point mutant mass after exposure to 6 confirmed the selectivity of FP warheads for catalytically active SHs.
Figure 3.

(A). Phosphonylation of the NocTE active site serine by FP substrate analogue 6. (B). Left: Mass spectral analysis of wild type NocTE (MW = 27575 Da) and NocTE incubated with 6 (MW = 27992 Da) displaying corresponding N-glucuronidated masses (27753 and 28171 Da, respectively). Right: Mass spectral analysis of NocTE mutant (MW = 27560 Da) and NocTE mutant incubated with 6 (MW = 27560 Da) displaying the corresponding N-glucuronidated mass (27739 Da). The lack of covalent adduct mass shift suggests modification occurs selectively at the active site serine residue of the wild type enzyme.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Craig A. Townsend (ctownsend@jhu.edu).
METHOD DETAILS
Safety Statement
All reactions were performed in an efficient chemical fume hood observing necessary safety precautions. Diazomethane was safely prepared and co-distilled with diethyl ether using an Aldrich Diazald® kit with Clear-Seal® joints. Some fluorophosphonate compounds are documented nerve agents and require the use of appropriate personal protective equipment (gloves, goggles, labcoat, chemical fume hood) during their handling and preparation.
Protein Expression and Purification
Wild type NocTE and NocTE*S1779A proteins were prepared as previously reported (Gaudelli and Townsend, 2014). Briefly, pET28b plasmids containing nocTE and nocTE*S1179 genes were transformed into Rosetta 2 (DE3) cells by electroporation and plated on LB agar containing 50 μg/mL kanamycin and 50 μg/mL chloramphenicol at 37 °C overnight. A single colony was selected and used to inoculate 2 × 35 mL overnight starter cultures in LB supplemented with 50 μg/mL kanamycin and 50 μg/mL chloramphenicol at 37 °C. 6 × 1 liter of 2XYT medium containing 50 μg/mL kanamycin and 50 μg/mL chloramphenicol was inoculated with 10 mL of starter culture and allowed to grow to an optical density (A600) of 0.6–0.7 at 37 °C, 200 r.p.m. before being cooled to 4 C for 1–2 h. IPTG was added to a final concentration of 1 mM and the induced cultures were shaken at 18 °C, overnight at 200 r.p.m. Cells were harvested by centrifugation (4000 × g, 30 min, 4 °C) and stored at −80 °C until purification.
Cells were thawed in cold lysis buffer (50 mM Na/PO4, 300 mM NaCl, pH 8.0, 3 mL/g cells) prior to sonication (60% amplitude, 9.9 s on/off, 7 min) on ice. The lysate was cleared by centrifugation (25,000 × g, 30 min, 4 °C) before the debris-free supernatant was incubated with 12 mL of 50% TALON cobalt affinity-resin (2 mL/L culture) for 1–2 h, 4 °C in a batch-binding format. The resin was pelleted (750 × g, 5 min, 4 °C) and the supernatant discarded. The resin was resuspended in lysis buffer and loaded onto a gravity column before being washed with lysis buffer containing 20 mM imidazole (2 × 12 mL). The desired protein was next eluted with lysis buffer containing 200 mM imidazole (3 × 12 mL). Fractions containing purified protein, as determined by SDS-PAGE with Coomassie staining, were combined and concentrated using an Amicon Ultra 0.5 mL 10 kMWCO filtration device to 8 mL (4,000 × g, 10 min, 4 °C) before being dialyzed twice against 2 L of 10 mM HEPES, 25 mM NaCl, pH 7.5. Protein concentration was determined by Bradford assay. See Figure S1.
General Synthetic Methods and Instrumentation
All chemicals were used without further purification unless indicated. THF and DCM were distilled from 4 Å MS before use, while acetone was distilled from boric anhydride (1 g/mL). Flash-column chromatography was performed using Sorbtech Silica Gel 60Å 40–75 μm particle size.
1H NMR spectra were recorded on 400 MHz or 300 MHz Bruker (Billerica, MA) Advance NMR spectrometers as specified. All 13C NMR and 31P NMR spectra were recorded on a 400 MHz Bruker Advance NMR spectrometer. All 19F NMR spectra were recorded on a 300 MHz Bruker Advance NMR spectrometer. 1H and 13C Chemical shifts are reported relative to the reference shift for the solvent used relative to TMS, 31P to phosphoric acid.
Small-molecule preparative HPLC purifications were carried out on a Teledyne ISCO (Lincoln, NE) CombiFlash® EZ Prep system equipped with a reverse-phase Phenomenex (Torrance, CA) Luna 10μ C18(2) 100 Å column (250 × 21.20 mm ID). Preparative method: solvent A (water + 0.1% TFA), solvent B (acetonitrile + 0.1% TFA), 0–35 min gradient, 10–40% B; 35–45 min gradient, 40–50% B. Fractions found to contain desired products by UPLC-HRMS were combined, frozen, and lyophilized to dryness.
Ultra-performance liquid chromatography-high resolution mass spectrometry (UPLC-HRMS) experiments were carried out on a Waters Acquity/Xevo G2 UPLC-MS system outfitted with a multi-wavelength UV-Vis diode array detector at the Johns Hopkins University Department of Chemistry Mass Spectrometry Facility. Small molecule samples were separated with a Waters Acquity BEH UPLC column, packed with an ethylene-bridged hybrid C-18 stationary phase (2.1 mm × 50 mm, 1.7 um) in tandem with HRMS analysis by a Waters Xevo G2 Q-ToF ESI mass spectrometer. The raw data were processed with MassLynx v4.1 (Waters Inc.). Mobile phase: 0–1 min 100% water + 0.1% formic acid, 1 – 7.5 min 80% ACN + 0.1% formic acid, S3 7.5 min – 8.4 min isocratic 80% ACN + 0.1 % formic acid, 8.4 – 10 min 100% water + 0.1 % formic acid. Flow rate = 0.3 mL/min. Protein samples were separated with a Waters Acquity BEH UPLC column, packed with an ethylene-bridged hybrid C-4 stationary phase (2.1 mm × 50 mm, 1.7 μm) in tandem with HRMS analysis by a Waters Xevo G2 Q-ToF ESI mass spectrometer. Mobile phase: 0 – 1 min 100% water + 0.1% formic acid, 1 – 7.5 min 80% ACN + 0.1% formic acid, 7.5 min −8.4 min isocratic 80% ACN + 0.1 % formic acid, 8.4 – 12 min 100% water + 0.1 % formic acid. Flow rate = 0.3 mL/min.
Synthesis of DPP and FP Substrate Analogue Inhibitors
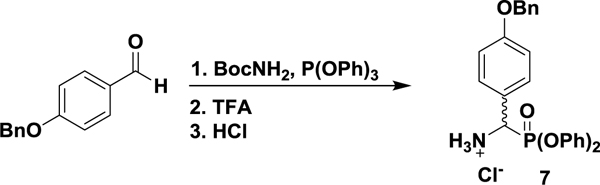
(+/−)-p-Benzyloxyphenylglycine diphenylphosphonate hydrochloride (7).
In a 250 mL round bottom flask fitted with an efficient reflux condenser were suspended p-benzyloxybenzaldehyde (5.34 g, 25.1 mmol), t-butyl carbamate (4.00 g, 26.5 mmol), and triphenylphosphite (7.63 mL, 29.1 mmol) in glacial acetic acid (4 mL), at room temperature. The mixture was heated to 60 °C for 6 h before being concentrated under reduced pressure to a viscous oil. The resulting residue was taken up in 1:1 TFA/DCM (100 mL) and allowed to stir at room temperature for 1 h. Concentration in vacuo produced a dark oil, which was partitioned into Et2O (100 mL), washed with saturated aqueous NaHCO3 (2 × 100 mL) and brine (1 × 100 mL) before being dried over anhydrous Na2SO4 and concentrated to yield the crude free base as an off-white solid. Suspension of the solid in 1:1 HCl/THF (1 M, 100 mL) for 20 min, followed by vacuum filtration and washes with Et2O (3 × 25 mL) produced the desired racemic hydrochloride salt as a white solid (9.63 g, 75%). 1H NMR (300 MHz; d6-DMSO): δ 9.57 (bs, 3 H), 7.64 (dd, J = 8.8, 2.1 Hz, 2 H), 7.47–7.10 (m, 15 H), 6.92 (d, J = 7.6 Hz, 2 H), 5.49 (bd, J = 17.2 Hz, 1 H), 5.16 (s, 2 H). 13C NMR (100 MHz; d6-DMSO): 159.55, 150.02, 149.89, 137.25, 131.04 (d, J = 5.4 Hz), 130.44, 130.31, 128.93, 128.36, 128.12, 126.13 (d, J = 10.3 Hz), 122.56 (d, J = 5.6 Hz), 120.92 (d, J = 4.0 Hz), 120.79 (d, J = 4.0 Hz), 115.60, 69.69, 50.35 (d, J = 158.0 Hz). δ 31P NMR (121 MHz; d6-DMSO): δ 11.65. UPLC-HRMS (ESI) calculated for C26H25NO4P [M+1]+, 446.1516; found, 446.1571.
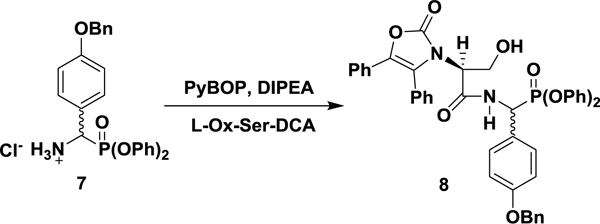
l-N-Ox-Ser-p-O-Bn-PGP(OPh)2: diphenyl (S)-((4-(benzyloxy)phenyl)(3-hydroxy-2-(2-oxo-4,5-diphenyloxazol-3(2H)-yl)propanamido)methyl)phosphonate (8).
In a 250 mL round bottom flask was suspended 7 (4.00 g, 8.30 mmol) and the dicyclohexylammonium salt of l-Ox-serine (Sheehan and Guziec, 1973) (4.20 g, 8.30 mmol) in anhydrous DMF (60 mL) at room temperature, under argon. The mixture was cooled to 0 °C before PyBOP (4.75 g, 9.13 mmol) was added in one portion. Stirring at 0 °C was allowed for 5 min before dropwise addition of DIPEA (1.60 mL, 9.13 mmol), resulting in a yellow solution. The reaction mixture was allowed to warm to room temperature over 3 h prior to dilution with EtOAc (50 mL) and washing with saturated aqueous NH4Cl (2 × 100 mL). The aqueous phase was re-extracted with EtOAc (50 mL) and the combined organics were further washed with saturated aqueous NaHCO3 (2 × 100 mL), brine (100 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to a yellow foam. The desired dipeptide was isolated as a mixture of diastereomers by flash chromatography (45% EtOAc/hexanes, isocratic) as a white solid (4.24 g, 68%). A diastereomerically pure analytical sample was produced by crystallization from EtOAc and hexanes. 1H NMR (400 MHz; CDCl3): δ 8.37 (d, J = 9.3 Hz, 1 H), 7.46–7.25 (m, 10 H), 7.22–7.07 (m, 15 H), 6.96 (d, J = 8.4 Hz, 2 H), 6.67 (dd, J = 8.4 Hz, 1.2 Hz, 2 H), 5.80 (dd, J = 20.9 Hz, 9.6 Hz, 1 H), 5.10 (s, 2 H), 4.07 (t, J = 4.9 Hz, 1 H), 3.99–3.89 (m, 2 H). 13C NMR (100 MHz; CDCl3): δ 167.13 (d, J = 9.3 Hz), 158.97 (d, J = 2.6 Hz), 155.25, 150.09 (d, J = 10.3 Hz), 149.97 (d, J = 9.7 Hz), 136.80, 135.47, 130.68, 130.25, 129.92 (d J = 0.8 Hz), 129.82 (d, J = 6.6 Hz), 129.62 (d, J = 0.9 Hz), 129.51, 128.64, 128.45, 128.03, 127.96, 127.40, 127.35, 126.02, 125.71, 125.60 (d, J = 0.7 Hz), 125.38 (d, J = 0.7 Hz), 124.48, 123.77, 120.73 (d, J = 4.2 Hz), 120.30 (d, J = 4.2 Hz), 115.30 (d, J = 1.8 Hz), 70.01, 60.83, 59.49, 49.89 (d, J = 159.2 Hz). 31P NMR (162 MHz; CDCl3): δ 13.77. UPLC-HRMS (ESI) calculated for C44H38N2O8P [M+1]+, 753.2360; found, 753.2366. The alternative diastereomer could not be separated or enriched and is reported from a ~1:1 stereoisomeric mixture. 1H NMR (400 MHz; CDCl3): δ 8.09 (dd, J = 9.5, 4.1 Hz, 1 H), 7.46–7.10 (m, 25 H), 6.97 (m, 2 H), 6.85 (m, 2 H), 5.87 (dd, J = 20.5, 9.8 Hz, 1 H), 5.07 (s, 2 H), 4.19 (t, J = 7.5 Hz, 1 H), 4.08 (m, 2 H). 31P NMR (162 MHz; CDCl3): δ 13.57. UPLC-HRMS (ESI) calculated for C44H38N2O8P [M+1]+, 753.2360; found, 753.2354.
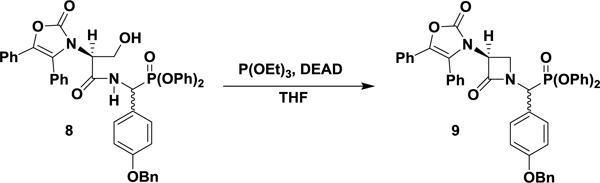
Diphenyl (S)-((4-(benzyloxy)phenyl)(2-oxo-3-(2-oxo-4,5-diphenyloxazol-3(2H)-yl)azetidin-1-yl)methyl)phosphonate (9).
To a solution of 8 (1.28 g, 1.70 mmol) in freshly distilled THF (65 mL) was added triethylphosphite (0.44 mL, 2.5 mmol) at room temperature, under argon. The reaction flask was covered in aluminum foil before DEAD (0.40 mL, 2.5 mmol) was added dropwise, resulting in a bright yellow solution. The mixture was stirred at room temperature for 1.5 h before being diluted with EtOAc (50 mL) and washed with 1 M HCl (50 mL). The acidic aqueous layer was re-extracted with EtOAc (25 mL) and the combined organic were further washed with a saturated aqueous solution of NaHCO3 (2 × 100 mL) and brine (100 mL) before being dried over anhydrous Na2SO4. Concentration under reduced pressure produced the crude product as a yellow foam. The desired β-lactam diphenylphosphonate was isolated as a diastereomeric mixture by flash column chromatography (35% EtOAc/hexanes, isocratic) as a white foam (0.81 g, 65 %). A diastereomerically pure analytical sample was obtained by crystallization from chloroform/hexanes. 1H NMR (400 MHz; CDCl3): δ 7.51–7.32 (m, 16 H), 7.28–7.18 (m, 8 H), 7.10 (t, J = 7.4 Hz, 1 H), 6.95 (m, 4 H), 5.54 (d, J = 21.4 Hz, 1 H), 5.06 (s, 2 H), 4.69 (dd, J = 5.7, 2.9 Hz, 1 H), 4.47 (ddd, J = 5.7, 2.7, 2.7 Hz, 1 H), 3.77 (ddd, J = 5.7, 5.7, 0.7 Hz, 1 H). 13C NMR (100 MHz; CDCl3): δ 163.43 (d, J = 7.9 Hz), 159.28, 152.71, 150.20 (d, J = 9.2 Hz), 149.79 (d, J = 10.7 Hz), 136.57, 135.20, 130.72, 130.61 (d, J = 7.6 Hz), 130.47, 129.87 (d, J = 1.1 Hz), 129.80, 129.54 (d, J = 0.5 Hz), 128.64, 128.51, 128.10, 128.05, 127.42, 127.39, 126.07, 125.60 (d, J = 1.5 Hz), 125.21 (d, J = 1.2 Hz), 124.57, 123.86 (d, J = 1.1 Hz), 122.67, 121.16 (d, J = 4.1 Hz), 120.55 (d, J = 4.1 Hz), 115.43 (d, J = 0.7 Hz), 70.04, 57.43, 52.68 (d, J = 159.9 Hz), 46.18. 31P NMR (162 MHz; CDCl3): δ 10.65. UPLC-HRMS (ESI) calculated for C44H36N2O7P [M+1]+, 735.2255; found, 735.2260. The alternative diastereomer was enriched (5:1) by flash column chromatography for characterization. 1H NMR (400 MHz; CDCl3): δ 7.52–7.06 (m, 25 H), 7.00 (d, J = 8.5 Hz, 2 H), 6.87 (d, J = 8.5 Hz, 2 H), 5.55 (d, J = 20.8 Hz, 1 H), 5.12 (s, 2 H), 4.53 (ddd, J = 5.6, 2.8, 2.8, 1 H), 3.80 (ddd, J = 5.7, 5.7, 2.7 Hz, 1 H), 3.66 (m, 1 H). 13C NMR (100 MHz; CDCl3): δ 163.96 (d, J = 6.7 Hz), 159.33 (d, J = 1.7 Hz), 152.97, 150.40 (d, J = 9.2 Hz), 149.76 (d, J = 10.2 Hz), 136.71, 135.15, 134.91, 130.78, 130.71, 130.42, 129.73, 129.69 (d, J = 0.7 Hz), 129.48, 128.68, 128.49, 128.14, 127.51, 127.27, 125.66, 125.51 (d, J = 1.2 Hz), 125.49 (d, J = 1.4 Hz), 124.50, 122.45, 122.30, 120.40 (d, J = 4.2 Hz), 120.16 (d, J = 4.2 Hz), 115.40, 70.03, 56.94, 52.75 (d, J = 160.7 Hz), 45.91. 31P NMR (162 MHz; CDCl3): δ 9.82. UPLC-HRMS (ESI) calculated for C44H36N2O7P [M+1]+, 735.2255; found, 735.2258.
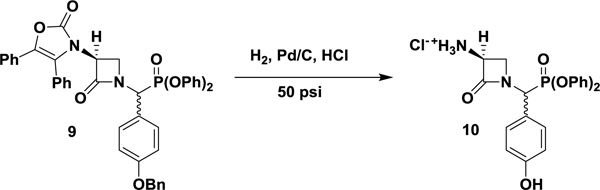
Diphenyl (S)-((3-amino-2-oxoazetidin-1-yl)(4-hydroxyphenyl)methyl)phosphonate hydrochloride (10).
In a 250 mL hydrogenation flask was dissolved diphenylphosphonate 9 (0.74 g, 1.00 mmol) in EtOAc (3 mL) and absolute EtOH (2 mL). To the clear solution was added 10% palladium on carbon (0.50 g) and a freshly prepared aqueous solution of 2 M HCl (0.53 mL, 1.05 mmol) dropwise at room temperature, open to air. The flask was placed on a Parr hydrogenation apparatus, degassed and backfilled with 30 psi of H2 three times before being pressurized to 50 psi and allowed to shake for 72 h at room temperature. The Pd/C was filtered over Celite, which was washed with EtOH (2 × 10 mL). The organic solvents were removed under reduced pressure to yield an oily solid, which was triturated with Et2O (5 mL) to produce a white precipitate. The resulting solid was further washed with Et2O (2 × 5 mL) to yield the desired diastereomeric (~1:1) hydrochloride salt as a white solid (0.28 g, 62%). A diastereomerically pure analytical sample was isolated by preparative-HPLC. 1H NMR (400 MHz; d6-DMSO): δ 9.73 (bs, 1 H), 8.87 (bs, 3 H), 7.43–7.17 (m, 10 H), 6.86 (d, J = 8.8 Hz, 2 H), 6.79 (d, J = 8.4 Hz, 2 H), 5.71 (d, J = 21.8 Hz, 1 H), 4.65 (dd, J = 5.3, 2.5 Hz, 1 H), 3.83 (t, J = 5.7 Hz, 1 H), 3.70 (d, J = 6.0 Hz, 1 H). 13C NMR (100 MHz; d6-DMSO): δ 162.59 (d, J = 7.8 Hz), 158.45 (d, J = 2.5 Hz), 150.12 (d, J = 9.5 Hz), 150.00 (d, J = 10.6 Hz), 131.15 (d, J = 7.42 Hz), 130.47 (d, J = 0.82 Hz), 130.23 (d, J = 1.0 Hz), 126.10 (d, J = 0.8 Hz), 125.90 (d, J = 1.4 Hz), 121.98, 121.10 (d, J = 4.1 Hz), 120.84 (d, J = 4.0 Hz), 116.03, 54.34, 53.70 (d, J = 158.4 Hz), 46.20. 31P NMR (162 MHz; d6-DMSO): δ 12.08. UPLC-HRMS (ESI) calculated for C22H22N2O5P [M+1]+, 425.1261; found, 425.1262. The alternative diastereomer was enriched (3:1) by preparative-HPLC for characterization. 1H NMR (400 MHz; d6-DMSO): δ 9.79 (bs, 1 H), 8.78 (bs, 3 H), 7.46–7.39 (m, 4 H), 7.32–7.25 (m, 3 H), 7.19–7.15 (m, 3 H), 6.86 (d, J = 8.4 Hz, 2 H), 6.82 (d, J = 8.4 Hz, 2 H), 5.80 (d, J = 21.1 Hz, 1 H), 4.61 (dd, J = 5.4, 2.7 Hz, 1 H), 3.92 (t, J = 5.7 Hz, 1 H), 3.44 (d, J = 5.8 Hz, 1 H). 13C NMR (100 MHz; d6-DMSO): δ 162.60 (d, J = 6.5 Hz), 158.60 (d, J = 2.4 Hz), 150.10 (d, J = 10.0 Hz), 150.00 (d, J = 9.6 Hz), 131.53 (d, J = 7.4 Hz), 130.53 (d, J = 0.7 Hz), 130.30 (d, J = 0.6 Hz), 126.11, 125.97, 121.17, 120.86 (d, J = 4.0 Hz), 120.76 (d, J = 4.0 Hz), 116.10, 54.05, 52.99 (d, J = 159.0 Hz), 45.71. 31P NMR (162 MHz; d6-DMSO): δ 11.79. UPLC-HRMS (ESI) calculated for C22H22N2O5P [M+1]+, 425.1261; found, 425.1258.
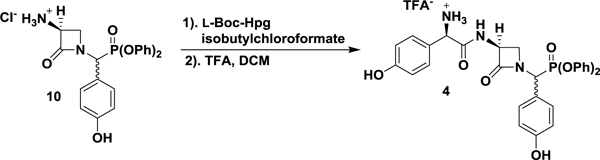
(R)-2-(((S)-1-((Diphenoxyphosphoryl)(4-hydroxyphenyl)methyl)-2-oxoazetidin-3-yl)amino)-1-(4-hydroxyphenyl)-2-oxoethan-1-aminium trifluoroacetate (nocGP(OPh)2) (4).
To a flame dried 10 mL round bottom flask was dissolved N-Boc-l-Hpg (Gaudelli and Townsend, 2013) (0.25 g, 0.93 mmol) in freshly distilled acetone (2.0 mL, over B2O3) at room temperature, under argon. To the solution were added 2,6-lutidine (100 μL, 0.86 mmol) and N-methylmorpholine (20 μL, 0.19 mmol) before being cooled to −50 °C. Isobutylchloroformate (113 μL, 0.86 mmol) was then added dropwise before the mixture was allowed to warm to −30 °C over 30 min, during which a white precipitate began to form. The suspension was transferred to a 0 °C ice bath for 20 min, while in another 5 mL round bottom flask, a solution of 10 (0.28 g, 0.62 mmol) in anhydrous DMF (2.7 mL) supplemented with 2,6-lutidine (100 μL, 0.86 mmol) at 0 °C was prepared. The neutralized DMF solution of 10 was then added dropwise to the in situ prepared mixed anhydride, where the reaction was stirred at 0 °C for 30 min before being allowed to warm to room temperature over 3 h. The mixture was diluted with EtOAc (10 mL) and washed with saturated aqueous NH4Cl (20 mL) before the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were next washed with 10% aqueous AcOH (2 × 20 mL) to remove the excess 2,6-lutidine, neutralized with saturated aqueous NaHCO3 (30 mL), and washed with brine (30 mL) before being dried over anhydrous Na2SO4 and concentrated under reduced pressure to provide the crude N-Boc-nocGP(OPh)2 used in the next reaction without further purification. The crude tripeptide product was dissolved in DCM (5 mL) before trifluoroacetic acid (5 mL) was added dropwise resulting in a pale yellow solution. The reaction was allowed to stir at room temperature for 1.5 h before being concentrated in vacuo. The resulting oily residue was azeotroped with cyclohexane (3 × 15 mL) under vacuum to produce a light brown foam containing the desired tripeptide as a ~1:1 mixture of diastereomers (0.20 g, 69% over two steps). A diastereomerically pure analytical sample was isolated by preparative-HPLC. 1H NMR (400 MHz; d6-DMSO): δ 9.80 (bs, 1 H), 9.68 (bs, 1 H), 9.23 (d, J = 7.2 Hz, 1 H), 8.50 (bs, 3 H), 7.40–7.10 (m, 12 H), 6.87 (d, J = 8.6 Hz, 2 H), 6.80 (d, J = 8.6 Hz, 2 H), 6.77 (d, J = 8.7 Hz, 2 H), 5.64 (d, J = 21.6 Hz, 1 H), 4.95 (m, 1 H), 4.83 (m, 1 H), 3.80 (t, J = 5.2 Hz, 1 H), 3.53 (m, 1 H). 13C NMR (100 MHz; d6-DMSO): δ 168.61, 165.78, 58.83, 158.29, 150.12 (d, J = 10.7 Hz), 150.01 (d, J = 11.5 Hz), 130.99, 130.93, 130.46, 130.24, 129.92, 125.99, 125.85, 124.15, 122.42, 121.09 (d, J = 4.3 Hz), 120.88 (d, J = 4.2 Hz), 115.99, 56.24, 55.54, 53.07 (d, J = 158.1 Hz), 48.33. 31P NMR (162 MHz; d6-DMSO): δ 12.51. UPLC-HRMS (ESI) calculated for C30H29N3O7P [M+1]+, 574.1738; found, 574.1746. The alternative diastereomer was isolated by preparative-HPLC for characterization. 1H NMR (400 MHz; d6-DMSO): δ 9.78 (bs, 1 H), 9.72 (bs, 1 H), 9.00 (d, J = 8.2 Hz, 1 H), 8.45 (bs, 3 H), 7.43–7.15 (m, 12 H), 6.89 (d, J = 8.6 Hz, 2 H), 6.78 (m, 4 H), 5.70 (d, J = 21.0 Hz, 1 H), 4.76 (m, 2 H), 3.85 (m, 1 H), 3.34 (m, 1 H). 13C NMR (100 MHz; d6-DMSO): δ 168.46, 166.08, 158.80, 158.39, 150.26 (d, J = 9.9 Hz), 150.00 (d, J = 9.6 Hz), 131.23, 131.16, 130.46, 130.31, 129.94, 125.99, 125.94, 124.07, 121.69, 120.80 (d, J = 3.0 Hz), 120.77 (d, J = 3.3 Hz), 116.03 (d, J = 11.2 Hz), 55.80, 55.58, 52.69 (d, J = 159.0 Hz), 47.38. 31P NMR (162 MHz; d6-DMSO): δ 12.07. UPLC-HRMS (ESI) calculated for C30H29N3O7P [M+1]+, 574.1738; found, 574.1740.
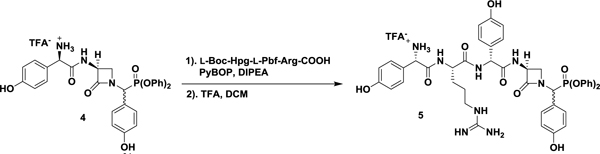
(S)-2-(((S)-1-(((R)-2-(((S)-1-((Diphenoxyphosphoryl)(4-hydroxyphenyl)methyl)-2-oxoazetidin-3-yl)amino)-1-(4-hydroxyphenyl)-2-oxoethyl)amino)-5-guanidino-1-oxopentan-2-yl)amino)-1-(4-hydroxyphenyl)-2-oxoethan-1-aminium trifluoroacetate (pro-nocGP(OPh)2) (5).
To a 10 mL round bottom flask was dissolved 4 (0.019 g, 0.027 mmol) in anhydrous DMF (1.0 mL). DIPEA (9.4 μL, 0.054 mmol) was added before cooling to 0 °C under argon. To a separate 5 mL round bottom flask was dissolved L-Boc-Hpg-L-Pbf-Arg (Gaudelli and Townsend, 2013) (0.021 g, 0.031 mmol) in anhydrous DMF (1.0 mL) prior to the addition of DIPEA (5.2 μL, 0.030 mmol) and cooling to 0 °C under argon. To the 5 mL flask containing the carboxylic acid solution was added PyBOP (0.016 g, 0.031 mmol) at once, resulting in a light yellow solution. After 1 min, the solution of 4 was added dropwise at 0 °C and stirred for 30 min before being allowed to warm to room temperature over 3 h. The reaction mixture was diluted with EtOAc (15 mL) and washed with saturated aqueous solutions of NH4Cl (10 mL), NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 before being concentrated under reduced pressure to a light brown oil. The crude residue was taken up in DCM (1.0 mL) before trifluoroacetic acid (1.0 mL) was added dropwise, darkening the solution. The mixture was stirred for 1 h at room temperature, during which UPLC-HRMS analysis indicated complete conversion to product. The reaction mixture was azeotroped with toluene (3 × 5 mL) to produce a dark brown solid. Diastereomerically enriched samples were acquired by preparative-HPLC (3.1 mg, 12 % and 3.3 mg, 13 % respectively). Diastereomer 1; 1H NMR (400 MHz; d6-DMSO): δ 9.71 (s, 1 H), 9.66 (s, 1 H), 9.44 (s, 1 H), 9.12 (d, J = 7.4 Hz, 1 H), 8.59 (d, J = 7.6 Hz, 2 H), 8.43 (bs, 3 H), 7.38–7.10 (m, 14 H), 6.87 (d, J = 8.3 Hz, 2 H), 6.77 (m, 4 H), 6.68 (d, J = 8.6 Hz, 2 H), 5.63 (d, J = 21.8 Hz, 1 H), 5.40 (d, J = 8.3 Hz, 1 H), 4.95 (m, 1 H), 4.88 (m, 1 H), 4.51 (m, 1 H), 3.77 (t, J = 3.7 Hz, 1 H), 3.44 (m, 1 H), 3.05 (m, 2 H), 1.60 (m, 2 H), 1.44 (m, 2 H). 31P NMR (162 MHz; d6-DMSO): δ 12.51. UPLC-HRMS (ESI) calculated for C44H48N8O10P [M+1]+, 879.3226; found, 879.3225. Diastereomer 2; 1H NMR (400 MHz; d6-DMSO): δ 9.76 (bs, 2 H), 9.46 (bs, 1 H), 8.96 (d, J = 8.1 Hz, 1 H), 8.55 (d, J = 7.9 Hz), 1 H), 8.54 (d, J = 8.1 Hz, 1 H), 8.45 (bs, 3 H), 7.58 (bs, 1 H), 7.42–7.13 (m, 13 H), 7.03 (d, J = 8.6 Hz, 2 H), 6.88 (d, J = 8.4 Hz, 2 H), 6.79 (m, 4 H), 6.65 (d, J = 8.6 Hz, 2 H), 5.68 (d, J = 21.2 Hz, 1 H), 5.26 (d, J = 8.2 Hz, 1 H), 4.87 (m, 1 H), 4.79 (m, 1 H), 4.48 (m 1 H), 3.84 (m, 1 H), 3.29 (m, 1 H), 3.03 (m, 2 H), 1.57 (m, 1 H), 1.42 (m, 3 H). 31P NMR (162 MHz; d6-DMSO): δ 12.04. UPLC-HRMS (ESI) calculated for C44H48N8O10P [M+1]+, 879.3226; found, 879.3218.
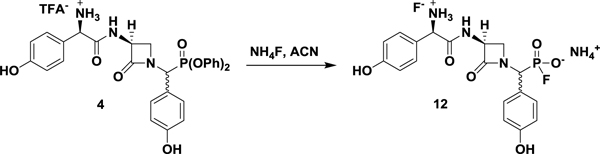
(((S)-3-((R)-2-ammonio-2-(4-hydroxyphenyl)acetamido)-2-oxoazetidin-1-yl)(4-hydroxyphenyl)methyl)-phosphonofluoridate ammonium fluoride salt (12).
In a 5 mL oven dried, pear-shaped flask fitted with a reflux condenser was suspended 6 (10.5 mg, 0.015 mmol) in freshly distilled acetonitrile (1.0 mL) under argon. Ammonium fluoride (5.6 mg, 0.15 mmol) was added in one portion and the mixture heated at 60 °C for 14 h. After cooling to room temperature, the remaining acetonitrile was removed in vacuo to produce an off-white solid. Trituration with diethyl ether (3 × 3 mL) produced the desired fluorophosphonate salt 12 (~1:1 diastereomeric mixture) as a white solid, used without further purification (quantitative by 31P NMR). Diastereomer 1; 31P NMR (162 MHz; d6-DMSO): δ 8.76 (d, J = 990.6 Hz). Diastereomer 2; 31P NMR (162 MHz; d6-DMSO): δ 9.22 (d, J = 995.7 Hz). UPLC-HRMS (ESI) calculated for C18H20FN3O6P [M+1]+, 424.1068; found, 424.1063. 1H and 19F NMR characterization was impeded by dramatic line broadening likely due to high salt, multiple charge-states and slow ion-pair exchange.
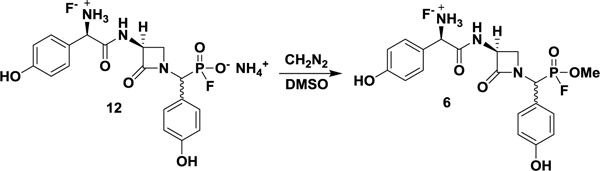
(1R)-2-(((3S)-1-((fluoro(methoxy)phosphoryl)(4-hydroxyphenyl)methyl)-2-oxoazetidin-3-yl)amino)-1-(4-hydroxyphenyl)-2-oxoethan-1-aminium (6).
To a round bottom flask (5 mL) containing crude 12 (0.020 g, 0.044 mmol) was added dry (ampoule) d6-DMSO (400 μL) before transferring the solution to a microcentrifuge tube (1.5 mL). To the anhydrous DMSO solution (~110 mM) was slowly added a diluted ethereal solution of diazomethane prepared from Diazald® (Sigma-Aldrich) (6 × 180 μL, 0.034 mmol, ~0.03 M, measured spectrophotometrically: A410, ε = 7.2 M−1 cm−1) (Gassman and Greenlee, 1973) resulting in a biphasic mixture. The progress of the reaction was monitored by UPLC-HRMS. The ethereal layer was removed under reduced pressure to yield an ~80% solution of 6, which was immediately used to modify NocTE or frozen over ice. Note: it is critical to use ampoule-DMSO or another source of anhydrous DMSO to prevent hydrolytic inactivation of 6 after it is formed. If stored frozen, a stock may be reliably kept for 1 week. UPLC-HRMS (ESI) calculated for C19H22FN3O6P [M+1]+, 438.1225; found, 438.1236.
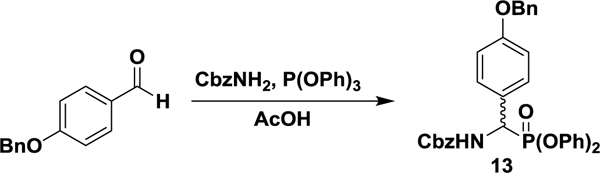
Benzyl ((4-(benzyloxy)phenyl)(diphenoxyphosphoryl)methyl)carbamate (13).
In a 100 mL round bottom flask were suspended p-benzyloxybenzaldehyde (6.37 g, 30.0 mmol), benzylcarbamate (3.02 g, 20.0 mmol), and triphenylphosphite (5.76 mL, 22.0 mmol) in glacial acetic acid (3 mL), at room temperature. The mixture was heated to 80 °C for 1.5 h before being concentrated under reduced pressure to a light yellow solid. The solid was suspended in cold chloroform and filtered. The remaining solid was recrystallized from hot chloroform and MeOH to yield the pure, racemic diphenylphosphonate as a white solid (8.64, 74%). 1H NMR (400 MHz; d6-DMSO): δ 8.83 (d, J = 10.1 Hz, 1 H), 7.54 (dd, J = 8.7, 2.0 Hz, 2 H), 7.44–7.29 (m, 14 H), 7.18 (t, J = 7.4 Hz, 2 H), 7.04 (d, J = 6.9 Hz, 2 H), 7.02 (d, J = 8.5 Hz, 2 H), 6.94 (d, J = 8.3 Hz, 2 H), 5.52 (dd, J = 21.7, 10.6 Hz, 1 H), 5.13 (d, J = 12.4 Hz, 1 H), 5.11 (s, 2 H), 5.04 (d, J = 12.4 Hz, 1 H). 13C NMR (100 MHz; d6-DMSO): δ 158.72 (d, J = 2.8 Hz), 156.42 (d, J = 8.4 Hz), 150.59 (d, J = 10.0 Hz), 150.29 (d, J = 9.9 Hz), 137.43, 137.13, 130.35, 130.28, 130.24, 128.90, 128.82, 128.40, 128.38, 128.28, 128.09, 126.83, 125.73, 125.64, 120.80 (d, J = 3.6 Hz), 120.74 (d, J = 4.0 Hz), 115.21, 69.69, 66.60, 52.71 (d, J = 158.7 Hz). 31P NMR (162 MHz; d6-DMSO): δ 15.01. UPLC-HRMS (ESI) calculated for C34H31NO6P [M+1]+, 580.1884; found, 580.1880.
Enzyme/Inhibitor Incubation Experiments
Incubation of NocTE with DPP 4
To a 1.5 mL microcentrifuge tube containing NocTE (1.1 μL, 0.91 mM stock) in reaction buffer (83.0 μL, 50 mM HEPES, 25 mM NaCl, DMSO 3%, pH 7.5) was added a DMSO solution of 4 (6.9 μL, 14.5 mM stock) at room temperature. Final concentrations: [NocTE] = 10 μM, [4] = 1 mM. After being allowed to stand overnight, a 10 μL aliquot was removed, diluted to 100 μL with water, and acidified to 0.1% TFA before UPLC-HRMS analysis indicated no formation of a covalent complex with NocTE compared to a control experiment replacing the addition of 4 with an equivalent volume of water. The same experiment was carried out at 30 °C and 37 °C degrees without change in result. See Figure S2, A.
Incubation of NocTE with DPP 5
To a 1.5 mL microcentrifuge tube containing NocTE (1.1 μL, 0.91 mM stock) in reaction buffer (94.1 μL, 50 mM HEPES, 25 mM NaCl, DMSO 5%, pH 7.5) was added a DMSO solution of 5 (4.8 μL, 20.8 mM stock) at room temperature. Final concentrations: [NocTE] = 10 μM, [5] = 1 mM. After being allowed to stand overnight, a 10 μL aliquot was removed, diluted to 100 μL with water, and acidified to 0.1% TFA before UPLC-HRMS analysis indicated no formation of a covalent complex with NocTE compared to a control experiment replacing the addition of 5 with an equivalent volume of water. The same experiment was carried out at 30 °C and 37 °C degrees without change in result. See Figure S2, B.
Incubation of NocTE with 12
To a 1.5 mL microcentrifuge tube containing NocTE (1.1 μL, 0.91 mM stock) in reaction buffer (96.9 μL, 50 mM HEPES, 25 mM NaCl, pH 7.5) was added a DMSO solution of 12 (2.0 μL, 50.0 mM stock) at room temperature. Final concentrations: [NocTE] = 10 μM, [12] = 1 mM. After being allowed to stand overnight, a 10 μL aliquot was removed, diluted to 100 μL with water, and acidified to 0.1% TFA before UPLC-HRMS analysis indicated no formation of a covalent complex with NocTE compared to a control experiment replacing the addition of 12 with an equivalent volume of water. See Figure S2, C.
Incubation of NocTE with FP 6
To a 1.5 mL microcentrifuge tube containing NocTE (1.1 μL, 0.91 mM stock) in reaction buffer (89.1 μL, 50 mM HEPES, 25 mM NaCl, pH 7.5) was added a DMSO solution of 6 (9.8 μL, 10.2 mM stock) at room temperature. Final concentrations: [NocTE] = 10 μM, [6] = 1 mM. After 15 min, a 10 μL aliquot was removed, diluted to 100 μL with water, and acidified to 0.1% TFA before UPLC-HRMS analysis indicated complete consumption of unmodifed NocTE and formation of the 417 Da adduct mass. A control experiment replacing the addition of 6 with an equivalent volume of water was carried out and found no modification of NocTE. See Figure 3.
Incubation NocTE*S1179A with FP 6
To a 1.5 mL microcentrifuge tube containing NocTE*S1179 (2.5 μL, 0.40 mM stock) in reaction buffer (87.7 μL, 50 mM HEPES, 25 mM NaCl, pH 7.5) was added a DMSO solution of 6 (9.8 μL, 10.2 mM stock) at room temperature. Final concentrations: [NocTE] = 10 μM, [6] = 1 mM. After 15 min, a 10 μL aliquot was removed, diluted to 100 μLwith water, and acidified to 0.1% TFA where UPLC-HRMS analysis indicated no modification of the protein compared to a no-inhibitor control, supporting the selective reactivity of 6 for active site seryl residues. See Figure 3.
Stability of NocTE–6 Covalent Adduct in Buffer
The NocTE–6 complex formed by the procedure outlined above was allowed to remain in buffer (50 mM HEPES, 25 mM NaCl, pH 7.5) at room temperature for 1 week. At the beginning and end of this time-period a 10 μL aliquot of the reaction was removed, diluted to 100 μL with water, and acidified to 0.1% TFA and analyzed by UPLC-HRMS to confirm the permanence of the 417 Da covalent adduct. See Figure S2, D.
Fluorinative Hydrolysis Mechanism Experiments
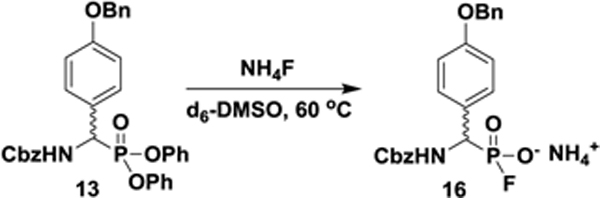
31P NMR and UPLC-HRMS Monitoring of the Fluorinative Hydrolysis Reaction
In a 5 mm NMR tube were dissolved 13 (0.020 g, 0.034 mmol) and NH4F (0.012 g, 0.34 mmol) in dry (ampoule) d6-DMSO (600 μL). The tube was heated to 60 °C in a water bath and allowed to cool to room temperature before 31P NMR acquisition was undertaken on a 400 MHz Bruker Advance NMR spectrometer. After 1 h, a 1 μL aliquot of the reaction mixture was analyzed by UPLC-HRMS. An additional identical experiment was supplemented with 15NH4Cl (0.012 g, 0.34 mmol) and monitored analogously. See Figure S3 and S4.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli: Rosetta 2 (DE3) | Novagen | Cat#: 71397 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TALON® Metal Affinity Resin | Takara (Clontech) | Cat#: 635503 |
| Bio-Rad Protein Assay Kit I (Bradford Reagent) | Bio-Rad | Cat#: 5000001 |
| kanamycin monosulfate | Gold Bio | Cat#: K-120–100 |
| chloramphenicol | Gold Bio | Cat#: C-105–100 |
| IPTG (isopropyl β-D-1-thiogalactopyranoside) | Gold Bio | Cat#: I2481C100 |
| Triphenylphosphite | Sigma-Aldrich | Cat#: T84654–500G |
| p-benzyloxybenzaldehyde | Sigma-Aldrich | Cat#: 123714–100G |
| t-butylcarbamate (Boc-NH2) | Oakwood Chemicals | Cat#: 043378–100g |
| trifluoroacetic acid | Sigma-Aldrich | Cat#: T6508–500ML |
| PyBOP | Oakwood Chemicals | Cat#: 009017–100g |
| anhydrous DMF | Fisher Scientific | Cat#: AC610941000 |
| diisopropylethylamine (DIPEA) | Sigma-Aldrich | Cat#: D125806–100ML |
| triethylphosphite | Sigma-Aldrich | Cat#: T61204–100ML |
| diethylazodicarboxylate (DEAD) | Alfa Aesar | Cat#: L19348–14 |
| 10% palladium on activated charcoal | Strem Chemicals | Cat#: 46–1900 |
| isobutylchloroformate | Sigma-Aldrich | Cat#: 177989–25G |
| 2,6-lutidine | Sigma-Aldrich | Cat#: 336106–100ML |
| N-methylmorpholine | Sigma-Aldrich | Cat#: M56557–100ML |
| ammonium fluoride | Fischer Scientific | Cat#: 60–003-36 |
| benzylcarbamate | TCI America | Cat#: C0590 |
| Diazald® | Sigma-Aldrich | Cat#: D28000–25G |
| Recombinant DNA | ||
| nocTE(wt) pET28b | Gaudelli,Townsend, 2014 | N/A |
| nocTE*S1779A pET28b | Gaudelli, Townsend, 2014 | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| MassLynx V4.1 | Waters | http://www.waters.com/waters/en_US/MassLynx-MS-Software/ |
| Other | ||
| Diazald® kit with Clear-Seal® joints | Sigma-Aldrich | Cat#: Z100250 |
| Ultrasonic Processor (Sonicator) | Cole-Palmer | Cat#: EW-04711–70 |
| Amicon Ultra 0.5 mL 10 kMW centrifugal filter | Millipore | Cat#: UFC501096 |
| Silica Gel 60Å 40–75 μm | Sorbtech | Cat#: 52500 |
SIGNIFICANCE.
To gather meaningful structural information on enzyme mechanism, substrate specificity or conformational changes, small molecule–enzyme complexes must faithfully represent native structures. In cases where substrate-mimicking DPP inhibitors have failed to react (e.g. 4 and 5 in this study), investigators have historically been forced to resort to simpler FP inhibitors and enzyme adducts of diminished structural merit (Xue et al., 2012). Late-stage conversion of the classic DPP moiety into a more reactive FP electrophile should be of great utility when confronted by an enzyme recalcitrant to modification because two prospective inhibitors are effectively provided from one synthetic effort. The straightforward DPP→FP transformation mechanistically characterized here efficiently couples the synthetic accessibility of structurally diverse DPP analogues with the potent reactivity of the FP warhead to produce complex substrate mimics capable of forming covalent SH-substrate complexes with high structural fidelity. Such an approach opens the door to the incorporation of the established FP warhead on previously synthetically intractable substrate classes and may find further application in the development of more enzyme-specific affinity probes for the discovery and characterization of diverse serine hydrolases.
ACKNOWLEDGEMENTS
We are grateful to the National Institutes of Health research grant AI121072 for financial support, and to the Greer and Dean’s Research Awards (JHU) to F. B. D. We thank Dr. I. P. Mortimer at the Johns Hopkins University Department of Chemistry Mass Spectrometry Facility for UPLC-HRMS support and Dr. J. A. Tang for NMR guidance. We are also grateful to Drs. N. M. Gaudelli and D. H. Long for expression plasmids and advice, and E. K. Sinner for helpful insights and discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, and Smith JL (2006). Structural basis for macrolactonization by the pikromycin thioesterase. Nat. Chem. Biol. 2, 537–542. [DOI] [PubMed] [Google Scholar]
- Bartlett PA, and Lamden LA (1986). Inhibition of chymotrypsin by phosphonate and phosphonamidate peptide analogs. Bioorg. Chem. 14, 356–377. [Google Scholar]
- Benschop HP, van den Berg GR, and Kraay GW (1970). Organophosphorus compounds. Part IX: Decomposition of ω-dimethylaminoalkyl methylphosphonofluoridates and isomerization of 2-dipropylaminoethyl methylphosphonofluoridothionate. Recl. Des Trav. Chim. Des Pays-Bas 89, 1025–1037. [Google Scholar]
- Bisogno T, Mahadevan A, Coccurello R, Chang JW, Allarà M, Chen Y, Giacovazzo G, Lichtman A, Cravatt B, Moles A, et al. (2013). A novel fluorophosphonate inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. Br. J. Pharmacol. 169, 784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller AR, and Townsend CA (2013). Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. 110, E653–E661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colović MB, Krstić DZ, Lazarević-Pašti TD, Bondžić AM, and Vasić VM (2013). Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr. Neuropharmacol. 11, 315–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR (2000). Thrombin signalling and protease-activated receptors. Nature 407, 258–264. [DOI] [PubMed] [Google Scholar]
- Davie EW, Fujikawa K, Kurachi K, and Kisiel W (2006). The Role of Serine Proteases in the Blood Coagulation Cascade. (John Wiley & Sons, Inc.), pp. 277–318. [DOI] [PubMed] [Google Scholar]
- Drag M, and Salvesen GS (2010). Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekici OD, Paetzel M, and Dalbey RE (2008). Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 17, 2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks LM, Stowe GN, De Lamo Marin S, Mayorov AV, Hixon MS, and Janda KD. (2011). Identification of α2 Macroglobulin as a Major Serum Ghrelin Esterase. Angew. Chemie Int. Ed. 50, 10699–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassman P., and Greenlee W. (1973). Dideuteriodiazomethane. Org. Synth. 53, 38. [Google Scholar]
- Gaudelli NM, and Townsend CA (2013). Stereocontrolled syntheses of peptide thioesters containing modified seryl residues as probes of antibiotic biosynthesis. J. Org. Chem. 78, 6412–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, and Townsend CA (2014). Epimerization and substrate gating by a TE domain in β-lactam antibiotic biosynthesis. Nat. Chem. Biol. 10, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Long DH, and Townsend CA (2015). β-Lactam formation by a non-ribosomal peptide synthetase during antibiotic biosynthesis. Nature 520, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldes JW, Akey DL, Kittendorf JD, Sherman DH, Smith JL, and Fecik RA (2006). Structural and mechanistic insights into polyketide macrolactonization from polyketide-based affinity labels. Nat. Chem. Biol. 2, 531–536. [DOI] [PubMed] [Google Scholar]
- Grzywa R, and Sienczyk M (2013). Phosphonic Esters and their Application of Protease Control. Curr. Pharm. Des. 19, 1154–1178. [DOI] [PubMed] [Google Scholar]
- Guarino C, Legowska M, Epinette C, Kellenberger C, Dallet-Choisy S, Sieńczyk M, Gabant G, Cadene M, Zoidakis J, Vlahou A, et al. (2014). New selective peptidyl di(chlorophenyl) phosphonate esters for visualizing and blocking neutrophil proteinase 3 in human diseases. J. Biol. Chem. 289, 31777–31791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsman ME, Hari TPA, and Boddy CN (2016). Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat. Prod. Rep. 33, 183–202. [DOI] [PubMed] [Google Scholar]
- Hudlický M (1988). Fluorination with Diethylaminosulfur Trifluoride and Related Aminofluorosulfuranes In Organic Reactions, (Hoboken, NJ, USA: John Wiley & Sons, Inc.), pp. 513–637. [Google Scholar]
- Joachimiak Ł, and Błażewska KM (2018). Phosphorus-Based Probes as Molecular Tools for Proteome Studies: Recent Advances in Probe Development and Applications. J. Med. Chem. acs.jmedchem.8b00249. [DOI] [PubMed] [Google Scholar]
- Kasperkiewicz P, Poreba M, Snipas SJ, Lin SJ, Kirchhofer D, Salvesen GS, and Drag M (2015). Design of a Selective Substrate and Activity Based Probe for Human Neutrophil Serine Protease 4. PLoS One 10, e0132818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B-S, Kim B-T, and Hwang K-J (2009). A Practical Method to Cleave Diphenyl Phosphonate Esters to Their Corresponding Phosphonic Acids in One Step. Bull. Korean Chem. Soc. 30, 1391–1393. [Google Scholar]
- Lamden LA, and Bartlett FA (1983). Aminoalkylphosphonofluoridate derivatives: Rapid and potentially selective inactivators of serine peptidases. Biochem. Biophys. Res. Commun. 112, 1085–1090. [DOI] [PubMed] [Google Scholar]
- Long JZ, and Cravatt BF (2011). The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease. Chem. Rev. 111, 6022–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai T, Takenaka T, Inaji S, and Tonomura Y (2008). Fluoride-ion-mediated Hydrolysis of Phosphoric Acid Esters, Amides, and Phosphorous Acid Esters Leading to Phosphorofluoridic, Phosphoramide Fluoridic, and Phosphonic Acid Monoester Salts. Chem. Lett. 37, 1198–1199. [Google Scholar]
- Murai T, Tonomura Y, and Takenaka T (2011). Phosphorofluoridic acid ammonium salts and acids: Synthesis, NMR properties, and application as acid catalysts. Heteroat. Chem. 22, 417–425. [Google Scholar]
- Newman AG, Vagstad AL, Storm PA, and Townsend CA (2014). Systematic Domain Swaps of Iterative, Nonreducing Polyketide Synthases Provide a Mechanistic Understanding and Rationale For Catalytic Reprogramming. J. Am. Chem. Soc. 136, 7348–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L-M, and Powers JC (1998). Synthesis and kinetic studies of an amidine-containing phosphonofluoridate: a novel potent inhibitor of trypsin-like enzymes. Bioorg. Med. Chem. 6, 1767–1773. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J, and Powers JC (1991). Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 30, 485–493. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J, and Subotkowska L (1979). Diphenyl 1-Aminoalkanephosphonates. Synthesis (Stuttg). 985–986. [Google Scholar]
- Ortega C, Anderson LN, Frando A, Sadler NC, Brown RW, Smith RD, Wright AT, and Grundner C (2016). Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell Chem. Biol. 23, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JC, Asgian JL, Ekici ÖD, and James KE (2002). Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev. 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- Reynolds MA, Gerlt JA, Demou PC, Oppenheimer NJ, and Kenyon GL (1983). Nitrogen-15 and oxygen-17 NMR studies of the proton binding sites in imidodiphosphate, tetraethyl imidodiphosphate, and adenylyl imidodiphosphate. J. Am. Chem. Soc. 105, 6475–6481. [Google Scholar]
- Salituro GM, and Townsend CA (1990). Total syntheses of (−)-nocardicins A-G: a biogenetic approach. J. Am. Chem. Soc. 112, 760–770. [Google Scholar]
- Sampson NS, and Bartlett PA (1991). Peptidic phosphonylating agents as irreversible inhibitors of serine proteases and models of the tetrahedral intermediates. Biochemistry 30, 2255–2263. [DOI] [PubMed] [Google Scholar]
- Serim S, Mayer SV, and Verhelst SHL (2013). Tuning activity-based probe selectivity for serine proteases by on-resin ‘click’ construction of peptide diphenyl phosphonates. Org. Biomol. Chem. 11, 5714. [DOI] [PubMed] [Google Scholar]
- Sheehan JC, and Guziec FS (1973). Amino group protection in peptide synthesis. The 4,5-diphenyl-4-oxazolin-2-one group. J. Org. Chem. 38, 3034–3040. [DOI] [PubMed] [Google Scholar]
- Shields DJ, Niessen S, Murphy EA, Mielgo A, Desgrosellier JS, Lau SKM, Barnes LA, Lesperance J, Bouvet M, Tarin D, et al. (2010). RBBP9: a tumor-associated serine hydrolase activity required for pancreatic neoplasia. Proc. Natl. Acad. Sci. U. S. A. 107, 2189–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RP, and Shreeve JM (2002). Recent Advances in Nucleophilic Fluorination Reactions of Organic Compounds-Using Deoxofluor and DAST. Synthesis (Stuttg). 2002, 2561–2578. [Google Scholar]
- Skoreński M, and Sieńczyk M (2013). Viral proteases as targets for drug design. Curr. Pharm. Des. 19, 1126–1153. [DOI] [PubMed] [Google Scholar]
- Süssmuth RD, and Mainz A (2017). Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chemie Int. Ed. 56, 3770–3821. [DOI] [PubMed] [Google Scholar]
- Wagner S, Accorsi M, and Rademann J (2017). Benzyl Mono-P-Fluorophosphonate and Benzyl Penta-P-Fluorophosphate Anions Are Physiologically Stable Phosphotyrosine Mimetics and Inhibitors of Protein Tyrosine Phosphatases. Chem. Eur. J. 23, 15387–15395. [DOI] [PubMed] [Google Scholar]
- Waters WA, and de Worms CGM (1949). 198. A kinetic study of the hydrolysis of diisopropyl fluorophosphonate. J. Chem. Soc. 926. [Google Scholar]
- Whitcomb DC, and Lowe ME (2007). Human Pancreatic Digestive Enzymes. Dig. Dis. Sci. 52, 1–17. [DOI] [PubMed] [Google Scholar]
- Xue Y, Chowdhury S, Liu X, Akiyama Y, Ellman J, and Ha Y (2012). Conformational Change in Rhomboid Protease GlpG Induced by Inhibitor Binding to Its S′ Subsites. Biochemistry 51, 3723–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.